

Abstract
The function of Ric-3, which is required for nicotinic acetylcholine receptor (nAChR) expression in C. elegans, is unclear. Here we found that Ric-3 can promote or inhibit cell-surface delivery of α-bungarotoxin-binding nAChRs (BgtRs) composed of α7 subunits. At low levels, Ric-3 promoted BgtR assembly, endoplasmic reticulum (ER) release, and cell-surface delivery without trafficking from the ER. At high Ric-3 levels, Ric-3 suppressed BgtR surface delivery, but not its assembly, and BgtRs were retained in the ER or in Ric-3-containing aggregates. In PC12 cells, native BgtRs trafficked to the cell surface from the ER where low levels of endogenous Ric-3 were observed. In cultured neurons, native Ric-3 levels were higher than in PC12 cells, and Ric-3 and α7 subunits were found in somata and dendrites, but not axons, of inhibitory interneurons. Ric-3 trafficked with α7 subunits in rapidly moving vesicles to dendrites, where it was restricted to the ER subcompartment. We conclude that Ric-3 has two potential functions. At low levels, Ric-3 interactions are short-lived and promote BgtR assembly and ER release. At higher levels, Ric-3 interactions are longer-lived and mediate ER retention. In neurons, Ric-3 ER retention appears to promote transport within the dendritic ER subcompartment, thereby restricting α7 trafficking to dendrites and preventing axonal transport.
Introduction
Nicotinic acetylcholine receptors (nAChRs) are members of the cys-loop family of neurotransmitter-gated ion channels (Karlin and Akabas, 1995; Albuquerque et al., 2009). The first neuronal nAChR subtype was identified by its high-affinity α-bungarotoxin (Bgt) binding (Betz et al., 1982; Conti-Tronconi et al., 1985; Kemp et al., 1985; Whiting and Lindstrom, 1987). Bgt-binding nAChRs (BgtRs) are found on neurons throughout the nervous system at presynaptic terminals, postsynaptic sites, extrasynaptic sites, and on other non-neuronal cell types (Grando et al., 1996; Sekhon et al., 1999, Wang et al., 2003). BgtRs are nAChRs composed of a single subunit subtype, α7 subunits (Chen and Patrick, 1997; Rangwala et al., 1997; Drisdel and Green, 2000). Additionally, BgtRs are assembled only in certain cell types (Blumenthal et al., 1997; Cooper and Millar, 1997; Rangwala et al., 1997), suggesting that they require cell-specific conditions to properly fold and assemble. Indeed, the palmitoylation of α7 subunits occurs during BgtR assembly in the endoplasmic reticulum (ER) and is absent in cells unable to assemble BgtRs (Drisdel et al., 2004). However, cells unable to assemble BgtRs can palmitoylate other proteins, indicating that additional factors help regulate BgtR assembly.
Ric-3 was first identified in C. elegans (Nguyen et al., 1995; Miller et al., 1996) and is required for nAChR biogenesis and cell-surface expression (Halevi et al., 2002). With reports that Ric-3 promoted (Cheng et al., 2005; Lansdell et al., 2005) or inhibited (Halevi et al., 2003; Castillo et al., 2005) 5HT3 receptors and α4β2 nAChR surface expression, there is uncertainty about how Ric-3 affects these receptors. There is also no consensus about the subcellular localization of Ric-3, with most studies finding Ric-3 at ER membranes (Castillo et al., 2005; Cheng et al., 2005; Wang et al., 2009) but others also finding it at the cell surface (Williams et al., 2005) and Golgi apparatus (Castillo et al., 2005). Studies showing that Ric-3 and receptor subunits coimmunoprecipitate (Cheng et al., 2005; Lansdell et al., 2005; Williams et al., 2005; Wang et al., 2009) suggest that Ric-3 chaperones receptor assembly in the ER through long-lived interactions with the subunits.
Here, we find that Ric-3 activity depended on its expression levels. At low levels, it acted in the ER to promote α7 subunit assembly and surface delivery. At higher levels, Ric-3 suppressed α7 surface delivery and retained it in the ER. In PC12 cells, endogenous Ric-3 expression levels are low and appear to promote α7 subunit assembly and surface delivery. In cultured neurons, Ric-3 levels are higher than in PC12 cells and Ric-3 is found in the ER of somata and the ER subcompartment of dendrites, where it colocalizes with α7 subunits. Our results indicate that at low levels short-lived interactions with α7 subunits predominated and Ric-3 facilitated BgtR assembly, ER-release, and cell-surface delivery. At higher levels, longer-lived interactions predominated that retained BgtRs in the ER. In neurons, Ric-3 retained α7 subunits in the ER subcompartment of dendrites, where they were transported together in vesicles within the ER subcompartment. In this way, Ric-3 long-lived interactions preferentially traffic α7 subunits synthesized in the soma to dendrites instead of axons.
Materials and Methods
Cell culture and constructs.
The human embryonic kidney (HEK) cell line stably transfected with the large T antigen (tSA201 cells) was from Dr. J. Kyle (University of Chicago, Chicago, IL) and maintained in DMEM supplemented with 10% calf serum (Hyclone). The PC12 cell subclone, PC12 N21 or PC12-C cells (Blumenthal et al., 1997), was from Dr. Richard Burry (Ohio State University, Columbus, OH). The cells were cultured in DMEM containing 5% heat-inactivated horse serum and 10% fetal bovine serum (Hyclone). Primary hippocampal neurons were prepared in the University of Chicago Neuronal Tissue Core using Sprague Dawley rats at embryonic day 18 (E18). As previously described (Marks et al., 2000), hippocampi from day 18 embryos were separated from the diencephalon, dissected free of meninges, and diced. Hippocampi were digested in 0.1% (w/v) trypsin and 0.015% DNase. After dissociation by trituration, cells were counted, suspended in plating medium consisting of Neuralbasal-A supplemented with 2% B-27 (both from Invitrogen), 0.5 mm l-glutamine, and 0.025 mm l-glutamate. Cells were plated on poly-l-lysine-coated 12 mm coverslips at a density of 8 × 104 cells/cm2. For the live imaging studies, E18 hippocampi were purchased from BrainBits and prepared per manufacturer's instructions. All cultures were maintained at 37°C and 5% CO2. Chick α7 and chick/mouse α7/5HT3A subunit plasmids were cloned into pMT3 (Rakhilin et al., 1999) and the human Ric-3 cDNA in pcDNA3 was a gift from Dr. M. Treinin (Hebrew University, Jerusalem, Israel). The fusion of cyan fluorescent protein (CFP) to the N terminus of Ric-3 was generated by subcloning Ric-3 into the pECFP-C1 vector (Clontech). An EcoRI site was inserted just 5′ to the start codon of Ric-3 using the Quikchange site-directed mutagenesis kit (Qiagen). The resulting Ric-3 cDNA was inserted between the EcoRI and SmaI sites of pECFP-C1. The FLAG epitope (DYKDDDDK) was inserted just after the start codon of Ric-3 using the extension-overlap method (Ho et al., 1989). The ER marker, DsRed-ER, was purchased from Clontech.
Cell transfections.
HEK cell transient transfections (5 μg of α7 or α7/5HT3A plasmid, varying amounts of Ric-3 plasmid) were performed using the calcium phosphate method (Eertmoed et al., 1998). The total plasmid per transfection was 10 μg for a 6 cm plate (p60), with levels adjusted with the pcDNA3 plasmid. The pMT3 and pcDNA3 plasmids without cDNA insert were used for control transfections without cDNAs (sham). Neuronal transfections were performed on 14–18 d in vitro cultures using Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions. Briefly, plasmid and Lipofectamine 2000 reagent were mixed in a one-to-one ratio and complexed in Neuralbasal-A media for 30 min. The complex was drop-wise added to neuronal cultures in 12-well plates and returned to the incubator for 24 h before processing for immunofluorescence.
125I-Bgt binding assays.
Intact cells were incubated with 125I-Bgt for 2 h at room temperature in PBS with 1 mm EDTA and 10 mm HEPES pH 7.4. For cells expressing α7, 4 nm 125I-Bgt (140–170 cpm/fmol; PerkinElmer) was used. For cells expressing α7/5HT3A, 125I-Bgt was diluted 10-fold with cold Bgt and 100 nm total Bgt was used to avoid depletion of the Bgt. Intact cells were collected, washed three times with PBS, and counted with a LKB-Wallac 1470 automatic gamma counter. Cells were lysed in lysis buffer with Triton (LBT) containing as follows: 150 mm NaCl, 5 mm EDTA, 50 mm Tris, pH 7.4, 1% Triton X-100 and supplemented with protease inhibitors (PIs) [2 mm phenylmethylsulfonyl fluoride; 10 μg/ml each of chymostatin, pepstatin, leupeptin, and tosyl-lysine chloromethylketone; and 50 nm N-ethylmaleimide (NEM)]. Lysates were centrifuged at 16,000 × g and supernatants incubated with 50 μl of concanavalin A (ConA)-Sepharose slurry (Sigma-Aldrich) for 3 h at room temperature, which quantitatively precipitates all (surface plus internal) BgtRs. To measure internal 125I-Bgt binding, 125I-Bgt binding to cell-surface BgtRs was estimated by ConA-Sepharose precipitation after 125I-Bgt binding to intact cells and solubilization. 125I-Bgt binding to all BgtRs (surface plus internal) was estimated by incubating the ConA-Sepharose precipitated BgtRs again with 125I-Bgt for 2 h at room temperature. 125I-Bgt binding to internal BgtRs was calculated by subtracting the cell-surface from the total 125I-Bgt binding. A final estimate of internal 125I-Bgt binding, adjusted for the loss with ConA pulldown, was made by multiplying by the ratio of surface 125I-Bgt binding to intact cells to surface 125I-Bgt binding from ConA pulldown. We determined protein concentrations (mg/ml) from an aliquot of supernatant using the BCA protein assay (Pierce).
Western blots.
Cultures were rinsed three times with PBS and lysed in LBT with PIs. Insoluble material was pelleted via centrifugation at 16,000 × g and the supernatant transferred to a new tube. Protein concentration was determined by the BCA protein assay (Pierce), and all samples were diluted to 1 mg/ml. Samples were then reduced with 50 mm tris[2-carboxyethyl]phosphine and alkylated with 10 mm NEM for 30 min, then denatured for SDS-PAGE with 5× Laemmli sample buffer. Each lane was loaded with 200 μg of total protein, which was the maximum amount of protein that did not distort SDS-PAGE, and was analyzed on an 8% SDS-PAGE gel that was transferred to a polyvinylidene difluoride membrane and probed with the following antibodies: rabbit anti-actin (1:1000; Sigma) and chicken anti-Ric3 (1:100) (Castelán et al., 2008), or mouse anti-FLAG-M2 (1:1000; Sigma), goat anti-α7 (1:100; Santa Cruz Biotechnology), and mouse anti-FLAG-M2 (1:1000; Sigma) overnight at 4°C. Membranes were washed and probed with appropriate fluorescent secondary antibodies (Jackson ImmunoResearch or Invitrogen) for 1 h at room temperature. Fluorescence was detected using a Biorad molecular imager pharos-FX, which allowed for quantification using ImageJ (Rasband, 2009).
Flow cytometry.
For surface BgtR labeling, transiently transfected cultures were incubated with 100 nm Alexa-647-Bgt (Invitrogen) for 1 h at room temperature. The cells were washed 2× with PBS and incubated in PBS with 10 mm HEPES pH 7.4 and 1 mm EDTA for 15 min to gently lift the cells off the plate. Sterile fluorescence-assisted cell sorting (FACS) was performed with the BD FACSAria using a 405 nm violet and 633 red diode laser. Nonspecific Alexa-647-Bgt binding or autofluorescence was determined by measuring the fluorescence from sham-transfected cultures. Sorting thresholds (gates) were set to collect cells with fluorescence intensities above those measured from sham-transfected cells. Collected cells were gently centrifuged at 100 × g for 5 min and resuspended in regular culture media. Cells (1 × 105) were seeded onto collagen-coated coverslips (10 μg/ml) and allowed to adhere for 12 h. Cultures were then processed for immunocytochemistry as described below. Analysis of FACS data was performed using the FlowJo software.
Whole-cell patch clamp recording.
