

Abstract
Organophosphorus compounds include many synthetic, neurotoxic substances that are commonly used as insecticides. The toxicity of these compounds is due to their ability to inhibit the enzyme acetylcholine esterase. Some of the most toxic organophosphates have been adapted for use as chemical warfare agents; the most well known are GA, GB, GD, GF, VX and VR. All of these compounds contain a chiral phosphorus center with the SP-enantiomers being significantly more toxic than the RP-enantiomers. Phosphotriesterase (PTE) is an enzyme capable of detoxifying these agents, but the stereochemical preference of the wild-type enzyme is for the RP-enantiomers. A series of enantiomerically pure chiral nerve agent analogues has been developed containing the relevant phosphoryl centers found in GB, GD, GF, VX and VR. Wild-type and mutant forms of PTE have been tested for their ability to hydrolyze this series of compounds. Mutant forms of PTE with significantly enhanced, as well as relaxed or reversed stereoselectivity, have been identified. A number of variants showed dramatically improved kinetic constants for the catalytic hydrolysis of the more toxic SP-enantiomers. Improvements of up to three orders of magnitude relative to the wild type enzyme were observed. Some of these mutants were tested against racemic mixtures of GB and GD. The kinetic constants obtained with the chiral nerve agent analogues accurately predict the improved activity and stereoselectivity against the authentic nerve agents used in this study.
Organophosphorus compounds have been utilized for more than 50 years as insecticides for the protection of agricultural crops (1) and similar compounds have been developed as chemical warfare agents (2). The structures of these latter compounds are presented in Scheme 1 and include tabun (GA), sarin (GB), soman (GD), cyclosarin (GF), VX and VR. GA has a cyanide leaving group, the three remaining G-agents (GB, GD, and GF) have a fluoride leaving group, and the two versions of VX have a thiolate leaving group. The toxicity of these organophosphonates is due to the inactivation of acetylcholinesterase (AChE), an enzyme that catalyzes the hydrolysis of acetylcholine at neural synapses, through the phosphonylation of an active site serine residue (3). GA, GB, GF, VX, and VR contain a chiral phosphorus center and thus each of these nerve agents has two stereoisomers, while soman has four stereoisomers because of an additional chiral center within the pinacolyl substituent. The enantiomers are differentially toxic; the SP-stereoisomer of sarin reacts with AChE approximately ~104 times faster than the RP-stereoisomer and the two SP-stereoisomers of soman react ~105 times faster than the two RP-isomers. Similarly, the SP-stereoisomer of VX is ~100-fold more toxic than is the RP- stereoisomer (2).
Scheme 1.

Structures of chemical warfare agents.
The detoxification of organophosphorus nerve agents can be catalyzed by organophosphate degrading enzymes such as human paraoxonase 1 (PON1), squid DFPase, organophosphorus acid anhydrolase (OPAA), and phosphotriesterase (PTE). Human paraoxonase is capable of hydrolyzing GB and GD but the overall catalytic activity is relatively low (4). The DFPase from Loligo vulgaris is able to hydrolyze GA, GB, GD, GF, and DFP (diisopropyl fluorophosphate). The value of kcat/Km for the hydrolysis of DFP is ~1.3 × 106 M−1 s−1, but the catalytic activities for the hydrolysis of GB and GD are significantly lower (5, 6). Organophosphorus acid anhydrolase from Alteromonas sp. JD6.5 is capable of hydrolyzing a wide variety of organophosphorus compounds, including GB, GD and GF but not VX (7). Phosphotriesterase was first isolated from soil microbes (8). The best substrate identified to date for this enzyme is the agricultural pesticide paraoxon and the value of kcat/Km approaches the diffusion controlled limit of ~108 M−1 s−1 (9). The enzymatic reaction for the hydrolysis of paraoxon to p-nitrophenol and diethyl phosphate is shown in Scheme 2. The substrate specificity of PTE is quite broad and this enzyme is capable of hydrolyzing GA, GB, GD, GF, VR and VX (10).
Scheme 2.

Hydrolysis of paraoxon by phosphotriesterase.
PTE is a homodimeric protein that contains a binuclear metal center embedded within a (α/β)8-barrel structure and is a member of the amidohydrolase superfamily (11). The native enzyme contains two Zn2+ ions and these metal ions can be substituted with Cd2+, Co2+, Ni2+, or Mn2+ without loss of catalytic activity (9). Previous investigations have identified three subsites within the active site of PTE that help to define the substrate specificity for this enzyme (12). The small pocket is defined by the side chains of Gly-60, Ile-106, Leu-303, and Ser-308. The large pocket is formed by the interactions of His-254, His-257, Leu-271, and Met-317. The leaving group pocket is surrounded by four aromatic residues: Trp-131, Phe-132, Phe-306, and Tyr-309. The substrate binding site of PTE is graphically presented in Figure 1.
Figure 1.
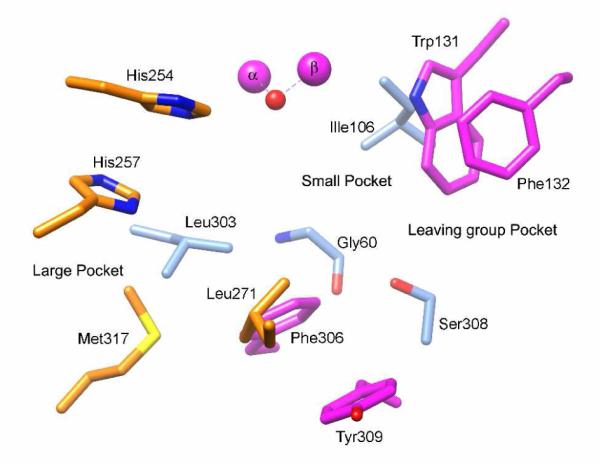
Graphic representation of the binding pockets within the active site of PTE. The small pocket consists of Gly-60, Ile-106, Leu-303, and Ser-308. The large pocket consists of His-254, His-257, Leu-271, and Met-317. The leaving group pocket is surrounded by Trp-131, Phe-132, Phe-306, and-Tyr 309.
Wild-type PTE is stereoselective for the hydrolysis of chiral organophosphates (13) and the degree of stereoselectivity depends on the substituents attached to the central phosphorus core (14, 15). For example, wild-type PTE hydrolyzes the RP-enantiomer of phenyl 4-acetylphenyl methylphosphonate approximately two orders of magnitude faster than the SP-enantiomer (15). The stereoselectivity of PTE can be manipulated through the mutation of residues that contact the substrate prior to catalytic turnover. Thus, the G60A mutant hydrolyzes the RP-enantiomer of phenyl 4-acetylphenyl methylphosphonate about 5 orders of magnitude faster than the SP-enantiomer (15). Unfortunately, the inherent stereoselectivity of wild-type PTE does not match that of AChE since PTE preferentially hydrolyzes the least toxic enantiomer of chiral organophosphates (16). However, the stereoselectivity of PTE can be inverted through multiple mutations to the small and large pockets in the active site (15). For example, the I106G/F132G/H257Y mutant catalyzes the hydrolysis of the SP-enantiomer of phenyl 4-acetylphenyl methylphosphonate about 3 orders of magnitude faster than the RP-enantiomer (15). This represents a change in stereoselectivity of nearly 8 orders of magnitude relative to the G60A mutant with only four amino acid changes.
