

Summary
Human cytomegalovirus (HCMV) continues to be a significant cause of morbidity and mortality in organ transplant recipients despite the availability of antiviral therapy. Considerable controversy exists regarding the use of granulocyte-colony stimulating factor (G-CSF) mobilized blood products from HCMV seropositive donors during stem cell transplantation (SCT) and in patients receiving granulocyte transfusions to treat neutropenia. In order to understand mechanisms of HCMV transmission to patients receiving G-CSF mobilized blood products, we generated a novel NOD-scid IL2Rγcnull humanized mouse model in which HCMV establishes a latent infection in human hematopoietic lineage cells. In this model, G-CSF induces the reactivation of latent HCMV in monocytes/macrophages that have migrated into organ tissues. These results suggest that the use of G-CSF mobilized blood products from seropositive donors pose an elevated risk for HCMV transmission to recipients.
Introduction
Bone marrow-derived myeloid precursor cells are considered to be the latent source of HCMV in seropositive hosts. The differentiation of these latently infected myeloid precursors into migrating macrophages is the proposed mechanism for organ dissemination and viral reactivation followed by subsequent disease(Sinclair and Sissons, 2006; Smith et al., 2004a; Smith et al., 2004b; Smith et al., 2007; Soderberg-Naucler et al., 1997). Consistent with the idea that myeloid precursors are the site of HCMV latency, reactivation of HCMV following allogeneic SCT in the absence of preventative measures occurs in 69% of HCMV-seropositive recipients and 36% of HCMV-seronegative recipients of cells from seropositive donors(Meyers et al., 1986). Although pre-emptive drug treatment reduces the risk of HCMV morbidity and mortality in SCT recipients, these therapies are associated with a higher frequency of late onset HCMV disease and a mortality rate of up to 50%(Asano-Mori et al., 2008; Castagnola et al., 2004).
Currently, G-CSF is commonly used to mobilize stem cells in donors for peripheral blood stem cell transplantation (PBSCT)(Korbling and Anderlini, 2001). Considerable controversy exists regarding the use of peripheral blood stem cells derived from G-CSF-mobilized donors and the risk of late-onset HCMV disease in transplant recipients. In particular, several studies indicate that the use G-CSF-mobilized blood products for PBSCT doubles the risk for both late-onset HCMV disease and chronic graft-versus-host disease when compared to bone marrow transplantation(Anderson et al., 2003; Blaise et al., 2000; Champlin et al., 2000; Kalayoglu-Besisik et al., 1998). The etiology of the increased risk of late-onset HCMV disease associated with PBSCT is unknown. Another controversy concerning HCMV and G-CSF-mobilized blood products exists with conflicting reports on the impact of donor HCMV sero-status and the development of HCMV disease in patients receiving G-CSF-mobilized granulocyte transfusions to treat prolonged neutropenia(Meyer-Koenig et al., 2004; Narvios et al., 2002; Nichols et al., 2002; Vij et al., 2003). The controversies in clinical studies surrounding HCMV and G-CSF-mobilized blood products likely exist due to differences in screening donors for HCMV, monitoring HCMV viral load in recipients, and prophylactic or pre-emptive HCMV treatment strategies, thus underscoring a need for animal studies evaluating the effects of G-CSF on HCMV transmission.
Since HCMV replication is strictly limited to cells of human origin, an animal model has not been available to examine mechanisms of HCMV latency and reactivation. Previous severe combined immunodeficient (SCID) mouse models for HCMV have utilized small human tissue implants, where the virus replicates locally and does not spread beyond the implanted tissue(Bidanset et al., 2001; Mocarski et al., 1993). Due to the absence of human bone marrow-derived myeloid precursor cells and mature myeloid lineage cells, SCID mice with human implants do not support systemic infection nor do these mice develop viral latency(Bidanset et al., 2004; Mocarski et al., 1993). Non-obese diabetic (NOD)/SCID mice can be stably engrafted with human (hu)CD34+ hematopoietic stem cells (HSCs) following sub-lethal irradiation due to a lack of an adaptive immune system and innate immunity defects. In this xenograft model, huCD34+ HSCs self-renew in the BM and provide long-term repopulation of mice with multi-lineage human myeloid and lymphoid cell populations (Legrand et al., 2006). NOD/SCID mice engrafted with huCD34+ HSCs have been shown to be useful tools to examine human viruses that involve hematopoietic cells in their life cycles(Islas-Ohlmayer et al., 2004; Sun et al., 2007; Wu et al., 2006). Therefore, we hypothesized that huCD34+ engrafted NOD/SCID mice would support a systemic HCMV infection and HCMV latency due to the critical role of myeloid progenitors, monocytes, and macrophages in HCMV latency, reactivation, and spread.
Results
G-CSF Promotes HCMV Dissemination to Organ Tissues in huCD34+ Engrafted NOD/LtSz-scid/IL2Rγcnull mice
For the development of a HCMV mouse model, we utilized NOD-scid IL2Rγcnull mice, which have higher huCD34+ stem cell engraftment potential compared to NOD/SCID mice due to additional defects in innate immunity resulting from IL2Rγc deficiency(Ito et al., 2002; Legrand et al., 2006). In testing multiple routes of HCMV infection, we found that intraperitoneal (IP) injection of HCMV-infected human fibroblasts was the only means of infection leading to detectable viral DNA in organ tissues by nested PCR (data not shown). To determine the organ distribution of HCMV, huCD34+ engrafted NOD-scid IL2Rγcnull mice were injected IP with HCMV-infected fibroblasts and sacrificed at 4 weeks post-infection. Nested PCR analysis revealed that engrafted, HCMV-infected mice had detectable HCMV DNA in the bone marrow (BM; 3/3 mice), spleen (2/3 mice), and kidneys (1/3 mice; Table 1). Viral DNA was not detected in any organ tissue tested from huCD34+ engrafted, mock-infected mice or non-engrafted, HCMV-infected mice (Table 1), indicating that the stem cell source was not contaminated with HCMV and that HCMV did not persist in murine cells, respectively.
Table 1.