Sterile sorted cells were shipped live via priority overnight delivery to collaborators at Cornell University. The whole-cell currents were recorded with use of an Axopatch 200A integrating patch-clamp amplifier and Clampex 9 data acquisition software (both from Molecular Devices), as described by Hamill et al. (1981). The composition of the buffer in the recording electrode was 140 mm KCl, 2 mm MgCl2, 1 mm CaCl2, 11 mm EGTA, and 25 mm HEPES, adjusted to pH 7.4 with KOH. The composition of the extracellular (bath) buffer was 145 mm NaCl, 3 mm KCl, 2 mm MgCl2, 1 mm CaCl2, and 25 mm HEPES, adjusted to pH 7.4 with NaOH. Recording electrodes were pulled from borosilicate glass capillaries on a PIP5 two-stage vertical pipette puller (HEKA Elektronik) and the electrode resistance was typically 2–3 MΩ. The series resistance was typically 3–5 MΩ, and was up to 70% compensated electronically. All measurements were performed at room temperature (22−24°C) and a membrane potential of −60 mV. ACh-Cl (1 mm) dissolved in the extracellular buffer was applied for 0.2 s using cell-flow technique (Udgaonkar and Hess, 1987). Data were low-pass filtered at 2 kHz (4-pole Bessel filter) and digitized at 5 kHz.
Immunocytochemistry.
For cell surface BgtR labeling, live cultures were incubated with 100 nm fluorescent Bgt (Invitrogen) for 30 min at 37°C. To block surface BgtRs for labeling of internal BgtRs, live cultures were incubated with 100 nm unlabeled Bgt for 30 min at 37°C. Cultures were washed three times with culture medium, then processed as follows. Cells were fixed in 2% paraformaldehyde with 75 mm lysine and 10 mm periodate in PBS for 10 min, quenched with 100 mm glycine for 10 min, and permeabilized with 0.5% Triton X-100 for 10 min. Blocking of nonspecific sites was performed with 5% normal donkey serum and 0.1% fish gelatin in PBS for 1 h before incubating with primary antibodies in blocking solution overnight at 4°C. Antibodies and dilutions used were as follows: anti- protein disulfide isomerase (PDI; 1:2500; Thermo Fisher Scientific), anti-GM-130 (1:300; BD Transduction Laboratories), fluorescent bungarotoxin (100 nm;> Invitrogen), anti-α7 (1:100; Santa Cruz Biotechnology), anti-Ric-3 (1:100) (Castelán et al., 2008), anti-microtubule-associated protein 2 (MAP2) (1:1000; Cell Signaling Technology), anti-neurofilament heavy chain (NFH; 1:1000; Aves Labs), anti-glutamic acid decarboxylase (GAD; 1:1000; Millipore Bioscience Research Reagents). Cells were then incubated in appropriate secondary antibodies (1:1000), as described in the figure legends, for 1 h at room temperature. In some cases, nuclei were labeled with Hoechst dye for 5 min. Cells were mounted in ProLong gold and allowed to cure for 24 h before imaging on either the Olympus DSU spinning disk or Leica SP-5 scanning laser confocal microscope (Integrated Microscopy Facility, University of Chicago). To facilitate quantitative fluorescence measurements, care was taken so that microscope settings were set below saturating fluorescence intensities. To estimate background fluorescence, samples were prepared without primary antibody or, in the case of Bgt, by including 1 mm nicotine for 5 min before and during fluorescently labeled (Fl)-Bgt labeling in the presence of 1 mm nicotine.
Image analysis.
In supplemental Figure 2 (available at www.jneurosci.org as supplemental material), sorted HEK cells were judged as containing CFP-Ric-3 aggregates if the cells contained one or more large aggregates or two or more smaller aggregates. To estimate the amount of Ric-3 or Fl-Bgt labeling in PC12 cells and neurons, preference was given to isolated cells. Each cell was circled using ImageJ software and the total fluorescence for each slice of a confocal stack was estimated and summed. To estimate the intensity of Fl-Bgt puncta, we measured the fluorescence intensity of n puncta, averaged the data, and compared it to fluorescence intensity measurements of a same-size region within 1 μm of the puncta. To estimate the size of Fl-Bgt puncta, the number of pixels through the center of the puncta was determined and the corresponding pixel size was used to calculate the diameter (0.16 μm per pixel at 100×). Sorted HEK cells were judged as containing Fl-Bgt puncta if they contained four or more Fl-Bgt puncta. In all cases, background fluorescence was determined by performing an identical analysis on cells labeled in the absence of primary antibody or, for Fl-Bgt imaging, labeled in the presence of nicotine as described above. When necessary, randomly sampled images were acquired by choosing fields of view using differential interference contrast microscopy (DIC).
Degree of colocalization estimations.
For colocalization analysis of confocal stacks, we used the ImageJ intensity correlation analysis plug-in (Li et al., 2004). Images were first deconvolved using the Huygens Pro deconvolution software (SVI). Individual cells were carefully outlined (including dendrites if analyzing neurons) and cropped, pixel intensities normalized to maximum intensity for each fluorophore, and the resulting confocal stack was then processed by the intensity correlation analysis plug-in. The frequency scatter plot generated is used to illustrate the extent of colocalization and the Mander's overlap coefficient as was determined from at least 10 cells used to test for significance.
Statistical analysis.
Results are expressed as mean ± SEM of n samples. Statistical significance was assessed by a two-tailed Student's t test or ANOVA as appropriate using Microsoft Excel.
Results
Ric-3 promotes and inhibits BgtR cell-surface delivery
To investigate the effects of Ric-3 on BgtR formation in more detail, we varied the amount of human Ric-3 plasmid transfected with either the α7 subunit or a chimeric subunit composed of the N-terminal half of α7 and the C-terminal half of 5HT3A subunits (α7/5HT3 subunits) (Fig. 1). Unlike the α7 subunit, the α7/5HT3 subunit does not require Ric-3 to form BgtRs in HEK cells (Eiselé et al., 1993; Rangwala et al., 1997). We compared the effects of Ric-3 on BgtR formation and cell-surface expression by measuring intracellular and surface 125I-Bgt binding to α7 (Fig. 1A) or α7/5HT3 BgtRs (Fig. 1B). Intracellular 125I-Bgt binding to α7 BgtRs increased fivefold over the whole range of transfected Ric-3 plasmid. In contrast, cell-surface expression peaked with a 40-fold increase at much lower levels of Ric-3 plasmid and declined to a 12-fold increase at higher levels of Ric-3 plasmid. Levels of intracellular 125I-Bgt binding to α7/5HT3 chimeric BgtRs were independent of transfected Ric-3 plasmid and cell-surface expression was unaffected at lower levels of Ric-3 plasmid and showed a steep decline at the higher levels of Ric-3 plasmid (Fig. 1B). Ric-3 protein levels were assayed by immunoblots using FLAG-tagged Ric-3 (Fig. 1C) and there was no difference between FLAG-tagged and untagged Ric-3 with respect to how varying Ric-3 plasmid affected BgtR cell-surface expression (data not shown). Ric-3 levels rose linearly with increasing transfected-plasmid amounts in the range in which FLAG-Ric-3 protein was detectable (corresponding to 0.01–2.5 μg of Ric-3 plasmid). Ric-3 protein levels are a linear function of transfected plasmid <2.5 μg, whereas with >2.5 μg of transfected plasmid, Ric-3 protein levels saturated. We examined a generic membrane protein, CD8α, and found its surface expression was unaltered by high Ric-3 levels (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), which indicates that the decline in surface 125I-Bgt binding we observed at high Ric-3 levels was specific to α7 and α7/5HT3 BgtRs and not a general effect of Ric-3 on membrane protein trafficking.
Figure 1.

The dependence of BgtR expression on Ric-3 levels. Surface and internal BgtR expression were assayed by 125I-Bgt binding, which is displayed as pmol 125I-Bgt per milligrams total cellular protein. A, B, 125I-Bgt binding assays were performed using transiently transfected HEK cells with set amounts of α7 (A) or α7/5HT3A (B) subunits (5 μg of plasmid) and varying amounts of Ric-3 plasmid. as shown. C, Immunoblots and quantification of FLAG-tagged Ric-3 protein levels at the indicated plasmid levels. D, Displayed is the cell surface to total ratio of 125I-Bgt binding sites.
We have previously observed that Bgt binding sites form on α7 and α7/5HT3 chimeric BgtRs during subunit assembly in the ER and that intracellular 125I-Bgt binding is a measure of the amount of newly assembled BgtRs before surface insertion (Rakhilin et al., 1999; Drisdel et al., 2004). The increased number of intracellular α7 BgtRs with Ric-3 levels (Fig. 1A) thus indicates that Ric-3 enhances Bgt site formation and α7 BgtR assembly in the ER. There was no effect of varying Ric-3 levels on the levels of α7 subunit protein, which remained constant over the range of Ric-3 tested (data not shown). In contrast to α7 BgtRs, there was no change in intracellular α7/5HT3 BgtRs with Ric-3 levels (Fig. 1B), from which we conclude that Ric-3 has little to no effect on α7/5HT3 BgtR assembly. The lack of a Ric-3 effect on α7/5HT3 Bgt site formation could be explained if the levels of α7/5HT3 BgtRs assembled were already at saturating levels in the absence of Ric-3. This is consistent with the observation that ∼50% of the total α7/5HT3 BgtRs are expressed on the cell surface in the absence of Ric-3 and that at peak surface expression of α7 BgtRs with Ric-3 coexpression, 45% of the total α7 BgtRs are also expressed on the cell surface (Fig. 1D), suggesting that the cell is at its maximum capacity for expressing surface BgtRs in relation to the total number available.
Ric-3 appears to have two separate actions on α7 subunits. The first two effects are observed at low Ric-3 levels (0–0.1 μg of plasmid), where there is an increase in subunit assembly into BgtRs as well as increased α7 BgtR forward trafficking. These effects are caused by increased BgtR assembly that, in turn, increased the rate of the forward trafficking. These effects occur at such low levels of Ric-3 plasmid (Fig. 1A) and protein (Fig. 1D) that the interactions between Ric-3 and α7 subunits are very far from stoichiometric. For such a small amount of Ric-3 to alter much larger amounts of α7 subunits, the data indicates that the interaction between Ric-3 and α7 subunits causing these effects is short lived. The third effect is observed at higher Ric-3 levels (0.1–5.0 μg of plasmid), where Ric-3 blocked BgtR insertion at the cell surface for α7 and α7/5HT3 BgtRs. Because Ric-3 only inhibited cell-surface and not intracellular BgtR levels, Ric-3 appears to block BgtR forward trafficking to the cell surface without altering its actions on BgtR assembly. Because these effects occur when Ric-3 and subunit plasmid levels are approximately equal, interactions between Ric-3 and the subunits may be much longer lived and may act to sterically retain the subunits within a compartment in the cells. A longer-lived interaction between Ric-3 and the subunits is indicated by our (data not shown) and others' (Cheng et al., 2005; Williams et al., 2005; Wang et al., 2009) ability to coimmunoprecipitate Ric-3 and the subunits at the higher Ric-3 levels.
Ric-3 alters BgtR cellular distribution and localization
To further address how Ric-3 affects BgtR expression, we examined their distribution at the single-cell level using fluorescence microscopy. From the outset, we found that the cell-to-cell variations resulting from transient transfection of the constructs made it difficult to analyze the experiments. Also, antibodies (Abs) generated against the Ric-3 C-terminal cytoplasmic domain (Castelán et al., 2008) could not reliably detect Ric-3 transfected with 0.01 μg of plasmid or less. To address these problems, we generated a fluorescent Ric-3 construct tagged with CFP at its N terminus (CFP-Ric-3) and used fluorescence-based flow cytometry to select cells for immunofluorescence microscopy. This strategy isolated more uniform populations of cells in terms of surface BgtR and Ric-3 expression. To assay BgtRs on the cell surface with cell sorting, BgtRs were stained with Fl-Bgt. With the addition of the CFP tag, Ric-3 reduced α7 subunit synthesis when transfected higher then at 0.1 μg of plasmid (data not shown). We avoided reduced α7 subunit synthesis by using a mixture of CFP-tagged (no more than 0.1 μg) and untagged Ric-3 for transfections with Ric-3 plasmid levels >0.1 μg of (data not shown). The CFP-tagged Ric-3 had the same cellular distribution as the untagged Ric-3 when cotransfected at both low and high levels of total Ric-3 (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material).