In this paper, we describe the synthesis of chiral analogues of GB, GD, GF, VX, and VR with a 4-acetylphenol leaving group (Scheme 3). We have utilized these compounds to test a series of mutant forms of PTE that have been previously shown to have stereochemical preferences differing from the wild-type enzyme (14, 15, 17). This analysis has allowed the identification of variants enhanced in their ability to hydrolyze the more toxic SP-enantiomers, and selected mutants were tested with GB and GD to access the utility of the analogues in predicting the properties against authentic nerve agents.
Scheme 3.

Structures of organophosphate substrates.
MATERIALS and METHODS
Materials
Escherichia coli BL21(DE3) and XL1-blue cells were purchased from Stratagene. The expression plasmid pET20b (+) was obtained from Invitrogen. The bacterial phosphotriesterase and the site-directed mutants (G60A, I106G, F132G, S308G, I106G/H257Y, I106G/F132G/H257Y, I106A/F132A/H257Y, I106A/H257Y/S308A, and H254G/H257W/L303T) were purified to homogeneity as previously described (13, 14, 18). The H254Q/H257F mutant was isolated from a H254X/H257X library (18, 19) and the H257Y/L303T mutant was identified within a H254X/H257X/L303T library (17). In each case H254 and H257 were completely randomized to generate 400 possible permutations with 20 different amino acids at each of the two targeted sites. Nearly 1000 colonies from each library were isolated and screened for catalytic activity. The H254Q/H257F mutant was initially identified as beneficial by screening with Demeton-S, an analogue of VX. The H257Y/L303T mutant was isolated by screening with the SP-enantiomers of compounds 2 and 4.
Synthesis of Racemic Substrates
The corresponding alcohol (1 equivalent) was dissolved in ethyl ether (~100 mL) and cooled in a dry ice/acetone bath. Butyl lithium (1 equivalent) was added to the solution while the mixture was stirred. A solution of methylphosphonic dichloride (1 equivalent) in ethyl ether (100 mL) was cooled in a dry ice/acetone bath for 10 minutes, added to the alcohol solution and then stirred at room temperature for another 4 hours. One equivalent of 4-acetylphenol and triethylamine (1.2 equivalents) were added to the reaction mixture and stirred for 8 hours. The solution was washed with water (2 × 50 mL and 1 × 10 mL) and subsequently dried with anhydrous magnesium sulfate. After removal of the solvent, the crude product was purified by silica gel chromatography to yield the 4-acetylphenyl methylphosphonates (compounds 1 - 5) in yields of 38 - 87%. The structures of compounds 1 – 5 were verified by mass spectrometry, 1H-NMR and 31P NMR spectroscopy. The 1H- and 31P-NMR spectra were obtained on a Varian Unity INOVA 300 spectrophotometer with residual CDCl3 as the internal reference for 1H-NMR, and H3PO4 (85% in water) as the external reference for 31P-NMR. Exact mass measurements were made on a PE Sciex APJ Qstar Pulsar mass spectrometer by the Laboratory for Biological Mass Spectrometry at Texas A&M University.
Isolation of SP-Enantiomers
The SP-enantiomers of compounds 1 - 5 were obtained by kinetic resolution of the corresponding racemic compounds using the G60A mutant of PTE. The reactions were carried out at room temperature in a methanol-water solution (20%) with 50 mM CHES buffer, pH 9.0. The concentration of the racemic phosphonate was approximately 1.0 g/L. The reactions were monitored by measuring the formation of 4-acetylphenol at 294 nm and were stopped when no further increase in the absorbance was observed. The SP-enantiomers were extracted from the reaction mixture with chloroform. The chloroform solutions were dried with sodium sulfate and condensed to dryness to obtain the final product. The SP-enantiomer of 1 was obtained in a yield of 83% and the SP-enantiomers of 2 - 5 were obtained in yields greater than 91%. The enantiomeric excess (ee) was determined using the relative intensities of the 31P-NMR signals in the presence of Fmoc-Trp(Boc)-OH (19). The ee value for the isolation of the SP-enantiomer of 1 was 95% and for compounds 2 - 5 the ee values were 99% (20).
Isolation of Rp-Enantiomers
The Rp-enantiomers of compounds 1 - 5 were obtained by kinetic resolution of the racemic compounds using various PTE mutants. The reactions were carried out at room temperature in a solution of methanol-water (20%) containing 50 mM CHES buffer, pH 9.0. The concentrations of the racemic phosphonates were ~1.0 g/L. The RP-enantiomer of compound 1 was obtained using 4.2 mg of the PTE mutant, H257Y/L303T and the isolated yield was 52% with an ee value of 98%. The RP-enantiomer of compound 2 was obtained using 4.2 mg of the PTE mutant, H257Y/L303T. The yield was 74% and the ee value was 98%. The RP-enantiomer of compound 3 was obtained using 7.2 mg of the mutant, I106A/H257Y/S308A. The yield was 48% and the ee value was 99%. The RPRC- and RPSC-enantiomers of compound 4 were obtained using 6.3 mg of the mutant H257Y/L303T. The yields were 98% and 99%, respectively. The ee values were 99% for both the RPRC- and RPSC-enantiomers of compound 4. The RP-enantiomer of compound 5 was obtained using 1.8 mg of the mutant I106G/F132G/H257Y. The yield was 48%, and the ee value was 97%. The ee values were determined based on the relative intensities of the 31P-NMR signals in the presence of Fmoc-Trp(Boc)-OH (20).
Expression, Growth, and Preparation of Wild-Type and Mutant Enzymes
The gene for the expression of PTE was cloned between the NdeI and EcoRI sites of a pET20b (+) plasmid. The plasmids for the expression of the wild-type and mutant enzymes were transformed into BL-21 cells (DE3) (9). The cells were inoculated in Luria-Bertani (LB) broth and grown overnight at 37 °C. The overnight cultures were incubated in Terrific Broth (TB) containing 100 g/mL ampicillin and 1.0 mM CoCl2 at 30 °C. The expression of PTE was induced by the addition of 1.0 mM IPTG when the OD600 reached 0.4. The cells were harvested at 4 °C by centrifugation after allowing the culture to reach stationary phase after 36-42 hours at 30 °C. All subsequent steps were carried out at 4 °C. The isolated cells were suspended with 10 mM HEPES, pH 8.5, and lysed with a 5-s pulsed sonication for 24 minutes at 0 °C at medium power setting using a Heat System-Ultrasonics, Inc. (Farmington, NY) model W830 ultrasonic processor. The lysed cell suspensions were combined and centrifuged at 13,000 × g for 15 minutes. The supernatant fluid was decanted and then a solution of protamine sulfate (2% w/v) was added dropwise over a 20 minute period while stirring until the protamine sulfate concentration reached 0.4%. The supernatant solution was subjected to ammonium sulfate fractionation by the addition of solid ammonium sulfate to 60% saturation (371 mg/mL) while stirring for 30 minutes and then kept stirring for an additional 45 minutes. The pellet was collected by centrifugation at 13,000 × g for 30 minutes and the protein was recovered by dissolving the precipitate in 3 mL of buffer. The protein was loaded onto a 5.0 × 150-cm gel filtration column containing Ultrogel AcA 54 (IBF, Columbia, MD) and eluted at a flow rate of 1.0 mL/minute. The fractions were pooled based on enzymatic activity and absorbance at 280 nm and then applied to a 5 × 25-cm column, containing DEAE-Sephadex A-25 previously equilibrated with 10 mM HEPES, pH 8.5. SDS-polyacrylamide gel electrophoresis demonstrated that all of the mutant enzymes were the same size as the wild-type PTE and that the purity of all proteins was greater than 95%.
