

Abstract
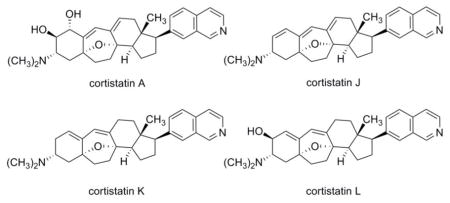
The cortistatins are a recently identified class of marine natural products characterized by an unusual steroidal skeleton and have been found to inhibit differentially the proliferation of various mammalian cells in culture by an unknown mechanism. We describe a comprehensive route for the synthesis of cortistatins from a common precursor, which in turn is assembled from two fragments of similar structural complexity. Cortistatins A and J, and for the first time, K and L, have been synthesized in parallel processes from like intermediates prepared from a single compound. With the identification of facile laboratory transformations linking intermediates in the cortistatin L synthetic series with corresponding intermediates to cortistatins A and J, we have been led to speculate that somewhat related paths might occur in nature, offering potential sequencing and chemical detail for cortistatin biosynthetic pathways.
Since the structure of cortistatin A was elucidated by Kobayashi and coworkers in 2006 more than 10 natural cortistatins have been described comprising a common modified steroidal skeleton with varying substitution of the A- and D-rings,1–3 many of these exhibiting profound effects upon cultured mammalian cells and human umbilical vein endothelial cells (HUVECs) in particular.1–4 Baran and coworkers reported the first laboratory synthetic route to cortistatin A,5 from prednisone, in 2008, and subsequent to this a number of different approaches to the synthesis of cortistatins have been pursued, with focus on the most highly functionalized (and potent) member of the family, cortistatin A (1). No fewer than three independent routes to cortistatin A have been reported to date,5–7 as well as a number of synthetic studies8–18 and formal routes.19,20 In addition, the Nicolaou–Chen group has described a synthesis of cortistatin J.21
Structure-activity relationships among natural cortistatins and synthetic analogs have suggested that the 7-substituted isoquinoline appendage is a key determinant of the phenotypic effects of cortistatins upon cells grown in culture.4,21–23 The Nicolaou–Chen group recently reported that cortistatin A inhibits the function of a number of different kinases in vitro and proposed that the natural product may occupy the ATP-binding site of at least one of the enzymes they identified.24 Prior to this, Kobayashi and coworkers had shown that the addition of cortistatin A to cultured HUVECs led to a marked decrease in levels of an unidentified 110 kDa phosphoprotein.4
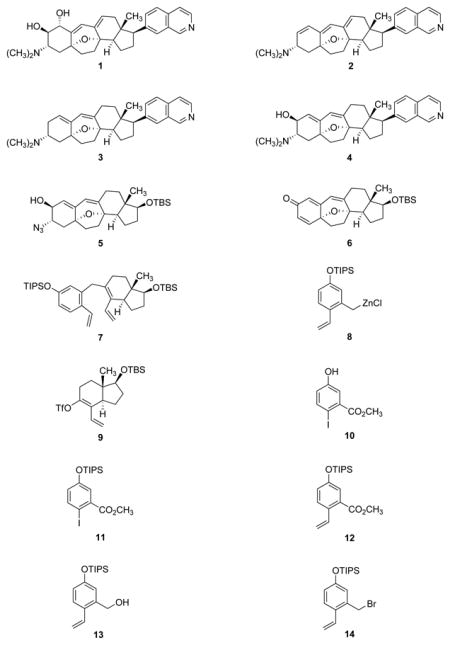
We envisioned that the naturally occurring cortistatins A, J, K, and L (1–4, respectively) and a diverse array of cortistatin analogs, including compounds suitable for use as biological probes in target identification experiments, could be prepared from the single azido alcohol intermediate 5, depicted in Figure 2.
Figure 2. A key precursor to cortistatins and its retrosynthetic disconnection.

The azido alcohol intermediate 5 serves as a common intermediate for the synthesis of cortistatin A, J, K, and L as well as cortistatin analogs. This intermediate arises in turn from the cyclohexadienone 6 and the triene 7. The latter intermediate is formed by coupling of the o-vinyl benzylzinc reagent 8 and the enol triflate intermediate 9. TBS = tert-butyldimethylsilyl; TIPS = triisopropylsilyl; Tf = trifluoromethanesulfonyl.
We considered it likely that one or more pathways linking the azido alcohol 5 to the cyclohexadienone 6 could be developed. The oxabicyclic core of the latter precursor could then be disconnected by an oxidative cyclization transform, a strategy independently conceived and previously demonstrated by Sarpong and coworkers, as well as others.8,15,16,18 To allow for a convergent assembly process, we imagined an earlier disconnection of the seven-membered B-ring by an olefin metathesis reaction, giving rise to the triene precursor 7, which we envisioned could be synthesized by coupling of the o-vinyl benzylzinc reagent 8 and the enol triflate intermediate 9, structures of similar complexity, but less than certain viability as stable chemical entities.
Results
The o-vinyl benzylzinc reagent 8, the A-ring precursor, was derived from the benzyl bromide 14, which was prepared in amounts up to 47-g by the four-step sequence shown in Figure 3 (via intermediates 10–13). Direct insertion of zinc25 into the carbon-bromine bond of the benzyl bromide 14 was attempted only once, without success; however, a two-stage process for the preparation of organozinc reagent 8 proved to be both highly reliable and readily scalable. Addition of benzyl bromide 14 to activated magnesium turnings26 in ether at 23°C led to rapid (<30 min) formation of a deep green solution of the corresponding Grignard reagent, contaminated with <5% of the product of Wurtz coupling. In contrast, formation of the Grignard reagent in tetrahydrofuran as solvent was accompanied by as much as 65% of the Wurtz coupling by-product. Addition of the freshly prepared ethereal Grignard product to a solution of anhydrous zinc chloride in tetrahydrofuran at 23 oC led to the formation of a cloudy white suspension of the presumed o-vinyl benzylzinc reagent 8, which was used directly for coupling with the enol triflate 9 (vide infra).