We performed cell sorting on three sets of transfected cells. One set of cells was sorted from cells expressing α7 subunits and no Ric-3 (Fig. 2A–C). Results from the cell sorting are displayed in Figure 2A where each point represents single-cell fluorescence values for surface Fl-Bgt (y axis) and, in this case, CFP (x axis). Very few cells had significant Fl-Bgt staining on the cell surface, consistent with the 125I-Bgt staining of the cells (Fig. 1A). This result was verified using immunofluorescence microscopy on the sorted cells (Fig. 2B). Because of a lack of Fl-Bgt staining, cells were cotransfected with a CFP plasmid to help select for cells that contained α7 subunits without Fl-Bgt staining. There was a high degree of α7 and CFP coexpression (92.3 ± 0.3%, n = 3 fields of view, 119 cells total) in cultures transfected with α7 and CFP plasmid (Fig. 2B,C). Another set of cells was sorted from cells expressing α7 subunits and low levels of Ric-3 (Fig. 2D–F). As expected, large numbers of these cells were stained for Fl-Bgt (89.5 ± 1.5%, n = 3 fields of view, 137 cells total) yet most cells lacked levels of CFP-Ric-3 fluorescence detectable by the flow cytometer (Fig. 2D). Because so few cells with α7 alone expressed detectable levels of surface BgtR, the cells cotransfected with CFP-Ric-3 exhibiting surface Bgt staining must express some CFP-Ric-3, even though most of them (95%) had CFP fluorescence undetectable by the flow cytometer. CFP-Ric-3 fluorescence was detectable in a higher percentage of the cells using a fluorescent scanning microscope (19.5 ± 3.1% of n = 3 fields of view, 190 cells total), as shown in Figure 2, E and F. Displayed in Figure 2E are two cells that are surface stained with Fl-Bgt. One cell was positive for CFP-Ric-3 fluorescence and in the other cell CFP-Ric-3 fluorescence was undetectable, as observed for most cells. These results provide further evidence that the Ric-3 levels that mediate α7 BgtR assembly and surface delivery are extremely low, such that they are often undetectable by immunoblot (Fig. 1C), flow cytometry (Fig. 2D), or fluorescent microscopy measurements (Fig. 2E). In the cells positive for Fl-Bgt and CFP-Ric-3 fluorescence, there was no detectable overlap of CFP-Ric-3 and surface Fl-Bgt fluorescence (Fig. 2E, inset). This finding indicated that little to no Ric-3 trafficked with α7 BgtRs to the cell surface or that Ric-3 was rapidly internalized once it arrived at the cell surface. In cells with detectable levels of CFP-Ric-3 fluorescence, Ric-3 was diffusely distributed throughout much of the cell with a perinuclear and reticulated pattern typical of the ER.
Figure 2.
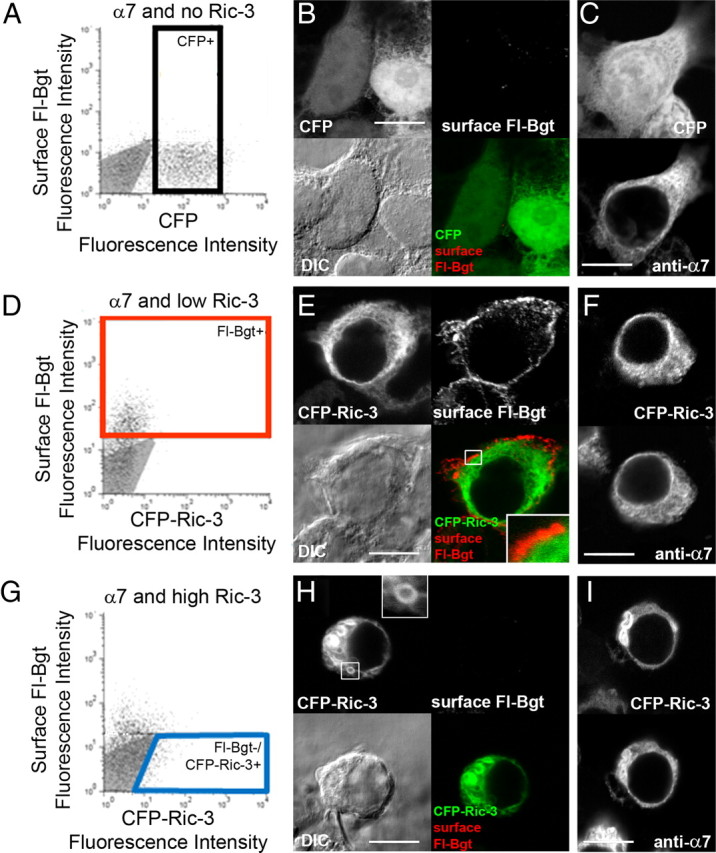
The cellular distribution of BgtRs and Ric-3 at different Ric-3 levels. We used flow cytometry to isolate representative cells for immunofluorescence analysis. To identify cells expressing BgtRs and different levels of Ric-3, cells were sorted using surface Fl-Bgt fluorescence and CFP or CFP-Ric-3 fluorescence. We performed three separate transfections, each with a set amount of α7 subunit plasmid and varying amounts of Ric-3 plasmid. A–C, No Ric-3, where α7 subunit expression was monitored using cotransfection with a CFP plasmid. D–I, Low Ric-3 (0.01 μg of CFP-Ric-3 plasmid; D–F) or high Ric-3 (0.1 μg of CFP-Ric3 and 4.9 μg of Ric-3 plasmids; G–I). A, D, G, Surface BgtRs were labeled with Fl-Bgt and points on the graphs represent single-cell fluorescence values for CFP or CFP-Ric-3 (x axis) and Fl-Bgt (y axis). The gray areas represent the level of nonspecific Fl-Bgt staining and/or autofluorescence of sham transfected cells. The horizontal line in each panel marks the maximum level of nonspecific Fl-Bgt staining of sham transfected cells. A, Cells expressing α7 subunits without Ric-3 were selected (boxed group, CFP+) only using CFP fluorescence because there was no significant Fl-Bgt fluorescence. D, Cells expressing significant amounts of surface BgtRs were selected (boxed group, Fl-Bgt+) only using Fl-Bgt because there was no significant CFP-Ric-3 fluorescence at the low Ric-3 levels. G, Cells expressing high Ric-3 levels without surface BgtRs were selected (boxed group, Fl-Bgt−/CFP-Ric-3+) using CFP-Ric-3 fluorescence and an absence of any significant Fl-Bgt fluorescence. B, C, E, F, H, I, Examples of sorted cells typical of the three different transfections. B, E, H, Images of the same cell set. Upper right, Cell surface BgtRs labeled with Fl-Bgt; upper left, CFP or CFP-Ric-3; Lower left, DIC; lower right, merged image of upper right and upper left. The boxed area in E is magnified in the inset to show the lack of overlap between surface BgtRs and CFP-Ric-3. The boxed area in H is magnified in the inset to show the features of the Ric-3 aggregates. C, F, I, Two images of the same cell set. Top, CFP or CFP-Ric-3; bottom, all α7 subunits labeled with anti-α7 Abs. Scale bars, 10 μm.
A final set of cells was sorted from cells expressing α7 subunits and high levels of Ric-3 (Fig. 2G–I). At high Ric-3 levels, the number of Fl-Bgt-positive cells was reduced (Fig. 2G), consistent with the decrease in surface 125I-Bgt binding observed at high Ric-3 levels (Fig. 1A). Cells lacking surface Fl-Bgt tended to have significant CFP-Ric-3 fluorescence and we sorted for the cells that were positive for CFP-Ric-3 and lacked Fl-Bgt (Fig. 2G, boxed area). An example typical of this cell population is displayed in Figure 2, H and I. The distribution of Ric-3 and α7 subunits in these cells was different from cells with lower Ric-3 expression. In addition to a diffuse distribution of Ric-3 and α7 subunits throughout the cells, there were round, aggregated structures with hollow centers (Fig. 2H, inset). Similar aggregated structures were observed previously with heterologous expression of Ric-3 (Castillo et al., 2005; Cheng et al., 2005; Wang et al., 2009). To further analyze Ric-3's cellular distribution, we determined the number of cells with aggregates, as in Figure 2, H and I, or free of aggregates, as in Figure 2, E and F. We did not find any aggregates at low Ric-3 levels when cells were positive for surface Fl-Bgt staining (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). At high Ric-3 levels, 70–80% of the Bgt-positive cells had aggregates and 20–30% of the cells lacked them (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material). Thus, aggregates of Ric-3 and α7 subunits were observed in cells with high Ric-3 levels in which BgtRs were transported to the cell surface, suggesting that the aggregates alone are not totally responsible for inhibiting surface delivery of BgtRs or that the aggregates formed subsequent to surface delivery of BgtRs. In cells with no significant Bgt staining (supplemental Fig. 2D, available at www.jneurosci.org as supplemental material), Ric-3 aggregates were observed at all levels of Ric-3 expression, together with a diffuse distribution of Ric-3. Ric-3 aggregates were even observed at the lower levels of Ric-3 expression, suggesting that in a small population of these cells, Ric-3 levels were high enough for aggregates to form.
Ric-3 regulation of α7 BgtR function
Previous studies have found increased numbers of functional α7-containing BgtRs with the addition of Ric-3 to the cells but did not test whether the increase varied with changes in Ric-3 levels. To address this question, we performed whole-cell patch-clamp recordings on sorted cells transfected with α7 and varying levels of Ric-3 plasmid (Fig. 3). Sorted cells positive for Fl-Bgt staining generally displayed rapidly desensitizing ACh-evoked currents, a feature of α7 nAChRs. An example is displayed in the inset in Figure 3. We observed no ACh-evoked currents from cells without significant Fl-Bgt staining. As shown in Figure 3, the frequency of observing a response (19 of 25 cells) and mean amplitude was significantly larger for cells with low Ric-3 levels than for cells with high levels of Ric-3 (8 of 15 cells). Cells with α7 subunits but no Ric-3 did have ACh-evoked currents, but the frequency of the responses (9 of 16 cells) and their mean amplitude was significantly smaller than for cells at the two Ric-3 levels. From these measurements, we conclude that the number of functional receptors has the same dependence on Ric-3 levels as the number of cell-surface BgtRs, as determined in Figures 1 and 2 using 125I-Bgt or Fl-Bgt.
Figure 3.

Effect of Ric-3 mediated on ACh-evoked currents of BgtRs. We performed whole-cell patch-clamp recordings on cells expressing α7 subunits and no Ric-3, low levels of Ric-3, and high levels of Ric-3 as in Figure 2. Cells were sorted as in Figure 2 for CFP fluorescence (CFP+), Fl-Bgt fluorescence (Fl-Bgt+), or for Ric-3 fluorescence and no Fl-Bgt fluorescence (Fl-Bgt−/CFP-Ric-3+). Displayed are mean current amplitudes in response to the application of saturating ACh (1 mm) for cells sorted by CFP fluorescence expressing α7 and no Ric-3 (n = 16), cells sorted by Fl-Bgt fluorescence with low levels of Ric-3 (n = 28), or high levels of Ric-3 (n = 15) when compared with sham transfected cells (n = 5). A current response recorded after application of saturating ACh (1 mm; denoted by line above the current trace) is displayed in the inset. The spikes corresponding to the beginning and end of ACh application were artifacts caused by opening and closing of the solenoid valve. Cells sorted by having a significant amount of Fl-Bgt fluorescence had significantly higher current amplitudes than α7-expressing cells with no Ric-3 (p < 0.05). Cells transfected with high amounts of Ric-3 had significantly smaller current amplitudes than those cells transfected with low amounts of Ric-3 (p < 0.05). There were no ACh-evoked currents from sham transfected cells or cells sorted with no significant Fl-Bgt fluorescence and high levels of Ric-3 (n = 6) and only one cell sorted with no significant Fl-Bgt fluorescence and low levels of Ric-3 exhibited a current response (n = 5).
