

Abstract
Tumor progression locus 2 (Tpl2, also known as Map3k8 and Cot) is a serine-threonine kinase critical in innate immunity, linking toll-like receptors (TLRs) to tumor necrosis factor (TNF) production through its activation of extracellular signal-regulated kinase (ERK). Tpl2−/− macrophages have abrogated TNF production but overproduce interleukin (IL)-12 in response to TLR ligands. Despite enhanced IL-12 production, Tpl2−/− T cells have impaired interferon (IFN)-γ production. Therefore, the role of Tpl2 in a bona fide bacterial infection where all of these cytokines are important in host defense is unclear. To address this issue, we infected Tpl2−/− mice with the model pathogen Listeria monocytogenes. We found that Tpl2−/− mice infected intravenously with L. monocytogenes had increased pathogen burdens compared to wild type mice, and rapidly succumbed to infection. Enhanced susceptibility correlated with impaired signaling through TLR2 and Nucleotide-binding oligomerization domain 2 (Nod2), two receptors previously shown to mediate Listeria recognition. Surprisingly, TNF production in response to infection was not significantly impaired, even though Tpl2 has been implicated in the regulation of TNF. We found that the role of Tpl2 has cell-type specific effects in regulating TNF and transduces signals from some, but not all, pattern recognition receptors (PRR). In contrast to the cell-type- and receptor-specific regulation of TNF, we found that Tpl2 is essential for IL-1β production from both macrophages and dendritic cells. These studies implicate Tpl2 as an important mediator for collaboration of PRR with danger-associated molecular patterns to induce TNF and IL-1β production and optimal host defense.
Keywords: cytokine, dendritic cell, macrophage, pattern recognition receptors, toll-like receptors
INTRODUCTION
The innate immune system uses pattern recognition receptors (PRRs) to recognize various pathogens based on their molecular structures. This leads to cellular activation and inflammatory cytokine production required for proper host defense (1). Cytokines produced by macrophages, dendritic cells (DC), neutrophils and natural killer (NK) cells in response to ligation of PRRs in the early phases of infection serve to control pathogen replication and initiate adaptive responses. The toll-like receptors (TLRs) are a large family of evolutionarily conserved PRRs that recognize a range of microbial products (1, 2). TLR1, 2, 4, 5, and 6 are located at the plasma membrane, whereas nucleic acid sensing TLRs (TLR3, 7, 8 and 9) are located intracellularly, where they are thought to signal from acidic endosomes (2). In addition to TLRs, other PRR exist including nucleotide-binding and leucine-rich repeat containing molecules (NLRs). Nod2, a member of the NLR family, is expressed in the cell cytoplasm and senses muramyl dipeptide (MDP), a component of peptidoglycan common to both gram-positive and gram-negative bacteria (3-7). Other PRRs include C-type lectin receptors (CLRs), such as DC-associated C-type lectin 1 (dectin-1), which recognizes the β-glucan component commonly present in fungal cell walls (8).
Tpl2 is a serine-threonine kinase expressed in hematopoietic cells that was originally identified as a proto-oncogene (9-11). Early studies demonstrated that Tpl2 is critical for the production of TNF due to its role in ERK activation downstream of TLR4 (12, 13). TNF production occurs in response to infection, and in cases of sepsis, can result in toxic shock. The criticality of Tpl2 in TNF regulation is supported by the finding that Tpl2−/− mice are resistant to LPS-induced endotoxic shock, a model for gram negative bacterial infection (13). The abrogation of TNF production in Tpl2-deficient macrophages has been reported to occur by multiple mechanisms, including transcriptional and post-translational control (13-15). In addition, we have recently shown that Tpl2 is important for CD4+ T cell IFN-γ production (16). Of note, Tpl2 does not serve as an essential, positive regulator of all TLR-induced cytokines. For example, Tpl2-deficiency is associated with increased IL-12 production in response to TLR4 and 9 stimulation; however the molecular mechanism has not been fully determined (14).
While many studies have focused on the role of Tpl2 in response to single TLR ligands (13, 14, 17), microbial pathogens activate multiple TLRs along with alternative PRR pathways (2). Listeria monocytogenes is an intracellular gram-positive bacterium capable of activating both TLRs and non-TLR PRRs, but its detection by the host depends primarily on TLR2, Nod1 and Nod2 (18). The role of Tpl2 in response to gram-positive bacterial infection is unclear, but TNF, IFN-γ and IL-12 are all produced during the early course of L. monocytogenes infection. These cytokines help to control replication of bacteria and activate adaptive immune responses, and mice unable to produce these cytokines are more susceptible to infection with this pathogen (19). Since Tpl2 is important for the production of TNF, IFN-γ and IL-12, we investigated the role of Tpl2 in L. monocytogenes infection and other settings in which multiple PRRs are activated. We found that Tpl2−/− mice infected with L. monocytogenes displayed significantly increased mortality rates and bacterial burdens. Enhanced susceptibility to infection in Tpl2-deficient mice correlated with impaired signaling through TLR2 and Nod2, two receptors previously shown to mediate Listeria recognition; however, contrary to expectations, there was no deficit in serum TNF production in mice infected with L. monocytogenes. To gain insight into why TNF production was unaffected in this model, we analyzed innate immune responses to a variety of model ligands for several classes of PRR. We observed cell-type specific effects for Tpl2 in TLR signaling and TNF production, with macrophages being more dependent upon Tpl2 than DCs for TNF production. Importantly, in contrast to TNF, we show that Tpl2 is essential for IL-1β production from all cell types analyzed, regardless of the PRR stimulus. These findings demonstrate that Tpl2 transduces signals by multiple PRRs but has complex, cell-specific functions in cytokine production. Consequently, Tpl2 has a critical function in integrating host immune responses to complex pathogens like L. monocytogenes.
MATERIALS AND METHODS
Mice and in vivo infections
The National Institute of Arthritis and Musculoskeletal and Skin Diseases animal care and use committee approved all animal experiments. Wild-type C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Tpl2−/− mice were bred at the National Institutes of Health (Bethesda, Maryland) (13). Tpl2−/− mice and age- and sex-matched control mice were infected via the tail vein with varying doses of L. monocytogenes strain EGD, obtained originally from ATCC (#BAA-679). Bacterial stocks were prepared by growth in Luria broth (LB) to mid-log phase, aliquoted, frozen at −70°C, and thawed immediately before use. The approximate intravenous LD50 in adult male C57BL/6J mice is 5 × 104 colony forming units (CFU). Spleens were collected and digested with Liberase RI (Roche) according to the manufacturer's protocol. Livers were mechanically digested. CFU were determined by culture on LB agar plates. Female Tpl2−/− mice and littermate controls were treated intraperitoneally with 6 mg of zymosan (Sigma) for 3h or 100 μg of LPS (Salmonella enterica, Sigma) for 30 min or 3h.
Bone marrow DC and macrophage culture
BMDCs were produced by culture of bone marrow cells from femurs and tibias of 6-12-week-old mice 1 × 106 cells/mL in complete RPMI (RPMI 1640 containing 10% fetal calf serum (FCS) (Biosource), 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine (Invitrogen) and 50 μM 2-mercaptoethanol (Sigma) at 37°C and 5% CO2), supplemented with 40 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF) (Peprotech) and 20 ng/mL IL-4 (Peprotech). On days 3 and 5, fresh medium equal to half of the initial culture volume containing 40 ng/mL GM-CSF and 20 ng/mL IL-4 was added to culture. On day 7 non-adherent cells were collected, incubated with anti-mouse CD11c labeled microbeads (Miltenyi Biotech) and CD11c+ cells were selected using an AUTOMACS (Miltenyi Biotech) according to the manufacturer's instructions. The purity of the cell population was determined to be more than 90% CD11c+ by flow cytometry. Macrophage differentiation was induced by culture of mouse bone marrow in complete DME (Dulbecco's Modified Eagles media containing 10% fetal calf serum (FCS) (Biosource), 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine (Invitrogen) at 37°C and 5% CO2), on non tissue culture treated plates supplemented with macrophage colony-stimulating factor (M-CSF) at 10 ng/ml for 7 days at 37°C and 5% CO2. On day 7 macrophages were washed with phosphate buffered saline (PBS) and incubated with cell dissociation buffer (Invitrogen) for 10 min at 37°C to remove macrophages from the culture dish. The cells were counted, re-plated in complete DME with 10 ng/ml of M-CSF and allowed to adhere overnight before stimulation or infection.