Figure 3. Preparation of the o-vinyl benzylzinc reagent 8.

The functionalized benzylzinc reagent 8 is prepared from the corresponding Grignard reagent and anhydrous zinc chloride. The Grignard reagent is readily formed from the benzyl bromide 14, which is prepared in multi-gram amounts by a four-step sequence. THF = tetrahydrofuran; Ph = phenyl; DIBAL-H = diisobutylaluminium hydride.
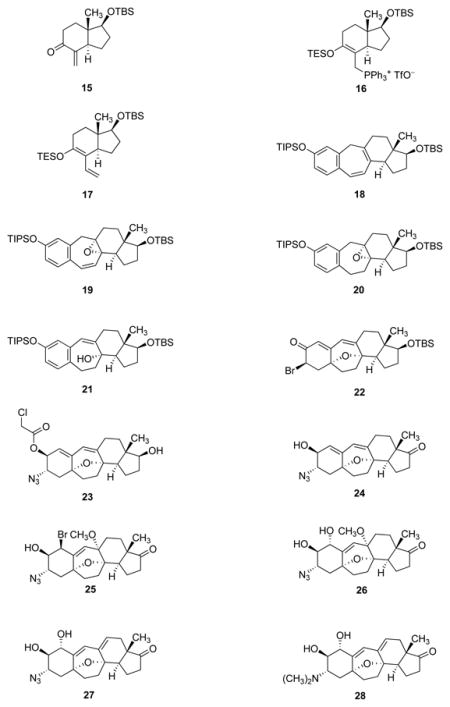
The enol triflate coupling partner 9, the CD-ring precursor, was synthesized in a three-step sequence from the known α-methylene ketone 15 (Figure 4). The latter substance was first prepared in its antipodal form by Danishefsky and coworkers27 (from the Hajos–Parrish enedione)28 in the context of their synthesis of taxol, and was also selected as a starting material by the Nicolaou-Chen6 and Sorensen18 groups in their research directed toward the synthesis of cortistatins. As illustrated in Figure 4, phosphoniosilylation29,30 of α-methylene ketone 15 with triphenylphosphine (1.02 equiv) and triethylsilyl trifluoromethanesulfonate (1.02 equiv) at −40°C in chloroform, uniquely effective as a solvent in this particular transformation, afforded the phosphonium salt 16 (containing a tetrasubstituted triethylsilyl enol ether), which was observed to be stable toward aqueous workup and extractive isolation. Heretofore, phosphoniosilylation-Wittig sequences have been conducted without isolation of the intermediate phosphonium salts;30 this was not an option in the present case, for chloroform was not compatible with formation of the Wittig reagent. The oily phosphonium salt obtained upon extractive isolation and concentration was dissolved in tetrahydrofuran, and after cooling to −78°C, the resulting solution was treated sequentially with n-butyllithium (1.1 equiv) and paraformaldehyde (5 equiv), affording the α-vinyl triethylsilyl enol ether 17 in 75% yield over the two steps from the α-methylene ketone 15. Methodology for triflation developed by Corey and coworkers was used to transform the triethylsilyl enol ether 17 into the corresponding enol triflate derivative 9 (92% yield after purification by flash-column chromatography).31 This product, a colorless oil, was stored frozen as a solution in benzene for not longer than a few days; decomposition was observed to occur over longer periods of time.
Figure 4. Synthesis of the azido alcohol 5 from the α-methylene ketone 15.

The α-methylene ketone 15 is transformed in three steps to the enol triflate intermediate 9. Coupling with the functionalized benzylzinc reagent 8 affords the triene 7, which undergoes efficient ring-closing metathesis and oxidation (in situ, with dimethyldioxirane) to form the tetracyclic diene monoepoxide 19. A sequence of hydrogenation, eliminative epoxide opening, silyl ether cleavage, and oxidative cyclization provides the cyclohexadienone 6, which is converted stereoselectively in three steps to the key azido alcohol intermediate 5. TES = triethylsilyl; DME = 1,2-dimethoxyethane; Pd2dba3 = tris(dibenzylideneacetone)dipalladium(0); S-Phos = 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl; DMDO = dimethyl dioxirane; TBAF = tetra-n-butylammonium fluoride; Py = pyridine; NBS = N-bromosuccinimide; TMG azide = tetramethylguanidinium azide; CBS = Corey-Bakshi-Shibata.
