

Abstract
We report the synthesis and DNA incorporation of a novel C-5 thiopropyne-substituted thymidine derivative which can be used to bring about covalent crosslinks between two noncomplementary DNA strands. This modified thymine pairs normally with adenine in duplex DNA and is shown not to be destabilizing to DNA double helices. Placement of the thiol-nucleotide near the center of opposing pyrimidine strands in pyr·pur·pyr triple helices results in crosslinking of the pyrimidine strands under aerobic conditions. Thermal melting studies at neutral pH show that such crosslinked ligands bind complementary purine strands with higher affinity than is possible with simple Watson–Crick recognition alone. In addition, we describe the construction of a triplex-forming circular oligonucleotide which contains a similar disulfide link across the center. This macrobicyclic ligand binds with extremely high affinity and sequence selectivity to a complementary purine DNA strand. The formation of crosslinks across two noncomplementary strands represents a new strategy for increasing affinity and selectivity of DNA recognition.
Introduction
Recent advances in the design of nucleic acid-binding ligands have resulted in novel structures which can bind target sequences with affinities higher than is possible with standard Watson–Crick pairing alone. High binding affinity can be useful in inhibition of biological processing of the specific nucleic acid sequences, with potential utility in the study of gene expression and in gene-directed disease therapies.
One of the most successful new strategies for the binding of single-stranded DNA has been the combined use of two oligonucleotide binding domains which form a triple helical complex with the target strand.1–20 In some cases this has been carried out using two physically separate strands,4,16,18,19 and this can result in a binding advantage if the resulting triplex acts cooperatively. Examples of this behavior have been seen for peptide-derived nucleic acid backbones4 and more recently for methylphosphonate DNA backbones.18
In most cases the physical linking of two triplex-forming binding domains gives a significant advantage in binding affinity relative to the use of two separate strands, since the binding is entropically favored and thus usually has greater cooperativity.1–3 In one such approach, a double-length oligonucleotide can fold back to form a triplex bridge by a loop composed of nucleotides or of nonnucleotide linkers.3,7–11,14,15,19 Ligands of this type have been shown to bind a complementary target strand more tightly than standard Watson-Crick complements can. Such a binding advantage has been shown to result in improved inhibition of enzymatic DNA synthesis.9
In another variant of this strategy, we have shown that double linking of two binding domains can result in even greater binding advantages. Circular oligonucleotides, in which opposing pyrimidine binding domains are bridged on both ends by loops, have been shown to bind with very high affinity1,6,13,17 and very high sequence selectivity2 relative to simple Watson–Crick complements. An additional advantage of the cyclic structure is that circular oligodeoxynucleotides are quite resistant to degradation in human serum.5 It is clear from all the studies that the linking of two binding domains by loops at the ends can very significantly improve nucleic acid binding properties.
One of the most successful approaches to the linking of two oligonucleotide strands is the introduction of thiol groups which can form a covalent disulfide bond.21–27 Such crosslinks between two complementary strands can be useful in the study of mechanisms of enzymatic processing of DNA21,23 and in the stabilization of folded structures for structural studies.22,24–26 The juxtaposition of thiols in two complementary strands makes formation of disulfide crosslinks a simple procedure under oxidative conditions. Thiol modification within base-pairing domains of oligonucleotides can be accomplished by the synthesis of DNA bases carrying protected thiol groups. In this vein, adenine, guanine, and cytosine bases modified in the hydrogen-bonding groups have been shown to form crosslinks in duplex DNA.21,23 Thymines have also been modified in the hydrogen-bonding domain.22 Although such structural modifications are useful, they have the disadvantage that they can significantly destabilize helices by inhibiting normal Watson–Crick pairing of the DNA. An alternative approach was described in a recent report, in which a thymine base modified at the C-5 position carries a thiol at the end of an alkyl chain.24 That strategy has the advantage of not interfering directly with base pairing, although it may indirectly destabilize pairing, since C-5 saturated alkyl substitution of thymines is known to lower binding affinity in duplex DNA.28
In the present study we wished to investigate whether disulfide bonds could be used to crosslink two noncomplementary strands of DNA. As part of this strategy we wished to develop a new thiol-carrying nucleotide which was not destabilizing to helices. Since alkynyl groups at the C-5 position of uracil are well-documented to be significantly stabilizing to DNA duplexes,28,29 we decided to investigate a thiopropyne modification for uracil in DNA. We now report the successful synthesis and DNA incorporation of such a thiopropyne-modified nucleotide and its crosslinking in triplex-forming oligonucleotides. This thiol modification does not destabilize DNA helices, and we find that crosslinks between pyrimidine strands containing this group can strongly stabilize complexes with purine target strands. Finally, a bicyclic ligand containing such a crosslink is characterized; this ligand binds with extremely high affinity and sequence selectivity to its DNA complement.
