

Until recently, the large majority of studies of the origins of DNA replication fidelity have concluded that complementarity of hydrogen bonding is the chief source of energetic selectivity between the four nucleotides at the transition state for initial insertion.[1,2] As part of this field of study, a wide number of modified nucleoside analogues have been incorporated into DNA in an effort to study the origins of mutagenesis and mechanisms for the fidelity of DNA replication.[3] Virtually all of these differed from natural nucleosides both in their hydrogen bonding arrangement and in their size and shape. While such approaches have led to valuable insights, with such analogues it is extremely difficult to distinguish between steric and hydrogen bonding effects as sources of observed differences in replication properties.[4,5]
To address this problem we proposed the structures of four nucleoside analogues designed to mimic as closely as possible the structure of natural nucleosides, but lacking standard polar hydrogen bonding functionality. For example, compound 1, a difluorotoluene deoxynucleoside, was designed to mimic the shape of the natural deoxynucleoside thymidine (2). Its difluorotoluene “base” is isoelectronic with thymine, and through the replacement of the carbonyl groups with C–F and the polar N–H group with C–H the two compounds are isosteric as well.[4]
Studies of the properties of nucleoside 1 have indicated that the compound is quite nonpolar and that the difluorotoluene moiety shows no measurable hydrogen bonding tendency in aqueous and organic solutions. For example, titration of 1 with adenine derivatives in chloroform does not lead to measurable complex formation. Further, replacement of thymine with difluorotoluene in the center of a DNA duplex 12 base pairs in length causes strong destabilization, and the difluorotoluene shows no preferential pairing with adenine over the other three bases; in contrast, thymine shows a 3–4 kcal mol−1 preference.[6,7] Based on the hydrogen bonding complementarity model of DNA replication fidelity, it was expected that during replication this analogue would serve as a very poor enzyme substrate and would show very little selectivity for inserting any one of the natural bases over another.
Despite this expectation, a recent study, in which the difluorotoluene analogue replaces thymidine in template DNA strands, has shown that it serves as an excellent and highly specific template for replication. The Klenow fragment (KF) of E. coli DNA polymerase I accepts this compound in the template with efficiency nearly matching that of thymine, and the enzyme specifically inserts adenine opposite it [5] Thus, despite the poor hydrogen bonding abilities of the nucleoside isostere, the enzyme treats it almost as if it were thymidine. If the size, shape, and conformation of the analogue are assumed to be the same as those of thymidine, then one is led to conclude that hydrogen bonding may be less important in the fidelity of replication than previously believed, and that steric effects may play a more direct role. This argument depends strongly, however, on the similarity of the size, shape, and conformation of 1 and thymidine. We now report the results of detailed structural studies of compound 1, with comparison to natural thymidine. We have obtained X-ray structural data for 1 and have carried out solution studies of it and thymidine by 1H NMR spectroscopy as well.
We were able to obtain X-ray quality crystals of deoxy-nucleoside 1 from ethanol. Analysis of the solid-state structure reveals that the nucleoside analogue is a remarkably good structural mimic of thymidine. The C-nucleoside crystallized in the P21 space group and has three molecules in the asymmetric unit. All three molecules have the anti-glycosidic conformation; two have S-type (C2′-endo) puckers in the sugar moiety; the third has an N/S intermediate conformation (C4′-exo). The average bond lengths for 1 and published structural data for thymidine are given in Figure 1.[8,9] In addition, dihedral torsion angles for one of the molecules in the asymmetric unit are given in Table 1 (C6-C(N)1-C1′-C2′ is referred to as C6-C2′), again along with the analogous angles for thymidine (C(N)1-C1′). Examination of the data shows that the conformations of the five-membered rings in the two compounds are essentially identical. The glycosidic torsion angles are also similar, only 18° apart. Bond lengths for the thymine base and difluorotoluene base mimic are nearly all within 0.07 Å of each other. The chief difference is the C=O bond lengths (1.23 and 1.21 Å) as compared to the analoguous C–F bond lengths (averaging 1.37 Å and 1.37 Å, respectively).
Figure 1.

Single-crystal X-ray structure of difluorotoluene deoxynucleoside 1 compared to the published structure of thymidine 2[8,9]. Top: Structures of the isolated nucleosides. Note that 1 has three different molecules in the asymmetric unit. Bottom: Bond lengths [Å] for 1 (averaged over the three molecules in the asymmetric unit) and thymidine.
Table 1.