Cellular localization of Ric-3 in the absence of α7 subunits
Contrary to a previous report (Williams et al., 2005), we did not observe colocalization between Ric-3 and surface BgtRs (Fig. 2E). Ric-3 was also reported in the Golgi complex (Castillo et al., 2005) as well as in the ER (Castillo et al., 2005; Cheng et al., 2005; Wang et al., 2009). In Figure 4, we tested whether Ric-3, in the absence of α7 subunits, is predominantly in the ER or at other sites along the secretory pathway, such as the Golgi complex. Cells with high or low Ric-3 levels and no α7 subunits were sorted using CFP-Ric-3 fluorescence to isolate a more homogenous population of cells containing different Ric-3 levels. These cells were stained with an ER marker (PDI-specific Ab) or a Golgi marker (GM-130-specific Ab). At the low Ric-3 levels, CFP-Ric-3 colocalized strongly with the ER marker and little to none with the Golgi marker (Fig. 4A). We gauged the degree of colocalization by comparing the intensities of the two fluorophores pixel by pixel in the confocal images (supplemental Fig. 3, available at www.jneurosci.org as supplemental material for details). The degree of linear correspondence between the CFP-Ric-3 fluorescence and that of the ER marker was determined (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), from which we obtained a Manders coefficient r (Manders et al., 1992) with values from 1 (perfect overlap) to 0 (no overlap). Eighty-six percent of the CFP-Ric-3 pixels colocalized with those of the ER marker (r = 0.66 ± 0.1, n = 10 cells) (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material), indicating strong colocalization. In contrast, no significant colocalization between CFP-Ric-3 and the Golgi marker was found, as shown by the lack of correspondence between the CFP-Ric-3 fluorescence and that of the Golgi marker (Fig. 4C). Only 2% of the CFP-Ric-3 pixels overlapped with the Golgi marker and 6% of the Golgi marker pixels overlapped with CFP-Ric-3 pixels (r = 0.12 ± 0.03, n = 10 cells) (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material). Thus at low Ric-3 levels, Ric-3 is predominantly localized in the ER and not in the Golgi or at the cell surface.
Figure 4.
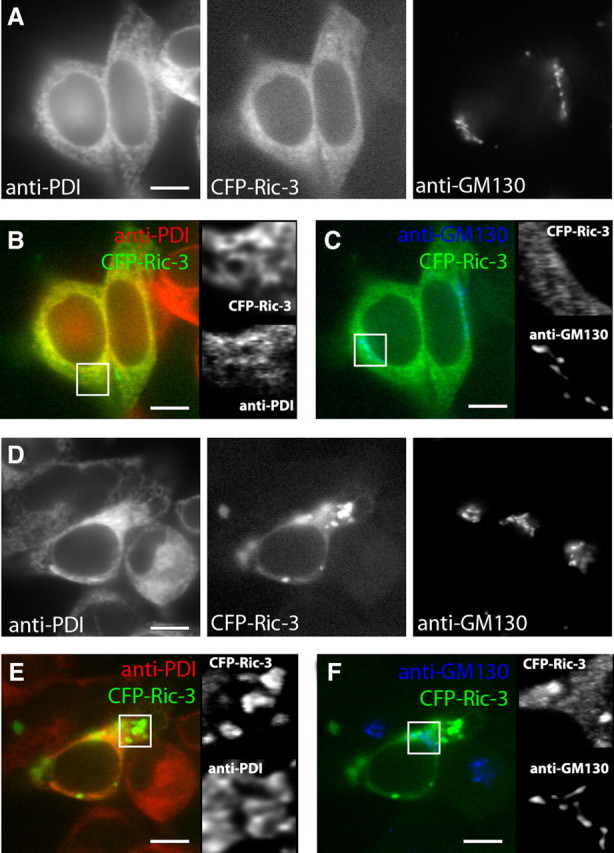
Cellular localization of Ric-3 in the absence of α7 subunits. Cells expressing low or high levels of Ric-3 without α7 subunits were sorted as in Figure 2. A, Sorted cells with low levels of Ric-3 were stained with Abs specific for an ER marker (anti-PDI) and a Golgi marker (anti-GM-130). B, C, Merged images of the cells from A showing the degree of colocalization between CFP-Ric-3 and the ER marker (B) or Golgi marker (C). To the right are enlarged views of the boxed regions. D, Sorted cells with high levels of Ric-3 were stained with Abs specific for an ER marker (anti-PDI) and a Golgi marker (anti-GM-130). E, F, Merged images of the indicated two images from D showing the degree of colocalization between CFP-Ric-3 and the ER marker (E) or Golgi marker (F). To the right are enlarged views of the boxed regions in the panels. Scale bars, 10 μm.
At high levels of Ric-3 (Fig. 4D–F), the Ric-3 distribution changed and we observed aggregated structures just like when Ric-3 was coexpressed with α7 subunits (Fig. 2F,I). Together with a lack of α7 subunit aggregates in the absence of Ric-3, this finding demonstrates that Ric-3 mediates aggregate formation. The Ric-3 that was not within aggregates colocalized with the ER marker (Fig. 4D). Many of the aggregated Ric-3 structures, however, did not colocalize with the ER marker whereas others colocalized partially or completely (Fig. 4E). Consistent with this observation, we measured less (65%, r = 0.47 ± 0.05, n = 10 cells) of the CFP-Ric-3 overlap with the ER marker (supplemental Fig. 3F, available at www.jneurosci.org as supplemental material) compared with 86% overlap with low levels of Ric-3. Again, there was no significant overlap of CFP-Ric-3 with the Golgi marker (1%, r = 0.07 ± 0.01, n = 10 cells) (Fig. 4F and supplemental Fig. 3F, available at www.jneurosci.org as supplemental material). The results suggest that the aggregation initially occurs in the ER and later departs.
Effects of Ric-3 levels on α7 subunit and BgtR localization
We performed additional experiments to examine whether Ric-3 altered the subcellular distribution of α7 subunits or vice versa. Cells were transfected with α7 subunit plasmid and no, low (0.01 μg), or high (5 μg) levels of Ric-3 plasmid and sorted using flow cytometry as in Figure 2. The distribution of α7 subunits in the absence of Ric-3 was examined using anti-α7 Ab staining (Fig. 5A) and found to colocalize with the ER marker, as has been previously observed (Cooper and Millar, 1997). The distribution of α7 coexpressed with low levels of Ric-3 was assayed using CFP-Ric-3 and is displayed in Figure 5C. There was strong overlap between Ric-3, α7, and the ER marker consistent with ER localization when Ric-3 increases BgtR surface delivery (Fig. 1A). As in Figure 2, we did not observe any significant colocalization of Ric-3 with the Golgi marker (data not shown), suggesting that Ric-3 again does not traffic with BgtRs out of the ER.
Figure 5.

Cellular localization of Ric-3 and α7 subunits. A–F, As in Figure 2, cells expressing α7 subunits and no Ric-3 (A, B), low levels of Ric-3 (C, D), and high levels of Ric-3 (E, F) were sorted. Images of typical cells are displayed for the different levels of Ric-3. The left half of each panel is divided into four sections. Upper left, The CFP fluor used for sorting; upper right, the ER marker (anti-PDI Ab); lower left; the α7 subunit-specific marker (anti-α7 Ab or Fl-Bgt); lower right, the DIC image. On the right half are merged images of the upper right and lower left quarters of the left half. A, Cells were stained for α7 subunits and the ER. B, Cells were stained for intracellular BgtRs and the ER. C, Cells were stained for α7 subunits and the ER. A higher magnification of the boxed region in the merged image is displayed in the inset and is a merged image of the CFP-Ric-3 and anti-α7 Ab fluorescence. D, Cells were stained for intracellular BgtRs and the ER. With low Ric-3 levels, we observed bright puncta stained with Fl-Bgt. A higher magnification of the puncta in the boxed region in the merged image is displayed in the inset and is a merged image of the CFP-Ric-3 and Fl-Bgt fluorescence. E, Cells were stained for α7 subunits and the ER marker. With high Ric-3 levels, we observed Ric-3 aggregates stained with anti-α7 and Fl-Bgt. A higher magnification of the boxed region in the merged image is displayed in the inset and is a merged image of the CFP-Ric-3 and anti-α7 Ab fluorescence. F, Cells were stained for intracellular BgtRs and the ER. G, Cells were sorted and stained as in D or F and we determined the percentage of sorted cells that had four or more Fl-Bgt-stained puncta. On the y axis, we plotted the percentage of total cells with four or more Fl-Bgt-stained puncta at the indicated levels of Ric-3 for cells sorted for Fl-Bgt fluorescence (Fl-Bgt+) or for cells lacking Fl-Bgt fluorescence but positive for CFP-Ric-3 fluorescence (Fl-Bgt−/CFP-Ric-3+). Scale bars, 5 μm.
We used Fl-Bgt staining to assay the more mature, Bgt-binding pool of intracellular receptors. Surface BgtRs were excluded by binding surface BgtRs on intact cells with unlabeled Bgt before permeabilizing the cells. Bgt binding sites form on α7 subunits as they assemble into functional BgtRs in the ER (Rakhilin et al., 1999; Sweileh et al., 2000; Drisdel et al., 2004). Thus, Fl-Bgt should stain the pool of BgtRs assembling in the ER and a second pool of BgtRs that are released from the ER and trafficked to the cell surface. As expected, we did not detect Fl-Bgt staining in cells expressing α7 subunits without Ric-3 (Fig. 5B), conditions in which few to no BgtRs are assembled (Fig. 1A). At low levels of Ric-3, some of the intracellular Fl-Bgt staining coincided with the ER marker (Fig. 5D), just like the staining with anti-α7 Ab (Fig. 5C). These Fl-Bgt-stained receptors were diffuse and of relatively weak intensity, consistent with a pool of intracellular BgtRs that are still assembling in the ER. In addition, Fl-Bgt stained punctate structures throughout the cytoplasm (Fig. 5D,G). The size and shape of the punctate structures (diameter, 0.3–0.7 mm, n = 20) were consistent with transport vesicles, in which the BgtRs had a higher fluorescence intensity (2.9 ± 0.3-fold, n = 7 cells, 32 puncta) than the Fl-Bgt in the ER and, thus, 3-fold higher density (Fig. 5D). There was little colocalization between the punctate Fl-Bgt-stained structures and CFP-Ric-3, the ER (Fig. 5D), or Golgi markers (data not shown), consistent with these BgtR-containing structures being transport vesicles released from the ER and/or Golgi with BgtRs at a higher density.
To further test whether the Bgt-stained puncta had features expected of transport vesicles, we analyzed whether the Bgt-stained puncta correlated with the surface delivery of BgtRs as a function of varying Ric-3 levels. In Figure 5G, we plotted the number of sorted cells with four or more Bgt-stained puncta at the different levels of Ric-3. Significant numbers of cells with Bgt-stained puncta were only observed in cells sorted for surface Fl-Bgt staining (Fl-Bgt+) at the different Ric-3 levels. Puncta were not observed in the absence of Ric-3 expression or for cells with no significant surface Bgt staining and positive for CFP-Ric-3 fluorescence. We previously reported the presence of Bgt-stained puncta in cells with α7/5HT3 BgtRs (Drisdel et al., 2004). Examples of these α7/5HT3 BgtR puncta are also displayed in supplemental Figure 4, A and B (available at www.jneurosci.org as supplemental material). In contrast to α7 BgtRs, α7/5HT3 BgtRs form and traffic to the cell surface very efficiently in the absence of Ric-3. Additionally, the surface and intracellular levels of α7/5HT3 BgtRs are much higher than that obtained with α7 BgtRs and low levels of Ric-3 do not affect the surface expression of α7/5HT3 BgtRs (Fig. 1B). We performed cell sorting and immunostaining experiments in which α7/5HT3 was transfected with or without low levels of CFP-Ric-3 (supplemental Fig. 4A,B, available at www.jneurosci.org as supplemental material). We found that in the absence or presence of low levels of CFP-Ric-3, virtually every cell contained Bgt-stained puncta. These findings are different from that with α7 BgtRs, where no puncta were observed in the absence of Ric-3 and at best only ∼40% of the cells had Bgt-stained puncta (Fig. 5G). Thus, for both α7 and α7/5HT3 BgtRs, the number of cells positive for Bgt-stained puncta correlates well with the levels of surface BgtR expression, another indication that the puncta are transport vesicles. Altogether the data are consistent with Ric-3, at low levels, facilitating the formation of BgtRs in the ER. Ric-3 may also help mediate the entry and increased density of BgtRs at ER release sites, where their exit in transport vesicles occurs. However, Ric-3 does not appear to enter the vesicles with the BgtRs as they traffic to the cell surface.