Results
Design Considerations
Models indicated that a thiopropyne substituted at the C-5 position of deoxyuridine would be structurally well suited to geometries required for crosslinking (see Figures 1–3). Substituted of this nucleotide at various positions on opposite pyrimidine strands in model pyr·pur·pyr triple helices indicated that formation and stability of crosslinks would clearly depend on the geometry between the two thiopropynes. Because of the right-handed twist of the helix, the closest approach of two such thiols does not occur when they are in the same base step. In fact, the models indicated that two thiols would come in closest proximity (within ∼2–3 Å) when one of them is one (termed here +1) or two (+2) bases in the 5′ direction on the opposite strand relative to the other. Placement either in the same base step (position 0) or 3 bases in the 5′ direction (+3) gave a closest approach of only ∼6 Å, and positions further along the helix in either direction (−1, +4) placed the thiols at least 9 Å apart. A close approach of the thiols is important both in aiding the crosslinking reaction in the triple helical complex and in allowing formation of a bond which does not strain the geometry of the complex.
Figure 1.
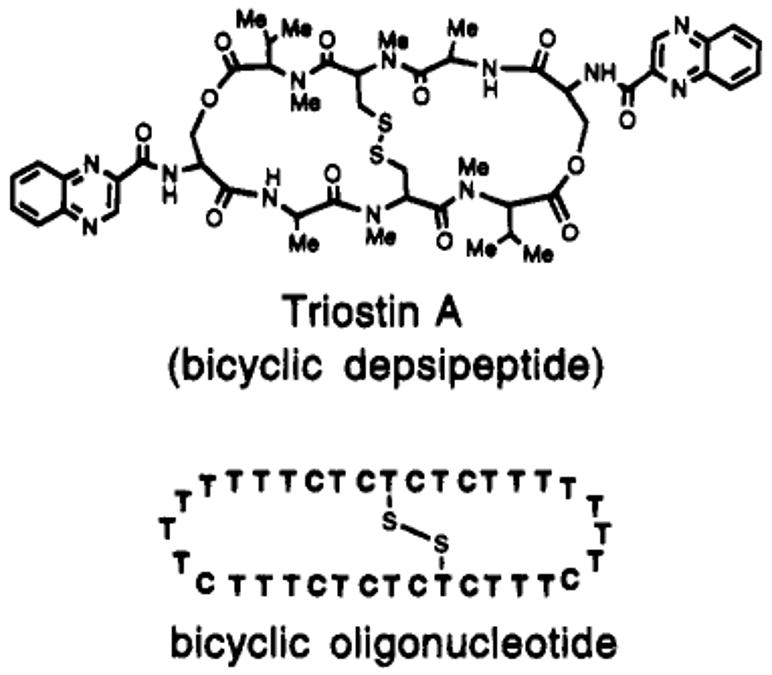
Structures of two disulfide-cross-linked DNA-binding macrocycles. The antibiotic Triostin A binds duplex DNA; the bicyclic oligonucleotide described herein binds with very high affinity to single-stranded DNA.
Figure 3.
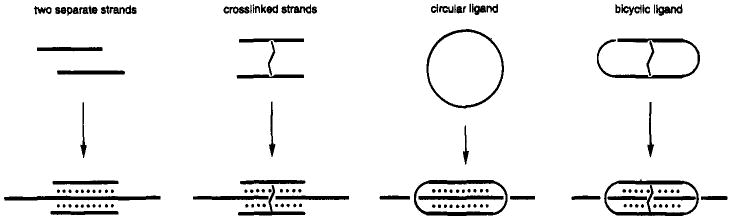
Four strategies for binding single-stranded DNA by triplex formation. Increasing the number of links between the binding domains (as shown from left to right) results in increased binding affinity.