Spectrophotometric Enzyme Assays
The kinetic constants for each substrate were determined by monitoring the formation of p-acetylphenol at 294 nm (ε = 7710 M−1 cm−1) at 30 °C using a SpectraMax plate reader (Molecular Devices Inc., Sunnyvale CA). The assay mixtures contained 50 mM CHES, pH 9.0, 100 μM CoCl2, and various concentrations of substrates. The reactions were initiated by the addition of enzyme. Due to the limited solubility of compounds 4 and 5, 10% methanol (v/v) was added to the assay mixtures.
Enzymatic Assays for GB and GD
Wild-type PTE and the variants G60A, S308G, H257Y/L303T, and H254G/H257W/L303T were purified to homogeneity and stored at −80 °C prior to use. The time courses for the enzymatic hydrolysis of GB and GD were obtained by following the heat of reaction using a MicroCal iTC200 (Northampton, MA) isothermal titration calorimeter (ITC), with a 200 μL reaction cell. All reactions were conducted by injection of the phosphonofluoridate into the enzyme solution at 25 °C with a reference offset of 5 μcal/s, syringe stirring speed of 1000 rpm, pre-injection delay of 180 seconds, and a 5 second recording interval. The enzymes were diluted into 10 mM potassium phosphate (pH 7.4) for GB or 30 mM potassium phosphate (pH 7.4) for GD. The final concentration of GB for all experiments was 250 μM. The final concentration of GD for all experiments was 300 μM. The total heat of hydrolysis for GB (50 nmol) was determined by following the reaction catalyzed by 20 nM G60A and 80 nM H254G/H257W/L303T to completion. The total heat of hydrolysis of GD (60 nmol) was determined by following the reaction catalyzed by 200 nM wild-type PTE, 200 nM G60A, and 600 nM H254G/H257W/L303T to completion.
To isolate the deferential hydrolysis of individual enantiomers with PTE variants, the hydrolysis reactions were conducted with high and low concentrations of enzyme. The hydrolysis of racemic GD with each variant was also analyzed by gas chromatography with a chiral separation column as previously described (21). Reaction solutions were removed from the ITC cell and mixed with an equal volume of dry ethyl acetate to extract the remaining agent. One μL aliquots of the organic phase were injected into the gas chromatograph for analysis.
Data Analysis
The kinetic constants (kcat and kcat/Km) were obtained from a fit of the data to equation 1, where v is the initial velocity, kcat is the turnover number, [A] is the substrate concentration, and Km is the Michaelis constant. Estimates of kcat/Km for racemic GB and GD were obtained by transformation of the rate of heat applied for each ITC experiment. The ITC instrument records the instantaneous rate of heat applied to the reference cell to maintain thermal equilibrium at 5 second intervals. The rate of heat released during the enzymatic hydrolysis of these substrates was obtained by subtracting the experimental values from the baseline value at each time point. The total heat given off as a function of time was obtained by manual integration using a geometric approximation method according to equation 2 where Hi is the total heat observed at time i, dUi is the instantaneous rate of heat produced by the reaction at each time period. The heat of complete hydrolysis of GB (−730 μcal) or GD (−930 μcal) was determined according to equation 2 as the value of Hi as the reaction catalyzed by variants of PTE went to completion. The fractional hydrolysis catalyzed by individual variants as a function of time was determined by dividing the total heat observed at each time period by the total expected heat. Plots of the fraction GB or GD hydrolyzed over time appear as exponential time courses. Depending on the agent or variant used one or two phases could be observed and fit to equation 3 or 4, respectively, where F is the fraction hydrolyzed, a and b are the magnitude of the exponential phases, t is time, k1 and k2 are the exponential components for each phase. The magnitude of the exponential components was allowed to float. Assuming the concentration of substrate is less than Km, the value of kcat/Km was estimated from the exponentials according to equation 5 where kx is the exponential component and Et is the enzyme concentration.
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
Results
Selection of Mutants
Specific residues in the substrate binding pocket of PTE were targeted for mutagenesis. The smallest residue in the small subsite of PTE, Gly-60, was mutated to alanine to reduce the size of the small pocket. The small pocket and adjacent areas were also systematically enlarged by mutation of Ile-106, Ser-308, and Phe-132 to glycine or alanine. Other mutations combined alterations to the small and large subsites simultaneously. The catalytic activities of the wild-type and mutant enzymes with the compounds presented in Scheme 3 were determined and the kinetic parameters are provided in Tables 1 and 2. The stereoselectivities of these enzymes using the isolated chiral enantiomers are presented in Table 3.