G-CSF promotes HCMV spread in engrafted NOD/LtSz-scid/IL2Rgcnull mice
| Treatment | PBMC Pre-G-CSF |
PBMC Post-G-CSF |
Bone Marrow |
Spleen | Liver | Kidney | Lung | SMG | Bladder |
|---|---|---|---|---|---|---|---|---|---|
| Engrafted Mock + G-CSF |
0/2 | 0/2 | 0/2 | 0/2 | 0/2 | 0/2 | 0/2 | 0/2 | 0/2 |
| Non-Engrafted HCMV + G-CSF |
0/2 | 0/2 | 0/2 | 0/2 | 0/2 | 0/2 | 0/2 | 0/2 | 0/2 |
| Engrafted HCMV − G-CSF |
0/3 | NA | 3/3 | 2/3 | 0/3 | 1/3 | 0/3 | 0/3 | 0/3 |
| Engrafted HCMV + G-CSF |
0/3 | 3/3 | 3/3 | 3/3 | 3/3 | 3/3 | 2/3 | 1/3 | 1/3 |
CD34+ HSC-engrafted NOD-scid IL2Rgcnull mice were mock-infected or HCMV-infected at 4 weeks post-engraftment. At 4 weeks post-infection, engrafted mice were administered G-CSF by osmotic pump for 7 days and sacrificed (Engrafted Mock + G-CSF; Engrafted HCMV + G-CSF). 3 mice did not receive G-CSF treatment and were sacrificed at 5 weeks post-infection (Engrafted HCMV − G-CSF). As a negative control, non-engrafted mice were HCMV infected, administered G-CSF for 7 days at 4 weeks post-infection, and sacrificed (Non-Engrafted HCMV + G-CSF). Peripheral blood mononuclear cells, bone marrow mononuclear cells, and organ tissue was tested for the presence or absence of HCMV genomic DNA by nested PCR. Shown are the ratios of mice testing positive for HCMV DNA out of total mice tested. PBMC Pre-G-CSF and PBMC Post-G-SCF indicate peripheral blood mononuclear cells tested before and after mobilization respectively. NA, not applicable; PBMC, peripheral blood mononuclear cells; SMG, submandibular salivary gland.
In vitro studies implicate monocytes as the cells responsible for hematogenous dissemination of HCMV to organ tissue(Bentz et al., 2006; Smith et al., 2004a; Smith et al., 2007; Soderberg-Naucler et al., 2001). Therefore, we hypothesized that G-CSF mobilization of myeloid cells from the BM would increase the distribution and frequency of organ tissue positive for HCMV DNA. To test this hypothesis, huCD34+ engrafted mice were administered G-CSF by osmotic pump for 7 days at 4 weeks post-infection. To assess the efficacy of G-CSF treatment, peripheral blood mononuclear cells (PBMCs) were analyzed. Although the relative ratios of mouse and human hematopoietic cells (CD45+) remained similar to that observed in pre-mobilized mice (Fig. 1a), the total number of huCD45+ cells/µl in the peripheral blood was significantly increased from pre-mobilization levels following 7 days of G-CSF treatment (Fig. 1b). Moreover, the percentage of human monocytic cells (huCD45+/CD33+/CD14+) increased from 2.6% (±0.9%) pre-mobilization to an average of 26.94% (±6.3%) on day 7 of mobilization (Fig. 1a and data not shown). Total huCD33+/CD14+ cells/µl in the peripheral blood was significantly increased from pre-mobilization levels at days 3 and 7 of G-CSF treatment (Fig. 1c). By contrast, PBS administered by osmotic pump had no effect on human PBMC populations in huCD34+ engrafted, HCMV-infected mice (data not shown).
Fig. 1.
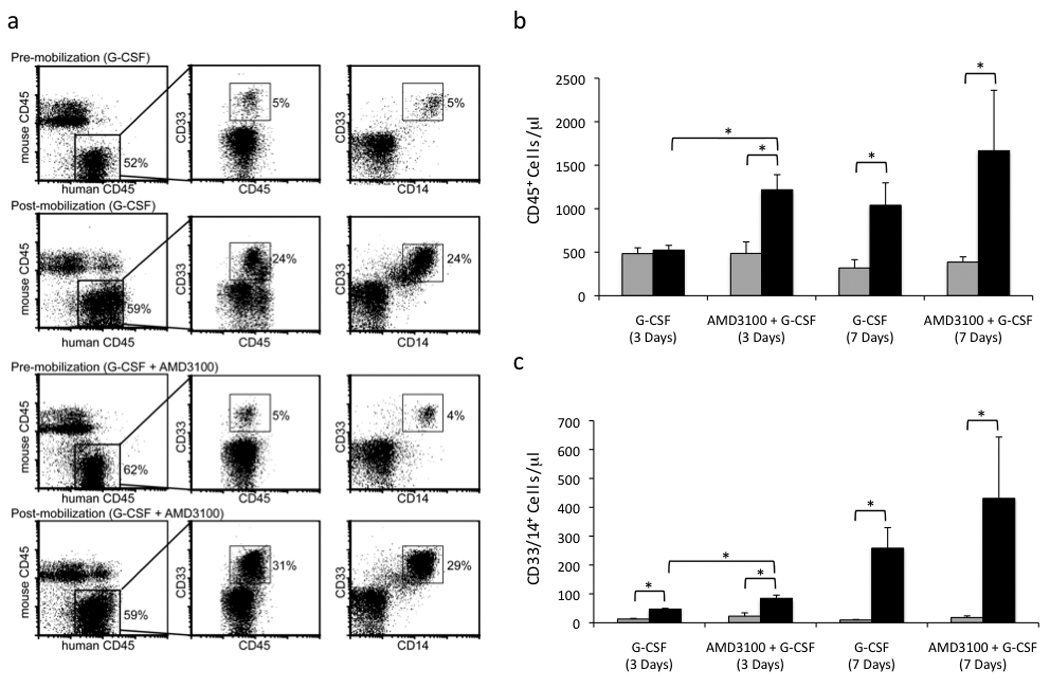
Mobilization of monocytes to the peripheral blood in HCMV-infected, huCD34+ engrafted mice with G-CSF and AMD3100. a, Peripheral blood mononuclear cells from HCMV-infected, engrafted mice were analyzed for murine CD45 (Ly5.2), huCD45, huCD33, and huCD14 expression by flow cytometry before and after 7 days of mobilization with either G-CSF or G-CSF + AMD3100. b,c, AMD3100 + G-CSF treatment resulted in an earlier and more robust mobilization of total huCD45+ PBMCs and huCD33+/huCD14+ monocytes in comparison to G-CSF treatment alone. Total huCD45+ cells/µl (b) or huCD33+/huCD14+ cells/µl (c) of peripheral blood in HCMV-infected engrafted mice were determined pre-mobilization and at days 3 and 7 of mobilization with either G-CSF or G-CSF + AMD3100 by normalizing the percent of cells positive for huCD45 expression by flow cytometry to total white blood cell counts.  , pre-mobilized;
, pre-mobilized;  , mobilized. Data represent mean ± s.e.m. (n=4). *, P < 0.05.
, mobilized. Data represent mean ± s.e.m. (n=4). *, P < 0.05.
Following G-CSF mobilization, a notable increase in HCMV organ dissemination was observed. All infected mice treated with G-CSF had HCMV DNA in BM, spleen, liver, kidney, and PBMCs (Table 1). Some HCMV-infected mice that received G-CSF also had viral DNA in lung, bladder, and submandibular salivary glands (Table 1). These findings suggest that G-CSF treatment of engrafted mice resulted in the mobilization of a persistently or latently infected population of hematopoietic cells from BM to the peripheral blood and organ tissues.