We also examined how high levels of Ric-3 altered BgtR localization (Fig. 5E,F). A significant change observed with high Ric-3 levels was the loss of surface delivery of BgtRs (Figs. 1 and 2) and loss of the BgtR-containing small puncta observed with low Ric-3 levels (Fig. 5G). Many of the aggregated structures observed with high levels of Ric-3 were stained with anti-α7 Abs (Fig. 5E) and Fl-Bgt (Fig. 5F). We observed a similar change in the distribution of α7/5HT3 BgtRs in the presence of high levels of Ric-3 (supplemental Fig. 4C, available at www.jneurosci.org as supplemental material). In addition to the aggregates, we observed significant levels of anti-α7 Ab and Fl-Bgt staining diffusely colocalized with Ric-3 in the ER of these cells. We estimated the total cellular Fl-Bgt and that portion that colocalized with the aggregates 36 h after transfection when surface 125I-Bgt binding peaked. At this time, we found that 67% of BgtRs were in the aggregates and 33% of BgtRs were not aggregated and were diffusely distributed with Ric-3 in the ER. One possibility is that BgtRs associate with aggregated Ric-3 and this association retains BgtRs in the ER, reducing the amount of BgtRs available for surface transport. Because at high Ric-3 levels so few of the intracellular BgtRs arrive on the cell surface (∼10% of α7 BgtRs) (Fig. 1), despite a significant pool of diffuse BgtRs, another possibility is that the diffuse Ric-3, not the aggregated Ric-3, retains BgtRs in the ER. Many of the aggregated structures that contain Ric-3 and α7 subunits do not colocalize with the ER marker (Fig. 4D,E), consistent with the structures moving out of the ER. These data suggest that Ric-3 at high levels prevents BgtRs assembled in the ER from entering transport vesicles and retains BgtRs in the ER. Retained BgtRs complexed with Ric-3 subsequently aggregate and the aggregates somehow depart the ER.
Native Ric-3 in PC12 cells
To further investigate the relationship between Ric-3 levels and BgtR expression, we assayed Ric-3 in cells with endogenous Ric-3 and BgtRs. We first examined the rat pheochromocytoma cell line, PC12 (N21 variant) (Blumenthal et al., 1997; Rangwala et al., 1997), to assay the levels of endogenous Ric-3 and compare its distribution relative to that of endogenous α7 subunits. The levels of α7 BgtRs on the surface of PC12 cells (∼0.15 pmol/mg; data not shown) were somewhat less than the peak levels we observed in HEK cells coexpressed with α7 subunit and Ric-3 plasmids (Fig. 1A). Ric-3 mRNA is found in PC12 cells (Williams et al., 2005), but we were unable to detect Ric-3 by immunoblot analysis with similar levels of total cellular protein (data not shown). This finding indicates that the levels of Ric-3 in PC12 cells are less than what we obtained with heterologous expression in the range of 0.05–0.01 μg of Ric-3 plasmid (Fig. 1C). To confirm the presence of Ric-3 mRNA in our PC12 cells, we performed RT-PCR and observed a band at the expected size for rat Ric-3 (data not shown). We observed immunostaining of the PC12 cells with Ric-3-specific Abs (Fig. 6A) not observed with a preimmune serum control (data not shown). The pattern and levels of staining were similar to that observed in 20% of HEK cells with low levels of Ric-3 (Fig. 2E,H), consistent with the presence of the Ric-3 protein in the PC12 cells. As assayed by immunofluorescence, Ric-3 was present in virtually all PC12 cells (Fig. 6A), colocalized with α7 subunits, and varied in expression levels over a twofold range (Fig. 6B). The ability of the Ric-3 Ab to stain the PC12 cells but not the corresponding immunoblots suggests that the Ric-3 Ab affinity is dependent on the epitope conformation.
Figure 6.

Native Ric-3 in PC12 cells. A, PC12 cells were immunostained with Abs specific for Ric-3 (left) and anti-α7 subunits (right). B, Relation between intracellular and surface BgtR levels as determined by Fl-Bgt labeling (y axis) and Ric-3 levels as determined by staining with Ric-3-specific Abs (x axis). C, PC12 cells were labeled with Fl-Bgt on the cell surface, permeabilized, then stained using the anti-Ric-3 Ab. D, Staining with anti-Ric-3, anti-α7, and anti-PDI (ER marker) Abs. E, Intracellular Fl-Bgt staining of PC12 cells together with anti-Ric-3 and anti-PDI (ER marker) Ab staining. Scale bars, 5 μm.
Based on several observations, the levels and distribution of Ric-3 in PC12 cells were similar to that seen in HEK cells expressing low levels of Ric-3. First, increasing levels of Ric-3 correlated with increased levels of surface and internal BgtRs as assayed by Fl-Bgt staining (Fig. 6B), similar to what we observed with low levels of Ric-3, but not for high levels of Ric-3, in the HEK cells (Fig. 1A). As observed in the HEK cells, none of the Ric-3 staining in the PC12 cells colocalized with the surface Fl-Bgt staining (Fig. 6C). Second, Ric-3 had a diffuse, perinuclear distribution in the PC12 cells that was highly colocalized with ER markers as well as with α7 subunits (Fig. 6D), consistent with an ER localization. None of the Ric-3 aggregation observed with high levels of Ric-3 in HEK cells was observed in the PC12 cells. Third, with Fl-Bgt intracellular staining, we also observed two pools of intracellular BgtRs (Fig. 6E), similar to that observed in HEK cells. The first pool was diffuse, of weaker intensity and highly colocalized with Ric-3 staining. The second pool was in puncta with the features of transport vesicles, in that the Fl-Bgt staining did not colocalize with Ric-3 or ER staining and had a threefold higher fluorescence intensity than the first pool (3.2 ± 0.3-fold, n = 10 cells, 61 puncta) (Fig. 6E).
Ric-3 and α7 subunits in neurons
We also examined native Ric-3 in primary cultures of rat hippocampal neurons and compared its distribution to that of α7 subunits. Previous studies found that 60–100% of the GABAergic interneurons in neuronal cultures contain α7 subunits (Liu et al., 2001; Kawai et al., 2002), which are 7–15% of the total neurons. These finding are consistent with results from slices and sections taken from adult hippocampi where it was observed that 125I-Bgt binding sites and functional BgtRs are found predominantly on GABAergic interneurons (Jones and Yakel, 1997; Frazier et al., 1998; Albuquerque et al., 2009). We observed a similar distribution of α7 subunits and GABAergic neurons in our cultures using staining for GAD (Table 1). As shown in Figure 7A and Table 1, there is a strong correspondence between neurons that stain for Fl-Bgt, Ric-3, and anti-GAD. Furthermore, like α7 subunits (Kawai et al., 2002; Xu et al., 2006), Ric-3 is restricted to somata and dendrites, as shown by its colocalization with MAP2, and is not found in axons, as shown by a lack of colocalization with NFH in axons (Fig. 7B). As in PC12 cells, Ric-3 closely colocalized with the ER marker. In somata (Fig. 7C), the staining was similar to the perinuclear distribution seen in PC12 cells and HEK cells with low levels of Ric-3. There was also a high degree of colocalization between Ric-3 and α7 (Fig. 7D) in the ER as in PC12 and HEK cells. Much of the Fl-Bgt in somata was diffuse and colocalized with Ric-3 (Fig. 7E) in the ER. Fl-Bgt puncta with threefold higher fluorescence intensity (3.3 ± 0.3-fold, n = 6 cells, 27 puncta) (Fig. 7E, inset) were also observed. There may have been some colocalization between the Fl-Bgt puncta and Ric-3 in the perinuclear region, but we believe that this was caused by the close proximity of the Fl-Bgt puncta, which appear to be transport vesicles. Fl-Bgt puncta that we observed further from the ER in somata clearly showed no colocalization with Ric-3 (Fig. 7E, inset).
Table 1.
Distribution of Ric-3, α7, and GAD in cultured hippocampal neurons
| Ab epitope | Anti-GAD | Anti-Ric-3 | Anti-α7 |
|---|---|---|---|
| Total neurons | 12 ± 2% (1279 neurons) | 17 ± 0.7% (1129 neurons) | 16 ± 1% (833 neurons) |
| GAD + neurons | 100% | 92 ± 3% (126 neurons) | 90 ± 8% (89 neurons) |
| Ric-3 + neurons | 84 ± 3% (140 neurons) | 100% | 92 ± 5% (87 neurons) |
| α7 + neurons | 79 ± 6% (107 neurons) | 97 ± 2% (81 neurons) | 100% |
n = 3 experiments.
Figure 7.
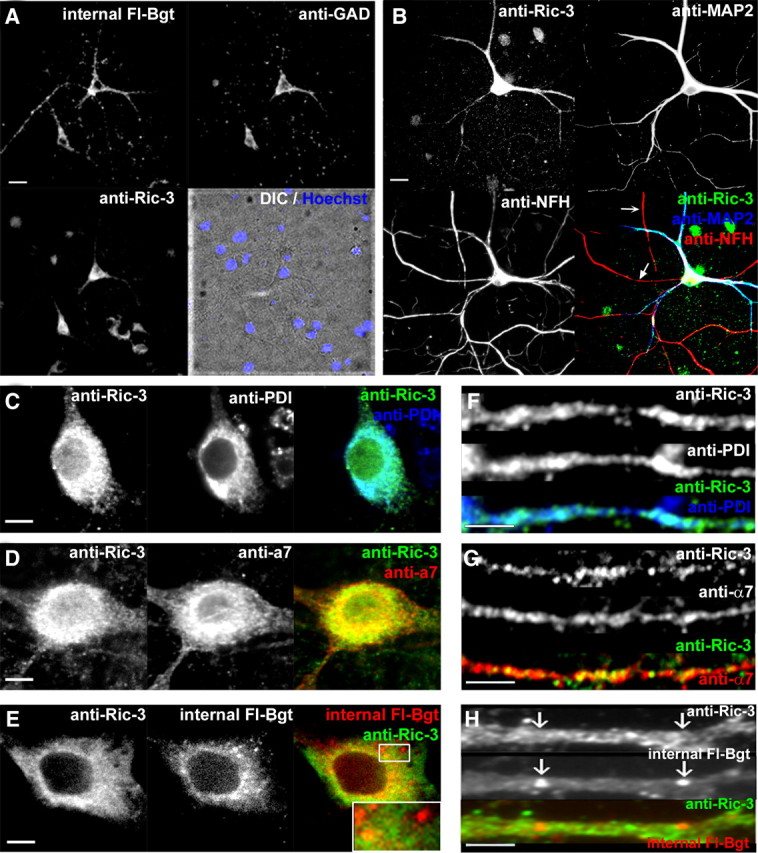
Native Ric-3 and α7 subunits in hippocampal neurons. Cultured hippocampal neurons (18–24 d in vitro) were stained to examine the distribution of Ric-3, α7 subunits, Bgt binding sites, and ER structures. A, Cultured neurons were stained with Fl-Bgt to internal sites (upper left), anti-GAD Ab (upper right), and anti-Ric-3 Ab (lower left). Lower right, Stained nuclei overlaid with a DIC image. B, Cultured neurons were stained with anti-Ric-3 Ab (upper left), anti-MAP2 Ab (upper right), and anti-NFH chain Ab (lower left). Lower right, Merged image. C, Somata were stained with anti-Ric-3 Ab (left) and the ER marker (anti-PDI Ab; middle). Right, Merged image. D, Somata were stained with anti-Ric-3 Ab (left) and anti-α7 Ab (middle). Right, Merged image. E, Somata stained with anti-Ric-3 Ab (left) and stained with Fl-Bgt to internal sites (middle). Right, Merged image. Examples of bright Fl-Bgt-stained puncta are highlighted in the box and magnified in the inset. F, Dendrites were stained with anti-Ric-3 Ab (top) and the ER marker (anti-PDI Ab; middle). Bottom, Merged image. G, Dendrites were stained with anti-Ric-3 Ab (top) and anti-α7 Abs (middle). Bottom, Merged image. H, Dendrites were stained with anti-Ric-3 Ab (top) and with Fl-Bgt to internal sites (middle). Bottom, Merged image. Scale bars, A, B, 50 μm; C–E, G–I, 5 μm.