Cell stimulation, in vitro infection and measurement of cytokines
BMDC, bone marrow macrophages, or mouse spleen cells were plated at 1×106/mL. Cells were stimulated with lipopolysaccharide (LPS) from Escherichia coli 1 μg/mL (Sigma), zymosan A from Saccharomyces cerevisiae 10 μg/mL (Sigma), cytosine-phosphate-guanosine (CpG) DNA ODN2395 10 μg/mL (Invivogen), polyinosine-polycytidylic acid (poly IC) 5 μg/mL (Sigma), R848 1 μg/mL (Alexis Biochemicals), Pam3CSK4 (50 ng/mL), Curdlan (10 μg/mL), IFN-γ 10 ng/mL (Peprotech), L-18-Muramyl dipeptide (MDP) 50 ng/mL (Invivogen) and ATP 5 mM (Alexis Biochemicals). Bone marrow macrophages were infected for 2h with L. monocytogenes at a ratio of 1 bacterium to 40 macrophages. Cells were treated with gentamicin for 45 min, washed with fresh media, and cultured for a further 18h. Cell culture supernatants were collected 18h after stimulation or infection for analysis of cytokine levels. For detection of IL-1β in response to PRR ligands, cells were stimulated with PRR ligands for 4 h followed by ATP stimulation for 20 min. Cytometric bead array (BD Pharmingen) or luminex bead array (Bio Rad) was used to detect TNF, IL-1β and IL-12(p70) from cell culture supernatants and serum. ELISA (R&D systems) was used for detection of IL-27.
Analysis of IL-1β mRNA expression
Following stimulation, cells were washed with PBS, and RNA was extracted using RNeasy kit (Qiagen) per the manufacturers instructions. Reverse transcription was performed using Taqman reverse transcription reagents (Roche) followed by real-time PCR using ABI PRISM7500 Sequence Detection System (Applied Biosystems). Analysis of IL-1β and GAPDH mRNA levels was performed using commercially available primer/probe sets (Applied Biosystems). Relative levels of IL-1β were determined by normalization to GAPDH levels and are presented as relative to the un-stimulated control which was arbitrarily designated a value of 1.
Flow Cytometry
Spleen cells were stimulated with PRR ligands in the presence of IFN-γ and golgi plug (BD Pharmingen) for 4h. Cells were stained with anti-CD11b-PerCP and anti-CD11c-APC (BD Pharmingen). Cells were washed with PBS/2%FCS and fixed with CytoFix/Perm buffer (BD Pharmingen) for 30 min. Intracellular staining was performed in Perm buffer (BD Pharmingen) for 1h with anti-TNF-FITC or anti-IL-12(p40)-PE (BD Pharmingen). Spleen cells from mice infected with L. monocytogenes were cultured with Golgi Plug for 4h and stained for CD11b, CD11c, Gr1 and intracellular TNF and IL-12 p40 (BD Pharmingen). Acquisition was performed using a BD FACSCanto or FACSCalibur flow cytometer (BD Pharmingen).
Western blotting
Cell lysates were separated on 4-12% gradient gels (Invitrogen) and probed with antibodies to detect phospho-ERK1/2 (Th4202/Tyr204) and ERK1/2 (Cell Signaling Technology), IκBα (Santa Cruz Biotechnology), phospho-Jnk and Jnk (Cell Signaling), IL-1β (R&D Systems), phospho-Raf-1 (ser 338) and Raf-1 (Cell Signaling) and Actin (Chemicon International). Image J software was used to quantitate band intensity.
RESULTS
Tpl2 regulates cytokine production in response to L. monocytogenes
Tpl2 is essential for TNF production by macrophages in response to TLR ligation in a murine model of septic shock, although it also negatively regulates macrophage IL-12 production (13, 14). Therefore we sought to determine the role of Tpl2 in response to a bacterial infection where production of both IL-12 and TNF participate in successful resolution of disease. Mice were infected intravenously (i.v.) with 2 × 103 CFU L. monocytogenes, and sera from wild-type and Tpl2−/− mice were analyzed at 48h. Surprisingly, TNF levels were comparable between wild-type and Tpl2−/− mice (Figure 1A). IL-12 was increased in sera of Tpl2−/− mice compared to wild-type controls (Figure 1A), but the IL-12-related cytokine IL-23 was not detected (data not shown). IL-27 is another IL-12 family member that plays an essential role in host defense (20, 21), which in contrast to IL-12 and IL-23, has critical anti-inflammatory effects (20, 21). Compared to wild-type controls, increased levels of IL-27 were detected in sera from Tpl2−/− mice (Figure 1A); however, there was no difference between wild-type and Tpl2−/− mice in the levels of other cytokines, such as IL-10, IL-6 and IFN-γ (data not shown).
Figure 1. Tpl2 is important for host defense against Listeria monocytogenes.
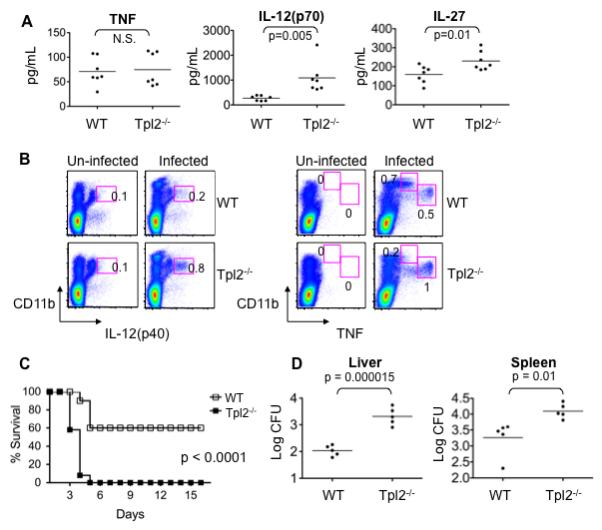
(A) Normal TNF production in serum of Tpl2-deficient mice infected with Listeria monocytogenes. Wild-type (WT) and Tpl2−/− mice were infected i.v. with 2×103 CFU L. monocytogenes for 48 h. Sera were collected and analyzed for TNF, IL-12(p70) and IL-27. (B) L. monocytogenes-induced TNF production is dependent on Tpl2 in some, but not all innate immune cells. Spleens were removed from infected mice and splenocytes were fixed and permeabilized. Intracellular cytokine levels were assessed by flow cytometry. (C) Tpl2-deficient mice have enhanced lethality in response to L. monocytogenes. WT and Tpl2−/− mice were infected i.v. with 2×104 CFU L. monocytogenes, and survival was monitored. (D) Lethality in Tpl2−/− mice results from impaired host defense. WT and Tpl2−/− mice were infected i.v. with 2×103 CFU L. monocytogenes for 48 h. Spleens and livers were homogenized and bacterial burden was measured by plating serial dilutions of homogenates on LB agar plates. Results are representative of 3 – 4 independent experiments. Error bars correspond to standard deviations and * denotes p<0.05 as determined by Student's t-test. Abbreviations: not significant (N.S.).