Coupling of the enol triflate 9 (1 equiv) with the benzylzinc reagent 8 (~1.2 equiv, a solution in ether–tetrahydrofuran) in the presence of tris(dibenzylideneacetone)dipalladium (0.045 equiv) and the ligand 2-dicyclohexylphosphino-2′,6′-dimethoxy-biphenyl (S-Phos, 0.18 equiv)32 in N-methylpyrrolidone at 70°C (20 h) afforded the metathesis substrate 7 in 70% yield on a 16-g scale. Warming of a 0.2 M solution of the latter product with the second-generation Grubbs catalyst (0.025 equiv)33 in dichloromethane at 45°C for 5 h led to highly efficient ring-closing metathesis, forming the tetracyclic diene 18 (>95% yield, 11-g scale). In our initial route design we had planned to hydrogenate selectively the disubstituted alkene within this product and then epoxidize the remaining (tetra-substituted) alkene; however, we were not able to achieve selective 1,2-reduction of the diene. Sarpong and coworkers have reported the successful execution of this exact two-step sequence, albeit using a substrate with different A-ring substituents (1-methoxyl and 2-p-methoxybenzyl groups).8
We found that we could achieve the desired transformation (18→20) by reversing the order of the two operations and in so doing we were able to avoid an isolation step. Thus, direct addition of a solution of dimethyldioxirane (1.5 equiv) in acetone34 to the unpurified metathesis product solution at 0°C led to stereoselective epoxidation of the tetrasubstituted alkene, affording the acid-sensitive tetracyclic diene monoepoxide 19. Without purification, the epoxide 19 was hydrogenated in benzene under 500 psi hydrogen in the presence of a mixture of Wilkinson’s catalyst (0.15 equiv) and suspended solid sodium bicarbonate (1.5 equiv), the latter added as an acid scavenger. The saturated product, epoxide 20, then underwent selective eliminative opening with lithium diethylamide (2 equiv) in tetrahydrofuran at −15°C,35 furnishing the conjugated allylic alcohol 21, a sufficiently polar intermediate to allow for facile chromatographic isolation (>95% purity, 1H NMR analysis, 50% yield over the four-step sequence). Cleavage of the triisopropylsilyl protective group with tetra-n-butylammonium fluoride (1.1 equiv) in tetrahydrofuran and oxidative cyclization of the resulting phenol with [bis(trifluoroacetoxy)iodo]benzene (1.8 equiv) in a mixture of dichloromethane and hexafluoroisopropanol provided the cyclohexadienone 6 in 50% yield.8,18,36
The synthesis of the key azido alcohol intermediate 5 was achieved in three steps from the cyclohexadienone 6, a sequence initiated by hydrosilylation with triethylsilane (2 equiv) in the presence of Wilkinson’s catalyst (0.05 equiv).37 The intermediate triethylsilyl enol ether was not isolated; rather, pyridine was added (14% by volume) followed by N-bromosuccinimide (2 equiv), forming the (3R)-bromo ketone 22 as a single diastereomer in 70% yield. In the absence of pyridine, or with lesser quantities of pyridine, the bromination reaction was much less stereoselective, which evidence suggests was related to the presence of Wilkinson’s catalyst during the bromination (bromination of the triethylsilyl enol ether generated in the absence of Wilkinson’s catalyst was highly diastereoselective). Bromide displacement with tetramethylguanidinium azide (2 equiv) proceeded with clean inversion of C3-stereochemistry and this was followed by direct reduction of the resulting (3S)-α-azido ketone with catecholborane (2 equiv) in the presence of the (R)-Corey–Bakshi–Shibata catalyst38 (0.2 equiv, −40°C) and tetramethylguanidine (1 equiv), producing a 15:1 mixture of the (2S)- and (2R)-configured alcohols, respectively, in 85% yield after chromatographic isolation. Similar stereocontrol of the C3-center could not be achieved with other reductants examined (lithium tri-tert-butoxyaluminum hydride was the next most selective reductant, affording a 5:1 mixture of products favoring the (2S)-isomer).
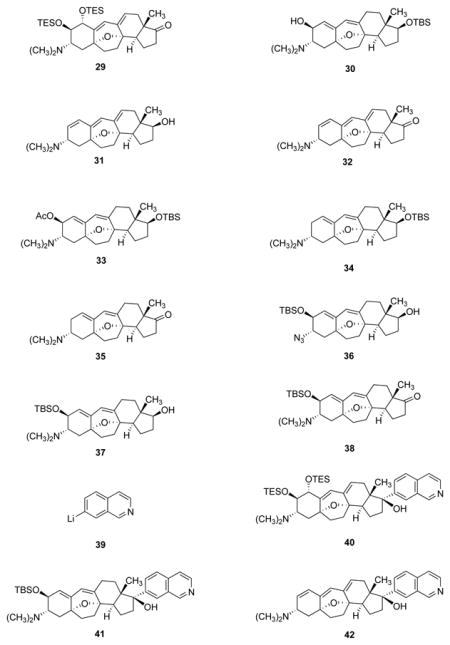
Shown in parallel in Figure 5 are the sequential pathways by which the key intermediate 5 was transformed into the corresponding 17-keto precursors to cortistatins A, J, K, and L. The synthetic route to the 17-keto precursor to cortistatin A (29) is depicted first, and was initiated by protection of the 2-hydroxyl substituent of intermediate 5 with excess chloroacetyl chloride in pyridine. Cleavage of the tert-butyldimethylsilyl ether within the resulting product occurred upon exposure to aqueous hydrofluoric acid in a 1:1 mixture of acetonitrile and tetrahydrofuran at 0°C, affording the alcohol 23 in 80% yield. Significantly, we observed that if during the latter transformation the reaction mixture was allowed to warm from 0 to 23°C, 1,6-elimination of the chloroacetate ester accompanied deprotection of the tert-butyldimethylsilyl ether, an observation later used to advantage in the synthesis of the 17-keto precursor to cortistatin J (32). Oxidation of alcohol 23 with the Dess–Martin periodinane (2.4 equiv) in dichloromethane at 0 oC followed by addition of methanol and solid potassium carbonate gave in a single operation the keto alcohol 24, in 85% yield. Addition of N-bromosuccinimide (1.02 equiv) to a solution of keto alcohol 24 in a 4:1 mixture of acetonitrile and methanol, respectively, at 23°C led to stereoselective (trans-diaxial) 1,4-bromoetherification of the conjugated diene, forming the bromide 25 which, without purification, was directly subjected to nucleophilic displacement at 0°C with potassium superoxide (2.1 equiv) in a 3:1 mixture of toluene and dimethyl sulfoxide, respectively.39,40 After extractive isolation and chromatography, the trans-diol methyl ether 26 was obtained in diastereomerically pure form (41% yield, 100-mg scale, two steps). When combined with scandium triflate (0.03 equiv) and trifluoroacetic acid (9.4 equiv) in dioxane at 23°C, this substance was found to undergo efficient 1,2-elimination of methanol, affording the azido diol 27 (94% yield). The methoxyl substituent of substrate 26 is axially oriented and therefore stereoelectronically disposed toward elimination; none of the other allylic C–O bonds of substrate 26 (nor product 27) is so oriented. Reductive (di)methylation of the azido group and diol protection with chlorotriethylsilane then furnished the protected 17-keto cortistatin A precursor (29) in 65% yield for the two steps (75-mg scale).