Table 1.
Turnover numbers (kcat, s−1) for the wild type and mutant forms of PTE.
| Compound | WT | G60A | I106G | F132G | S308G | H254Q/ H257F |
H257Y/ L303T |
I106G/ H257Y |
I106G/ F132G/ H257Y |
I106A/ F132A/ H257Y |
I106A/ H257Y/ S308A |
H254G/ H257W/ L303T |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rp/Sp-1 | 3.9e2 | 1.6e2 | 1.8e2 | 1.8e2 | - | 3.8e1 | 1.7e2 | 4.8e2 | 1.5e2 | 7.9e2 | 2.3e3 | 2.7e2 |
| Rp-1 | 1.5e2 | 1.3e2 | 6.8e1 | 6.8e1 | 1.1e2 | 1.7e2 | 7.3e0 | 8.9e1 | 3.4e1 | N/D | 2.0e2 | 1.4e1 |
| Sp-1 | 6.7e2 | 1.1e2 | 3.0e2 | 3.0e2 | 2.9e2 | 3.2e1 | 4.1e2 | 6.8e2 | 3.5e2 | 9.0e2 | 1.4e3 | 1.9e2 |
| Rp/Sp-2 | 9.7e1 | 1.1e2 | 3.9e1 | 2.9e1 | 1.8e2 | 1.2e1 | 3.2e2 | 5.9e1 | 5.3e1 | 1.6e2 | 6.5e2 | 1.2e2 |
| Rp-2 | 1.0e2 | 1.2e2 | 4.0e1 | 3.9e1 | 2.5e2 | 4.8e1 | 1.8e1 | 4.7e1 | - | 4.5e1 | 1.3e2 | - |
| Sp-2 | 4.0e1 | 7.4e0 | 2.8e1 | 1.6e1 | 1.5e1 | 7.2e0 | 3.7e2 | 1.0e2 | 4.8e1 | 5.1e2 | 3.4e2 | 9.2e1 |
| Rp/Sp-3 | 5.4e1 | 4.7e1 | 4.9e1 | 3.5e1 | 1.2e2 | 1.1e1 | 1.3e2 | 4.7e2 | - | 6.3e2 | 6.7e2 | 4.7e1 |
| Rp-3 | 9.3e1 | 3.7e1 | 3.1e1 | 2.8e1 | 1.1e2 | 7.0e1 | 5.1e1 | 8.4e0 | - | - | 2.2e1 | 2.0e1 |
| Sp-3 | 2.2e1 | - | 4.8e1 | 2.5e1 | 1.2e1 | 6.3e0 | 1.0e2 | 2.1e2 | 2.3e2 | 2.3e2 | 5.3e2 | 5.0e1 |
| RpRc/RpSc /SpRc/SpSc-4 |
2.1e0 | 4.1e0 | 1.2e0 | - | 1.1e0 | 1.3e0 | 2.4e0 | 2.4e-1 | 2.9e-2 | 4.3e-1 | 1.1e0 | 1.8e1 |
| RpRc/SpRc-4 | 2.5e0 | 3.9e0 | - | - | 2.5e0 | 8.2e-1 | 3.0e0 | 4.8e-1 | 4.2e-2 | 5.0e-1 | 7.8e-1 | 1.1e1 |
| RpSc/SpSc-4 | 6.1e-1 | 1.3e0 | 9.0e-2 | 1.3e-1 | 4.2e-1 | 4.2e-1 | 8.0e-1 | 1.6e-2 | 4.1e-2 | 1.1e-1 | 1.4e-1 | 4.6e0 |
| RpRc-4 | 3.4e0 | 8.5e0 | 3.3e0 | 1.2e1 | 2.2 e0 | 5.5e-1 | 4.1e-1 | 7.6e-2 | 4.9e-2 | 5.0e-2 | 1.7e-1 | 2.0e0 |
| RpSc-4 | 4.5e-1 | 1.5e0 | 1.5e-1 | 1.5e-1 | 6.2e-1 | 1.7e-1 | - | 6.5e-2 | 5.5e-3 | - | 4.2e-2 | 2.1e-1 |
| SpRc-4 | 7.7e-1 | 9.5e2 | 5.0e-2 | 3.2e-1 | 5.0e-1 | 6.3e-1 | 6.3e0 | 3.7e-2 | 1.6e-1 | 7.4e-1 | 1.0e0 | 1.2e1 |
| SpSc-4 | 1.6e-2 | - | - | 1.2e-1 | 1.2e-1 | 3.3e-1 | 2.1e0 | 2.2e-2 | 5.3e-2 | 2.4e-1 | 3.5e-1 | 2.9e0 |
| Rp/Sp-5 | 2.8e1 | 1.9e1 | 1.1e1 | 5.4e0 | 8.4e1 | 3.8e1 | 4.5e0 | 7.8e0 | 1.6e1 | 2.4e0 | - | 1.3e1 |
| Rp-5 | - | 9.3e1 | - | 1.4e1 | 1.5e2 | 3.8e1 | 5.9e0 | - | - | - | 4.2e0 | 8.1e-1 |
| Sp-5 | - | - | 5.1e0 | 3.3e-1 | 1.4e-1 | - | 5.1e0 | 1.8e1 | 1.3e1 | 4.7e0 | 4.5e0 | 1.9e1 |
Table 2.
Values of kcat/Km (M−1 s−1) for the wild type and mutant forms of PTE.
| Compound | WT | G60A | I106G | F132G | S308G | H254Q/ H257F |
H257Y/ L303T |
I106G/ H257Y |
I106G/ F132G/ H257Y |
I106A/ F132A/ H257Y |
I106A/ H257Y/ S308A |
H254G/ H257W/ L303T |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rp/Sp-1 | 7.2e5 | 2.7e5 | 9.4e4 | 2.2e5 | 9.8e4 | 1.9e6 | 7.6e4 | 1.8e5 | 8.3e4 | 1.6e5 | 2.6e5 | 7.8e4 |
| Rp-1 | 4.9e5 | 5.2e5 | 6.2e4 | 3.5e4 | 7.5e4 | 1.5e6 | 2.4e3 | 3.3e4 | 6.3e3 | 6.9e3 | 6.0e4 | 1.5e3 |
| Sp-1 | 1.2e6 | 1.3e5 | 7.7e4 | 3.3e5 | 1.5e5 | 7.1e6 | 1.6e5 | 3.8e5 | 1.1e5 | 3.5e5 | 7.2e5 | 2.2e5 |
| Rp/Sp-2 | 2.4e5 | 2.5e5 | 3.3e4 | 2.7e4 | 5.4e4 | 8.4e5 | 3.9e4 | 1.2e4 | 4.9e3 | 3.0e4 | 3.1e4 | 4.0e4 |
| Rp-2 | 5.8e5 | 5.2e5 | 5.8e4 | 5.9e4 | 1.1e5 | 8.2e5 | 2.5e3 | 8.9e3 | 1.5e3 | 2.3e3 | 1.9e4 | 1.8e3 |
| Sp-2 | 2.7e4 | 6.2e3 | 3.3e3 | 5.5e3 | 3.1e3 | 1.2e6 | 1.1e5 | 3.0e4 | 6.8e3 | 5.0e4 | 6.7e4 | 5.9e4 |
| Rp/Sp-3 | 2.7e5 | 5.2e5 | 5.4e4 | 8.3e4 | 1.1e5 | 5.2e5 | 5.3e4 | 6.0e4 | 3.6e4 | 5.9e4 | 1.7e5 | 9.9e4 |
| Rp-3 | 8.5e5 | 7.6e5 | 6.2e4 | 1.2e5 | 1.8e5 | 3.1e5 | 1.3e4 | 2.3e3 | 4.5e2 | 4.9e2 | 9.5e3 | 1.5e3 |
| Sp-3 | 3.4e4 | 9.9e1 | 4.4e4 | 3.6e4 | 9.5e3 | 1.6e6 | 7.3e4 | 1.5e5 | 9.1e4 | 1.3e5 | 3.9e5 | 1.8e5 |
| RpRc/RpSc /SpRc/SpSc-4 |
3.8e2 | 8.2e2 | 7.1e1 | 1.6e2 | 5.0e2 | 2.8e2 | 7.3e2 | 2.6e1 | 7.6e0 | 6.2e1 | 1.2e2 | 1.9e3 |
| RpRc/SpRc-4 | 5.4e2 | 1.6e3 | 1.5e2 | 1.7e2 | 7.4e2 | 5.5e2 | 9.3e2 | 3.1e1 | 8.8e0 | 1.3e2 | 1.7e2 | 3.3e3 |
| RpSc/SpSc-4 | 1.2e2 | 2.6e2 | 2.8e1 | 2.3e1 | 2.1e2 | 6.3e1 | 3.1e2 | 3.9e0 | 2.8e0 | 1.7e1 | 2.3e1 | 7.7e2 |
| RpRc-4 | 1.3e3 | 3.0e3 | 3.8e2 | 3.3e2 | 2.3e3 | 1.9e2 | 5.8e1 | 1.7e1 | 8.4e0 | 2.3e1 | 5.9e1 | 2.2e2 |
| RpSc-4 | 2.0e2 | 5.9e2 | 5.0e1 | 4.5e1 | 4.9e2 | 5.5e1 | 1.6e1 | 2.3e0 | 1.1e0 | 1.2e0 | 4.5e0 | 1.3e2 |
| SpRc-4 | 1.1e2 | 1.7e1 | 1.4e1 | 8.4e1 | 1.1e2 | 1.6e3 | 1.8e3 | 8.0e0 | 1.3e1 | 1.5e2 | 2.4e2 | 8.1e3 |
| SpSc-4 | 3.2e0 | 1.1e0 | 1.0e1 | 5.2e0 | 6.7e0 | 6.2e1 | 2.5e2 | 2.1e0 | 4.5e0 | 2.2e1 | 3.0e1 | 1.7e3 |
| Rp/Sp-5 | 7.4e3 | 9.6e3 | 3.6e3 | 1.9e3 | 1.4e4 | 3.2e3 | 2.9e3 | 1.1e3 | 1.5e3 | 4.4e2 | 4.0e2 | 1.5e4 |
| Rp-5 | 1.6e4 | 2.1e4 | 4.9e3 | 2.9e3 | 2.8e4 | 5.2e3 | 1.9e3 | 3.8e2 | 5.2e1 | 7.5e1 | 8.4e2 | 2.5e2 |
| Sp-5 | 2.1e1 | 9.4e-1 | 8.4e2 | 8.1e1 | 1.0e2 | 3.3e2 | 5.8e3 | 2.6e3 | 4.6e3 | 1.0e3 | 5.7e2 | 2.8e4 |
Table 3.
