

Abstract
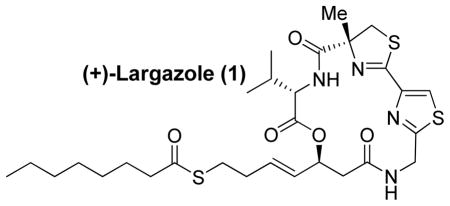
An enantioselective total synthesis of cytotoxic natural product, (+)-largazole (1) is described. It is a potent histone deacetylase inhibitor. Our synthesis is convergent and involves the assembly of thiazole 3-derived carboxylic acid with amino ester 4 followed by cycloamidation of the corresponding amino acid. The synthesis features an efficient cross metathesis, an enzymatic kinetic resolution of a β-hydroxy ester, a selective removal of a Boc-protecting group, a HATU/HOAt-promoted cycloamidation reaction, and synthetic manipulations to a sensitive thioester functional group.
In January 2008, Luesch and co-workers reported the isolation of largazole, a novel 16-membered depsipeptide from Floridian marine cyanobacterium Symploca sp.1 Largazole’s structure was elucidated by extensive NMR studies and through chemical degradation. It has shown impressive growth inhibitory activity of transformed mammary epithelial cells (MDA-MB-231) in a dose dependent manner with a GI50 value of 7.7 nM. In addition, it has shown excellent selectivity over nontransformed murine mammary epithelial cells (NMuMG) with a GI50 of 122 nM. More recently, Luesch, Hong, and co-workers reported the first total synthesis of largazole. Their synthesis featured a macrocyclization at C6 and a late stage addition of the thioester using cross metathesis. Subsequently, they determined that histone deacetylase (HDAC) is the molecular target for largazole.2 This is very significant as HDAC inhibitors are emerging as a new and exciting class of antineoplastic agents for the treatment of solid and nematological malignancies.3 Incidentally, a number of depsipeptides are undergoing clinical trials for treatment of various cancers.4 Largazole’s important biological activity, its selectivity for cancer cells, and its unique structural features led to considerable interest in its chemistry and biology. To establish structure-activity relationships and design novel structural variants, we sought a convergent route to largazole. Herein, we report an enantioselective synthesis of (+)-largazole.
As shown in Figure 1, our synthetic strategy involves a late stage cycloamidation of a sterically less demanding carboxylic acid and an amine to form the 16-membered ring from the corresponding amino acid derived from 2. Ester derivative 2 could be obtained by the formation of an amide bond between the acid arising from thiazole methyl ester 3 and the 4-derived amine. Our plan is to carry out the remainder of the synthesis with the sensitive thioester functional group attached. The synthesis of thiazole 3 can be achieved from a 5-derived thiazole acid and protected (R)-2-methyl cysteine 6.5 Amino ester 4 could be accessed by a cross metathesis reaction between thioester 8 and optically active allylic alcohol 7 followed by a Yamaguchi esterification with the appropriately protected L-valine. Alcohol 7 could be prepared by a lipase mediated kinetic resolution of racemic β-hydroxy ester.
Figure 1.

Retrosynthetic analysis of largazole
As shown in Scheme 1, our synthesis starts with the known azido amide 96 which was treated with Lawesson’s reagent in THF for 12 h to provide the corresponding thioamide in 67% yield.7 The resulting thioamide was then reacted with ethyl bromopyruvate in refluxing ethanol for 1 h which provided thiazole 5 in 82% yield.8 Saponification of ester 5 with 1 M aqueous LiOH gave the acid. The resulting acid was then coupled with trityl- protected α-methyl cysteine9 under EDC/HOBt conditions in the presence of diisopropylethylamine to furnish amide 10 in 96% yield over 2 steps. The conversion to thiazole-thiazoline fragment 11 was achieved following a procedure reported by Kelly and co-workers.10 Accordingly, amide 10 was reacted with 3 equiv. of triphenylphosphine oxide and 1.5 equiv. of Tf2O in CH2Cl2 at 0 °C for 10 min to provide ester 11 in 89% yield. The azide group in 11 was reduced using PPh3 in refluxing methanol11 to give the amine which was then exposed to Boc2O to furnish fragment 3 in 95% yield over 2 steps.
Scheme 1.

Synthesis of segment 3
Optically active synthesis of β-hydroxy ester and its conversion to ester 15 is shown in Scheme 2. Racemic aldol product 12 was prepared by LDA deprotonation of t-butyl acetate followed by reaction of the resulting enolate with acrolein at −78 °C to provide 12 in 81% yield. The racemic alcohol was then exposed to lipase PS-30 in pentane in the presence of excess vinyl acetate at 23 °C for 12 h to provide enantio enriched alcohol 13 and acetate derivative 14 in 45% and 42% yields respectively. Selective removal of the acetate was carried out by exposure of 14 to potassium carbonate in methanol at −30 °C to afford optically active β-hydroxy ester 7 in high enantiomeric purity (93% ee). The enantiomeric excess was determined by formation of the corresponding Mosher ester of alcohol 15 followed by analysis of 19F NMR.12 The kinetic resolution of β-hydroxy ester has provided a convenient access to optically active esters.13 For preparation of alcohol 15 we planned a cross metathesis of alcohol 7 and thioester 8. The requisite thioester was prepared by reaction of 3-butene thiol14 and octanoyl chloride in the presence of DMAP. A cross metathesis reaction of alcohol 7 and thioester 8 in the presence of 3 mol% Grubbs’ 2nd generation catalyst afforded E-olefin 15 exclusively in 67% yield.15
Scheme 2.