As with PC12 cells, the expression levels of Ric-3 in the somata of the neurons varied over a twofold range. Levels of Ric-3 in neurons were significantly higher than in PC12 cells, yet there were no signs of Ric-3 aggregation as was observed with higher levels in HEK cells. Compared with the levels of Ric-3 in PC12 cells (1.0 ± 0.06-fold, n = 67 cells), Ric-3 levels in neurons were approximately threefold higher (2.8 ± 0.11-fold, n = 94 cells, p < 0.001). Because Ric-3 is restricted to the ER as in HEK and PC12 cells, we were initially surprised by the levels of Ric-3 in the dendrites. However, in dendrites, Ric-3 colocalizes with the ER marker in the shafts (Fig. 7F), consistent with Ric-3 present in the ER subcompartment that exists throughout dendrites (Bannai et al., 2004; Mironov and Symonchuk, 2006). α7 subunits are found throughout the dendrites and colocalized with Ric-3 (Fig. 7G) in the ER subcompartment. To label intracellular receptors with Fl-Bgt, surface BgtRs were blocked with unlabeled Bgt. We found brightly stained Fl-Bgt puncta throughout the dendrites (Fig. 7H), which did not colocalize with Ric-3 or the ER marker. There was also weak and diffuse Fl-Bgt staining in the shafts that appeared to colocalize with Ric-3.
To better visualize Ric-3 in neurons, we transfected neurons with CFP-Ric-3 (Fig. 8). Most transfections of CFP-Ric-3 did not significantly increase total Ric-3 above normal levels, and CFP-Ric-3 showed a diffuse reticulated pattern in the somata similar to endogenous Ric-3 and colocalized in the ER (Fig. 8A, left). When CFP-Ric-3 was transfected at higher levels, we occasionally observed aggregates indicating that we were overexpressing Ric-3 as observed in HEK at high levels (Fig. 8A, right). At the lower levels, CFP-Ric-3 was distributed in the somata and dendrites but not in axons (Fig. 8B), indicating that at these levels CFP-Ric-3 had the expected distribution. Furthermore, transfection of CFP-Ric-3 did not significantly interfere with the assembly and surface transport of native BgtRs, as shown by strong surface Fl-Bgt staining of endogenous BgtRs in transfected neurons (Fig. 8C). As observed in PC12 and HEK cells, none of the Ric-3 colocalized with surface Fl-Bgt staining (Fig. 8C, inset).
Figure 8.
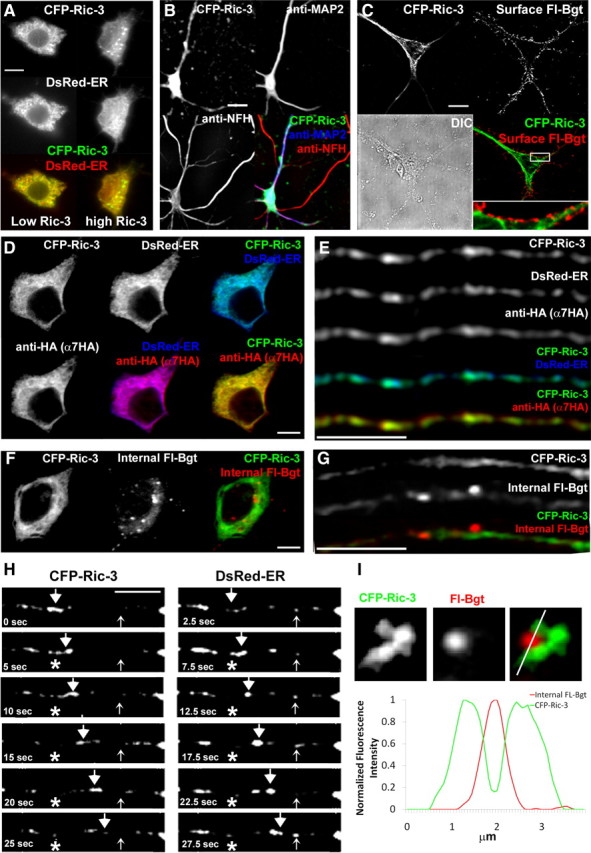
Transfected Ric-3 and α7 subunits in hippocampal neurons. Cultured hippocampal neurons transfected with the CFP-Ric-3, DsRed-ER, and epitope-tagged α7 constructs. A, Somata imaged for CFP-Ric-3 (top) and the ER marker, DsRed-ER (middle). Bottom, Merged image. Left, Soma transfected with low levels of CFP-Ric-3; right, soma transfected with high levels of CFP-Ric-3 (right). B, Transfected neurons imaged for CFP-Ric-3 (upper left), anti-MAP2 Ab (upper right), and anti-NFH Ab (lower left). Lower right, Merged image. Image contrast has been enhanced to show distribution of CFP-Ric-3 in dendrites, which results in overexposed somata. C, Transfected neurons imaged for CFP-Ric-3 (upper left) and surface FL-Bgt (upper right). Lower right, Merged image; lower left, DIC image. D, A soma from a transfected neuron imaged for CFP-Ric-3 (upper left), DsRed-ER (upper middle), and α7-HA subunits (lower left). Upper right, lower middle, and lower right, Corresponding merged images, as indicated. E, A dendrite from a transfected neuron imaged for CFP-Ric-3 (top row), DsRed-ER (second row), and α7-HA subunits (third row). Fourth and fifth rows, Merged images, as indicated. F, A soma from a transfected neuron imaged for CFP-Ric-3 (left) and internal Fl-Bgt (middle). The merged image is in the right panel. G, A dendrite from a transfected neuron imaged for CFP-Ric-3 (top) and internal Fl-Bgt (middle). Bottom, Merged image. H, Time lapse images of CFP-Ric-3 and ER-DsRed transfected neurons showing an example of a fast moving puncta (large arrowheads) and static structures (small arrowheads) that contain both CFP-Ric-3 and ER-DsRed. The asterisk denotes the start location of the moving puncta. Images acquired alternated between the CFP channel and the DsRed channel with 2.5 s between acquisitions, starting with CFP-Ric-3 at time 0. I, Top, An example of an ER subcompartment structure imaged with CFP-Ric-3 (right) and a bright internal Fl-Bgt stained puncta (middle) in a dendrite. Right, Merged image. Bottom, A line profile analysis of the Fl-Bgt (red) and CFP-Ric-3 (green) structures illustrating the Fl-Bgt puncta juxtaposed to the CFP-Ric-3 structure. Scale bars, A–C,10 μm; D–H, 5 μm.
In the somata, CFP-Ric-3 colocalized with transfected α7-HA subunits in the ER labeled by DsRed-ER (Fig. 8D). We only analyzed transfected GABAergic interneurons by anti-GAD immunostaining (data not shown). In dendrites, CFP-Ric-3, α7-HA subunits, and the ER subcompartment labeled with DsRed-ER were better resolved than endogenous α7 subunits (Fig. 8E). We determined the degree of colocalization and the coefficients of colocalization, r, and found that α7 subunits and Ric-3 in dendrites were highly colocalized with the ER subcompartment, as was observed with endogenous Ric-3. Eighty-three percent of the ER marker pixels colocalized with α7 subunits (r = 0.74 ± 0.03, n = 10 neurons) and 80% with CFP-Ric-3 (r = 0.67 ± 0.04, n = 12 neurons), indicating strong colocalization. There was also strong colocalization between α7 and CFP-Ric-3 (r = 0.71 ± 0.04, n = 13 neurons). When we stained for intracellular BgtRs, we observed two distributions of Fl-Bgt staining in the somata similar to what we observed in HEK and PC12 cells: a diffuse pool that colocalized with CFP-Ric-3 and bright Fl-Bgt puncta (Fig. 8F). In the dendrites, we clearly resolved two different types of staining that contained α7 subunits. The first type contained α7-HA subunits, CFP-Ric-3, DsRed-ER, and either no staining or light staining with Fl-Bgt (Figs. 8E,G). This type of staining, while generally diffuse and reticulated as in the somata, also consisted of smaller puncta (Fig. 8H). The second type of α7 subunit structures in dendrites, although fewer in number than the first type, contained Fl-Bgt at higher intensity but no Ric-3 or DsRed-ER (Fig. 8G). These structures were similar in appearance to the Fl-Bgt transport vesicles found in the somata (Fig. 8F). We often observed these structures in close apposition to ER subcompartment membranes that contained Ric-3 (Fig. 8I) and α7 subunits (data not shown), which suggests that the BgtR containing puncta budded from the ER subcompartment membranes.
Why do Ric-3 and α7 subunits colocalize to the ER subcompartment of dendrites? One possibility is that Ric-3 and/or α7 subunit mRNA are transported to sites in dendrites where they are translated via local protein synthesis (Martin and Zukin, 2006) and synthesized at the ER subcompartment. To test for this, we transfected cultured neurons with different combinations of the plasmids for CFP-Ric-3, hemagglutinin (HA)-tagged α7 subunits (α7-HA), and the ER marker, DsRed-ER, which colocalizes well with the dendritic ER subcompartment (Bannai et al., 2004). Previous studies of transfected α7 subunits (Xu et al., 2006) and DsRed-ER (Bannai et al., 2004) found that their basic properties in terms of their expected distribution in the neurons were not altered. Virtually all of the transfected CFP-Ric-3, α7-HA subunits, and DsRed-ER appeared first in the somata before we observed their staining in the dendrites (data not shown). This finding does not rule out the possibility that some local dendritic synthesis is occurring but does indicate that the bulk of these proteins is synthesized in the somatic ER and then transported out to the dendrites.
Another possible explanation for why Ric-3 and α7 subunits colocalize to the ER subcompartment of dendrites is that ER subcompartment is the compartment in which α7 subunits are trafficked along dendrites. The ER subcompartment in dendrites has a dedicated transport system composed of small vesicles that traffic proteins synthesized in the somatic ER to the dendritic ER subcompartment (Bannai et al., 2004; Mironov and Symonchuk, 2006). NMDA-type glutamate receptors (NMDARs) are trafficked via this pathway and bypass the Golgi in somata if associated with SAP97 (Jeyifous et al., 2009). Thus, Ric-3 could play a similar role as SAP97 and α7 subunits could be trafficked from the ER in the soma via the ER subcompartment in dendrites when associated with Ric-3. To test for this possibility, we performed additional experiments to determine whether α7 subunits, when complexed with Ric-3, are trafficked from the ER in the soma via the ER subcompartment in dendrites. If Ric-3 is synthesized in the somatic ER and trafficked to dendrites via the ER subcompartment, we would predict that Ric-3, which is highly colocalized with α7 subunits in dendrites, should be observed in rapidly moving puncta (0.2–0.3 μm/s) (Bannai et al., 2004). We would also predict that the Ric-3 puncta should contain ER markers as previously observed (Bannai et al., 2004). To test these predictions, we performed time-lapse live imaging of neurons transfected with CFP-Ric-3 and the fluorescent ER marker DsRed-ER. A large number of the puncta containing both constructs was seen rapidly moving (0.22 ± 0.04 μm/s, n = 17 puncta), as shown in Figure 8H. The high degree of colocalization between CFP-Ric-3, ER-DsRed, and α7HA immunostaining strongly suggests that the moving CFP-Ric-3 and ER-DsRed puncta contain α7 subunits consistent with their transport via the ER subcompartment. Another prediction is that changes in Ric-3 levels in the neurons should alter the trafficking of α7 subunits from the soma to dendrites. Ric-3 knockdown experiments would be difficult to interpret because at levels below detection by immunoblotting and immunofluorescence (Figs. 1 and 2), Ric-3 promotes BgtR assembly and release from the ER.