Figure 5. Synthetic pathways to 17-keto precursors to cortistatins A, J, K, and L from the azido alcohol 5.

a, Synthesis of the 17-keto precursor to cortistatin A (29) by an eight-step sequence. b, A three-step sequence to the 17-keto precursor to cortistatin J (32). c, Synthesis of the 17-keto precursor to cortistatin K (35) by a five-step sequence. d, A four-step sequence to the 17-keto precursor to cortistatin L (38). DMP = Dess-Martin periodinane;18-Cr-6 = 18-crown-6; TFA = trifluoroacetic acid; DMAP = 4-dimethylaminopyridine; Ac = acetyl; DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene.
Syntheses of the 17-keto precursors to cortistatins J, K, and L were substantially more direct (Figure 5). The 17-keto cortistatin J precursor (32) was synthesized in just three steps from the azido alcohol 5. Following reductive (di)methylation to form the dimethylamino alcohol 30 (85% yield), 1,6-elimination of water and deprotection of the tert-butyldimethylsilyl ether occurred simultaneously upon stirring the dimethylamino alcohol 30 with a biphasic mixture of chloroform and concentrated hydrochloric acid (20 min, 23°C). Without purification, the product of the latter transformation (31) was subjected to oxidation with the Dess–Martin periodinane, providing the 17-keto cortistatin J precursor (32) as a white solid (77% yield following chromatographic purification, two steps).
The synthetic route to the 17-keto precursor to cortistatin K (35) also proceeded through the dimethylamino alcohol 30 (derived from the azido alcohol 5, as described in the paragraph above), in a four-step sequence that was initiated by acetylation at 0°C with acetic anhydride (a large excess) and scandium triflate as catalyst (0.05 equiv) in a 1:1 mixture of acetonitrile and dichloromethane (93% yield).41 Regioselective reductive cleavage of the allylic C–O bond of the resulting acetate occurred in the presence of lithium borohydride (excess) and tetrakis(triphenylphosphine)-palladium (0.2 equiv).42 This afforded the dimethylamino diene 34 in complex with borane; decomplexation was achieved by treatment of this intermediate with Raney nickel in methanol,43 providing the free amine as a pale yellow solid (90% yield after chromatographic purification over Davisil® silica gel). Silyl ether cleavage (tetra-n-butylammonium fluoride, 87% yield) and oxidation (Dess–Martin periodinane, 90%), then completed the route to the 17-keto cortistatin K precursor (35), also obtained as a pale yellow solid.
Lastly, the 17-keto precursor to cortistatin L (38) was synthesized from the azido alcohol 5 by a straightforward four-step sequence involving silyl ether deprotection (conducted at 0°C, as in the cortistatin A series, to avoid 1,6-elimination), selective protection,44 reductive (di)methylation, and oxidation (Figure 5, 60% yield overall).
Depicted in Figure 6 are the parallel paths by which each advanced 17-keto cortistatin precursor was transformed into the corresponding cortistatin. The method we used to introduce stereoselectively the 7-isoquinolyl substituent differs from earlier reported syntheses of cortistatin A, where in each case a variation of the sequence 17-keto→vinyl iodide5,7 or vinyl triflate6 followed by cross-coupling with a 7-metalloisoquinoline derivative, then stereoselective hydrogenation was pursued. The hydrogenation step in particular has been established to be challenging to execute with intermediates containing the conjugated diene function of cortistatin A5,7 and we gauged it as likely that this would be even more problematic with precursors to cortistatins J, K, and L. As a general alternative, we developed the three-step 17-keto addition–reductive deoxygenation sequence shown in Figure 6. While our work was in progress, Hirama and coworkers reported a conceptually similar but differentially executed approach to introduce the isoquinolyl substituent in their formal synthesis of cortistatin A.19 We observed that lithiation of 7-iodoisoquinoline (5 equiv) with an equimolar amount of n-butyllithium in tetrahydrofuran at −78°C followed by sequential addition of N, N, N′, N′-tetramethylethylenediamine (TMEDA, 15 equiv) and a 17-keto cortistatin precursor (1 equiv) reproducibly afforded the corresponding (17S)-1,2-addition product in 52–62% yield in all cases.45 In the absence of TMEDA, 1,2-addition did not occur, undoubtedly a consequence of competing enolization of the 17-keto substituent. Enolization likely occurs, but to a much lesser extent, in our optimized conditions as well (in the presence of TMEDA) for we typically recover between 34–40% of each 17-keto precursor. The Hirama group developed a very different protocol for metalation (employing n-butyllithium and cerium (III) chloride with 1-chloro-7-iodoisoquinoline as substrate) and with this organometallic reagent observed highly efficient addition to the carbonyl group of the Nicolaou–Chen 17-keto cortistatin A precursor. They then transformed the resulting tertiary alcohol into a thionocarbamate derivative and the latter was deoxygenated using tri-n-butyltin hydride.19
Figure 6. Syntheses of cortistatins A, L, J, and K.

Parallel paths of 17-keto addition of 7-lithioisoquinoline followed by reductive deoxygenation transform each 17-keto cortistatin precursor into the corresponding cortistatin. TMEDA = N, N, N′, N′-tetramethylethylenediamine; TFAA = trifluoroacetic anhydride; AIBN = 2,2′-azobis(2-methylpropionitrile).