G-CSF-Induced HCMV Dissemination Correlates with Increased Viral DNA Levels in Organ Tissues
Since G-CSF robustly mobilized monocytes in huCD34+ engrafted mice and monocytes are the proposed vehicle of viral spread, we hypothesized that G-CSF treatment would increase viral DNA levels in organ tissue. To quantitatively evaluate differences in viral DNA pre- and post-G-CSF mobilization, real time quantitative PCR was performed on organ tissue. Briefly, engrafted mice were injected IP with mock-infected, HCMV-infected, and UV-inactivated HCMV-treated fibroblasts. HCMV-infected, non-engrafted mice were again included as an additional negative control. G-CSF mobilization increased viral DNA in the spleen (Fig. 2a), BM (Fig. 2b), and liver (Fig. 2c) of engrafted, HCMV-infected mice at 5 weeks post-infection compared to non-mobilized HCMV-infected mice and controls, thereby demonstrating that mobilization of hematopoietic cells results in increased HCMV dissemination. The lack of detectable HCMV DNA in UV-inactivated HCMV control mice indicated that a live HCMV infection was required for viral spread to occur.
Fig. 2.
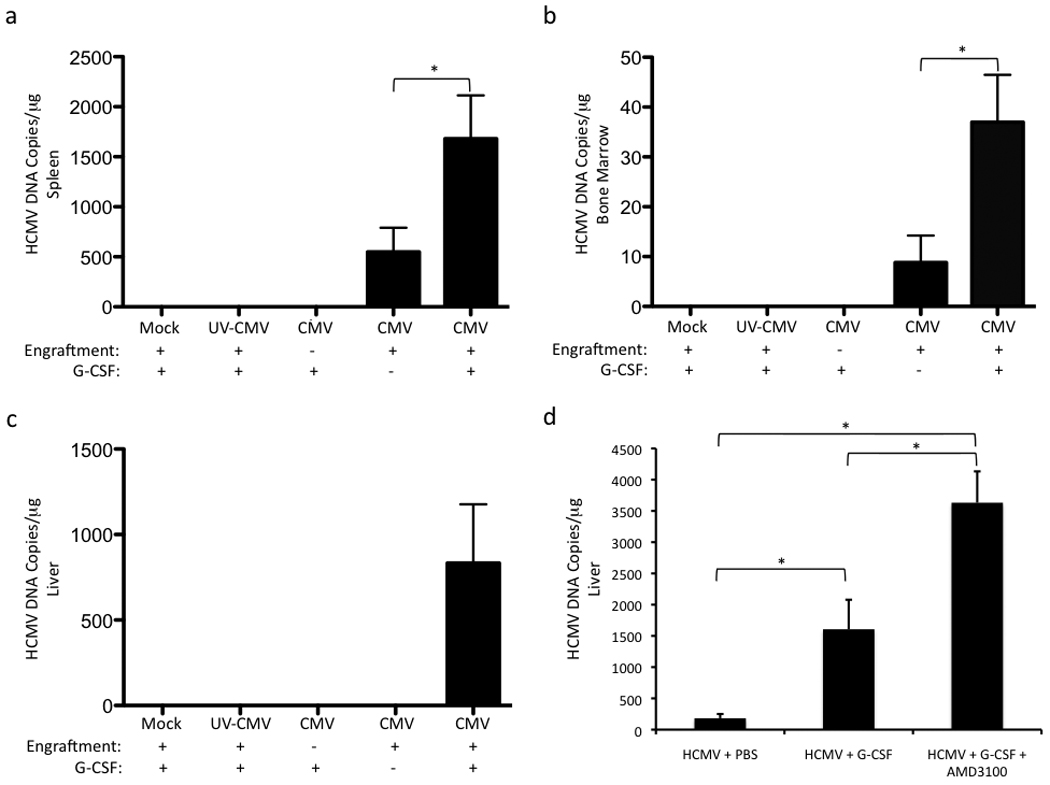
HSC mobilization increases viral genomic DNA in organ tissues of huCD34+ engrafted mice infected with HCMV. a–c, G-CSF treatment of HCMV-infected engrafted mice for 7 days results in increased HCMV genomic DNA in spleen (a), bone marrow mononuclear cells (b), and liver (c). Engrafted mice were injected IP with mock-infected fibroblasts (engrafted mock), UV-inactivated HCMV-treated fibroblasts (engrafted UV-HCMV), or HCMV-infected fibroblasts (engrafted HCMV). At 4 weeks post-infection, engrafted mice were treated with G-CSF (+ G-CSF) for 7 days or left untreated (− G-CSF) and sacrificed. As an additional negative control, non-engrafted mice were injected IP with HCMV-infected fibroblasts, treated with G-CSF for 7 days at 4 weeks post-infection, and sacrificed (Non-engrafted HCMV + G-CSF). Total DNA was harvested from organ tissue and analyzed for HCMV genomic DNA by quantitative real time PCR. Data represent mean copies/µg ± s.d. (n=5). *, P < 0.05. d, Enhanced HSC mobilization with AMD3100 + G-CSF correlates with a further increase in HCMV viral load in liver. HCMV-infected engrafted mice were administered PBS, G-CSF, or AMD3100 + G-CSF for 7 days and sacrificed. Total DNA was isolated from liver and analyzed by quantitative real time PCR for HCMV genomic DNA. Data represent mean copies/µg ± s.d. (n=3, HCMV + PBS; n=4, HCMV + G-CSF, HCMV + G-CSF + AMD3100). *, P < 0.05.
Enhanced Hematopoietic Cell Mobilization with G-CSF and AMD3100 Results in Increased Viral DNA Compared to G-CSF Treatment Alone
Stromal cell-derived factor-1 (SDF-1), which is constitutively expressed in BM, is essential for the retention of undifferentiated HSCs within the BM(Aiuti et al., 1997; Ma et al., 1999; Shen et al., 2001). Inhibition of the SDF-1 receptor CXCR4 with the CXCR4 antagonist AMD3100 increases hematopoietic stem cell mobilization in humans(Broxmeyer et al., 2005) and in NOD/SCID mice(Pitchford et al., 2009), particularly when administered with G-CSF(Flomenberg et al., 2005). To further demonstrate the link between hematopoietic cell mobilization and increased HCMV spread, we treated engrafted, HCMV-infected mice with G-CSF alone and G-CSF + AMD3100 via osmotic pump for 7 days at 4 weeks post-infection. Although at day 7 of treatment, all mobilized mice had similar ratios of mouse and human CD45+ cells and a similar percentage of huCD33+/CD14+ cells (Fig. 1a), we observed a significant increase in total huCD45+ cells/µl (Fig. 1b) and huCD33+/CD14+ cells/µl (Fig. 1c) after 3 days of mobilization in G-CSF + AMD3100-treated mice compared to G-CSF-treated mice. The earlier kinetics of hematopoietic cell mobilization seen with G-CSF + AMD3100 correlated with a greater than two-fold increase in viral DNA in the liver compared to G-CSF treatment alone (Fig. 2d). Altogether, these data demonstrate a direct link between increased mobilization of myeloid lineage cells from the BM and increased viral load in organ tissue.