Ratios of kcat/Km for the hydrolysis of chiral substrates by the wild-type and mutant forms of PTE.
| Compound | (Rp:Sp) | (Rp:Sp) | (Rp:Sp) | (RpRc:RpSc:SpRc:SpSc) | (Rp:Sp) |
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| wild-type | 1:2 | 22:1 | 25:1 | 400:63:35:1 | 760:1 |
| G60A | 4:1 | 84:1 | 7600:1 | 3600:700:20:1 | 23000:1 |
| I106G | 1:1 | 18:1 | 1:1 | 38:5:1.5:1 | 6:1 |
| F132G | 1:9 | 11:1 | 3:1 | 64:8:16:1 | 35:1 |
| S308G | 1:2 | 34:1 | 19:1 | 340:73:17:1 | 280:1 |
| H254Q/H257F | 1:5 | 1:2 | 1:5 | 3:1:29:1 | 16:1 |
| H257Y/L303T | 1:64 | 1:44 | 1:6 | 4:1:120:16 | 1:3 |
| I106G/H257Y | 1:12 | 1:3 | 1:64 | 8:1:3:1 | 1:7 |
| I106G/F132G/H257Y | 1:18 | 1:5 | 1:200 | 7:1:11:4 | 1:89 |
| I106A/F132A/H257Y | 1:51 | 1:22 | 1:270 | 19:1:120:19 | 1:14 |
| I106A/H257Y/S308A | 1:12 | 1:4 | 1:41 | 13:1:54:7 | 2:1 |
| H254G/ H257W/ L303T | 1:140 | 1:33 | 1:120 | 2:1:65:14 | 1:110 |
Catalytic Activity and Stereoselectivity of Wild-Type PTE
The values of kcat and kcat/Km for wild-type PTE with compounds 2 - 5 demonstrate that this enzyme preferentially hydrolyzes the RP-enantiomers of these organophosphonates. There is, however, a slight preference for the SP-enantiomer of compound 1. As the size of the substituent attached to the phosphorus center becomes larger, the preference for the RP-enantiomer becomes greater. For example, with compound 2 the ratio for R/S(kcat/Km) is 22, whereas for compound 5, the enantiomeric preference increases to 760.
Enhancement of Stereoselectivity
Gly-60 was originally mutated to an alanine residue in an attempt to decrease the size of the cavity for the binding of substrates in the active site of PTE. The values of kcat and kcat/Km for the slow SP-enantiomers with G60A are decreased up to 340-fold relative to the wild-type enzyme. In addition, the kinetic constants for the RP-enantiomers of 4 and 5 are increased relative to the wild-type enzyme. The highest values of kcat/Km were obtained for the two RP-enantiomers of 4. The G60A mutant has an enhanced stereoselectivity relative to the wild-type enzyme. The most extreme case is for compound 5 where the RP-enantiomer is preferred by a factor of 2 × 104.
Relaxation of Stereoselectivity
The mutation of single residues in the small or leaving group pockets of PTE to amino acids with smaller side chains results in an active site that is somewhat larger than the wild type enzyme. In general, this perturbation to the active site results in the relaxation of stereoselectivity through an enhancement in the rate of the initially slower SP-enantiomers. For example, the I106G mutant diminishes the stereoselectivity for compounds 1, 3, 4, and 5 relative to wild-type PTE. The F132G mutant relaxes the stereoselectivity for compounds 2 - 5 compared with the wild-type PTE. Thus, single mutations in the small or leaving group pockets to glycine residues relaxes the stereoselectivity of the mutants relative to the wild-type PTE.
Reversal of the Stereoselectivity
The mutants with a relaxed enantioselectivity contain an expanded small pocket. Modification of the large and small pockets simultaneously inverts the inherent stereoselectivity relative to the wild-type PTE. The mutants, H257Y/L303T, I106G/H257Y, I106G/F132G/H257Y, I106A/F132A/H257Y, and I106A/H257Y/S308A all have residues in the small pocket (Ile-106, Phe-132, and Ser-308) that are replaced with either glycine or alanine. In addition, these mutants include changes to His-257 in the large pocket. For example, wild-type PTE prefers the RP-enantiomer of compound 3 by a factor of 25, whereas the I106A/F132A/H257Y mutant inverts the stereoselectivity with compound 3 and prefers the SP-enantiomer by a factor of 260.
The H254G/H257W/L303T mutant contains an extended small pocket and a modified large pocket. This mutant has a more distinct effect on the reversal of stereoselectivity than the other mutants for the hydrolysis of compounds 1 and 5. For these compounds, the SP-enantiomers are preferred by factors of 140 and 110, respectively. In addition to an inversion in enantioselectivity relative to the wild type enzyme, the H254G/H257W/L303T mutant exhibits the highest overall catalytic activity for the more toxic SP-enantiomers of compounds 4 and 5. The values of kcat/Km for the hydrolysis of the SPRC- and SPSC-stereoismers of compound 4 are enhanced 74- and 540-fold relative to the wild-type enzyme. The most notable case is that for the SP-enantiomer of compound 5 with a kcat/Km that is more than three orders of magnitude higher than the wild-type enzyme. The reversal in stereoselectivity, relative to the G60A mutant, for the hydrolysis of racemic 5 is shown in Figure 2. The addition of H254G/H257W/L303T hydrolyses approximately 50% of the total material in about 5 minutes and then the addition of G60A hydrolyzes the remaining material.
Figure 2.
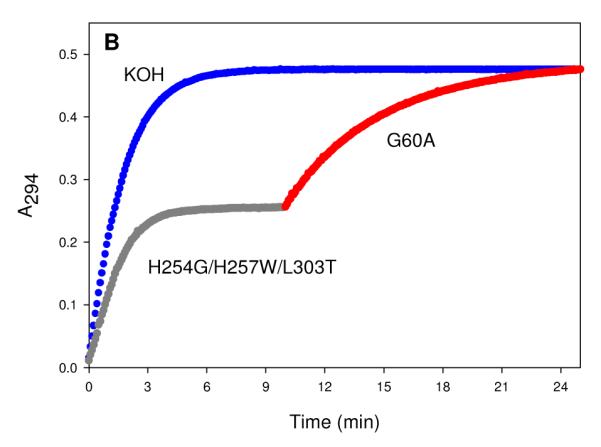
Time course for the hydrolysis of racemic 5 using two mutants of PTE. The total concentration of compound 5 was determined by the complete hydrolysis using 0.1 M KOH. The hydrolysis of the SP-enantiomer of racemic 5 was initiated with 165 nM H254G/H257W/L303T of PTE. After 10 minutes, the G60A mutant of PTE was added to hydrolyze the RP-enantiomer.
Hydrolysis of Racemic GB and GD
Wild-type PTE is known to prefer the less toxic RP-enantiomers of the nerve agents GB and GD (16). PTE mutants with differing catalytic properties with the analogues to these compounds (2 and 4) were selected for testing with racemic GB and GD. Due to regulatory considerations on the maximum allowable concentrations of these two nerve agents, the enzymatic reactions were followed via ITC for the complete hydrolysis at a single fixed concentration of agent. The PTE mutants were tested under identical reaction conditions for their ability to hydrolyze 250 μM GB and 300 μM GD in 200 μL reaction volumes. The values of kcat/Km for each mutant were determined from the exponential phases for hydrolysis of the racemic mixtures and reported in Table 4. Representative time courses for the hydrolysis of GB are given in Figure 3.