To explore the unexpected finding of normal TNF production in L. monocytogenes-infected Tpl2−/− mice, we analyzed TNF levels in sera from infected mice at earlier timepoints. We infected wild-type and Tpl2−/− mice with 2×103 CFU and assessed TNF levels in sera obtained 6 h and 24 h after infection. In two independent experiments, the TNF levels at these earlier time-points were very low, and even undetectable in some samples; nonetheless, levels were generally comparable between wild-type and Tpl2−/− mice (Supplemental Figure 1A). In addition, we assessed levels of TNF in sera obtained from wild-type and Tpl2−/− mice infected for 48 h with a higher dose of 2×104 CFU, which is a lethal dose for Tpl2−/− mice (see Supplementary Table 1); under these conditions, levels were readily detectable and comparable between the two groups (Supplemental Figure 1B).
We also considered the possibility that Tpl2 might have cell-specific functions in the regulation of TNF production. For example, Tpl2 was previously shown to differentially regulate signaling pathways in macrophages and fibroblasts (22). To this end, intracellular cytokine staining was performed for IL-12(p40) and TNF on spleen cells taken from wild-type and Tpl2−/− mice infected with 2×103 L. monocytogenes for 48 h. Cells were also stained with CD11b to identify the types of cells making cytokines 48 h after infection. Prior to infection, very few cells stained positive for IL-12p40 or TNF (Figure 1B); however following infection, IL-12p40 was detected in cells from both wild-type and Tpl2−/− mice. An increase in the proportion of p40+ cells was detected from Tpl2−/− spleen compared to wild-type spleen (Figure 1B). After L. monocytogenes infection of wild-type mice, two different populations of CD11b+ splenocytes (Figure1B) were noted to produce TNF (CD11bhigh and CD11bintermediate). Based on other cell surface markers and light scatter characteristics, the CD11bhigh population consisted of monocytes and neutrophils (data not shown), whereas the CD11bintermediate cells comprised so-called inflammatory DCs (23). Interestingly, we found that these populations have differential requirements for Tpl2 to induce TNF protein production; TNF production in monocytes and neutrophils (CD11bhigh) was completely dependent upon Tpl2, whereas TNF production by CD11bintermediate cells was Tpl2-independent (Figure 1B). Together, the normal production of TNF by CD11bintermediate cells, as assessed by intracellular staining, and the similar levels of TNF secretion in Figure 1A suggest that Tpl2 is not obligatory for all TNF production.
Tpl2 is important for host defense against L. monocytogenes
Based on the normal serum levels of TNF and hyper-production of IL-12 during infection, it was reasonable to expect that Tpl2−/− mice might readily survive infection with L. monocytogenes. To test this hypothesis, we initially infected wild-type and Tpl2−/− mice with various doses of L. monocytogenes ranging from 2×102 to 2×105 CFU. Surprisingly, at a dose of 2×104 CFU, there was a marked reduction in the survival of Tpl2−/− mice compared to wild-type controls (Supplemental Table I). We further explored the finding of increased susceptibility of Tpl2−/− mice to L. monocytogenes infection at this dose (Figure 1C). While most wild-type mice infected with 2×104 CFU survived, all Tpl2−/− mice rapidly succumbed to infection between days 3 and 5 (Figure 1C).
Infection with microbial pathogens can result in increased lethality due to either impaired or exaggerated immune responses. For instance, IL-12- and IFN-γ-deficient mice are more susceptible to Toxoplasma gondii because of impaired host defense, whereas IL-27- or IL-10-deficient mice infected with this pathogen die of overwhelming inflammatory disease (20, 21). To address why Tpl2−/− mice were more susceptible to infection with L. monocytogenes, we assessed bacterial burdens by removing livers and spleens from wild-type and Tpl2−/− mice 48 h after infection with a lower dose (2×103 CFU). Consistent with the idea that Tpl2-deficiency resulted in impaired host defense, we found no different in bacterial burdens at 6 or 24 h after infection with 2 × 103 CFU i.v. (data not shown), but we consistently noted significantly more bacteria in organs from Tpl2−/− mice by 48 h (Figure 1D). We conclude that the increased IL-12 production from Tpl2−/− mice does not confer a survival advantage; on the contrary, Tpl2 is important for optimal host defense to L. monocytogenes. However, equally surprising was that systemic TNF production was not impaired.
Distinct requirement for Tpl2 in response to different pattern recognition receptors in different cell types
Normal in vivo production of TNF from Listeria-infected Tpl2−/− mice (Figure 1A) was surprising to us, since Tpl2 has been reported to be essential for macrophage TNF production in response to a range of different TLR ligands, including those activated during Listeria infection (13, 14, 17). Our results indicate that the requirement for Tpl2 in TNF production was cell type-specific (Figure 1B). Therefore, to gain insight into how Listeria might be inducing TNF in a Tpl2-independent manner, both bone marrow-derived macrophages and bone marrow-derived dendritic cells (BMDCs) were stimulated with various TLR ligands, and TNF secretion was assessed. Ligands for TLR2 (zymosan) and TLR9 (CpG) have previously been shown to be involved in Listeria recognition, while ligands for TLR4 (LPS), TLR3 (Poly IC) and TLR7/8 (R848) have been shown to induce TNF from macrophages in a Tpl2-dependent manner (13, 17). In agreement with these published results, wild-type macrophages produced large amounts of TNF in response to LPS, whereas Tpl2−/− macrophages displayed a severe defect in TNF production (Figure 2A). Wild-type BMDCs produced even more TNF than macrophages (Figure 2A); however, in sharp contrast to Tpl2−/− macrophages in which TNF production was abrogated, Tpl2−/− BMDCs activated with LPS, Poly IC or CpG produced TNF, albeit at somewhat reduced levels (Figure 2A). Interestingly, there was no difference in the amount of TNF released from BMDCs stimulated with zymosan (Figure 2A). Thus, while Tpl2 is absolutely essential for the secretion of TNF by macrophages (13), these results show that Tpl2 is only partially required by DC to induce TNF.
Figure 2. Tpl2 regulates TNF production in a cell type- and PRR-specific manner.
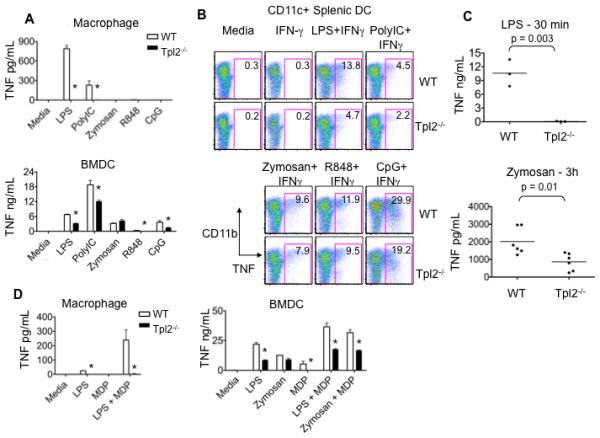
(A) Tpl2 is required for TLR-induced TNF production in macrophages. Bone marrow derived macrophages or BMDCs from WT or Tpl2−/− mice were stimulated with LPS, poly IC, zymosan, R848 or CpG for 18 h, and TNF was measured in cell supernatants. (B) Tpl2 is not required for TNF production by splenic DCs. Spleen cells from WT or Tpl2−/− animals were cultured with IFN-γ or IFN-γ plus LPS, poly IC, zymosan, R848 or CpG for 4 h. Cells were stained with anti- CD11c and CD11b antibodies, fixed, permeabilized and stained with anti-TNF antibody. Dot plots are gated on CD11c+ cells. (C) Tpl2 controls LPS and zymosan-induced TNF production in vivo. WT or Tpl2−/− mice were injected intraperitoneally with LPS (30 min) or zymosan (3h), sera were collected to measure TNF concentrations. (D) Tpl2 is important for Nod2 signaling in both macrophages and BMDCs. Bone marrow derived macrophages were stimulated with LPS, MDP or combinations of LPS and MDP for 18 h. BMDCs were stimulated with LPS, zymosan, MDP or combinations of these ligands for 18 h, and TNF was measured in cell supernatants. Results are representative of 3 independent experiments. Error bars correspond to standard deviations and * denotes p<0.05 as determined by Student's t-test.