Structural data for 1 and thymidine obtained by X-ray crystallographrc analysis.
| Nucleoside | Dihedral angles[°][d] | |||||
|---|---|---|---|---|---|---|
| C6-C2′ | O4′-C3′ | C1′-C4′ | C2′-C5′ | C(N)1-C3′ | C3′-C1′ | |
| 1[a] | −97.6 | +32.2 | −36.7 | −91.5 | + 153.2 | −9.0 |
| thymidine[b] | −79.5 | +28.4 | −37.2 | −87.3 | + 147.8 | − 16.8 |
| H1′-H2′ | H1′-H2″ | H2′-H3′ | H2″-H3′ | H3′-H4′ | C(N)1-H2′ | |
| 1[c] | +157.2 | +35.5 | −73.4 | +84.2 | −93.9 | +34.0 |
| thymidine[c] | + 149.2 | + 21.9 | − 41.7 | + 87.2 | − 88.5 | + 33.8 |
Data for one of the three molecules in the asymmetric unit (see Figure 1).
Data from ref.[9].
The H – H dihedral angles were obtained from the crystal structures with atoms added to give normal sp3 and sp2 geometries.
Only those atoms at the end of the sequence forming the dihedral angle are given. For example. C6-C2′ refers to C6-C(N)1-C1′-C2′.
Also of interest are the noncovalent interactions found in the crystal structure of difluorotoluene deoxynucleoside and those of thymidine (Figure 2). In both structures the 3′ and 5′ hydroxyl groups form intermolecular hydrogen bonds. For compound 1, the sugar hydroxyls are hydrogen bonded to other sugar hydroxyl groups. For thymidine, the carbonyl and N–H groups, which undergo Watson–Crick hydrogen bonding in DNA, are found to be hydrogen bonded to the sugar hydroxyl groups of neighboring molecules.[9] For deoxynucleoside 1, no hydrogen bonds or other close contacts[10] are observed with the fluorines on the aromatic ring (the nearest contacts are several F–H contacts of 2.49–2.79 Å).[10,11] In the crystal packing array, the nonpolar difluorotoluenes are segregated from the more polar, H-bonding furanose groups (Figure 2B). Finally, in the thymidine crystal structure the heterocyclic bases are stacked in a linear array; in contrast, the difluorotoluene rings do not form stacks.
Figure 2.
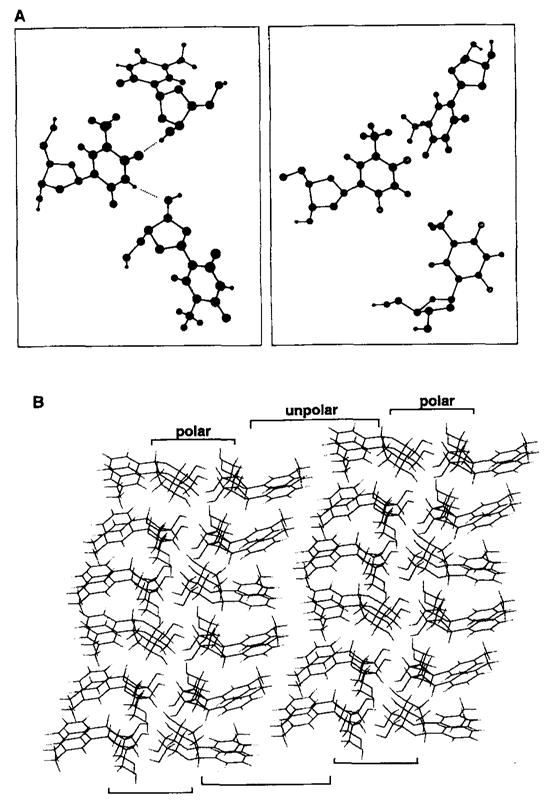
Intermolecular arrangement in the crystal structures of thymidine [9] and nucleoside 1. A) Local environment surrounding the aromatic “bases” of the nucleosides. Note the presence of two hydrogen bonds between the thymine bases and lack of such contacts for difluorotoluene. B) Overall packing arrangement for 1 showing segregation of polar deoxyribose units from nonpolar difluorotoluene rings.
To examine these structural details in solution we carried out a conformational analysis of the difluorotoluene deoxy-nucleoside and of thymidine in D2O by measuring the furanose coupling constants (Table 2).[12,13] The sugar ring protons for both molecules were completely assigned, and the coupling constants were measured at 500MHz. The chief difference in furanose resonances for the two compounds is found in the H1′ signals; that for thymidine is δ = 0.93 downfield of that for 1, consistent with the strong difference in glycosidic bond polarities.
Table 2.