Table 4.
Values of kcat/Km (M−1 s−1) for wild-type and mutant forms of PTE against the nerve agents GB and GD.
| compound | GB | GD | |||
|---|---|---|---|---|---|
| kcat/Km1a | kcat/Km2 | kcat/Km1 | kcat/Km2 | Enantiomeric Preferenceb |
|
| Wild-type | (9±3) × 104 | (2.6±0.2) × 103 | RpSc,RpRc,SpRc>>SpSc | ||
| G60A | (1.2±0.1) × 105 | (2.66±0.01) × 104 | (2.5±0.3) ×104 | (2.2±0.3) × 103 | RpSc,RpRc>SpSc,SpRc |
| S308G | (3.9±0.9) × 104 | (9.1±0.5) × 103 | (8±2) × 102 | RpRc,RpSc>SpRc>>SpSc | |
| H254G/ H257W/ L303T |
(1.5±0.1) × 104 | (9.7±0.1) ×102 | (2.2±0.2) × 103 | SpSc,SpRc>>RpSc,RpRc | |
| H275Y/ L303T |
(3±1) × 105 | (3.65±0.01) × 104 | (8.9±0.9) × 104 | (6.8±0.2) × 103 | SpSc,SpRc>RpSc,RpRc |
kcat/Km1 and kcat/Km2 were determined from the first and second exponential phases, respectively, during the reaction of GB or GD as followed by ITC.
first phase > second phase >> not observed.
Figure 3.

Hydrolysis of 250 μM racemic GB followed by ITC. All reactions were initiated by injection of 5 μL of GB into a volume of 200 μL and followed with a MicroCal iTC200 isothermal calorimeter. (A) Hydrolysis of racemic GB by 30 nM wild-type PTE as a function of time. The data are fit to single exponential. (B) Hydrolysis of GB by 60 nM G60A as function of time. The data are fit to the sum of two exponentials. (C) Hydrolysis of racemic GB by 10 nM H257Y/L303T mutant as function of time. The data are fit to the sum of two exponentials.
The fractional hydrolysis of GB catalyzed by wild-type PTE fits to a single exponential (Figures 3a, and Table 4). Similarly, the ITC traces for the S308G mutant with GB (data not shown) can be fit to a single exponential (Table 4). The time courses for three other mutants (G60A, H254G/H257W/L303T, and H257Y/L303T) could not be fit to a single exponential indicating that the two enantiomers were hydrolyzed at different rates. G60A has a value of kcat/Km for the first phase that is similar to that observed with the wild-type enzyme and a second phase that is about 5-fold slower (Figure 3b, and Table 4). The value of kcat/Km for the H254G/H257W/L303T mutant, obtained from the first phase, is reduced in value relative to the wild-type enzyme, and a second phase that is reduced by about two orders of magnitude (data not shown). H257Y/L303T has the highest value of kcat/Km observed for the hydrolysis of GB at 3 × 105 M−1 s−1and a clear second phase that was approximately 10-fold slower (Figure 3c).
The racemic GD used in these experiments contained four diastereomers which can be separated by chiral gas chromatography (Figure 4). In all of the enzymatic time courses only one or two phases could be clearly distinguished from one another. Representative plots are given in Figure 5. To determine which enantiomers were being degraded in each phase, the substrates remaining after selected ITC experiments were extracted with ethyl acetate and separated by gas chromatography to identify the remaining isomers. At high enzyme concentrations, wild-type PTE exhibited a single exponential phase with an estimated value of kcat/Km that is reduced from that obtained with GB (Figure 5a, and Table 4). The gas chromatographic separation indicated that the two RP-enantiomers were almost completely hydrolyzed along with the SPRC-enantiomer leaving only the SPSC-enantiomer in the reaction mixture (Figure 5b). The G60A variant exhibited two phases in the ITC experiment with the faster phase having a kcat/Km value nearly an order of magnitude higher than that obtained with wild-type PTE (Figure 5c). Gas chromatographic analysis revealed that the faster phase corresponded primarily to the degradation of the two RP-enantiomers while the two SP-enantiomers only begin to be degraded in the second phase (Figure 5d). The SPSC-diastereomer is hydrolyzed more slowly than the SPRC-stereoisomer by G60A. The H254G/H257W/L303T mutant exhibited a single exponential phase with a value of kcat/Km similar to that observed with wild-type PTE (Figure 5e). However the gas chromatographic separation indicates that the diastereomeric preference is inverted since the SPSC- and SPRC-diastereomers are hydrolyzed significantly faster than are the RPRC- and RPSC-stereoisomers (Figure 5f). The H257Y/L303T mutant has the highest value of kcat/Km values for the hydrolysis of GD with the hydrolysis of the two SP-enantiomers in the first phase and the two RP-enantiomers in the second phase (Figure 5g). The S308G mutant also exhibited two phases in time course experiments (data not shown) with the first phase having a value of kcat/Km value enhanced relative to the wild type enzyme and a second phase reduced about an order of magnitude.
Figure 4.
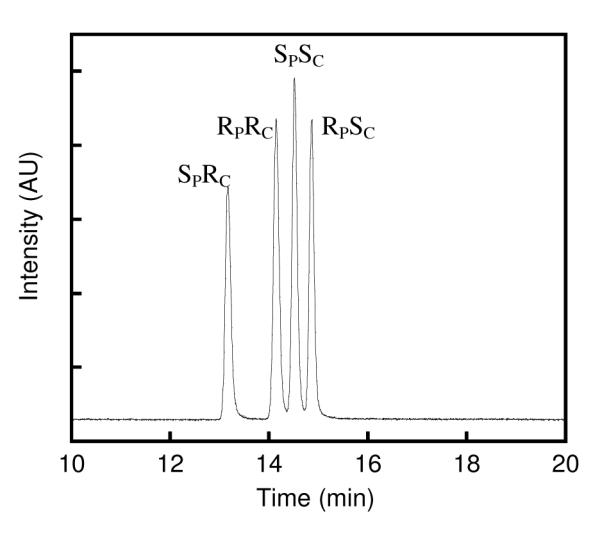
Separation of stereoisomers of racemic GD by gas chromatography.
Figure 5.

Hydrolysis of 300 μM GD. All reactions were conducted in a volume of 200 μL and initiated by the injection of 6 μL of GD into the enzyme solution. Hydrolysis of GD (panels A, C, E, and G) was followed by monitoring the heat liberated as a function of time. Following each experiment the unreacted fraction of GD was analyzed by gas chromatography with a chiral separation column (panels B, D, F, and H). Order of elution from the gas chromatography column is SPRC (13 min), RPRC (14.1 min), SPSC (14.5 min), RPSC (15 min). (A) Hydrolysis of GD by 800 nM wild-type PTE as a function of time. Line is fit to single exponential. (B) Analysis of GD remaining following reaction with wild-type PTE. (C) Hydrolysis of GD by 200 nM G60A as a function of time. Line is a fit to the sum of 2 exponentials. (D) Analysis of unreacted GD following reaction with G60A. (E) Hydrolysis of GD by 600 nM H254G/H257W/L303T as a function of time. The line represents a fit of the data to a single exponential. (F) Analysis of uneacted GD remaining following reaction with H254G/H257W/L303T. (G) Hydrolysis of GD by100 nM H257Y/L303T as function of time. The line is a fit to the sum of two exponentials. (H) Analysis of unreacted GD remaining following reaction with H257Y/L303T.