We considered the possibility that BMDCs might not reflect the functionality of freshly isolated primary DCs. To determine if similar effects were observed with splenic DCs, wild-type or Tpl2−/− spleen cells were stimulated in vitro with the same panel of TLR ligands in the presence of IFN-γ. Intracellular staining was performed to determine the level of TNF production. Consistent with secretion of TNF from in vitro cultured BMDCs, Tpl2−/− CD11c+ splenic DCs also displayed reduced, but not absent, TNF production (Figure 2B). Again, unlike other TLR ligands, zymosan induced relatively normal levels of TNF from Tpl2−/− cells (Figure 2B). In addition to ligands for TLR2, zymosan contains β-glucan, a component of yeast cell walls, which stimulates dectin-1 (8, 24, 25). In vivo treatment of mice with LPS induced TNF in wild-type mice, whereas TNF production was completely abrogated in the absence of Tpl2 (Figure 2C). In contrast, treatment of mice with zymosan resulted in only a partial reduction, and not an abrogation, of TNF production in Tpl2−/− compared to wild-type mice. Taken together, these results strongly argue that the ability of Tpl2−/− mice infected with L. monocytogenes to produce TNF (Figure 1A) can be explained by the finding that Tpl2-independent pathways exist in DCs to produce TNF. Furthermore, with respect to TNF production, some PRRs are more dependent upon Tpl2 than others.
Nod2 is an intracellular pathogen sensor capable of recognizing MDP, a component in the cell wall of L. monocytogenes (4-7). To test whether Tpl2 contributes to this alternative PRR signaling pathway, cells were stimulated with the Nod2 ligand, muramyl dipeptide (MDP). MDP alone did not induce measurable TNF in wild-type macrophages (26); however, LPS plus MDP acted synergistically to increase TNF production in wild-type cells (Figure 2D). In contrast, no TNF production was detected in Tpl2−/− macrophages (Figure 2D). Unlike macrophages, stimulation with MDP alone induced TNF production in BMDCs, and this was abrogated in Tpl2−/− BMDCs (Figure 2D). Combinations of MDP with LPS or zymosan acted to increase TNF production from wild-type cells, and this increase was diminished in the absence of Tpl2 (Figure 2D). Thus, while Nod2 requires Tpl2 for TNF production in both macrophages and DCs, most TLRs evidently have alternative means of inducing TNF in BMDCs.
Tpl2-dependent TNF production correlates with cell-type-specific coupling to NF-κB
Having observed distinct requirements for Tpl2 by different receptors and cell types in vitro and in the context of Listeria infection in vivo, we next sought to determine the role of this kinase in signaling by different PRRs. Our intent was to define correlates that might help explain the cell-type specific role of Tpl2 in regulating TNF. Wild-type and Tpl2−/− macrophages or BMDCs were stimulated with LPS for various times, and the activation of MAPKs (ERK and Jnk) were assessed by immunoblotting. LPS induced phosphorylation of ERK1/2 in wild-type macrophages and, consistent with previous reports (13, 14), failed to induce ERK1/2 phosphorylation in Tpl2−/− macrophages (Figure 3A). Interestingly, LPS also failed to induce phosphorylation of ERK1/2 in Tpl2-deficient BMDCs (Figure 3B). To compare the effect of Tpl2 on other signaling pathways commonly activated by PRRs, we next assessed the role of Tpl2 in the activation of Jnk. Using scanning densitometry to quantitate results, there was no consistent difference in the activation of Jnk between wild-type and Tpl2−/− macrophages (Figure 3A) and BMDC (Figure 3B) (data not shown).
Figure 3. Tpl2 controls the MAP kinase and NF-κB pathways in a cell type-specific and stimulus-specific manner.

(A-B) Enhanced activation of IκBα in Tpl2-deficient DCs. WT or Tpl2−/− (A) macrophages or (B) BMDCs were stimulated with LPS for 5, 15, 30, 60, or 120 min. Cell lysates were immunoblotted with antibodies against phospho-ERK1/2, phospho-Jnk or IκBα. Membranes were re-probed with antibodies against ERK1/2, Jnk or actin as loading controls. (C) Tpl2 links the Nod2 receptor to the ERK pathway. WT or Tpl2−/− macrophages were stimulated with MDP for 5, 15, 30, 60, or 120 min. The cell lysates were immunoblotted with anti-phospho-ERK1/2 or anti-ERK1/2. (D) Tpl2 is not required for zymosan-induced ERK activation. WT or Tpl2−/− BMDCs were stimulated with zymosan for the indicated times and cell lysates were probed with antibodies for phospho-ERK1/2. The membrane was stripped and re-probed for total ERK1/2 as a loading control. Results are representative of at least 2 independent experiments.
Activation of PRRs also results in the degradation of IκBα to release NF-κB subunits, which dimerize and translocate to the nucleus to initiate gene transcription. In response to LPS, IκBα was degraded by 30 min in both wild-type and Tpl2−/− macrophages (Figure 3A). On the other hand, IκBα degradation was accelerated in Tpl2−/− BMDCs compared to wild-type cells; LPS-dependent IκBα degradation occurred at 30 min in controls, but was nearly complete at 15 min in Tpl2−/− BMDCs (Figure 3B). To further investigate the apparent increase in NF-κB activation, we performed transcription factor ELISA to measure the nuclear levels of NF-κB-p65 before and after LPS stimulation of BMDC for 30 min. Tpl2−/− BMDCs displayed increased nuclear NF-κB-p65 both basally and after stimulation with LPS compared to wild-type controls (Supplemental Figure 2). While the fold-change was actually lower in the Tpl2-deficient cells upon LPS treatment, this was likely due to the already high level of NFκB activity observed even at basal conditions. The increased NF-κB activity observed in Tpl2−/− BMDCs measured both by IκB degradation and transcription factor ELISA may, in part, explain why these cells have residual TNF production, whereas Tpl2−/− macrophages are absolutely defective.
To characterize the mechanism by which Tpl2 might contribute to Nod2 signaling, wild-type and Tpl2−/− macrophages were stimulated with MDP. As shown in Figure 3C, MDP induced ERK1/2 phosphorylation in wild-type cells; however, ERK1/2 phosphorylation was abrogated in Tpl2−/− macrophages, suggesting that Tpl2 also regulates Nod2 signaling at the level of ERK activation.
As indicated above, Tpl2 was dispensable for zymosan-induced TNF production in BMDCs, but was important to induce normal levels of LPS-induced TNF. We therefore sought to determine if the ability to activate ERK correlated with cytokine production. Unlike LPS, zymosan induced strong phosphorylation of ERK1/2 in both wild-type and Tpl2−/− BMDCs (Figure 3D), consistent with the contention that pathways independent of Tpl2 are utilized in DCs.
An ITAM-containing PRR utilizes a distinct MAP3K
Because the complex TLR2 ligand zymosan induced TNF and activated ERK normally in Tpl2−/− DCs, we considered the possibility that ligation of non-TLR PRR by components of zymosan might activate alternative signaling pathways through dectin-1. Dectin-1 couples to the spleen tyrosine kinase (Syk) via an ITAM-like domain to induce NF-κB and ERK activation (8, 27-32). As signaling through TLR2 is required for optimal control of Listeria infection (18), it is important to determine whether the TLR2 and/or dectin-1 pathways are both intact in Tpl2-deficient mice. To delineate the potential contribution of Tpl2 to signaling via each receptor, we used model ligands that selectively utilize TLR2 or dectin-1. Pam3CSK4 (Pam3), a selective TLR2 ligand, induced production of TNF and strong ERK1/2 phosphorylation in wild-type BMDCs, which was abrogated in Tpl2−/− BMDCs (Figure 4A); however, curdlan, a selective dectin-1 ligand, generated equivalent levels of TNF and ERK phosphorylation in both wild-type and Tpl2−/− cells (Figure 4B). v-raf-1 murine leukemia viral oncogene homolog 1 (Raf-1), like Tpl2, is a MAP3K, which links to a variety of ITAM-containing immunoreceptors (33). Stimulation of wild type BMDCs with curdlan induced rapid Raf-1 phosphorylation at serine 338, consistent with another recent report (34), whereas stimulation with Pam3 failed to induce phosphorylation of Raf-1. Even in Tpl2−/− BMDCs, curdlan induced prominent Raf-1 phosphorylation (Figure 4C). These results suggest that dectin-1 utilizes the MAP3K Raf-1 to induce ERK activation independent of Tpl2.