Discussion
In this study, we addressed how Ric-3 functions with respect to α7 subunits. The initial genetic screen and characterization of Ric-3 mutations in C. elegans indicated that Ric-3 had a role in nAChR assembly and/or trafficking (Halevi et al., 2002). The introduction of Ric-3 into cultured mammalian cells or Xenopus oocytes altered cell surface and total expression of 5HT3 receptors and different nicotinic receptor subtypes but not other receptors such as AMPA receptors (Halevi et al., 2002, 2003; Castillo et al., 2005). Ric-3 either promoted (Cheng et al., 2005, 2007) or inhibited (Halevi et al., 2003) 5HT3A receptor surface levels. Results coexpressing Ric-3 with α4β2 nAChRs were also inconsistent. One study found that Ric-3 decreased whole-cell currents (Halevi et al., 2003) and another found that Ric-3 increased whole-cell epibatidine binding to nAChRs (Lansdell et al., 2005). The introduction of Ric-3 consistently enhanced surface expression of α7-containing BgtRs in a variety of expression systems (Halevi et al., 2002, 2003; Ben-Ami et al., 2005; Castillo et al., 2005, 2006; Lansdell et al., 2005; Williams et al., 2005; Wang et al., 2009), although the degree of the enhancement was variable. These discrepancies can be explained by our findings that Ric-3's function changes dependent on Ric-3 levels. In cases where the number of cell-surface receptors increased, our data indicate that Ric-3 levels were low enough that Ric-3 increased BgtR formation and ER release. In cases with decreased cell-surface expression, levels of Ric-3 high enough to cause receptor ER retention explain the results. In all studies, surface expression of α7-containing BgtRs was enhanced by Ric-3 because the number of surface BgtRs was not significant in the absence of Ric-3. Thus, even at the highest Ric-3 levels there was a net increase in the number of surface α7-containing BgtRs (Fig. 1A).
Differences were also reported in studies examining where Ric-3 resides in cells. Most studies found Ric-3 predominantly in the ER (Castillo et al., 2005; Cheng et al., 2005, 2007; Wang et al., 2009), where it is synthesized. However, Ric-3 has also been reported to colocalize with nAChRs at the cell surface (Williams et al., 2005) and in the Golgi apparatus (Castillo et al., 2005; Cheng et al., 2007). When we transfected low levels of Ric-3 into HEK cells, Ric-3 colocalized with markers of the ER but did not colocalize with markers of the Golgi apparatus or at the cell surface. Similar results were obtained for endogenous Ric-3 in PC12 cells and cultured neurons. In these cells, native Ric-3 again was localized to the ER and not to the Golgi apparatus or cell surface. In neurons, the localization in the ER extended from the soma into the dendrites, where we observed that Ric-3 trafficked in transport vesicles. These vesicles appeared to be a part of the ER subcompartment of dendrites (Spacek and Harris, 1997; Toresson and Grant, 2005) and did not localize to Golgi outposts or the cell surface. Although we only found Ric-3 in the ER, it is still possible that Ric-3 traffics out of the ER to other compartments in the secretory pathway, but such departures must be rapid under normal circumstances.
We initially thought that the ER retention of α7 and α7/5HT3 subunits observed at high Ric-3 levels in HEK cells was an artifact caused by overexpression. We based this conclusion on the correlation between ER retention and aggregation and the finding that aggregation was not observed for endogenous Ric-3 in PC12 cells and neurons. As discussed in more detail below, subsequent observations suggested that native Ric-3 in cultured neurons can act to retain α7 subunits in the ER and this retention occurs without aggregates forming. The lack of Ric-3 aggregates in the neurons indicates that it is the nonaggregated, or free, Ric-3 that mediates subunit ER retention. ER retention of the subunits appears to result from a change in the nature of the interaction between Ric-3 and the subunits transitioning from a transient, short-lived interaction at low levels of Ric-3 to a more stable, long-lived interaction at higher levels. This is suggested by the ability to coimmunoprecipitate Ric-3 and the subunits at the higher Ric-3 levels and the extremely low levels of Ric-3 that increased BgtR formation and trafficking to the surface (Fig. 1). Because Ric-3 is apparently an ER resident protein, long-lived interactions with Ric-3 could act to prevent interactions with COPII complex proteins that mediate release of membrane proteins from the ER. Ric-3 aggregates would form as Ric-3-subunit complexes accumulate in the ER and may involve a loss of ER-associated degradation (ERAD). The large, aggregated Ric-3 structures we observed outside the ER of HEK cells with or without the subunits have the appearance of autophagosomes that can form when ERAD is overwhelmed and somehow depart the ER (Kruse et al., 2006).
Ric-3 trafficked differently in neurons than in PC12 or HEK cells. In cultured neurons, Ric-3, along with α7 subunits, moved rapidly in puncta with the features of transport vesicles previously found to be part of the ER subcompartment in dendrites (Bannai et al., 2004). Ric-3-containing puncta were not observed in PC12 or HEK cells and Ric-3 was clearly absent from the α7 subunit-containing puncta stained by Fl-Bgt that appeared to be transporting mature BgtRs to the surface of PC12 and HEK cells. Bgt-stained puncta that lacked Ric-3 were also present in the somata and dendrites of neurons. Our finding that significantly higher levels of native Ric-3 in the somata of neurons than in PC12 cells, together with the colocalization of Ric-3 and α7 subunits in moving puncta that are part of the ER subcompartment of dendrites, suggests that the events depicted in Figure 9 are occurring in neurons. In the model, Ric-3 has two separate functions, regulated by the levels of Ric-3, available to interact with α7 subunits. At low levels (Fig. 9A,C), Ric-3 acts only to mediate BgtR folding and assembly. This appears to be how Ric-3 functions in PC12 cells and in neurons when BgtRs are released from the ER in the soma and traffic via the somatic Golgi to the cell membrane. At higher levels in neurons, Ric-3 retains α7 subunits in the ER in addition to mediating BgtR folding and assembly (Fig. 9B). When restricted to the ER, complexes of Ric-3 and α7 subunits together remain within the ER subcompartment and are transported on moving puncta in dendrites (Fig. 9C, red arrow pathway). We recently determined that NMDARs in hippocampal neurons are also trafficked through such an ER-restricted pathway in dendrites (Jeyifous et al., 2009). Transport of NMDARs through the ER subcompartment requires the presence of SAP97, which associates specifically with NMDAR NR2B subunits and, like Ric-3, acts to retain its binding partner in the ER of HEK cells. Thus, Ric-3 may have a role similar to SAP97 in retaining its binding partner in the ER subcompartment. In neurons, the ER-restricted pathway taken by NMDARs sorts NMDARs from AMPA-type glutamate receptors, which are trafficked through the conventional secretory pathway in the soma (Jeyifous et al., 2009). After departing the ER subcompartment, NMDA receptors traffic through Golgi outposts near spines (Jeyifous et al., 2009). Similarly, we assume that BgtRs are released from the ER subcompartment and traffic through Golgi outposts before inserting in the plasma membrane (Fig. 9C). Consistent with this idea are the BgtR-containing puncta we observed in close opposition to the ER subcompartment membranes in the dendrites (Fig. 8I). These puncta may traffic via Golgi outposts to the dendritic surface after release from the ER subcompartment membranes.
Figure 9.

Model of how Ric-3 function is altered by changes in Ric-3 levels in neurons. A, At low levels, Ric-3 interactions with α7 subunits are short-lived and mediate BgtR assembly and release from the ER. B, At higher levels, Ric-3 interactions with α7 subunits are long lived and retain BgtRs in the ER in addition to promoting BgtR assembly. C, The BgtR trafficking pathway is determined by the type of Ric-3 interaction with α7 subunits. At low levels, short-lived interactions with Ric-3 result in ER release of BgtRs in the soma and BgtRs traffic through the conventional secretory pathway through the ER-Golgi intermediate compartment (ERGIC), Golgi apparatus, and trans-Golgi network (TGN) in the soma to the plasma membrane (blue arrow pathway). At high levels, long-lived interactions with Ric-3 result in ER retention of BgtRs in the soma and BgtRs traffic through the atypical secretory pathway in transport vesicles that are part of the ER subcompartment in dendrites to Golgi outposts and then to the plasma membrane in dendrites (red arrow pathway).
As shown previously (Kawai et al., 2002; Xu et al., 2006), BgtRs are found on the somata and dendrites of cultured hippocampal neurons and do not traffic to axons. However, there is much evidence that BgtRs do traffic to axons in vivo and, in particular, to axon terminals where their activation can regulate the presynaptic release of neurotransmitters (Gray et al., 1996; Fabian-Fine et al., 2001). In cultured hippocampal neurons, BgtR axonal transport may be prevented in part by the relatively high levels of Ric-3 that restrict α7 subunits to the dendritic ER subcompartment. If true, mechanisms should exist to regulate Ric-3 levels in cells. Recent work indicates that levels of Ric-3 are tightly regulated by the ubiquitin proteosome system in C. elegans (Shteingauz et al., 2009). Mechanisms may also exist that regulate Ric-3 interactions with α7 subunits, thereby determining when BgtR is released from the ER subcompartment in dendrites. An example of other membrane proteins regulated this way is the ER retention of the proteins that mediate cholesterol homeostasis. The release of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG CoA) reductase and other proteins involved in cholesterol homeostasis from the ER is regulated by sterol levels (Espenshade and Hughes, 2007; Sun et al., 2007). Ric-3 and the proteins that regulate α7 subunit assembly and trafficking may have a similar role to the proteins SCAP and Insig-1 and -2 that regulate HMG CoA reductase and sterol regulatory element binding-protein assembly and trafficking.
Footnotes
This work was supported by grants from the Ministry of Science and Innovation of Spain (BFU2008-02160 and CSD2008-00005; The Spanish Ion Channel Initiative-Consolider Ingenio 2010) (to M.C.) and National Institutes of Health (NIH) Grants NS043782 and DA019695 and the Peter F. McManus Foundation (to W.N.G.). A.V.K. was supported by NIH Grant DA11643A, awarded to R. E. Oswald and G. P. Hess (Cornell University). We thank Okunola Jeyifous for comments on the manuscript, members of the Green Lab for discussion and comments on the work, and Dr. Vytas Bindokas for help with the microscopy.