DISCUSSION
Phosphotriesterase possesses broad substrate specificity and a tunable stereoselectivity that can be harnessed for the detoxification of chemical warfare agents such as GA, GB, GD, GF, VX, and VR. These nerve agents contain a chiral phosphoryl center and the toxicity of the individual stereoisomers differs substantially. Wild-type PTE preferentially hydrolyzes the less toxic RP-stereoismers of these organophosphorus nerve agents and the analogues prepared for this investigation (16, 22). Compounds 1-5 were developed as readily prepared analogues to the G- and V-agents that can be isolated with chiral purities of 95-99% ee. These compounds can also be used in high throughput screening trials for the rapid identification of enzyme mutants that are enhanced in the catalytic hydrolysis of the most toxic stereoisomers.
The inherent stereoselectivity of wild-type PTE for the RP-enantiomers of compounds 1-5 increases with the size of the large substituent attached directly to the phosphorus center. Site specific mutations to the active site of wild-type PTE have demonstrated that the kinetic constants for the hydrolysis of individual compounds can be significantly enhanced by the direct manipulation of the relative size of the large and small sub-pockets within the active site of this enzyme. For example, substitution of an alanine for a glycine residue in the small pocket significantly enhances the stereochemical preference, R/S(kcat/Km), for all of the compounds tested. With this mutant the stereochemical selectivity is enhanced while maintaining a relatively high turnover for the preferred RP-enantiomer.
A significant number of the variants tested for this investigation exhibited an enhanced ability to degrade the SP-stereoisomers of the nerve agent analogues. The dramatic improvements in the catalytic constant, kcat/Km, for the SP-enantiomers of compounds 2 - 5 are graphically illustrated in Figure 6. The best variant of PTE with the SP-enantiomers of compounds 2 and 3, H254Q/H257F, is enhanced by nearly 2 orders of magnitude relative to the wild-type enzyme. For compounds 4 and 5 the best mutant tested is H254G/H257W/L303T. With the SPRC-enantiomer of analogue 4 this mutant is enhanced by a factor of 74 and for the SPSC-isomer the enhancement, relative to the wild type enzyme, is 540. For compound 5, the enhancement is even greater; the H254G/H257W/L303T mutant hydrolyzes the SP-enantiomer of analogue 5 more than three orders of magnitude faster than the wild-type enzyme. For compounds 3 - 5 the second best mutant identified to date is the H257Y/L303T mutant.
Figure 6.

Bar graph illustrating changes in the value of kcat/Km for the SP-enantiomers of compounds 2, 3, 4, and 5 catalyzed by the wild-type and mutant forms of PTE.
The utilization of nerve agent analogues simplifies the identification of enzyme variants with the desired ability to hydrolyze the most toxic enantiomers of GB, GD and GF. However, it is important that the nerve agent stereoselectivity of mutant enzymes be determined by assessing hydrolysis of authentic nerve agent enantiomers. The kcat/Km values for the nerve agents GB and GD were determined for a select group of site-specific mutations to the active site of PTE. The values of kcat/Km reported in Table 4 were obtained from the time courses for the hydrolysis of the racemic mixtures at a single fixed concentration of GB or GD. It must be noted, however, that the values reported in some cases are derived from the composite hydrolysis of multiple enantiomers and thus represent a weighted average of the kinetic constants. It is also assumed in these studies that the initial substrate concentrations for these assays are less than the Km. However, all of the mutants were tested under identical conditions allowing for a direct comparison of the observed kinetic properties and thus this assumption will have relatively little influence on the overall conclusions.
Under the experimental conditions employed for this investigation only a single first-order rate constant for the hydrolysis of racemic GB could be obtained with wild-type PTE. The mutant G60A, which shows more stereochemical selectivity against the analogue of GB (compound 2), clearly shows differential rates of hydrolysis using racemic GB. The value of kcat/Km for the faster enantiomer (presumably the RP-enantiomer) is approximately 4-fold lower than the value of kcat/Km for the RP-enantiomer of compound 2. The mutant S308G did not display a measurable rate differential for hydrolysis of the two enantiomers of GB. The single mutation in this variant is located in the leaving group pocket and it is likely that the significant difference in the leaving group between analogue 2 and GB is a contributing cause for the differing characteristics. The remaining variants (H257Y/L303T, and H254G/H257W/L303T) show greater stereochemical selectivity with compound 2 than the wild-type enzyme. These two mutants also exhibited differential rates of hydrolysis of the two stereoisomers of GB that mirror the differences in the values of kcat/Km with analogue 2. The best enzyme identified for the hydrolysis of GB is H257Y/L303T. The value of kcat/Km for the fastest enantiomer of GB (3 × 105 M−1 s−1) matches quite well with the value of kcat/Km for the SP-enantiomer of the analogue 2 (1.1 × 105 M−1 s−1).
The experiments with GD are complicated by the presence of four stereoisomers. Under the conditions used, no more than two distinct phases could be observed in the time courses for the enzymatic hydrolysis of GD. Gas chromatography was utilized concurrently with the ITC measurements to determine the specific stereoisomers that were preferentially hydrolyzed in the early phases of these reactions. The time course for the hydrolysis of GD by wild-type PTE could be fit to a rate equation with a single exponential which appears to be due to the hydrolysis of three of the four stereoisomers. The stereoselective preferences measured with analogue 4 show only a 10-fold difference in the values of kcat/Km among the three fastest stereoisomers. The slowest stereoisomer of compound 4 (SPSC) is hydrolyzed by wild-type PTE ~35-fold slower than the next fastest stereoisomer (SPRC). The diasteromeric preference determined by gas chromatography indicated that the two RP-stereoisomers, along with the SPRC-enantiomer of GD, were the preferred substrates.
Differential rates of hydrolysis were detected in the hydrolysis of racemic GD by the mutant G60A. Hydrolysis of the two RP-enantiomers was preferred. The measured values of kcat/Km for the two RP-enantiomers with G60A were significantly improved over the wild-type enzyme as was also observed with analogue 4. The hydrolysis reaction catalyzed by S308G exhibited two phases in the time courses, with the first phase due to the two RP-stereoisomers and the second phase dominated by the hydrolysis of the SPRC-enantiomer. The kinetic measurements with the nerve agent analogues indicated that kcat/Km should be enhanced over wild-type PTE, which was observed. The H254G/H257W/L303T mutant exhibited a single phase during the hydrolysis of racemic GD. This phase is dominated by the hydrolysis of the two SP-stereoisomers. The observed value of kcat/Km was in rough agreement with that obtained for the hydrolysis of the racemic GD by the wild-type enzyme but this mutant is significantly enhanced in the hydrolysis of the two most toxic stereoisomers.
The reaction catalyzed by the H257Y/L303T mutant exhibited two exponential phases during the time courses for the hydrolysis of racemic GD, with the faster phase being the hydrolysis of the two SP-diastereomers and the slower phase being due to the hydrolysis of the two RP-diastereomers. The values of kcat/Km for the hydrolysis of racemic GD were significantly higher than what was predicted from the investigations with the nerve agent analogs.