Figure 4. ITAM-containing PRRs can utilize a distinct MAP3K8.

(A) Tpl2 is required for normal TNF production and ERK activation downstream of TLR2. WT or Tpl2−/− BMDCs were stimulated with Pam3 for 18 h to measure supernatant TNF levels. BMDCs were also stimulated with Pam3 for 15, 30, 60, or 120 min, cell lysates were probed for phospho-ERK1/2 and total ERK1/2. (B) Tpl2 is not required for dectin-1-induced TNF production or signal transduction. WT or Tpl2−/− BMDCs were stimulated with curdlan for 18h to measure supernatant TNF levels. Cells were also stimulated with curdlan for 15, 30, 60, or 120 min, western blot was performed and cell lysates were probed for phospho-ERK1/2 and total ERK1/2. (C) Raf-1 is activated in response to stimulation of dectin-1. Western blot was also performed on WT BMDCs stimulated with Pam3 or Curdlan for 15 or 30 min, cell lysates were probed with anti-phospho-Raf-1, the membrane was stripped and re-probed for total Raf-1 protein as a loading control. Results are representative of 2-3 independent experiments. Error bars are equal to standard deviation and * p<0.05 as determined by Student's t-test.
Tpl2 is critical for IL-1 production by both macrophages and dendritic cells
The above results indicate that Tpl2 is important in signaling in response to TLR and Nod2 ligands, but is dispensable when microbes, complex microbial products and dectin-1 ligands are used to induce TNF production. Therefore, despite its essential role in mediating LPS-induced septic shock (13), Tpl2 links some but not all PRRs to TNF production. Moreover, some cell types (macrophages) absolutely require Tpl2 expression to induce TNF, whereas others (DCs) only partially require Tpl2. While these findings explained some of the results we observed, the lethality seen in Tpl2−/− mice infected with L. monocytogenes remained enigmatic. We therefore hypothesized that Tpl2 might regulate production of other important pro-inflammatory cytokines. Previous reports have suggested that Tpl2 also regulates the production of IL-1β (13, 35); however, this has not been studied in detail in Tpl2−/− mice. IL-1β is prominently induced in innate responses to microbial pathogens and is known to contribute to resistance of Listeria infection (36-41). Like TNF, the production of active IL-1β is a two-step process. First, TLR ligation triggers pro-IL-1β production; second, a multimeric protein complex called the inflammasome assembles to promote caspase-1-dependent cleavage of pro-IL-1β into its mature, secreted form (42). This process is thought to be activated by danger-associated molecular patterns (DAMPs), which include endogenous danger or stress signals (43, 44). To explore whether Tpl2 plays a role in IL-1β production, we stimulated macrophages with LPS, poly IC or MDP for 4h to up-regulate pro-IL-1β. The cells were then stimulated with adenosine triphosphate (ATP) to trigger caspase-1-induced cleavage of pro-IL-1β to its active form. As expected, wild-type macrophages produced large amounts of IL-1β in response to LPS, poly IC and the combination of LPS and MDP. In contrast, Tpl2−/− macrophages produced significantly less IL-1β in response to all of these ligands (Figure 5A). We similarly explored the response of BMDCs (Figure 5B). Consistent with IL-1β production from macrophages, we observed a significant reduction in IL-1β secretion from Tpl2−/− BMDCs compared to wild-type controls, even when zymosan was used to stimulate cells (Figure 5B). These results show that IL-1β is not subject to the same cell type specific effects that control TNF production. Furthermore, they suggest that Tpl2 regulates IL-1β production via a mechanism independent of the ERK pathway, since ERK activation was normal in Tpl2-deficient BMDCs stimulated with zymosan (Figure 3D).
Figure 5. Tpl2 regulates IL-1β production in response to TLR stimulation.

(A-B) Tpl2 is essential for IL-1β production by macrophages and BMDCs. (A) Macrophages from WT or Tpl2−/− mice were cultured and stimulated with LPS, poly IC or MDP and the combination of LPS and MDP for 4 h. The cells were stimulated with ATP for 20 min, after which IL-1β was measured in cell supernatants. (B) WT or Tpl2−/− BMDCs were stimulated with LPS, poly IC, zymosan, MDP separately and LPS in combination with MDP for 4 h, followed by a second stimulation with ATP for 20 min. Cell supernatants were collected, and IL-1β was measured. (C) Tpl2 is critical for production of pro-IL-1β protein. Macrophages were stimulated as in (A) cells were lysed and western blotting was performed to measure IL-1β protein. (D) Tpl2 is essential for induction of IL-1β mRNA. WT or Tpl2−/− bone marrow macrophages were stimulated with LPS for 3, 6, 9 and 12 h. The cells were lysed and IL-1β mRNA expression was analyzed by real-time PCR relative to a GADPH control. Error bars indicate standard deviations and * denotes p<0.05 as determined by Student's t-test.
Tpl2 regulates IL-1β mRNA production
We have shown previously that Tpl2 regulates TNF production post-transcriptionally (13, 45). Others have alternatively proposed that Tpl2 regulates TNF at the level of transcription (14,15). To determine whether Tpl2 regulates IL-1β production by similar mechanism(s), we analyzed the production of IL-1β pro-protein or mRNA. Wild-type and Tpl2−/− macrophages were stimulated with LPS, poly IC, MDP or the combination of MDP and LPS for 4 h, followed by stimulation with ATP for 20 min. The cells were lysed and the pro- and active forms of IL-1β were detected by immunoblotting (Figure 5C). Pro-IL-1β is strongly induced in wild-type macrophages, while Tpl2−/− macrophages display significantly reduced levels of pro-IL-1β in response to simulation with LPS, poly IC and the combination of LPS and MDP (Figure 5C). As a result, the processed active form of IL-1β could not be detected in Tpl2−/− cells compared to wild-type macrophages (Figure 5C).
To determine if Tpl2 regulates production of IL-1β mRNA, we stimulated macrophages with LPS for 3-12 h (Figure 5D). IL-1β mRNA from wild-type macrophages increased 800-fold over un-stimulated controls after LPS treatment, while IL-1β mRNA from Tpl2−/− macrophages remained unchanged (Figure 5D). These results show that Tpl2 plays an important role in the transcriptional regulation of IL-1β and explains why cells lacking Tpl2 fail to produce normal levels of pro-IL-1β protein.
Tpl2 controls IL-1β production in response to L. monocytogenes
IL-1β production is typically transient and difficult to reliably measure in vivo in the context of infection. Therefore, to determine the role of Tpl2 in IL-1β production in response to infection with L. monocytogenes, we isolated bone marrow derived macrophages and infected them in vitro. This resulted in the production of IL-1β and other cytokines from wild-type macrophages (Figure 6A). Infection of Tpl2−/− macrophages resulted in more IL-27 and less TNF and IL-1β production compared to wild-type cells. IL-12 could not be detected. Notably, IL-1β production was even more dramatically impaired than TNF in Tpl2−/− macrophages.
Figure 6. Tpl2 controls IL-1β production in response to L. monocytogenes.

(A-B) Tpl2 is important for IL-1β production by macrophages and spleen cells. Bone marrow derived macrophages (A) or total spleen cells (B) from WT or Tpl2−/− mice were infected with L. moncytogenes at a ratio of 1 bacteria to 40 cells for 2 h. Cells were treated with gentamicin, washed with fresh media, cultured for an additional 18 h and cytokines were measured in cell supernatants. (C) Tpl2 is important in vivo for the production of IL-1β. WT and Tpl2−/− mice were injected i.p with LPS. Serum was collected 3 h later, and IL-1β was measured.