We briefly explored a similar method for hydroxyl activation in our deoxygenation studies but our attempts to form a thionocarbonyl ester intermediate were unsuccessful. Instead, we adapted methodology by Jang and coworkers for free-radical deoxygenation of alcohols via their trifluoroacetate esters.46,47 As shown in Figure 6, each tertiary alcohol intermediate was readily transformed into the corresponding trifluoroacetate ester with trifluoroacetic anhydride and pyridine–N, N-dimethylaminopyridine. When heated with tri-n-butyltin hydride (excess) and azobisisobutyronitrile in benzene in a sealed tube at 100°C for 1–2 h, each trifluoroacetate ester underwent efficient deoxygenation to provide the (17S)-configured reduction product. Attempted deoxygenation under the conditions originally reported by Jang et al. (Ph2SiH2, (t-BuO)2, 130°C)46,47 led primarily to elimination of the trifluoroacetate ester, a problem that was almost completely circumvented by the use of tri-n-butyltin hydride as reductant at 100°C.
In the cortistatin J and K series, deoxygenation led to these substances directly; spectroscopic data for the synthetic materials matched values reported for the natural products. In the cortistatin A and L series, cleavage of the silyl ether protective group(s) with tetra-n-butylammonium fluoride at 23°C following deoxygenation provided these targets; spectroscopic data obtained from the synthetic substances were found to be the same as those reported for the natural products. In all four series (A, J, K, and L), the yield for the two- or three-step deoxygenation (deprotection) procedure was 61–72% (1–20-mg amounts of final product).
In addition to spectroscopic characterization, synthetic cortistatins A, J, K, and L were evaluated for their ability to inhibit the growth of HUVECs in culture (96-h incubation). The GI50 values we measured are summarized in Figure 1 alongside literature values. For synthetic cortistatin A we observed growth inhibition of HUVECs consistent with reported values, while measurements for synthetic cortistatin J more closely matched reports from the Nicolaou–Chen group than initial reports, and GI50 values for synthetic cortistatins K and L fell within the range of those originally reported to slightly higher.1,3,21
Figure 1. Cortistatins A, J, K, and L and their IC50 values measured in cultured HUVECs.

Members of the cortistatin natural product family comprise a common modified steroidal skeleton with varying substitutions, primarily within the A, B, and C rings. These substitutional variations are found to modulate the potencies of natural cortistatins as growth inhibitors of human umbilical vein endothelial cells (HUVECs) in culture.
Discussion
In the course of our studies we observed that several compounds closely related to cortistatin L undergo facile 1,6-elimination to form the conjugated triene function of cortistatin J (see 30→31, Figure 5, discussion of intermediate 23 above, and the additional example of the transformation of dienyl alcohol 30 to triene 44, Figure 7). In an interesting and somewhat related transformation, we observed that reaction of diene 45 (cortistatin L oxidation state) with dimethyldioxirane afforded the highly sensitive epoxide 46 as primary product, which upon standing in benzene solution underwent spontaneous1,4-elimination to form a product (dienyl alcohol 47) with functionality characteristic of cortistatin A. Consideration of these findings has led us to speculate that cortistatins A and J may derive from cortistatin L or a precursor of the same oxidation state, as opposed to an alternative sequencing such as J→A (although this cannot be ruled out).2 Cortistatin L might in turn be derived by C2 oxidation of cortistatin K. We note that many sterols bearing 2β-hydroxy and 2β-sulfato groups have been isolated from marine sponges, but detailed biosynthetic routes to these substances have not been determined.48,49
Figure 7. Chemical interconversions among intermediates of cortistatin series A, J, and L and a hypothesized parallel biosynthetic sequence.

Intermediates of the cortistatin L series are found to undergo facile laboratory transformations to form corresponding intermediates of the A and J series, suggesting a plausible sequencing for the latter stages of cortistatin biosyntheses, which are unknown. Ms = methanesulfonyl.
We have shown that the protected azido alcohol intermediate 5, synthesized in a nine-step sequence beginning with the coupling of the benzylzinc reagent 8 and the enol triflate 9, is readily transformed into advanced 17-keto precursors to cortistatins A, J, K, and L. Each of these intermediates is in turn converted into the corresponding cortistatin final product by a three- or four-step sequence involving addition of a 7-isoquinolyl organometallic intermediate followed by deoxygenation (deprotection). The latter sequence appears to be a general route to cortistatins with divergent substitutions of the A, B, and C rings and it is anticipated that it will allow for late-stage introduction of diversely substituted isoquinoline groups and other heterocycles at position C17.
Supplementary Material
Acknowledgments
Financial support from the National Institutes of Health (Stimulus Grant No. CA047148-22S1) is gratefully acknowledged. A.N.F. acknowledges a scholarship from Eli Lilly and Company. We gratefully acknowledge Dr. Ge Zou for measuring the GI50 values of cortistatins A, J, K, and L. We acknowledge helpful discussions with Professor Christopher T. Walsh and Dr. Emily Balskus.