Hematopoietic Cell Mobilization Results in Increased HCMV Gene Expression and Antigen Expression in Human Tissue Macrophages
To determine if G-CSF-mediated viral spread correlated with increased viral mRNA expression, quantitative RT-PCR was performed on liver RNA isolated from PBS-treated and G-CSF-treated HCMV-infected mice at 2 weeks post-treatment. Transcripts for the late HCMV glycoprotein B (gB; UL55), the early HCMV tegument protein pp65 (UL83), and the viral-encoded CXC chemokine UL146 were detectable in all 5 G-CSF mobilized mice and absent in the 3 PBS-treated control mice (Fig. 3a). Furthermore, viral DNA increased in liver tissue of infected mice in comparison to PBS-treated control mice (Fig. 3c). The detection of early and late HCMV mRNA expression in the liver of G-CSF treated mice demonstrated that G-CSF-mobilized hematopoietic cells were permissive for HCMV replication and would likely contribute to viral spread in a fully permissive human host. BM mononuclear cells isolated from mobilized and non-mobilized HCMV-infected mice were also negative for all three transcripts (Fig. 3a) despite the presence of viral DNA (Fig. 3b), suggesting that viral reactivation occurs during the migration of infected cells to organ tissue.
Fig. 3.
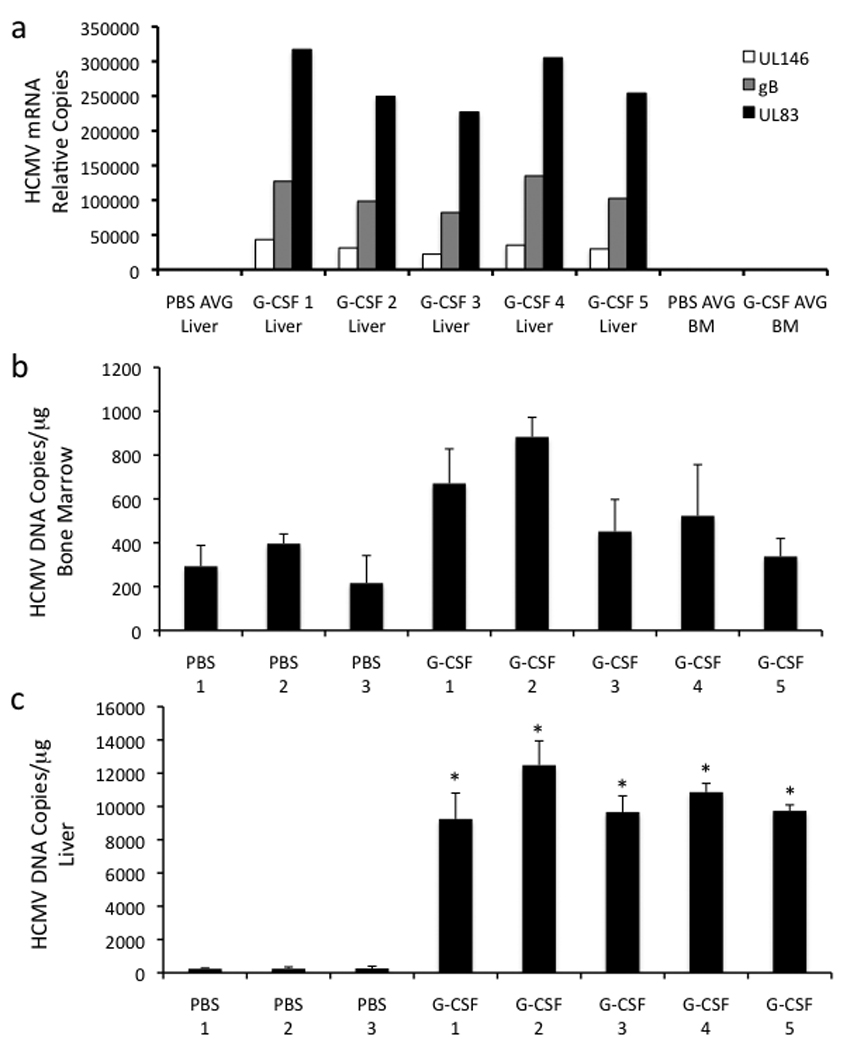
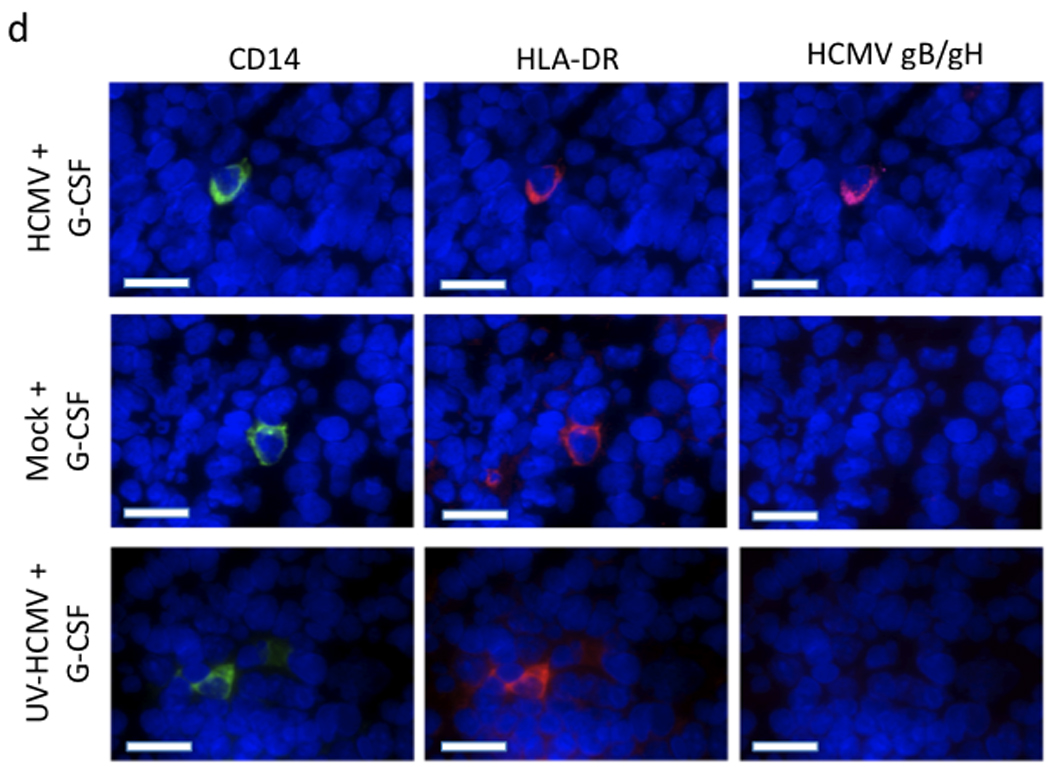
HCMV permissively infects monocytes/macrophages in huCD34+ engrafted mice. a, HCMV mRNAs are expressed in liver following HSC mobilization. Engrafted mice were infected with HCMV, treated with G-CSF (n=5) or PBS (n=3) for 7 days at 4 weeks post-infection, and sacrificed at 6 weeks post-infection. Levels of UL55 (gB, ), UL83 (pp65, ), and UL146 ( ) mRNAs were quantified by real-time PCR of total liver and bone marrow mononuclear cell mRNA. PBS AVG Liver, PBS AVG BM, and G-CSF AVG BM represent the mean values of transcripts for PBS-treated mouse liver tissue, PBS-treated mouse bone marrow mononuclear cells, and G-CSF-treated mouse bone marrow mononuclear cells respectively. b,c, HCMV genomic DNA is present in the bone marrow of G-CSF-mobilized and PBS-treated mice (b) and increases in the liver of HCMV-infected engrafted mice following G-CSF treatment (c). Engrafted mice were infected with HCMV, treated with G-CSF (n=5) or PBS (n=3) for 7 days at 4 weeks post-infection, and sacrificed at 6 weeks post-infection. Total DNA was harvested from organ tissue and analyzed for HCMV genomic DNA by quantitative real time PCR. Data represent mean copies/µg ± s.d. (n=5). *, P < 0.01. d, HCMV gB/gH expression is limited to CD14+ HLA-DR+ monocytes. Engrafted mice were injected IP with HCMV-infected fibroblasts, UV-inactivated HCMV-treated fibroblasts, or mock-infected fibroblasts. At 4 weeks post-infection, mice were administered G-CSF for 7 days and sacrificed. Liver tissue sections were stained with antibodies against huCD14, huHLA-DR, and HCMV gB/gH. 4-color immunofluorescence images of 8 µm liver cryosections were obtained for Hoechst-stained nuclei (blue; all panels), huCD14 (green; left), huHLA-DR (red; middle), and HCMV gB/gH (purple; right). HLA-DR+CD14+ monocytes co-expressing HCMV gB and gH were seen only in HCMV-infected mice treated with G-CSF. Scale bars = 25µm.