Since we observed cell type-specific effects of Tpl2-deficiency on TNF production, we sought to determine whether IL-1β production would have a similar requirement for Tpl2. We therefore cultured freshly isolated, un-fractionated spleen cells with L. monocytogenes and again measured cytokine production. As with bone marrow-derived macrophages, infection of Tpl2-deficient spleen cells resulted in significantly impaired IL-1β production (Figure 6B). Increased IL-12 production was also observed. However, Tpl2−/− spleen cells produced normal amounts of TNF (Figure 6B). These results indicate that Tpl2 is critical for multiple cell types to produce IL-1β; in contrast, Tpl2 is not required for TNF production from spleen cells infected with L. monocytogenes.
To ensure that differences observed were not related to different pathogen burdens, we measured bacterial numbers after initial phagocytosis and overnight culture. Bacterial counts revealed no significant difference in uptake or bacterial growth within wild-type or Tpl2−/− macrophages or spleen cells at these time points (data not shown). These results suggest that the increased bacterial burden observed in Tpl2−/− mice in vivo was not due to differences in macrophage uptake or bacterial survival within macrophages.
Finally, to confirm the in vivo relevance of our findings, we treated Tpl2−/− mice with LPS for 3 h and measure systemic IL-1β production in serum. IL-1β was strongly induced in wild-type control mice. In contrast, IL-1β was severely reduced in Tpl2−/− mice (Figure 6C). These results confirm a critical role for Tpl2 in IL-1β production and further establish a major role for Tpl2 in the activation and control of innate immune responses.
DISCUSSION
Tpl2 is essential for TNF production in models of septic shock (13); however, the role of Tpl2 in host defense against a gram positive organism such as Listeria monocytogenes has not been previously assessed. In this study, we report that Tpl2−/− mice are more susceptible to primary L. monocytogenes infection, due to a primary failure in innate host defense likely involving defects in both PRR signaling and cytokine production. Although Tpl2 is not required for systemic TNF production and has distinct roles in different innate immune cells, Tpl2 is critical for production of IL-1β. Furthermore, a range of structurally distinct PRRs evidently utilize Tpl2 for the induction of IL-1β. Thus, the present study establishes a new role for Tpl2 in host defense and immunoregulation.
We were very surprised to find that Tpl2−/− mice infected with L. monocytogenes produced abundant TNF. Upon further analysis, we found that Tpl2 is required in macrophages but is not essential in DCs for TNF induction. Moreover, production of this cytokine in response to some ligands was entirely independent of Tpl2. What might account for the cell type- and receptor-specific requirements for Tpl2 in TNF production? It is abundantly clear that Tpl2 is essential in macrophages for linking all TLRs to ERK, and our data are consistent with this conclusion (13, 17); however, other signaling pathways are activated by TLRs, and we found that in the absence of Tpl2, these pathways are differentially activated in macrophages and BMDCs. The absence of Tpl2 resulted in increased NF-κB activation in BMDCs but not in macrophages. Another PRR commonly activated by bacterial products is Nod2 (4). Our results show that Tpl2 is required for ERK activation in response to the Nod2 ligand, MDP. Tpl2 also plays a role in TNF production in response to MDP in both macrophages and BMDCs. It will be useful to ascertain whether Tpl2 is involved in signaling by other intracellular pathogen sensors and to dissect how Tpl2 is biochemically linked to these diverse PRRs. In contrast, we found that ligands that engage dectin-1 activate ERK and TNF production independent of Tpl2.
The data provided in the present study indicate that Tpl2 is also critical for IL-1β production – even more so than TNF. We show that Tpl2 is required for maximal production of IL-1β in macrophages and BMDCs, independent of the PRR engaged. Tpl2 regulates the transcription of IL-1β. Further work is needed to dissect where Tpl2 fits into the pathway leading to IL-1β production. It has previously been shown that inhibition of ERK interferes with TLR-induced IL-1β production (46). This might lead one to suspect that this would be the mechanism by which Tpl2 contributes to IL-1β induction; however, zymosan-dependent IL-1β production is also disrupted by the absence of Tpl2, even though ERK activation is normal. This implies that Tpl2 has an additional, ERK-independent role in linking PRRs to the regulation of IL-1β. Regulation of IL-12 production by Tpl2 is also thought to be independent of ERK (14). Clearly, identification of additional downstream targets of Tpl2 will be an important area for future investigation.
TNF is absolutely essential for defense against L. monocytogenes (47), and the fact that some cell types are not able to produce TNF in the absence of Tpl2 may contribute to susceptibility of infection. Importantly, responsiveness to TNF is also altered in Tpl2−/− antigen presenting cells (22). While IL-1 is not absolutely required for host defense against L. monocytogenes, it also clearly plays a role (37-41). Likely, the absence of IL-1β production and the loss of TNF in certain cell types in combination with diminished responsiveness to TNF by Tpl2−/− cells contribute to increased susceptibility to L. monocytogenes in Tpl2−/− mice. It is also possible that Tpl2 plays an important role for other cytokines as well.
Our findings have important implications in terms of signal transduction, but also have very practical implications. The treatment of rheumatoid arthritis (RA) and inflammatory bowel disease (IBD) has been revolutionized by the use of TNF antagonists (48). Because of its profound role in models of septic shock, Tpl2 has been considered an important target for the generation of new immunosuppressive drugs (49-53). Consistent with this notion, mice bearing a deletion in the 3′ AU-rich element of TNF mRNA (TnfΔARE), over-produce TNF and develop IBD with a histopathologic phenotype similar to Crohn's disease (54). When the TnfΔARE mice are crossed onto a Tpl2−/− background, onset of intestinal inflammation is significantly delayed (55); however, the work presented herein provides a cautionary note with respect to the idea that Tpl2 antagonism will necessarily block TNF production, even though TNF signal transduction might be impaired in some cell types (22). Our results clearly show that different PRRs have distinct dependence upon Tpl2 for TNF production; in fact, with some ligands, TNF production is entirely normal in the absence of Tpl2. Moreover, some cells, like DCs only display partial reductions in TNF in the absence of Tpl2 in response to TLR stimulation. Gut flora express multiple PRRs that are thought to play a role in disease pathogenesis of IBD (56). Clearly, it will be of interest to examine the role of Tpl2 in models of IBD and to determine if and how cytokine production is affected. Conversely, our results argue that inhibition of Tpl2 could be an effective means of antagonizing IL-1β. This could be useful in the spectrum of inflammatory diseases characterized by overproduction of IL-1β, like neonatal onset multisystem inflammatory disease and other cryopyrinopathies, Still's disease, and possibly diabetes (57-60).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Prof. Douglas J. Hilton for insights regarding this study. The authors would also like to thank Jim Simone and the flow cytometry core, as well as the animal care staff of the NIAMS Office of Science and Technology for excellent support.