References
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannai H, Inoue T, Nakayama T, Hattori M, Mikoshiba K. Kinesin dependent, rapid, bi-directional transport of ER subcompartment in dendrites of hippocampal neurons. J Cell Sci. 2004;117:163–175. doi: 10.1242/jcs.00854. [DOI] [PubMed] [Google Scholar]
- Ben-Ami HC, Yassin L, Farah H, Michaeli A, Eshel M, Treinin M. RIC-3 affects properties and quantity of nicotinic acetylcholine receptors via a mechanism that does not require the coiled-coil domains. J Biol Chem. 2005;280:28053–28060. doi: 10.1074/jbc.M504369200. [DOI] [PubMed] [Google Scholar]
- Betz H, Graham D, Rehm H. Identification of polypeptides associated with a putative neuronal nicotinic acetylcholine receptor. J Biol Chem. 1982;257:11390–11394. [PubMed] [Google Scholar]
- Blumenthal EM, Conroy WG, Romano SJ, Kassner PD, Berg DK. Detection of functional nicotinic receptors blocked by alpha-bungarotoxin on PC12 cells and dependence of their expression on post-translational events. J Neurosci. 1997;17:6094–6104. doi: 10.1523/JNEUROSCI.17-16-06094.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelán F, Castillo M, Mulet J, Sala S, Sala F, Domínguez Del Toro E, Criado M. Molecular characterization and localization of the RIC-3 protein, an effector of nicotinic acetylcholine receptor expression. J Neurochem. 2008;105:617–627. doi: 10.1111/j.1471-4159.2007.05169.x. [DOI] [PubMed] [Google Scholar]
- Castillo M, Mulet J, Gutiérrez LM, Ortiz JA, Castelán F, Gerber S, Sala S, Sala F, Criado M. Dual role of the RIC-3 protein in trafficking of serotonin and nicotinic acetylcholine receptors. J Biol Chem. 2005;280:27062–27068. doi: 10.1074/jbc.M503746200. [DOI] [PubMed] [Google Scholar]
- Castillo M, Mulet J, Gutiérrez LM, Ortiz JA, Castelán F, Gerber S, Sala S, Sala F, Criado M. Role of the RIC-3 protein in trafficking of serotonin and nicotinic acetylcholine receptors. J Mol Neurosci. 2006;30:153–156. doi: 10.1385/JMN:30:1:153. [DOI] [PubMed] [Google Scholar]
- Chen D, Patrick JW. The alpha-bungarotoxin-binding nicotinic acetylcholine receptor from rat brain contains only the alpha7 subunit. J Biol Chem. 1997;272:24024–24029. doi: 10.1074/jbc.272.38.24024. [DOI] [PubMed] [Google Scholar]
- Cheng A, McDonald NA, Connolly CN. Cell surface expression of 5-hydroxytryptamine type 3 receptors is promoted by RIC-3. J Biol Chem. 2005;280:22502–22507. doi: 10.1074/jbc.M414341200. [DOI] [PubMed] [Google Scholar]
- Cheng A, Bollan KA, Greenwood SM, Irving AJ, Connolly CN. Differential subcellular localization of RIC-3 isoforms and their role in determining 5-HT3 receptor composition. J Biol Chem. 2007;282:26158–26166. doi: 10.1074/jbc.M703899200. [DOI] [PubMed] [Google Scholar]
- Conti-Tronconi BM, Dunn SM, Barnard EA, Dolly JO, Lai FA, Ray N, Raftery MA. Brain and muscle nicotinic acetylcholine receptors are different but homologous proteins. Proc Natl Acad Sci U S A. 1985;82:5208–5212. doi: 10.1073/pnas.82.15.5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper ST, Millar NS. Host cell-specific folding and assembly of the neuronal nicotinic acetylcholine receptor alpha7 subunit. J Neurochem. 1997;68:2140–2151. doi: 10.1046/j.1471-4159.1997.68052140.x. [DOI] [PubMed] [Google Scholar]
- Drisdel RC, Green WN. Neuronal alpha-bungarotoxin receptors are alpha7 subunit homomers. J Neurosci. 2000;20:133–139. doi: 10.1523/JNEUROSCI.20-01-00133.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drisdel RC, Manzana E, Green WN. The role of palmitoylation in functional expression of nicotinic alpha7 receptors. J Neurosci. 2004;24:10502–10510. doi: 10.1523/JNEUROSCI.3315-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eertmoed AL, Vallejo YF, Green WN. Transient expression of heteromeric ion channels. Methods Enzymol. 1998;293:564–585. doi: 10.1016/s0076-6879(98)93034-8. [DOI] [PubMed] [Google Scholar]
- Eiselé JL, Bertrand S, Galzi JL, Devillers-Thiéry A, Changeux JP, Bertrand D. Chimaeric nicotinic-serotonergic receptor combines distinct ligand binding and channel specificities. Nature. 1993;366:479–483. doi: 10.1038/366479a0. [DOI] [PubMed] [Google Scholar]
- Espenshade PJ, Hughes AL. Regulation of sterol synthesis in eukaryotes. Annu Rev Genet. 2007;41:401–427. doi: 10.1146/annurev.genet.41.110306.130315. [DOI] [PubMed] [Google Scholar]
- Fabian-Fine R, Skehel P, Errington ML, Davies HA, Sher E, Stewart MG, Fine A. Ultrastructural distribution of the alpha7 nicotinic acetylcholine receptor subunit in rat hippocampus. J Neurosci. 2001;21:7993–8003. doi: 10.1523/JNEUROSCI.21-20-07993.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier CJ, Rollins YD, Breese CR, Leonard S, Freedman R, Dunwiddie TV. Acetylcholine activates an alpha-bungarotoxin-sensitive nicotinic current in rat hippocampal interneurons, but not pyramidal cells. J Neurosci. 1998;18:1187–1195. doi: 10.1523/JNEUROSCI.18-04-01187.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grando SA, Horton RM, Mauro TM, Kist DA, Lee TX, Dahl MV. Activation of keratinocyte nicotinic cholinergic receptors stimulates calcium influx and enhances cell differentiation. J Invest Dermatol. 1996;107:412–418. doi: 10.1111/1523-1747.ep12363399. [DOI] [PubMed] [Google Scholar]
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- Halevi S, McKay J, Palfreyman M, Yassin L, Eshel M, Jorgensen E, Treinin M. The C. elegans ric-3 gene is required for maturation of nicotinic acetylcholine receptors. EMBO J. 2002;21:1012–1020. doi: 10.1093/emboj/21.5.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halevi S, Yassin L, Eshel M, Sala F, Sala S, Criado M, Treinin M. Conservation within the RIC-3 gene family: effectors of mammalian nicotinic acetylcholine receptor expression. J Biol Chem. 2003;278:34411–34417. doi: 10.1074/jbc.M300170200. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Jeyifous O, Waites CL, Specht CG, Fujisawa S, Schubert M, Lin EI, Marshall J, Aoki C, de Silva T, Montgomery JM, Garner CC, Green WN. SAP97 and CASK mediate sorting of NMDA receptors through a novel secretory pathway. Nat Neurosci. 2009;12:1011–1019. doi: 10.1038/nn.2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Yakel JL. Functional nicotinic ACh receptors on interneurones in the rat hippocampus. J Physiol. 1997;504:603–610. doi: 10.1111/j.1469-7793.1997.603bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin A, Akabas MH. Toward a structural basis for the function of nicotinic acetylcholine receptors and their cousins. Neuron. 1995;15:1231–1244. doi: 10.1016/0896-6273(95)90004-7. [DOI] [PubMed] [Google Scholar]
- Kawai H, Zago W, Berg DK. Nicotinic alpha 7 receptor clusters on hippocampal GABAergic neurons: regulation by synaptic activity and neurotrophins. J Neurosci. 2002;22:7903–7912. doi: 10.1523/JNEUROSCI.22-18-07903.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp G, Bentley L, McNamee MG, Morley BJ. Purification and characterization of the alpha-bungarotoxin binding protein from rat brain. Brain Res. 1985;347:274–283. doi: 10.1016/0006-8993(85)90187-8. [DOI] [PubMed] [Google Scholar]
- Kruse KB, Brodsky JL, McCracken AA. Autophagy: an ER protein quality control process. Autophagy. 2006;2:135–137. doi: 10.4161/auto.2.2.2388. [DOI] [PubMed] [Google Scholar]
- Lansdell SJ, Gee VJ, Harkness PC, Doward AI, Baker ER, Gibb AJ, Millar NS. RIC-3 enhances functional expression of multiple nicotinic acetylcholine receptor subtypes in mammalian cells. Mol Pharmacol. 2005;68:1431–1438. doi: 10.1124/mol.105.017459. [DOI] [PubMed] [Google Scholar]
- Li Q, Lau A, Morris TJ, Guo L, Fordyce CB, Stanley EF. A syntaxin 1, Galpha(o), and N-type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. J Neurosci. 2004;24:4070–4081. doi: 10.1523/JNEUROSCI.0346-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Ford B, Mann MA, Fischbach GD. Neuregulins increase alpha7 nicotinic acetylcholine receptors and enhance excitatory synaptic transmission in GABAergic interneurons of the hippocampus. J Neurosci. 2001;21:5660–5669. doi: 10.1523/JNEUROSCI.21-15-05660.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci. 1992;103:857–862. doi: 10.1242/jcs.103.3.857. [DOI] [PubMed] [Google Scholar]
- Marks JD, Bindokas VP, Zhang XM. Maturation of vulnerability to excitotoxicity: intracellular mechanisms in cultured postnatal hippocampal neurons. Brain Res Dev Brain Res. 2000;124:101–116. doi: 10.1016/s0165-3806(00)00096-1. [DOI] [PubMed] [Google Scholar]
- Martin KC, Zukin RS. RNA trafficking and local protein synthesis in dendrites: an overview. J Neurosci. 2006;26:7131–7134. doi: 10.1523/JNEUROSCI.1801-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KG, Alfonso A, Nguyen M, Crowell JA, Johnson CD, Rand JB. A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc Natl Acad Sci U S A. 1996;93:12593–12598. doi: 10.1073/pnas.93.22.12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov SL, Symonchuk N. ER vesicles and mitochondria move and communicate at synapses. J Cell Sci. 2006;119:4926–4934. doi: 10.1242/jcs.03254. [DOI] [PubMed] [Google Scholar]
- Nguyen M, Alfonso A, Johnson CD, Rand JB. Caenorhabditis elegans mutants resistant to inhibitors of acetylcholinesterase. Genetics. 1995;140:527–535. doi: 10.1093/genetics/140.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhilin S, Drisdel RC, Sagher D, McGehee DS, Vallejo Y, Green WN. alpha-bungarotoxin receptors contain alpha7 subunits in two different disulfide-bonded conformations. J Cell Biol. 1999;146:203–218. doi: 10.1083/jcb.146.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangwala F, Drisdel RC, Rakhilin S, Ko E, Atluri P, Harkins AB, Fox AP, Salman SS, Green WN. Neuronal alpha-bungarotoxin receptors differ structurally from other nicotinic acetylcholine receptors. J Neurosci. 1997;17:8201–8212. doi: 10.1523/JNEUROSCI.17-21-08201.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband WS. ImageJ. Bethesda, Maryland: National Institutes of Health; 2009. [Google Scholar]
- Sekhon HS, Jia Y, Raab R, Kuryatov A, Pankow JF, Whitsett JA, Lindstrom J, Spindel ER. Prenatal nicotine increases pulmonary alpha7 nicotinic receptor expression and alters fetal lung development in monkeys. J Clin Invest. 1999;103:637–647. doi: 10.1172/JCI5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteingauz A, Cohen E, Biala Y, Treinin M. The BTB-MATH protein BATH-42 interacts with RIC-3 to regulate maturation of nicotinic acetylcholine receptors. J Cell Sci. 2009;122:807–812. doi: 10.1242/jcs.036343. [DOI] [PubMed] [Google Scholar]
- Spacek J, Harris KM. Three-dimensional organization of smooth endoplasmic reticulum in hippocampal CA1 dendrites and dendritic spines of the immature and mature rat. J Neurosci. 1997;17:190–203. doi: 10.1523/JNEUROSCI.17-01-00190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun LP, Seemann J, Goldstein JL, Brown MS. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proc Natl Acad Sci U S A. 2007;104:6519–6526. doi: 10.1073/pnas.0700907104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweileh W, Wenberg K, Xu J, Forsayeth J, Hardy S, Loring RH. Multistep expression and assembly of neuronal nicotinic receptors is both host-cell- and receptor-subtype-dependent. Brain Res Mol Brain Res. 2000;75:293–302. doi: 10.1016/s0169-328x(99)00302-2. [DOI] [PubMed] [Google Scholar]
- Toresson H, Grant SG. Dynamic distribution of endoplasmic reticulum in hippocampal neuron dendritic spines. Eur J Neurosci. 2005;22:1793–1798. doi: 10.1111/j.1460-9568.2005.04342.x. [DOI] [PubMed] [Google Scholar]
- Udgaonkar JB, Hess GP. Chemical kinetic measurements of a mammalian acetylcholine receptor by a fast-reaction technique. Proc Natl Acad Sci U S A. 1987;84:8758–8762. doi: 10.1073/pnas.84.24.8758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yao Y, Tang XQ, Wang ZZ. Mouse RIC-3, an endoplasmic reticulum chaperone, promotes assembly of the alpha7 acetylcholine receptor through a cytoplasmic coiled-coil domain. J Neurosci. 2009;29:12625–12635. doi: 10.1523/JNEUROSCI.1776-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting P, Lindstrom J. Purification and characterization of a nicotinic acetylcholine receptor from rat brain. Proc Natl Acad Sci U S A. 1987;84:595–599. doi: 10.1073/pnas.84.2.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams ME, Burton B, Urrutia A, Shcherbatko A, Chavez-Noriega LE, Cohen CJ, Aiyar J. Ric-3 promotes functional expression of the nicotinic acetylcholine receptor alpha7 subunit in mammalian cells. J Biol Chem. 2005;280:1257–1263. doi: 10.1074/jbc.M410039200. [DOI] [PubMed] [Google Scholar]
- Xu J, Zhu Y, Heinemann SF. Identification of sequence motifs that target neuronal nicotinic receptors to dendrites and axons. J Neurosci. 2006;26:9780–9793. doi: 10.1523/JNEUROSCI.0840-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]