) mRNAs were quantified by real-time PCR of total liver and bone marrow mononuclear cell mRNA. PBS AVG Liver, PBS AVG BM, and G-CSF AVG BM represent the mean values of transcripts for PBS-treated mouse liver tissue, PBS-treated mouse bone marrow mononuclear cells, and G-CSF-treated mouse bone marrow mononuclear cells respectively. b,c, HCMV genomic DNA is present in the bone marrow of G-CSF-mobilized and PBS-treated mice (b) and increases in the liver of HCMV-infected engrafted mice following G-CSF treatment (c). Engrafted mice were infected with HCMV, treated with G-CSF (n=5) or PBS (n=3) for 7 days at 4 weeks post-infection, and sacrificed at 6 weeks post-infection. Total DNA was harvested from organ tissue and analyzed for HCMV genomic DNA by quantitative real time PCR. Data represent mean copies/µg ± s.d. (n=5). *, P < 0.01. d, HCMV gB/gH expression is limited to CD14+ HLA-DR+ monocytes. Engrafted mice were injected IP with HCMV-infected fibroblasts, UV-inactivated HCMV-treated fibroblasts, or mock-infected fibroblasts. At 4 weeks post-infection, mice were administered G-CSF for 7 days and sacrificed. Liver tissue sections were stained with antibodies against huCD14, huHLA-DR, and HCMV gB/gH. 4-color immunofluorescence images of 8 µm liver cryosections were obtained for Hoechst-stained nuclei (blue; all panels), huCD14 (green; left), huHLA-DR (red; middle), and HCMV gB/gH (purple; right). HLA-DR+CD14+ monocytes co-expressing HCMV gB and gH were seen only in HCMV-infected mice treated with G-CSF. Scale bars = 25µm.
Since we observed a robust mobilization of huCD33+/CD14+ monocytes in the peripheral blood (Fig. 1), we hypothesized that monocytes/macrophages would be the primary site of HCMV persistence. Therefore, we evaluated the expression of viral antigens by tissue resident macrophages in the liver of these mice by immunofluorescence staining. Sections of liver were stained with mAbs against huCD14 and huHLA-DR in conjunction with a polyclonal Ab cocktail against the late HCMV glycoproteins gB and gH. HCMV gB and gH staining was seen in all HCMV-infected animals examined, while mock-infected mice and those mice receiving fibroblasts infected with UV-inactivated HCMV had no detectable gB and gH antigen expression (Fig. 3d). Furthermore, all cells expressing gB and gH in infected mice also expressed huCD14 and huHLA-DR, indicating that cells permissive for HCMV replication are human monocytes/macrophages (Fig. 3d). Additional antigen combinations were also examined. Cells co-expressing the HCMV immediate early-1 (IE-1) protein and pp65 were detected in liver sections of HCMV-infected, G-CSF-treated mice, but not in HCMV-infected, non-G-CSF-treated mice (Supplementary Fig. 1). Furthermore, IE-1 was only detected in cells positive for a human-specific nuclear antigen (Supplementary Fig. 1). Altogether, these data demonstrate that viral antigen expression is limited to human monocyte/macrophages.
Discussion
In summary, we have shown that huCD34+ engrafted NOD-scid IL2Rγcnull mice infected with HCMV can support a latent viral infection that reactivates in macrophages following G-CSF-induced mobilization of bone marrow hematopoietic cells. In addition, co-treatment of latently infected mice with AMD3100 (Plerixafor) and G-CSF significantly increased HCMV load in these animals in comparison to G-CSF alone. These results provide the first report of a humanized mouse model that can be used to examine mechanisms of HCMV latency and reactivation. To date, the most commonly used humanized model for HCMV involves the implantation of a human fetal conjoint thymus/liver implant under the kidney capsule of SCID mice(Bidanset et al., 2001, 2004; Mocarski et al., 1993). This SCID-hu model provides a limited reconstitution of human hematopoietic cells, with an average of 0.7% huCD45+ cells in the peripheral blood 3–5 months post-transplantation(Legrand et al., 2006). The human PBMCs are almost exclusively of T cell origin in this model, although myeloid lineage cells are contained within the engrafted tissue(Legrand et al., 2006). While this HCMV SCID-hu model does support replication in epithelial cells of the thymic medulla, HCMV does not infect myeloid lineage cells within the implant and does not spread systemically within the mouse(Mocarski et al., 1993). In contrast, the huCD34+ engrafted NOD-scid IL2Rγcnull mouse model provides significantly higher engraftment of human hematopoietic cells, with an average of 52% huCD45+ cells in the peripheral blood at 5 weeks post-engraftment. Furthermore, huCD34+ engrafted NOD-scid IL2Rγcnull mice provide long-term and systemic repopulation of multi-lineage human myeloid and lymphoid cell populations(Legrand et al., 2006). We have demonstrated that the superior engraftment and repopulation of human hematopoietic lineage cells in this mouse model support both a latent HCMV infection in the bone marrow and a systemic reactivation of virus in macrophages following GCSF treatment. Thus the huCD34+ engrafted NOD-scid IL2Rγcnull mouse model represents a significant advancement over the previously utilized SCID-hu model.