The use of the nerve agent analogues greatly simplifies the identification of PTE variants with improved ability to hydrolyze the more toxic stereoisomers of the authentic nerve agents GB and GD. While the results with analogues and the specific nerve agents are not identical, these results indicate that the findings with analogues accurately predict improvements against GB and GD. Comparison of the results obtained with the nerve agents GB and GD and their respective analogues indicates that these compounds may provide a more stringent test of the stereochemical preferences of PTE based on the phosphoryl center. The changes in kcat/Km obtained with the analogues are largely reflected in the values of kcat/Km found with the authentic nerve agents. These strategies have allowed the isolation of variants that are significantly improved for the hydrolysis of both GB and GD and with a stereochemical preference for the more toxic SP-stereoismers.
1Abbreviations
- PTE
phosphotriesterase
- AChE
acetylcholine esterase
- ITC
isothermal titration calorimetry
- PON1
human paraoxonase 1
- OPAA
organophosphorus acid anhydrolase
- DFP
diisopropyl fluorophosphate
Footnotes
This work was supported by the NIH (GM 68550).
REFERENCES
- 1.Ecobichon DJ. Casarett and Doull’s Toxicology: The basic science of poisons. 6th ed McGraw-Hill; New York: 2001. pp. 763–810. [Google Scholar]
- 2.Benschop HP, Dejong LPA. Nerve agent stereoisomers - analysis, isolation, and toxicology. Accounts of Chemical Research. 1988;21:368–374. [Google Scholar]
- 3.Worek F, Szinicz L, Eyer P, Thiermann H. Evaluation of oxime efficacy in nerve agent poisoning: Development of a kinetic-based dynamic model. Toxicology and Applied Pharmacology. 2005;209:193–202. doi: 10.1016/j.taap.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 4.Josse D, Xie W, Renault F, Rochu F, Schopfer LM, Masson P, Lockridge O. Identification of Residues Essential for Human Paraoxonase (PON1) Arylesterase/Organophosphatase Activities. Biochemistry. 1999;38:2816–2825. doi: 10.1021/bi982281h. [DOI] [PubMed] [Google Scholar]
- 5.Hartleib J, Ruterjans H. High-yield expression, purification, and characterization of the recombinant diisopropylfluorophosphatase from Loligo vulgaris. Protein Expression and Purification. 2001;21:210–219. doi: 10.1006/prep.2000.1360. [DOI] [PubMed] [Google Scholar]
- 6.Wang F, Xiao MZ, Mu SF. Purification and properties of a diisopropyl-fluorophosphatase from squid Todarodes-Pacificus-Steenstrup. Journal of Biochemical Toxicology. 1993;8:161–166. doi: 10.1002/jbt.2570080308. [DOI] [PubMed] [Google Scholar]
- 7.Hill CM, Li WS, Cheng TC, DeFrank JJ, Raushel FM. Stereochemical specificity of organophosphorus acid anhydrolase toward p-nitrophenyl analogs of soman and sarin. Bioorganic Chemistry. 2001;29:27–35. doi: 10.1006/bioo.2000.1189. [DOI] [PubMed] [Google Scholar]
- 8.Harper LL, Mcdaniel CS, Miller CE, Wild JR. Dissimilar plasmids isolated from Pseudomonas-diminuta Mg and a Flavobacterium Sp (ATCC-27551) contain identical Opd genes. Applied and Environmental Microbiology. 1988;54:2586–2589. doi: 10.1128/aem.54.10.2586-2589.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Omburo GA, Kuo JM, Mullins LS, Raushel FM. Characterization of the zinc binding site of bacterial phosphotriesterase. J. Biol. Chem. 1992;267:13278–13283. [PubMed] [Google Scholar]
- 10.Kolakowski JE, DeFrank JJ, Harvey SP, Szafraniec LL, Beaudry WT, Lai KH, Wild JR. Enzymatic hydrolysis of the chemical warfare agent VX and its neurotoxic analogues by organophosphorus hydrolase. Biocatalysis and Biotransformation. 1997;15:297–312. [Google Scholar]
- 11.Benning MM, Shim H, Raushel FM, Holden HM. High resolution X-ray structures of different metal-substituted forms of phosphotriesterase from Pseudomonas diminuta. Biochemistry. 2001;40:2712–2722. doi: 10.1021/bi002661e. [DOI] [PubMed] [Google Scholar]
- 12.Vanhooke JL, Benning MM, Raushel FM, Holden HM. Three-dimensional structure of the zinc-containing phosphotriesterase with the bound substrate analog diethyl 4-methylbenzylphosphonate. Biochemistry. 1996;35:6020–6025. doi: 10.1021/bi960325l. [DOI] [PubMed] [Google Scholar]
- 13.Chen-Goodspeed M, Sogorb MA, Wu FY, Hong SB, Raushel FM. Structural determinants of the substrate and stereochemical specificity of phosphotriesterase. Biochemistry. 2001;40:1325–1331. doi: 10.1021/bi001548l. [DOI] [PubMed] [Google Scholar]
- 14.Chen-Goodspeed M, Sogorb MA, Wu FY, Raushel FM. Enhancement, relaxation, and reversal of the stereoselectivity for phosphotriesterase by rational evolution of active site residues. Biochemistry. 2001;40:1332–1339. doi: 10.1021/bi001549d. [DOI] [PubMed] [Google Scholar]
- 15.Nowlan C, Li YC, Hermann JC, Evans T, Carpenter J, Ghanem E, Shoichet BK, Raushel FM. Resolution of chiral phosphate, phosphonate, and phosphinate esters by an enantioselective enzyme library. Journal of the American Chemical Society. 2006;128:15892–15902. doi: 10.1021/ja0658618. [DOI] [PubMed] [Google Scholar]
- 16.Li WS, Lum KT, Chen-Goodspeed M, Sogorb MA, Raushel FM. Stereoselective detoxification of chiral sarin and soman analogues by phosphotriesterase. Bioorganic & Medicinal Chemistry. 2001;9:2083–2091. doi: 10.1016/s0968-0896(01)00113-4. [DOI] [PubMed] [Google Scholar]
- 17.Lum KT. Department of Toxicology, Texas A& M University. 2004. Ph.D. dissertation, Directed evolution of phosphotriesterase: Towards the efficient detoxification of sarin and soman. [Google Scholar]
- 18.Hill CM, Li WS, Thoden JB, Holden HM, Raushel FM. Enhanced degradation of chemical warfare agents through molecular engineering of the phosphotriesterase active site. Journal of the American Chemical Society. 2003;125:8990–8991. doi: 10.1021/ja0358798. [DOI] [PubMed] [Google Scholar]
- 19.Reeves TE, Wales ME, Grimsley JK, Li P, Cerasoli DM, Wild JR. Balancing the stability and the catalytic specificities of OP hydrolases with enhanced V-agent activities. Protein Eng Des Sel. 2008;21:405–412. doi: 10.1093/protein/gzn019. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Raushel FM. Differentiation of chiral phosphorus enantiomers by P-31 and H-1 NMR spectroscopy using amino acid derivatives as chemical solvating agents. Tetrahedron-Asymmetry. 2007;18:1391–1397. doi: 10.1016/j.tetasy.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeung DT, Smith JR, Sweeney RE, Lenz DE, Cerasoli DM. Direct detection of stereospecific soman hydrolysis by wild-type human serum paraoxonase. FEBS J. 2007;274:1183–1191. doi: 10.1111/j.1742-4658.2006.05650.x. [DOI] [PubMed] [Google Scholar]
- 22.Lum KT, Huebner HJ, Li YC, Phillips TD, Raushel FM. Organophosphate nerve agent toxicity in Hydra attenuata. Chemical Research in Toxicology. 2003;16:953–957. doi: 10.1021/tx034047k. [DOI] [PubMed] [Google Scholar]