Abbreviations used in this paper
- BMDC
Bone marrow derived dendritic cell
- CpG
cytosine-phosphate-guanosine
- DC
dendritic cell
- dectin-1
DC-associated C-type lectin 1
- MDP
muramyl dipeptide
- Nod2
nucleotide-binding oligomerization domain 2
- Pam3
Pam3CSK4
- poly IC
polyinosine-polycytidylic acid
- PRR
pattern recognition receptor
- Tpl2
Tumor progression locus 2
References
- 1.O'Neill LA. When signaling pathways collide: positive and negative regulation of toll-like receptor signal transduction. Immunity. 2008;29:12–20. doi: 10.1016/j.immuni.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 4.Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 5.Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, Foster SJ, Moran AP, Fernandez-Luna JL, Nunez G. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J Biol Chem. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 6.Inohara N, Ogura Y, Chen FF, Muto A, Nunez G. Human Nod1 confers responsiveness to bacterial lipopolysaccharides. J Biol Chem. 2001;276:2551–2554. doi: 10.1074/jbc.M009728200. [DOI] [PubMed] [Google Scholar]
- 7.Girardin SE, Travassos LH, Herve M, Blanot D, Boneca IG, Philpott DJ, Sansonetti PJ, Mengin-Lecreulx D. Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J Biol Chem. 2003;278:41702–41708. doi: 10.1074/jbc.M307198200. [DOI] [PubMed] [Google Scholar]
- 8.Brown GD, Gordon S. Immune recognition. A new receptor for beta-glucans. Nature. 2001;413:36–37. doi: 10.1038/35092620. [DOI] [PubMed] [Google Scholar]
- 9.Patriotis C, Makris A, Bear SE, Tsichlis PN. Tumor progression locus 2 (Tpl-2) encodes a protein kinase involved in the progression of rodent T-cell lymphomas and in T-cell activation. Proc Natl Acad Sci U S A. 1993;90:2251–2255. doi: 10.1073/pnas.90.6.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erny KM, Peli J, Lambert JF, Muller V, Diggelmann H. Involvement of the Tpl-2/cot oncogene in MMTV tumorigenesis. Oncogene. 1996;13:2015–2020. [PubMed] [Google Scholar]
- 11.Ceci JD, Patriotis CP, Tsatsanis C, Makris AM, Kovatch R, Swing DA, Jenkins NA, Tsichlis PN, Copeland NG. Tpl-2 is an oncogenic kinase that is activated by carboxy-terminal truncation. Genes Dev. 1997;11:688–700. doi: 10.1101/gad.11.6.688. [DOI] [PubMed] [Google Scholar]
- 12.Waterfield MR, Zhang M, Norman LP, Sun SC. NF-kappaB1/p105 regulates lipopolysaccharide-stimulated MAP kinase signaling by governing the stability and function of the Tpl2 kinase. Mol Cell. 2003;11:685–694. doi: 10.1016/s1097-2765(03)00070-4. [DOI] [PubMed] [Google Scholar]
- 13.Dumitru CD, Ceci JD, Tsatsanis C, Kontoyiannis D, Stamatakis K, Lin JH, Patriotis C, Jenkins NA, Copeland NG, Kollias G, Tsichlis PN. TNF-alpha induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell. 2000;103:1071–1083. doi: 10.1016/s0092-8674(00)00210-5. [DOI] [PubMed] [Google Scholar]
- 14.Sugimoto K, Ohata M, Miyoshi J, Ishizaki H, Tsuboi N, Masuda A, Yoshikai Y, Takamoto M, Sugane K, Matsuo S, Shimada Y, Matsuguchi T. A serine/threonine kinase, Cot/Tpl2, modulates bacterial DNA-induced IL-12 production and Th cell differentiation. J Clin Invest. 2004;114:857–866. doi: 10.1172/JCI20014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rousseau S, Papoutsopoulou M, Symons A, Cook D, Lucocq JM, Prescott AR, O'Garra A, Ley SC, Cohen P. TPL2-mediated activation of ERK1 and ERK2 regulates the processing of pre-TNF alpha in LPS-stimulated macrophages. J Cell Sci. 2008;121:149–154. doi: 10.1242/jcs.018671. [DOI] [PubMed] [Google Scholar]
- 16.Watford WT, Hissong BD, Durant LR, Yamane H, Muul LM, Kanno Y, Tato CM, Ramos HL, Berger AE, Mielke L, Pesu M, Solomon B, Frucht DM, Paul WE, Sher A, Jankovic D, Tsichlis PN, O'Shea JJ. Tpl2 kinase regulates T cell interferon-{gamma} production and host resistance to Toxoplasma gondii. J Exp Med. 2008 doi: 10.1084/jem.20081461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banerjee A, Gugasyan R, McMahon M, Gerondakis S. Diverse Toll-like receptors utilize Tpl2 to activate extracellular signal-regulated kinase (ERK) in hemopoietic cells. Proc Natl Acad Sci U S A. 2006;103:3274–3279. doi: 10.1073/pnas.0511113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corr SC, O'Neill LA. Listeria monocytogenes infection in the face of innate immunity. Cell Microbiol. 2009 doi: 10.1111/j.1462-5822.2009.01294.x. [DOI] [PubMed] [Google Scholar]
- 19.Pamer EG. Immune responses to Listeria monocytogenes. Nat Rev Immunol. 2004;4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 20.Hunter CA, Villarino A, Artis D, Scott P. The role of IL-27 in the development of T-cell responses during parasitic infections. Immunol Rev. 2004;202:106–114. doi: 10.1111/j.0105-2896.2004.00213.x. [DOI] [PubMed] [Google Scholar]
- 21.Gaddi PJ, Yap GS. Cytokine regulation of immunopathology in toxoplasmosis. Immunol Cell Biol. 2007;85:155–159. doi: 10.1038/sj.icb.7100038. [DOI] [PubMed] [Google Scholar]
- 22.Das S, Cho J, Lambertz I, Kelliher MA, Eliopoulos AG, Du K, Tsichlis PN. Tpl2/cot signals activate ERK, JNK, and NF-kappaB in a cell-type and stimulus-specific manner. J Biol Chem. 2005;280:23748–23757. doi: 10.1074/jbc.M412837200. [DOI] [PubMed] [Google Scholar]
- 23.Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 24.Underhill DM, Ozinsky A, Hajjar AM, Stevens A, Wilson CB, Bassetti M, Aderem A. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 1999;401:811–815. doi: 10.1038/44605. [DOI] [PubMed] [Google Scholar]
- 25.Sato M, Sano H, Iwaki D, Kudo K, Konishi M, Takahashi H, Takahashi T, Imaizumi H, Asai Y, Kuroki Y. Direct binding of Toll-like receptor 2 to zymosan, and zymosan-induced NF-kappa B activation and TNF-alpha secretion are down-regulated by lung collectin surfactant protein A. J Immunol. 2003;171:417–425. doi: 10.4049/jimmunol.171.1.417. [DOI] [PubMed] [Google Scholar]
- 26.Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 27.Rogers NC, Slack EC, Edwards AD, Nolte MA, Schulz O, Schweighoffer E, Williams DL, Gordon S, Tybulewicz VL, Brown GD, Reis e Sousa C. Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity. 2005;22:507–517. doi: 10.1016/j.immuni.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 28.Underhill DM, Rossnagle E, Lowell CA, Simmons RM. Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood. 2005;106:2543–2550. doi: 10.1182/blood-2005-03-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuller GL, Williams JA, Tomlinson MG, Eble JA, Hanna SL, Pohlmann S, Suzuki-Inoue K, Ozaki Y, Watson SP, Pearce AC. The C-type lectin receptors CLEC-2 and Dectin-1, but not DC-SIGN, signal via a novel YXXL-dependent signaling cascade. J Biol Chem. 2007;282:12397–12409. doi: 10.1074/jbc.M609558200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slack EC, Robinson MJ, Hernanz-Falcon P, Brown GD, Williams DL, Schweighoffer E, Tybulewicz VL, Reis e Sousa C. Syk-dependent ERK activation regulates IL-2 and IL-10 production by DC stimulated with zymosan. Eur J Immunol. 2007;37:1600–1612. doi: 10.1002/eji.200636830. [DOI] [PubMed] [Google Scholar]