Importantly, our studies indicate that G-CSF is a trigger for the reactivation and dissemination of HCMV in myeloid cells to various tissues in the host. These observations may explain the increase in HCMV disease in patients who receive G-CSF-mobilized blood products(Anderson et al., 2003; Meyer-Koenig et al., 2004; Nichols et al., 2002). Specifically, G-CSF mobilized myeloid precursors and monocytes from donors may be primed for HCMV replication and may pose an elevated risk for HCMV transmission to G-CSF-mobilized blood product recipients. Future studies will compare HCMV transmission between mice receiving SCT with either bone marrow or G-CSF mobilized stem cells from huCD34+ engrafted NOD-scid IL2Rγcnull mice. Lastly, this HCMV mouse model will provide a valuable tool to examine mechanisms of HCMV latency and reactivation and will be a platform to test new HCMV antiviral therapies.
Experimental Procedures
Preparation of HCMV-infected Fibroblasts
Neonatal normal human dermal fibroblasts (NHDF) were cultured in minimum essential media (MEM; Cellgro) supplemented with 10% fetal bovine serum at 37°C with 5% CO2. NHDF were infected with low passage clinical HCMV isolate TRpM1A at a multiplicity of infection (M.O.I.) of 3. Mock infection was performed by addition of an equivalent volume of MEM. UV-inactivated HCMV (UV-HCMV) was prepared as previously described(Smith et al., 2004a) and used to treat fibroblasts at equivalent M.O.I. of 3. UV-inactivated HCMV was replication defective as determined by plaque assay.
Engraftment and infection of NOD/SCID mice
7–10 week old NOD-scid IL2Rγcnull mice were sub-lethally irradiated (250 cGy by 137Cs γ-irradiation) and engrafted with 150,000 human CD34+ cord blood cells (Stemcell Technologies, Vancouver, BC, Canada) via retro-orbital injection. Mice were maintained in an SPF facility according to procedures approved by the Institutional Animal Care and Use Committee of the OHSU. At 4 weeks post-engraftment, mice were injected IP with 1 × 106 NHDF that had been HCMV-infected, mock-infected, or UV-HCMV-treated. At 4 weeks post-infection, hematopoietic cells were mobilized by administering 100 µL G-CSF (300 µg/ml; Amgen) or G-CSF + AMD3100 (5 mg/kg) for 7 days via a sub-cutaneous micro-osmotic pump (1007D; Alzet).
Nested PCR analysis of HCMV genomic DNA
DNA was isolated from 0.8 g of mouse tissues using the DNAzol method (Life Technologies). As described previously(Soderberg et al., 1993), a 332 bp region of the HCMV genome was amplified by nested PCR using HCMV-specific forward and reverse primers for exons 1 and 2 of the major immediate early (MIE) gene. The outer primer pairs used were 5'-GAGTCCTCTGCCAAGAGAAA-3' and 5'-GAGTTCTGCCAGGACATCTTT-3', and the inner primer pairs were 5'-GAGTTCTGCCAGGACATCTTT-3' and 5'-CTCGGGGTTCTCGTTGCAAT-3'. The outer primer pair PCR reaction mixture (20 µl) consisted of 100 ng DNA, 1× ThermoPol buffer (New England BioLabs), 200 µM each deoxynucleoside triphosphate (DNTP), 1 µM of each primer, and 0.2 µl of vent DNA polymerase (New England BioLabs). 30 cycles of outer primer PCR were performed, with each cycle consisting of denaturation (20 sec. at 94°C), annealing (50 sec. at 62°C), and extension (20 sec. at 72°C). For the inner primer pair PCR reactions, the same reaction mixture was utilized with 2 µl of the outer primer pair PCR reaction diluted 1:100. 30 cycles of inner primer PCR were performed, consisting of denaturation (20 sec. at 94°C) and combined annealing and extension steps (1 min. at 60°C). Southern blot analysis was performed on PCR products using a MIE-specific 32P-3’-end labeled oligonucleotide probe (5'-ATATCTCCTGTATGTGACCC-3'). All nested PCR results were confirmed with a second independent nested PCR reaction.
Flow cytometry
Peripheral blood from host mice was erythrocyte depleted by sedimentation in 3% dextran (Amersham Pharmacia, Sweden) followed by hypotonic lysis. Bone marrow from host mice was obtained by flushing tibia and femora. Human hematopoietic engraftment was analyzed with antibodies (all from BD) to human CD45 conjugated to allophycocyanin (APC), mouse CD45.1-fluorescein isothiocyanate (FITC), and the lineage markers CD33- phycoerythrin (PE), CD14-Pacific Blue, CD3-PE, CD4-APC-H7, CD8-FITC, and CD19-PE. Dead cells were excluded by a combination of scatter gates and propidium iodide staining. Cells were analyzed on a BD LSRII (BD Biosciences) and data were analyzed using FCS express V3 (De Novo, Los Angeles, CA). Complete circulating blood analysis of peripheral blood was performed by IDEXX Laboratories (Westbrook, ME).
Quantitative PCR detection of HCMV genomic DNA
DNA was extracted from 0.8 g of mouse tissues (liver, spleen and bone marrow) using the DNAzol method (Life Technologies). A total of 1 µg of DNA was analyzed by TaqMan PCR techniques using a probe/primer set recognizing a HCMV UL146 sequence. Primers used were UL146 Forward (5’-CCGCCTGGACATGGAGTATG-3’) and UL146 Reverse (5’-GTCTCGGTTCTGGTGATTTCG-3’). The probe used was 5’-TTATCGCCCGATCACC-3’ labeled on the 5’ end with the reporter FAM and on the 3’ end with a non-fluorescent quencher (Applied Biosystems, Foster City, CA). PCR reactions were set up using the TaqMan Universal PCR Master Mix (Applied Biosystems) according to the manufacturer’s specifications. Following thermal activation of AmpliTaq Gold (10 min. at 95°C), a total of 40 cycles were performed (15 sec. at 95°C and 1 min. at 60°C) using StepOne-plus TaqMan apparatus (Applied Biosystems). DNA isolated from sucrose gradient purified HCMV was used to determine the standard curve. TaqMan results were analyzed using ABI StepOne Real-Time software. The sensitivity of detection for this assay was <50 copies.
Quantitative RT-PCR detection of HCMV gene expression
RNA was extracted from 0.8 g of liver tissue using the TRlzol reagent (Invitrogen) and following the manufacturer’s protocol. cDNA was synthesized from 1 µg of total RNA using 1.0 µM oligo dT primer (oligo dT12–18) and 200 U Superscript III reverse transcriptase (Invitrogen). Primer and probe sets were identified using Primer Express software (Applied Biosystems). The primers and probe sequences used were UL55 P1 (5’-CCGTCCGTCCAAAGAATCTG-3’), UL55 P2 (5’-TTACACCAACGAGCAGGCTTAC-3’), UL55 probe (5’-TGCTGCGCTCGCT-3’), UL83 P1 (5’-TGGAGAACGTGTCGGTCAAC-3’), UL83 P2 (5’-GGATGTTCAGCATCTTGAGCG-3’), UL83 probe (5’-AGCCAGGAGCCCATGTCGATCTATGTGTAC-3’), UL146 P1 (5’-CCGCCTGGACATGGAGTATG-3’), UL146 P2 (5’-GTCTCGGTTCTGGTGATTTCG-3’), and UL146 probe (5’-TTATCGCCCGATCACC-3’). PRT-PCR reactions were performed using specific TaqMan probes and Master Mix (Applied Biosystems). Following thermal activation of AmpliTaq Gold (10min. at 95°C), a total of 40 cycles was performed (15 sec. at 95°C and 1 min. at 60°C) using the ABI Prism Step One Plus Sequence Detection System (Applied Biosystems). Plasmid clones containing each gene fragment were used as positive controls and quantification standards. PCR results were analyzed using ABI Prism Step One Sequence Detection Software. The sensitivity of detection of this assay was <100 plasmid copies for all the tested HCMV genes.