| 1 | cortistatin A (1) |
| 2 | cortistatin J (2) |
| 3 | cortistatin K (3) |
| 4 | cortistatin L (4) |
| 5 | azido alcohol 5 |
| 6 | cyclohexatrienone 6 |
| 7 | triene 7 |
| 8 | o –vinyl benzylzinc reagent 8 |
| 9 | enol triflate 9 |
| 10 | methyl 5-hydroxy-2-iodobenzoate (10) |
| 11 | methyl 2-iodo-5-(triisopropylsilyloxy)benzoate (11) |
| 12 | methyl 5-(triisopropylsilyloxy)-2-vinylbenzoate (12) |
| 13 | (5-(triisopropylsilyloxy)-2-vinylphenyl)methanol (13) |
| 14 | (3-(bromomethyl)-4-vinylphenoxy)triisopropylsilane (14) |
| 15 | α-methylene ketone 15 |
| 16 | triethylsilyl enol ether phosphonium salt 16 |
| 17 | triethylsilyl dienol ether 17 |
| 18 | tetracyclic diene 18 |
| 19 | tetracyclic diene monoepoxide 19 |
| 20 | tetracyclic epoxide 20 |
| 21 | allylic alcohol 21 |
| 22 | bromo ketone 22 |
| 23 | chloroacetate ester 23 (cortistatin A series) |
| 24 | dienyl keto alcohol 24 (cortistatin A series) |
| 25 | allylic bromide 25 (cortistatin A series) |
| 26 | trans diol methyl ether 26 (cortistatin A series) |
| 27 | dienyl azido diol 27 (cortistatin A series) |
| 28 | dimethylamino keto diol 28 (cortistatin A series) |
| 29 | dimethylamino ketone 29 (cortistatin A series) |
| 30 | dimethylamino alcohol 30 (cortistatin J and K series) |
| 31 | trienyl alcohol 31 (cortistatin J series) |
| 32 | dimethylamino ketone 32 (cortistatin J series) |
| 33 | acetate 33 (cortistatin K series) |
| 34 | tert- butyldimethylsilyl ether 34 (cortistatin K series) |
| 35 | dimethylamino ketone 35 (cortistatin K series) |
| 36 | azido tert -butyldimethylsilyl ether 36 (cortistatin L series) |
| 37 | dimethylamino alcohol 37 (cortistatin L series) |
| 38 | dimethylamino ketone 38 (cortistatin L series) |
| 39 | 7-lithioisoquinoline (39) |
| 40 | isoquinolyl alcohol 40 (cortistatin A series) |
| 41 | isoquinolyl alcohol 41 (cortistatin L series) |
| 42 | isoquinolyl alcohol 42 (cortistatin J series) |
| 43 | isoquinolyl alcohol 43 (cortistatin K series) |
| 44 | trienyl tert- butyldimethylsilyl ether 44 |
| 45 | dienyl bromo triethylsilyl ether 45 |
| 46 | allylic epoxide 46 |
| 47 | dienyl alcohol 47 |
| 48 | 5-hydroxy-2-iodobenzoic acid (48) |
| 49 | 7-isoquinolyl trifluoromethylsulfonate (49) |
| 50 | 7-trimethylstannylisoquinoline (50) |
| 51 | 7-iodoisoquinoline (51) |
| 52 | cortistatin A bis(triethylsilyl) ether (52) |
| 53 | dimethylamino alcohol 53 (cortistatin K series) |
| 54 | dienyl diol 54 (cortistatin L series) |
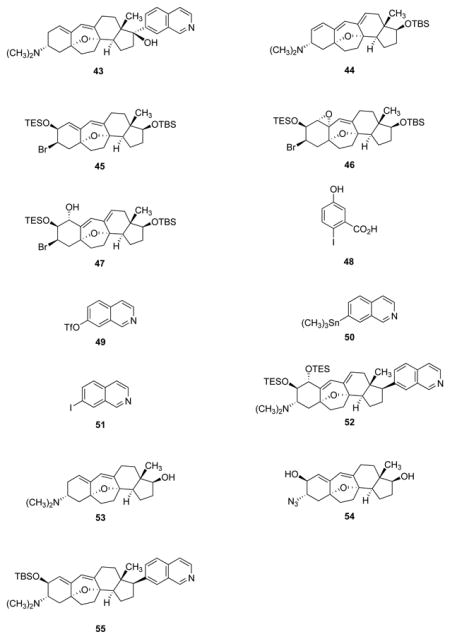
| 55 | cortistatin L tert -butyldimethylsilyl ether (55) |
Footnotes
Author contributions
A.N.F., C.S., and A.G.M conceived the synthetic route. A.N.F and C.S. conducted all experimental work and analyzed the results. A.N.F., C.S., and A.G.M. wrote the manuscript.
Additional information
Detailed experimental procedures and tabulated spectroscopic data (1H and 13C NMR, FT-IR, and HRMS) for all new compounds. This material is available free of charge via the Internet at www.nature.com/naturechemistry.
Contributor Information
Alec N. Flyer, Email: aflyer@fas.harvard.edu, Harvard Dept. of Chemistry and Chemical Biology,12 Oxford St, Box 408 Cambridge, MA 02138
Chong Si, Email: csi@fas.harvard.edu, Harvard Dept. of Chemistry and Chemical Biology,12 Oxford St, Box 485 Cambridge, MA 02138.