- 31.Adachi Y, Ishii T, Ikeda Y, Hoshino A, Tamura H, Aketagawa J, Tanaka S, Ohno N. Characterization of beta-glucan recognition site on C-type lectin, dectin 1. Infect Immun. 2004;72:4159–4171. doi: 10.1128/IAI.72.7.4159-4171.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown GD, Herre J, Williams DL, Willment JA, Marshall AS, Gordon S. Dectin-1 mediates the biological effects of beta-glucans. J Exp Med. 2003;197:1119–1124. doi: 10.1084/jem.20021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abram CL, Lowell CA. The expanding role for ITAM-based signaling pathways in immune cells. Sci STKE. 2007:re2. doi: 10.1126/stke.3772007re2. 2007. [DOI] [PubMed] [Google Scholar]
- 34.Gringhuis SI, den Dunnen J, Litjens M, van der Vlist M, Wevers B, Bruijns SC, Geijtenbeek TB. Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat Immunol. 2009;10:203–213. doi: 10.1038/ni.1692. [DOI] [PubMed] [Google Scholar]
- 35.Papoutsopoulou S, Symons A, Tharmalingham T, Belich MP, Kaiser F, Kioussis D, O'Garra A, Tybulewicz V, Ley SC. ABIN-2 is required for optimal activation of Erk MAP kinase in innate immune responses. Nature immunology. 2006;7:606–615. doi: 10.1038/ni1334. [DOI] [PubMed] [Google Scholar]
- 36.Dunne A, O'Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci STKE. 2003:re3. doi: 10.1126/stke.2003.171.re3. 2003. [DOI] [PubMed] [Google Scholar]
- 37.Deckert M, Virna S, Sakowicz-Burkiewicz M, Lutjen S, Soltek S, Bluethmann H, Schluter D. Interleukin-1 receptor type 1 is essential for control of cerebral but not systemic listeriosis. Am J Pathol. 2007;170:990–1002. doi: 10.2353/ajpath.2007.060507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glaccum MB, Stocking KL, Charrier K, Smith JL, Willis CR, Maliszewski C, Livingston DJ, Peschon JJ, Morrissey PJ. Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J Immunol. 1997;159:3364–3371. [PubMed] [Google Scholar]
- 39.Havell EA, Moldawer LL, Helfgott D, Kilian PL, Sehgal PB. Type I IL-1 receptor blockade exacerbates murine listeriosis. J Immunol. 1992;148:1486–1492. [PubMed] [Google Scholar]
- 40.Labow M, Shuster D, Zetterstrom M, Nunes P, Terry R, Cullinan EB, Bartfai T, Solorzano C, Moldawer LL, Chizzonite R, McIntyre KW. Absence of IL-1 signaling and reduced inflammatory response in IL-1 type I receptor-deficient mice. J Immunol. 1997;159:2452–2461. [PubMed] [Google Scholar]
- 41.Rogers HW, Tripp CS, Schreiber RD, Unanue ER. Endogenous IL-1 is required for neutrophil recruitment and macrophage activation during murine listeriosis. J Immunol. 1994;153:2093–2101. [PubMed] [Google Scholar]
- 42.Mariathasan S. ASC, Ipaf and Cryopyrin/Nalp3: bona fide intracellular adapters of the caspase-1 inflammasome. Microbes Infect. 2007;9:664–671. doi: 10.1016/j.micinf.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 43.Petrilli V, Dostert C, Muruve DA, Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Curr Opin Immunol. 2007;19:615–622. doi: 10.1016/j.coi.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 44.Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4:469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- 45.Skinner SJ, Deleault KM, Fecteau R, Brooks SA. Extracellular signal-regulated kinase regulation of tumor necrosis factor-alpha mRNA nucleocytoplasmic transport requires TAP-NxT1 binding and the AU-rich element. J Biol Chem. 2008;283:3191–3199. doi: 10.1074/jbc.M705575200. [DOI] [PubMed] [Google Scholar]
- 46.Maggi LB, Jr., Moran JM, Buller RM, Corbett JA. ERK activation is required for double-stranded RNA- and virus-induced interleukin-1 expression by macrophages. J Biol Chem. 2003;278:16683–16689. doi: 10.1074/jbc.M211744200. [DOI] [PubMed] [Google Scholar]
- 47.Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature. 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- 48.Wong M, Ziring D, Korin Y, Desai S, Kim S, Lin J, Gjertson D, Braun J, Reed E, Singh RR. TNFalpha blockade in human diseases: mechanisms and future directions. Clin Immunol. 2008;126:121–136. doi: 10.1016/j.clim.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Green N, Hu Y, Janz K, Li HQ, Kaila N, Guler S, Thomason J, Joseph-McCarthy D, Tam SY, Hotchandani R, Wu J, Huang A, Wang Q, Leung L, Pelker J, Marusic S, Hsu S, Telliez JB, Hall JP, Cuozzo JW, Lin LL. Inhibitors of tumor progression loci-2 (Tpl2) kinase and tumor necrosis factor alpha (TNF-alpha) production: selectivity and in vivo antiinflammatory activity of novel 8-substituted-4-anilino-6-aminoquinoline-3-carbonitriles. J Med Chem. 2007;50:4728–4745. doi: 10.1021/jm070436q. [DOI] [PubMed] [Google Scholar]
- 50.Gavrin LK, Green N, Hu Y, Janz K, Kaila N, Li HQ, Tam SY, Thomason JR, Gopalsamy A, Ciszewski G, Cuozzo JW, Hall JP, Hsu S, Telliez JB, Lin LL. Inhibition of Tpl2 kinase and TNF-alpha production with 1,7-naphthyridine-3-carbonitriles: synthesis and structure-activity relationships. Bioorg Med Chem Lett. 2005;15:5288–5292. doi: 10.1016/j.bmcl.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 51.Hall JP, Kurdi Y, Hsu S, Cuozzo J, Liu J, Telliez JB, Seidl KJ, Winkler A, Hu Y, Green N, Askew GR, Tam S, Clark JD, Lin LL. Pharmacologic inhibition of tpl2 blocks inflammatory responses in primary human monocytes, synoviocytes, and blood. J Biol Chem. 2007;282:33295–33304. doi: 10.1074/jbc.M703694200. [DOI] [PubMed] [Google Scholar]
- 52.Hu Y, Green N, Gavrin LK, Janz K, Kaila N, Li HQ, Thomason JR, Cuozzo JW, Hall JP, Hsu S, Nickerson-Nutter C, Telliez JB, Lin LL, Tam S. Inhibition of Tpl2 kinase and TNFalpha production with quinoline-3-carbonitriles for the treatment of rheumatoid arthritis. Bioorg Med Chem Lett. 2006;16:6067–6072. doi: 10.1016/j.bmcl.2006.08.102. [DOI] [PubMed] [Google Scholar]
- 53.Kaila N, Green N, Li HQ, Hu Y, Janz K, Gavrin LK, Thomason J, Tam S, Powell D, Cuozzo J, Hall JP, Telliez JB, Hsu S, Nickerson-Nutter C, Wang Q, Lin LL. Identification of a novel class of selective Tpl2 kinase inhibitors: 4-Alkylamino-[1,7]naphthyridine-3-carbonitriles. Bioorg Med Chem. 2007;15:6425–6442. doi: 10.1016/j.bmc.2007.06.054. [DOI] [PubMed] [Google Scholar]
- 54.Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- 55.Kontoyiannis D, Boulougouris G, Manoloukos M, Armaka M, Apostolaki M, Pizarro T, Kotlyarov A, Forster I, Flavell R, Gaestel M, Tsichlis P, Cominelli F, Kollias G. Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced Crohn's-like inflammatory bowel disease. J Exp Med. 2002;196:1563–1574. doi: 10.1084/jem.20020281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Flavell RA. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 57.Church LD, Cook GP, McDermott MF. Primer: inflammasomes and interleukin 1beta in inflammatory disorders. Nat Clin Pract Rheumatol. 2008;4:34–42. doi: 10.1038/ncprheum0681. [DOI] [PubMed] [Google Scholar]
- 58.Ehses JA, Boni-Schnetzler M, Faulenbach M, Donath MY. Macrophages, cytokines and beta-cell death in Type 2 diabetes. Biochem Soc Trans. 2008;36:340–342. doi: 10.1042/BST0360340. [DOI] [PubMed] [Google Scholar]
- 59.Farasat S, Aksentijevich I, Toro JR. Autoinflammatory diseases: clinical and genetic advances. Arch Dermatol. 2008;144:392–402. doi: 10.1001/archderm.144.3.392. [DOI] [PubMed] [Google Scholar]
- 60.Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517–1526. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.