Immunohistochemistry
Livers were frozen in O.C.T. compound (Tissue-Tek). 7 µm cryostat sections were fixed in Streck Tissue Fixative (Streck Laboratories) for 30 min. Antigen retrieval was performed with ficin (Digest-All 1 kit; Zymed) for 15 min. at 37°C. Sections were blocked with 10% normal rabbit serum and 10% normal goat serum for 30 min. Primary antibodies against huCD14 (mouse mAb; clone UCHM-1; Sigma-Aldrich), huHLA-DR (rabbit mAb; ab52478; Abcam), and HCMV gB/gH (goat polyclonal antibody; 0801; ViroStat) were incubated for 1 hr. Sections were stained with secondary antibodies (Alexa fluor 488 F(ab’)2 goat anti-mouse IgG, Alexa fluor 594 F(ab’)2 anti-goat IgG, Alexa fluor 647 F(ab’)2 goat anti-rabbit IgG; Molecular Probes) for 1 hr. Slides were stained with Hoechst for 10 min. All primary antibodies were pre-absorbed against mouse liver powder (Rockland), and all primary and secondary antibodies tested negative for background staining on non-engrafted mouse liver tissue sections.
Highlights.
The first report of an HCMV animal model supporting a systemic and latent infection
G-CSF and Plerixafor reactivate HCMV during hematopoietic cell mobilization
HCMV reactivation occurs in disseminating mobilized human monocytes/macrophages
Results may significantly impact protocols used for stem cell transplant procedures
Supplementary Material
Liver cryostat sections from HCMV-infected, engrafted mice treated which received G-CSF for 7 days (G-CSF) or left untreated (No-GCSF) were stained for either pp65 (green) and IE-1 (red) or IE-1 (green) and human nuclei (Hu Nuclei; red; MAB1281; Chemicon). All cells expressing pp65 co-expressed IE-1, and all cells expressing IE-1 co-expressed the human nuclei antigen. Scale bars = 10 µm.
Acknowledgements
This work was supported by research grants from the National Institutes of Health to J.A. Nelson (AI21640, HL65754, and HL71695), D.N. Streblow (HL083194), W. H. Fleming (HL077818 and HL069133), J. Victor Garcia (AI73146, AI71940 and AI39416), and D.N. Streblow (HL083194). D.N. Streblow is also supported by an AHA Scientist Development Grant. We thank Jamie Borden and Patricia Smith for their excellent technical assistance. We thank members of the Nelson lab for useful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aiuti A, Webb IJ, Bleul C, Springer T, Gutierrez-Ramos JC. The chemokine SDF-1 is a chemoattractant for human CD34+ hematopoietic progenitor cells and provides a new mechanism to explain the mobilization of CD34+ progenitors to peripheral blood. J Exp Med. 1997;185:111–120. doi: 10.1084/jem.185.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D, DeFor T, Burns L, McGlave P, Miller J, Wagner J, Weisdorf D. A comparison of related donor peripheral blood and bone marrow transplants: importance of late-onset chronic graft-versus-host disease and infections. Biol Blood Marrow Transplant. 2003;9:52–59. doi: 10.1053/bbmt.2003.50000. [DOI] [PubMed] [Google Scholar]
- Asano-Mori Y, Kanda Y, Oshima K, Kako S, Shinohara A, Nakasone H, Sato H, Watanabe T, Hosoya N, Izutsu K, et al. Clinical features of late cytomegalovirus infection after hematopoietic stem cell transplantation. Int J Hematol. 2008;87:310–318. doi: 10.1007/s12185-008-0051-1. [DOI] [PubMed] [Google Scholar]
- Bentz GL, Jarquin-Pardo M, Chan G, Smith MS, Sinzger C, Yurochko AD. Human cytomegalovirus (HCMV) infection of endothelial cells promotes naive monocyte extravasation and transfer of productive virus to enhance hematogenous dissemination of HCMV. J Virol. 2006;80:11539–11555. doi: 10.1128/JVI.01016-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidanset DJ, Rybak RJ, Hartline CB, Kern ER. Replication of human cytomegalovirus in severe combined immunodeficient mice implanted with human retinal tissue. J Infect Dis. 2001;184:192–195. doi: 10.1086/322015. [DOI] [PubMed] [Google Scholar]
- Bidanset DJ, Rybak RJ, Hartline CB, Kern ER. Efficacy of ganciclovir and cidofovir against human cytomegalovirus replication in SCID mice implanted with human retinal tissue. Antiviral Res. 2004;63:61–64. doi: 10.1016/j.antiviral.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Blaise D, Kuentz M, Fortanier C, Bourhis JH, Milpied N, Sutton L, Jouet JP, Attal M, Bordigoni P, Cahn JY, et al. Randomized trial of bone marrow versus lenograstim-primed blood cell allogeneic transplantation in patients with early-stage leukemia: a report from the Societe Francaise de Greffe de Moelle. J Clin Oncol. 2000;18:537–546. doi: 10.1200/JCO.2000.18.3.537. [DOI] [PubMed] [Google Scholar]
- Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, Liles WC, Li X, Graham-Evans B, Campbell TB, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005;201:1307–1318. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagnola E, Cappelli B, Erba D, Rabagliati A, Lanino E, Dini G. Cytomegalovirus infection after bone marrow transplantation in children. Hum Immunol. 2004;65:416–422. doi: 10.1016/j.humimm.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Champlin RE, Schmitz N, Horowitz MM, Chapuis B, Chopra R, Cornelissen JJ, Gale RP, Goldman JM, Loberiza FR, Jr., Hertenstein B, et al. Blood stem cells compared with bone marrow as a source of hematopoietic cells for allogeneic transplantation. IBMTR Histocompatibility and Stem Cell Sources Working Committee and the European Group for Blood and Marrow Transplantation (EBMT) Blood. 2000;95:3702–3709. [PubMed] [Google Scholar]
- Flomenberg N, Devine SM, Dipersio JF, Liesveld JL, McCarty JM, Rowley SD, Vesole DH, Badel K, Calandra G. The use of AMD3100 plus G-CSF for autologous hematopoietic progenitor cell mobilization is superior to G-CSF alone. Blood. 2005;106:1867–1874. doi: 10.1182/blood-2005-02-0468. [DOI] [PubMed] [Google Scholar]