Synthesis of thio ester 15
The final assembly of the largazole fragment is shown in Scheme 3. N-Boc-valine 16 was subjected to esterification with alcohol 15 using Yamaguchi’s protocol.16 Accordingly, reaction of 16 with 2,4,6-trichlorobenzoyl chloride in the presence of diisopropylethylamine gave the anhydride. Reaction of the resulting anhydride with alcohol 15 and DMAP furnished thioester 4 in 91% yield. Selective deprotection of the Boc group in the presence of a t-butyl ester was carried out by exposure of 4 to 30% trifluoroacetic acid in CH2Cl2 at 0 °C for 20 min to provide amine 17. For assembly of the largazole subunits, saponification of methyl ester 3 was carried out with 1 M aqueous LiOH to give acid 18. Coupling of acid 18 with amine 17 was accomplished by using HATU and HOAt in the presence of diisopropylethylamine to furnish the requisite protected amino ester 2 in 66% yield. Formation of the 16-membered cycloamide was carried out in a two-step sequence involving: (1) exposure of 2 to trifluoroacetic acid at 23 oC for 3 h to remove both the Boc and the t-butyl groups; (2) treatment of the resulting amino acid with 2 equiv. of HATU and 2 equiv. of HOAt in the presence of diisopropylethylamine under dilute conditions to provide synthetic (+)-largazole (1) in 40% isolated yield (2 steps). The spectral data (1H- and 13C-NMR) of synthetic (+)-largazole (1, [□]23D +24, c 0.13, MeOH; lit.1 [□]20D +22, c 0.1, MeOH) is identical with that reported for the natural (+)-largazole.1
Scheme 3.

Synthesis of Largazole
In summary, we have accomplished an enantioselective synthesis of (+)-largazole (1). The synthesis will provide a convenient access to a variety of largazole derivatives. Structural modifications are currently in progress.17
Supplementary Material
Acknowledgments
Financial support by the National Institute of Health is gratefully acknowledged. We thank Mr. David D. Anderson of Purdue University for his help with the HPLC analysis.
Footnotes
Supporting Information Available: Experimental procedures and 1H- and 13C-NMR spectra for compounds 1–5, 7–11, 15. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Taori K, Paul VJ, Luesch H. J Am Chem Soc. 2008;130:1806. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- 2.Ying Y, Taori K, Kim H, Hong J, Luesch H. J Am Chem Soc. 2008;130:8455. doi: 10.1021/ja8013727. [DOI] [PubMed] [Google Scholar]
- 3.(a) Viqushin DM, Coombes RC. 2002;13:1. doi: 10.1097/00001813-200201000-00001. [DOI] [PubMed] [Google Scholar]; (b) Marchion D, Munster P. Expert Rev Anticancer Ther. 2007;7:583. doi: 10.1586/14737140.7.4.583. [DOI] [PubMed] [Google Scholar]
- 4.Piekarz R, Bastes S. Curr Pharm Des. 2004;10:2289. doi: 10.2174/1381612043383980. [DOI] [PubMed] [Google Scholar]
- 5.Pattenden G, Thom SM, Jones MF. Tetrahedron. 1993;49:2131. [Google Scholar]
- 6.Dyke JM, Levita G, Morris A, Ogden JS, Dias AA, Algarra M, Santos JP, Costa ML, Rodrigues P, Barros MT. J Phys Chem A. 2004;108:5299. [Google Scholar]
- 7.Ozturk T, Ertas E, Mert O. Chem Rev. 2007;107:5210. doi: 10.1021/cr040650b. [DOI] [PubMed] [Google Scholar]
- 8.Boyce RJ, Pattenden G. Tetrahedron. 1995;51:7313. [Google Scholar]
- 9.For synthesis of the enantiomer see Hara S, Makino K, Hamada Y. Tetrahedron Lett. 2006;47:1081.
- 10.You SL, Razavi H, Kelly JW. Angew Chem Int Ed. 2003;42:83. doi: 10.1002/anie.200390059. [DOI] [PubMed] [Google Scholar]
- 11.Pal B, Jaisankar P, Giri VS. Synth Commun. 2004;34:1317. [Google Scholar]
- 12.Dale JA, Dull DL, Mosher HS. J Org Chem. 1969;34:2543. [Google Scholar]
- 13.(a) Vrielynck S, Vandewalle M, García AM, Mascareñas JL, Mouriño A. Tetrahderon Lett. 1995;36:9023. [Google Scholar]; (b) Pollini GP, Risi CD, Lumento F, Marchetti P, Zanirato V. Synlett. 2005:164. [Google Scholar]
- 14.Minozzi M, Nanni D, Walton JC. Org Lett. 2003;5:901. doi: 10.1021/ol034073+. [DOI] [PubMed] [Google Scholar]
- 15.(a) Scholl M, Ding S, Lee C, Grubbs RH. Org Lett. 1999;1:953. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]; (b) Chatterjee AK, Choi T, Sanders DP, Grubbs RH. J Am Chem Soc. 2003;125:11360. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 16.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Japan. 1990;55:7. [Google Scholar]
- 17.During the review of this manuscript, two other syntheses have appeared in the literature, see: Nasveschuk CG, Ungermannova D, Liu X, Phillips AJ. Org Lett. doi: 10.1021/ol8013478.Seiser T, Kamena F, Cramer N. Angew Chem. doi: 10.1002/anie.200802043.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.