Andrew G. Myers, Email: myers@chemistry.harvard.edu, Harvard Dept. of Chemistry and Chemical Biology,12 Oxford St, Naito 201 Cambridge, MA 02138, Work Telephone Number: 617-495-5718, Fax Number: 617-495-4976
References
- 1.Aoki S, Watanabe Y, Sanagawa M, Setiawan A, Kotoku N, Kobayashi M. Cortistatins A, B, C, and D, anti-angiogenic steroidal alkaloids, from the marine sponge Corticium simplex. J Am Chem Soc. 2006;128:3148–3149. doi: 10.1021/ja057404h. [DOI] [PubMed] [Google Scholar]
- 2.Watanabe Y, Aoki S, Tanabe D, Setiawan A, Kobayashi M. Cortistatins E, F, G, and H, four novel steroidal alkaloids from marine sponge Corticium simplex. Tetrahedron. 2007;63:4074–4079. [Google Scholar]
- 3.Aoki S, Watanabe Y, Tanabe D, Setiawan A, Arai M, Kobayashi M. Cortistatins J, K, L, novel abeo-9(10–19)-androstane-type steroidal alkaloids with isoquinoline unit, from marine sponge Corticium simplex. Tetrahedron Lett. 2007;48:4485–4488. [Google Scholar]
- 4.Aoki S, Watanabe Y, Tanabe D, Arai M, Suna H, Miyamoto K, Tsujibo H, Tsujikawa K, Yamamoto H, Kobayashi M. Structure activity relationship and biological property of cortistatins, anti-angiogenic spongean steroidal alkaloids. Bioorg Med Chem. 2007;15:6758–6762. doi: 10.1016/j.bmc.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 5.Shenvi RA, Guerrero CA, Shi J, Li CC, Baran PS. Synthesis of (+)-cortistatin A. J Am Chem Soc. 2008;130:7241–7243. doi: 10.1021/ja8023466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nicolaou KC, Sun YP, Peng XS, Polet D, Chen DYK. Total synthesis of (+)-cortistatin A. Angew Chem, Int Ed. 2008;47:7310–7313. doi: 10.1002/anie.200803550. [DOI] [PubMed] [Google Scholar]
- 7.Lee HM, Nieto-Oberhuber C, Shair MD. Enantioselective synthesis of (+)-cortistatin A, a potent and selective inhibitor of endothelial cell proliferation. J Am Chem Soc. 2008;130:16864–16866. doi: 10.1021/ja8071918. [DOI] [PubMed] [Google Scholar]
- 8.Simmons EM, Hardin AR, Guo X, Sarpong R. Rapid construction of the cortistatin pentacyclic core. Angew Chem, Int Ed. 2008;47:6650–6653. doi: 10.1002/anie.200802203. [DOI] [PubMed] [Google Scholar]
- 9.Yamashita S, Iso K, Hirama M. A concise synthesis of the pentacyclic framework of cortistatins. Org Lett. 2008;10:3413–3415. doi: 10.1021/ol8012099. [DOI] [PubMed] [Google Scholar]
- 10.Craft DT, Gung BW. The first transannular [4+3] cycloaddition reaction: synthesis of the ABCD ring structure of cortistatins. Tetrahedron Lett. 2008;49:5931–5934. doi: 10.1016/j.tetlet.2008.07.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dai MJ, Danishefsky SJ. A concise synthesis of the cortistatin core. Tetrahedron Lett. 2008;49:6610–6612. doi: 10.1016/j.tetlet.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dai MJ, Wang Z, Danishefsky SJ. A novel α,β unsaturated nitrone-aryne [3+2] cycloaddition and its application in the synthesis of the cortistatin core. Tetrahedron Lett. 2008;49:6613–6616. doi: 10.1016/j.tetlet.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kotoku N, Sumii Y, Hayashi T, Kobayashi M. Synthesis of CD-ring structure of cortistatin A, an anti-angiogenic steroidal alkaloid from marine sponge. Tetrahedron Lett. 2008;49:7078–7081. [Google Scholar]
- 14.Kurti L, Czako B, Corey EJ. A short, scalable synthesis of the carbocyclic core of the anti-angiogenic cortistatins from (+)-estrone by B-ring expansion. Org Lett. 2008;10:5247–5250. doi: 10.1021/ol802328n. [DOI] [PubMed] [Google Scholar]
- 15.Dai MJ, Danishefsky SJ. An oxidative dearomatization cyclization model for cortistatin A. Heterocycles. 2009;77:157–161. [Google Scholar]
- 16.Liu LZ, Gao YX, Che C, Wu N, Wang DZ, Li CC, Yang Z. A model study for the concise construction of the oxapentacyclic core of cortistatins through intramolecular Diels–Alder and oxidative dearomatization cyclization reactions. Chem Commun. 2009:662–664. doi: 10.1039/b817376a. [DOI] [PubMed] [Google Scholar]
- 17.Magnus P, Littich R. Intramolecular cyclopropene-furan [2+4] cycloaddition followed by a cyclopropylcarbinyl rearrangement to synthesize the BCD rings of cortistatin A. Org Lett. 2009;11:3938–3941. doi: 10.1021/ol901537n. [DOI] [PubMed] [Google Scholar]
- 18.Frie JL, Jeffrey CS, Sorensen EJ. A hypervalent iodine-induced double annulation enables a concise synthesis of the pentacyclic core structure of the cortistatins. Org Lett. 2009;11:5394–5397. doi: 10.1021/ol902168g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamashita S, Kitajima K, Iso K, Hirama M. Efficient and stereoselective installation of isoquinoline: formal total synthesis of cortistatin A. Tetrahedron Lett. 2009;50:3277–3279. [Google Scholar]
- 20.Simmons EM, Hardin-Narayan AR, Guo X, Sarpong R. Formal total synthesis of (±)-cortistatin A. Tetrahedron. 2010 doi: 10.1016/j.tet.2010.01.030. article in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicolaou KC, Peng XS, Sun YP, Polet D, Zou B, Lim CS, Chen DYK. Total synthesis and biological evaluation of cortistatins A and J and analogues thereof. J Am Chem Soc. 2009;131:10587–10597. doi: 10.1021/ja902939t. [DOI] [PubMed] [Google Scholar]