- Islas-Ohlmayer M, Padgett-Thomas A, Domiati-Saad R, Melkus MW, Cravens PD, Martin Mdel P, Netto G, Garcia JV. Experimental infection of NOD/SCID mice reconstituted with human CD34+ cells with Epstein-Barr virus. J Virol. 2004;78:13891–13900. doi: 10.1128/JVI.78.24.13891-13900.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, Ueyama Y, Koyanagi Y, Sugamura K, Tsuji K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002;100:3175–3182. doi: 10.1182/blood-2001-12-0207. [DOI] [PubMed] [Google Scholar]
- Kalayoglu-Besisik S, Budak-Alpdogan T, Nuri Yenerel M, Sargin D, Tangun Y. High risk of chronic graft-versus-host disease in unmanipulated allogeneic peripheral blood stem cell transplantation. Blood. 1998;92:2973–2975. [PubMed] [Google Scholar]
- Korbling M, Anderlini P. Peripheral blood stem cell versus bone marrow allotransplantation: does the source of hematopoietic stem cells matter? Blood. 2001;98:2900–2908. doi: 10.1182/blood.v98.10.2900. [DOI] [PubMed] [Google Scholar]
- Legrand N, Weijer K, Spits H. Experimental models to study development and function of the human immune system in vivo. J Immunol. 2006;176:2053–2058. doi: 10.4049/jimmunol.176.4.2053. [DOI] [PubMed] [Google Scholar]
- Ma Q, Jones D, Springer TA. The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity. 1999;10:463–471. doi: 10.1016/s1074-7613(00)80046-1. [DOI] [PubMed] [Google Scholar]
- Meyer-Koenig U, Hufert FT, Duffner U, Neumann-Haefelin D, Henschen M. G-CSF-mobilised granulocyte transfusion to an ALL patient complicated by cytomegalovirus transmission. Bone Marrow Transplant. 2004;34:1095–1096. doi: 10.1038/sj.bmt.1704644. [DOI] [PubMed] [Google Scholar]
- Meyers JD, Flournoy N, Thomas ED. Risk factors for cytomegalovirus infection after human marrow transplantation. J Infect Dis. 1986;153:478–488. doi: 10.1093/infdis/153.3.478. [DOI] [PubMed] [Google Scholar]
- Mocarski ES, Bonyhadi M, Salimi S, McCune JM, Kaneshima H. Human cytomegalovirus in a SCID-hu mouse: thymic epithelial cells are prominent targets of viral replication. Proc Natl Acad Sci U S A. 1993;90:104–108. doi: 10.1073/pnas.90.1.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narvios A, Pena E, Han XY, Lichtiger B. Cytomegalovirus infection in cancer patients receiving granulocyte transfusions. Blood. 2002;99:390–391. doi: 10.1182/blood.v99.1.390. [DOI] [PubMed] [Google Scholar]
- Nichols WG, Price T, Boeckh M. Cytomegalovirus infections in cancer patients receiving granulocyte transfusions. Blood. 2002;99:3483–3484. doi: 10.1182/blood.v99.9.3483. [DOI] [PubMed] [Google Scholar]
- Pitchford SC, Furze RC, Jones CP, Wengner AM, Rankin SM. Differential mobilization of subsets of progenitor cells from the bone marrow. Cell Stem Cell. 2009;4:62–72. doi: 10.1016/j.stem.2008.10.017. [DOI] [PubMed] [Google Scholar]
- Shen H, Cheng T, Olszak I, Garcia-Zepeda E, Lu Z, Herrmann S, Fallon R, Luster AD, Scadden DT. CXCR-4 desensitization is associated with tissue localization of hemopoietic progenitor cells. J Immunol. 2001;166:5027–5033. doi: 10.4049/jimmunol.166.8.5027. [DOI] [PubMed] [Google Scholar]
- Sinclair J, Sissons P. Latency and reactivation of human cytomegalovirus. J Gen Virol. 2006;87:1763–1779. doi: 10.1099/vir.0.81891-0. [DOI] [PubMed] [Google Scholar]
- Smith MS, Bentz GL, Alexander JS, Yurochko AD. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J Virol. 2004a;78:4444–4453. doi: 10.1128/JVI.78.9.4444-4453.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MS, Bentz GL, Smith PM, Bivins ER, Yurochko AD. HCMV activates PI(3)K in monocytes and promotes monocyte motility and transendothelial migration in a PI(3)K-dependent manner. J Leukoc Biol. 2004b;76:65–76. doi: 10.1189/jlb.1203621. [DOI] [PubMed] [Google Scholar]
- Smith MS, Bivins-Smith ER, Tilley AM, Bentz GL, Chan G, Minard J, Yurochko AD. Roles of phosphatidylinositol 3-kinase and NF-kappaB in human cytomegalovirus-mediated monocyte diapedesis and adhesion: strategy for viral persistence. J Virol. 2007;81:7683–7694. doi: 10.1128/JVI.02839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg C, Larsson S, Bergstedt-Lindqvist S, Moller E. Definition of a subset of human peripheral blood mononuclear cells that are permissive to human cytomegalovirus infection. J Virol. 1993;67:3166–3175. doi: 10.1128/jvi.67.6.3166-3175.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg-Naucler C, Fish KN, Nelson JA. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell. 1997;91:119–126. doi: 10.1016/s0092-8674(01)80014-3. [DOI] [PubMed] [Google Scholar]
- Soderberg-Naucler C, Streblow DN, Fish KN, Allan-Yorke J, Smith PP, Nelson JA. Reactivation of latent human cytomegalovirus in CD14(+) monocytes is differentiation dependent. J Virol. 2001;75:7543–7554. doi: 10.1128/JVI.75.16.7543-7554.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Denton PW, Estes JD, Othieno FA, Wei BL, Wege AK, Melkus MW, Padgett-Thomas A, Zupancic M, Haase AT, et al. Intrarectal transmission, systemic infection, and CD4+ T cell depletion in humanized mice infected with HIV-1. J Exp Med. 2007;204:705–714. doi: 10.1084/jem.20062411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vij R, DiPersio JF, Venkatraman P, Trinkaus K, Goodnough LT, Brown RA, Khoury HJ, Devine SM, Oza A, Shenoy S, et al. Donor CMV serostatus has no impact on CMV viremia or disease when prophylactic granulocyte transfusions are given following allogeneic peripheral blood stem cell transplantation. Blood. 2003;101:2067–2069. doi: 10.1182/blood-2002-07-2110. [DOI] [PubMed] [Google Scholar]
- Wu W, Vieira J, Fiore N, Banerjee P, Sieburg M, Rochford R, Harrington W, Jr., Feuer G. KSHV/HHV-8 infection of human hematopoietic progenitor (CD34+) cells: persistence of infection during hematopoiesis in vitro and in vivo. Blood. 2006;108:141–151. doi: 10.1182/blood-2005-04-1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Liver cryostat sections from HCMV-infected, engrafted mice treated which received G-CSF for 7 days (G-CSF) or left untreated (No-GCSF) were stained for either pp65 (green) and IE-1 (red) or IE-1 (green) and human nuclei (Hu Nuclei; red; MAB1281; Chemicon). All cells expressing pp65 co-expressed IE-1, and all cells expressing IE-1 co-expressed the human nuclei antigen. Scale bars = 10 µm.