1H NMR data for 1 and thymidine in D2O.
| Chemical shift | H1′ | H2′ | H2″ | H3′ | H4′ | H5′ |
|---|---|---|---|---|---|---|
| 1 (δ) | 5.42 | 2.21 | 2.33 | 4.48 | 4.08 | 3.75 |
| thymidine (δ) | 6.35 | 2.42 | 2.42 | 4.52 | 4.06 | 3.85 |
| Coupling constants | H1′-H2′ | H1′-H2″ | H2′-H3′ | H2″-H3′ | H3′-H4′ | H2′-H2″ |
| 1 J[Hz] | 10.37 | 5.80 | 5.79 | ≈ 1.0 | 2.44 | 13.58 |
| summed J values | Σ(23+23)- 3′+2″3′) | Σ(1′) | Σ (2′) | Σ (2″) | Σ (3′) | |
| 1 [Hz] | 6.80 | 16.17 | 29.74 | 20.88 | 9.73 | |
| thymidine [Hz] | 10.68 | 13.74 | [a] | [a] | 14.65 |
Not determined.
Despite the large difference in polarity of the glycosidic bonds, the sugar ring conformations of 1 and 2 are very similar. Analysis using published method[13] allowed to us to assign the overall conformation of the two deoxynucleosides in both cases as S type; thymidine is classified as 70% S, while the difluorotoluene compound is 90–100% S. In addition, NOE studies carried out on the two structures provided evidence as to the preferred glycosidic conformations in solution. Selective irradiation at the frequency of the H6 signal led to qualitatively larger enhancements of the C-5 methyl signal than of the H1′ signal, and the ratios of these two enhancements were similar for compound 1 and thymidine. This is consistent with predominantly anti- glycosidic conformations for both compounds in solution. Thus, we conclude that in solution the difluorotoluene nucleoside adopts an overall conformation virtually the same as that of thymidine, despite the replacement of the natural (and more polarized) C–N glycosidic bond by a C–C bond.
These structural studies have relevance to the question of how DNA is synthesized with high specificity. We have shown that the difluorotoluene nucleoside is replicated by KF polymerase with efficiency and specificity almost identical to that with thymidine when placed in synthetic DNA templates.[5] It is clear that the fluorines in 1, which replace carbonyl groups in thymidine, have very little (if any) tendency to form hydrogen bonds, especially in aqueous solution, and this is further supported by the lack of such noncovalent contacts in the crystal structure. Moreover, the thymine heterocycle and difluorotoluene have very different electrostatic charge localizations; octanol/water partioning studies showed 1 to be highly nonpolar while thymidine is very polar.[7] These data support the notion that the structural, rather than electrostatic, mimicry of 1 may lead to its specific replication.
As a result of these studies we are developing a new model for the physical origins of the fidelity of DNA synthesis. The KF enzyme initially selects an incorrect nucleotide in DNA synthesis only once in 103–104 times, and virtually the same selectivity is seen for insertion of A (rather than C, T, G) opposite compound 1.[5] We propose that the primary factor in this selectivity is steric exclusion of incorrect nucleotides. In this model the active site is a tightly restricted pocket defined by the surrounding enzyme and the DNA base in the template strand. The presence of thymine (or an isosteric replacement) in a template excludes guanine, cytosine, and thymine nucleotides by steric clashes with their DNA bases or with the molecules of water solvating them. Adenine is allowed because its shape and size fit perfectly into the active site cavity. This shape-selection hypothesis is currently being examined further in a number of ongoing studies.
In summary, we have analyzed the steric shape and conformation of the C-nucleoside 1 both in solution and in the solid state. The findings indicate that the difluorotoluene nucleoside is a nearly perfect isostere of the natural compound, both in bond lengths and in overall conformation. This nonpolar isostere may be therefore generally useful in the study of molecular recognition and catalytic events that involve the natural structure thymidine, such as protein – DNA recognition and enzymatic synthesis of nucleic acids.