- 22.Shi J, Shigehisa H, Guerrero CA, Shenvi RA, Li CC, Baran PS. Stereodivergent synthesis of 17-α and 17-β-aryl steroids: application and biological evaluation of D-ring cortistatin analogues. Angew Chem, Int Ed. 2009;48:4328–4331. doi: 10.1002/anie.200901116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Czako B, Kurti L, Mammoto A, Ingber DE, Corey EJ. Discovery of potent and practical antiangiogenic agents inspired by cortistatin A. J Am Chem Soc. 2009;131:9014–9019. doi: 10.1021/ja902601e. [DOI] [PubMed] [Google Scholar]
- 24.Cee VJ, Chen DYK, Lee MR, Nicolaou KC. Cortistatin A is a high-affinity ligand of protein kinases ROCK, CDK8, and CDK11. Angew Chem, Int Ed. 2009;48:8952–8957. doi: 10.1002/anie.200904778. [DOI] [PubMed] [Google Scholar]
- 25.Berk SC, Knochel P, Yeh MCP. General approach to highly functionalized benzylic organometallics of zinc and copper. J Org Chem. 1988;53:5789–5791. [Google Scholar]
- 26.Pearson DE, Cowan D, Beckler JD. A study of the entrainment method for making Grignard reagents. J Org Chem. 1959;24:504–509. [Google Scholar]
- 27.Isaacs RCA, Digrandi MJ, Danishefsky SJ. Synthesis of an enantiomerically pure intermediate containing the CD substructure of taxol. J Org Chem. 1993;58:3938–3941. [Google Scholar]
- 28.Hajos ZG, Parrish DR. Organic Syntheses. Vol. 63. Wiley & Sons; New York: 1985. (+)-(7aS)-7a-methyl-2,3,7,7a-tetrahydro-1H-indene-1,5-6H-dione; pp. 26–31. [Google Scholar]
- 29.Evans DA, Hurst KM, Takacs JM. New silicon-phosphorus reagents in organic synthesis: carbonyl and conjugate addition reactions of silicon phosphite esters and related systems. J Am Chem Soc. 1978;100:3467–3477. [Google Scholar]
- 30.Kozikowski AP, Jung SH. Phosphoniosilylation: an efficient and practical method for the β-functionalization of enones. J Org Chem. 1986;51:3400–3402. [Google Scholar]
- 31.Mi Y, Schreiber JV, Corey EJ. Total synthesis of (+)-α-onocerin in four steps via four-component coupling and tetracyclization steps. J Am Chem Soc. 2002;124:11290–11291. doi: 10.1021/ja027373f. [DOI] [PubMed] [Google Scholar]
- 32.Walker SD, Barder TE, Martinelli JR, Buchwald SL. A rationally designed universal catalyst for Suzuki-Miyaura coupling processes. Angew Chem, Int Ed. 2004;43:1871–1876. doi: 10.1002/anie.200353615. [DOI] [PubMed] [Google Scholar]
- 33.Scholl M, Ding S, Lee CW, Grubbs RH. Synthesis and activity of a new generation of ruthenium-based olefin metathesis catalysts coordinated with 1,3-dimesityl-4,5-dihydroimidazol-2-ylidene ligands. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 34.Murray RW, Singh M. Organic Syntheses. Vol. 74. Wiley & Sons; New York: 1997. Synthesis of epoxides using dimethyldioxirane: trans-stilbene oxide; pp. 91–100. [Google Scholar]
- 35.Thummel RP, Rickborn B. Base-induced rearrangement of epoxides. V Phenyl-substituted epoxides. J Org Chem. 1972;37:3919–3923. [Google Scholar]
- 36.Kita Y, Tohma H, Kikuchi K, Inagaki M, Yakura T. Hypervalent iodine oxidation of N-acyltyramines: synthesis of quinol ethers, spirohexadienones, and hexahydroindol-6-ones. J Org Chem. 1991;56:435–438. [Google Scholar]
- 37.Kogure T, Ojima I. Reduction of carbonyl compounds via hydrosilylation. 4 Highly regioselective reductions of α,β-unsaturated carbonyl compounds. Organometallics. 1982;1:1390–1399. [Google Scholar]
- 38.Corey EJ, Helal CJ. Reduction of carbonyl compounds with chiral oxazaborolidine catalysts: a new paradigm for enantioselective catalysis and a powerful new synthetic method. Angew Chem, Int Ed. 1998;37:1986–2012. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 39.San Filippo J, Chern CI, Valentine JS. Reaction of superoxide with alkyl halides and tosylates. J Org Chem. 1975;40:1678–1680. [Google Scholar]
- 40.Corey EJ, Nicolaou KC, Shibasaki M, Machida Y, Shiner CS. Superoxide ion as a synthetically useful oxygen nucleophile. Tetrahedron Lett. 1975;37:3183–3186. [Google Scholar]
- 41.Ishihara K, Kubota M, Kurihara H, Yamamoto H. Scandium trifluoromethanesulfonate as an extremely active acylation catalyst. J Am Chem Soc. 1995;117:4413–4414. doi: 10.1021/jo952237x. [DOI] [PubMed] [Google Scholar]
- 42.Hutchins RO, Learn K, Fulton RP. Reductive displacement of allylic acetates by hydride transfer via catalytic activation by palladium(0) complexes. Tetrahedron Lett. 1980;21:27–30. [Google Scholar]
- 43.Couturier M, Tucker JL, Andresen BM, Dube P, Negri JT. Palladium and Raney nickel catalyzed methanolic cleavage of stable borane–amine complexes. Org Lett. 2001;3:465–467. doi: 10.1021/ol006969+. [DOI] [PubMed] [Google Scholar]
- 44.Moon SS, Stuhmiller LM, McMorris TC. Synthesis of oogoniol. J Org Chem. 1989;54:26–28. [Google Scholar]
- 45.Foy N, Stephan E, Vessieres A, Salomon E, Heldt JM, Huche M, Jaouen G. Synthesis, receptor binding, molecular modeling, and proliferative assays of a series of 17α-arylestradiols. Chembiochem. 2003;4:494–503. doi: 10.1002/cbic.200200499. See also: Ref 21. [DOI] [PubMed] [Google Scholar]
- 46.Jang DO, Kim JG, Cho DH, Chung CM. Radical deoxygenation of alcohols via their trifluoroacetate derivatives with diphenylsilane. Tetrahedron Lett. 2001;42:1073–1075. [Google Scholar]
- 47.Kim JG, Cho DH, Jang DO. Radical deoxygenation of tertiary alcohols via trifluoroacetates. Tetrahedron Lett. 2004;45:3031–3033. [Google Scholar]
- 48.D’ Auria MV, Minale L, Riccio R. Polyoxygenated steroids of marine origin. Chem Rev. 1993;93:1839–1895. [Google Scholar]
- 49.Sarma NS, Krishna MSR, Rao SR. Sterol ring system oxidation pattern in marine sponges. Mar Drugs. 2005;3:84–111. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.