Experimental Section
Crystal structure analysis
Single crystals of the complex were grown from a concentrated ethanol solution. A colorless crystal of approximate dimensions 0.56 × 0.24 × 0.08 mm was mounted on a glass fiber with epoxy and placed on the X-ray diffractometer for data collection at room temperature. The X-ray intensity data were collected on a standard Siemens SMART CCD Area Detector System equipped with a normal focus molybdenum-target X-ray tube operated at 1.5 kW (50 kV, 30 mA). A total of 1321 frames of data (1.3 hemispheres) were collected using a narrow-frame method with scan widths of 0.3° in ω and exposure times of 10 s per frame; the detector-to-crystal distance was 5.094 cm (2θmax = 56.52°). Frames were integrated with the Siemens SAINT program to yield a total of 10400 reflections, of which 6606 were independent (Rint = 0.0425, Rsigma = 0.0614 with and . Crystal structure data: monoclinic, a = 13.9262(2), b = 6.4932(0), and c = 19.8156(5) Å, β = 104.574(1)°, and V = 1753.18(5) Å3 (T = 23°C); least-squares refinement of three-dimensional centroids of > 1000 reflections. (The integration program SAINT produces unreasonably cell constant errors, since systematic error is not included.) The space group was assigned as P21, on the basis of systematic absences and intensity statistics by using the XPREP program (Siemens, SHELXTL 5.04). The structure was solved with direct methods included in the SHELXTL program package and refined by full- matrix least-squares on F2. For Z = 6, there are three independent molecules in the asymmetric unit. Although the frames were integrated to a 2θmax = 56.52°, there were essentially no observed data >45°. Therefore these data were omitted from the refinement yielding a total of 7118 reflections, of which 4101 were independent (Rint = 0.0368, Rsigma = 0.0437) and 3548 reflections had >2σ(I). All non-hydrogen atoms were refined anisotropically with hydrogens included in idealized positions giving a data:parameter ratio of approximately 10:1. The structure refined to a goodness of fit (GOF) of 1.157 and final residuals of R1, = 0.0498 (I>2σ(I)), wR2 = 0.1073 (I>2σ(I)) ( , where n and p denote the number of data and parameters; R1 = (Σ||Fo| − |Fc||/Σ|Fo|; with and ).
Footnotes
This research was supported by the National Institutes of Health (GM52956). We thank Prof. T. R. Krugh for helpful discussions, Prof. W. D. Jones and Dr. R. Lachicotte for assistance with the crystallographic data, and Dr. J. Perlstein for assistance with the graphics.
References
- 1.a) Kornberg A, Baker TA. DNA Replication. 2. Freeman; New York: 1992. p. 113. [Google Scholar]; b) Loeb LA, Kunkel TA. Ann Rev Biochem. 1982;52:429. doi: 10.1146/annurev.bi.51.070182.002241. [DOI] [PubMed] [Google Scholar]; c) Goodman MF, Creighton S, Bloom LB, Petruska J. Crit Rev Biochem Mol Biol. 1993;28:83. doi: 10.3109/10409239309086792. [DOI] [PubMed] [Google Scholar]
- 2.See also discussions of replication fidelity in current textbooks: Watson JD, Hopkins NH, Roberts JW, Steitz JA, Weiner AM. Molecular Biology of the Gene. 4. Benjamin/Cummings; Menlo Park: 1987. p. 283.Stryer L. Biochemistry. 4. Freeman; New York: 1995. p. 89.
- 3.See, for example: Freese E. J Mol Biol. 1959;1:87.Sowers LC, Fazakerley GV, Eritja R, Kaplan BE, Goodman MF. Proc Natl Acad Sci USA. 1986;83:5434. doi: 10.1073/pnas.83.15.5434.Eritja R, Horowitz DM, Walker PA, Ziehler-Martin JP, Boosalis MS, Goodman MF, Itakura K, Kaplan BE. Nucleic Acids Res. 1986;14:8135. doi: 10.1093/nar/14.20.8135.Singer B, Spengler S. Biochemistry. 1981;20:1127. doi: 10.1021/bi00508a013.Strazewski P, Tamm C. Angew Chem. 1990;102:37.Angew Chem Int Ed Engl. 1990;29:36.Shibutani S, Takeshita M, Grollman AP. Nature. 1991;349:431. doi: 10.1038/349431a0.Ward DC, Reich E. J Biol Chem. 1972;247:705.
- 4.Schweitzer BA, Kool ET. J Org Chem. 1994;59:7238. doi: 10.1021/jo00103a013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moran S, Ren RX-F, Rumney S, Kool ET. J Am Chem Soc. 1997;119:2056. doi: 10.1021/ja963718g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schweitzer BA, Kool ET. J Am Chem Soc. 1995;117:1863. doi: 10.1021/ja00112a001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moran S, Ren RXF, Kool ET. Proc Natl Acad Sci USA. 1997;94:10506. doi: 10.1073/pnas.94.20.10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Young DW, Tollin P, Wilson HR. Acta Crystallogr Sect B. 1969;25:1423. [Google Scholar]
- 9.Chekhlov AN. J Struct Chem. 1995;36:155. [Google Scholar]
- 10.Close F–H contacts are defined here as contacts ≤2.35 Å [11].
- 11.Howard JAK, Hoy VJ, O’Hagan D, Smith GT. Tetrahedron. 1996;52:12613. [Google Scholar]
- 12.Wijmenga SS, Mooren MMW, Hilbers CW. In: NMR of MacromoIecules: A Practical Approach. Roberts GK, editor. Oxford University Press; Oxford: 1993. p. 258. [Google Scholar]
- 13.Rinkel LJ, Altona C. J Biomol Struct Dyn. 1987;4:621. doi: 10.1080/07391102.1987.10507665. [DOI] [PubMed] [Google Scholar]