The thiopropyne structure in particular would seem favorable because it is relatively rigid, having only two free rotations from base to sulfur. In addition, the chemistry of C-5 substitution of alkynes on uracil has been well developed.28–31 The thiopropyne nucleoside was unknown prior to the present study; after we began this project a report was published on the synthesis of a C-5 thiopropyl-derived uracil;24 this was derived by reduction of a C-5 propynyl group. Recent systematic studies of alkyl and alkynyl substituted uridines in DNA, however, have shown that saturated alkyl chains longer than methyl are destabilizing to helices.28 Interestingly, that same study concluded that alkynyl chains shorter than ∼6 carbons are significantly more stabilizing to DNA helices than is natural thymine.
Synthesis of the Thiol-Derivatized Nucleoside
The approach used for alkynyl derivatization of uridine at the C-5 position was that of Robins30 and Hobbs,31 who reported the introduction of alkynes at this position. Scheme 1 outlines the synthesis of our thiopropynyldeoxyuridine phosphoramidite. We synthesized the hydroxyproypne intermediate from 5-iododeoxyuridine following the procedure of Glick.24 Thiobenzoate was used to displace the mesylate derived from the hydroxypropyne in good yield. Subsequent deprotection and tritylation proceeded in good yields under standard conditions, and this was followed by phosphitylation at the secondary hydroxyl to give the desired phosphoramidite derivative.
Scheme 1a.
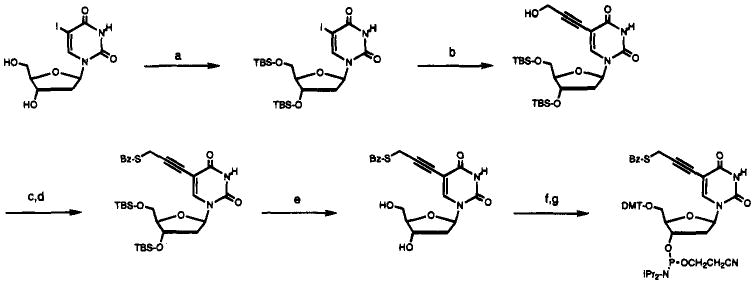
a (a) TBS-C1, imidazole, DMF, 90%; (b) hydroxypropyne, Pd(PPh3)4, CuI, Et3N, DMF, 67%; (c) MsCl, Et3N, CH2C12, −50 °C; (d) PhCOSH, CH2C12, −50 °C to 23 °C, 77%; (e) nBu4NF, pyr·HF, pyridine, 93%; (f) dimethoxytrityl chloride, DIPEA, CH2C12, 56%; (g) ClP(NiPr2)OCH2CH2CN, DIPEA, CH2C12, 94%.
Incorporation into Oligodeoxynucleotides
The thiopropyne modified phosphoramidite was first introduced at single sites in five 13mer and 14mer pyrimidine oligodeoxynucleotides having the sequence 5′-CTTCTTTTTCTTC or 5′-dCTTCTTTTTTCTTC (where underlined bases are each modified separately) (see Table 1). We used a commercially available N-acetyl deoxycytidine phosphoramidite for incorporation of C residues. The coupling procedure for the thiopropyne analog was the same as for unmodified bases, and the coupling proceeded generally with >97% efficiency as judged by trityl response. The deprotection of the protecting groups on the DNA, along with the S-benzoyl group, was carried out by treatment with triefhylamine while on the solid support followed by cleavage with aqueous NH4OH and methylamine (in the presence of dithiothreitol) for 1.5 h at room temperature.32 This rapid deprotection scheme was chosen after we found that the nucleoside appeared to be unstable to concentrated NH4OH at 55 °C for 8 h.
Table 1.
Thermal and Thermodynamic Stabilities of Triple Helical Complexes of Cross-Linked and Unlinked Pyrimidine Strands Binding Purine Sequencesa,b,d
| crosslink geometry | complex | pH = 7.0 | pH = 5.0 | ||
|---|---|---|---|---|---|
| Tm(°C) | -ΔG°37 (kcal/mol) | Tm(°C) | -ΔG°37 (kcal/mol) | ||
| no link (13mer) | 5′CTTCTTTTTCTTC | ||||
| 5′GAAGAAAAAGAAG | 13.3, 46.2c | -- | 48.0 | -- | |
| 3′CTTCTTTTTCTTC | |||||
| no link (14mer) | 5′CTTCTTTTTTCTTC | ||||
| 5′GAAGAAAAAAGAAG | 16.2, 47.9c | -- | 49.4 | -- | |
| 3′CTTCTTTTTTCTTC | |||||
| +0 link (13mer) |  |
-- no crosslinking observed -- | |||
| +1 link (14mer) |  |
51.9 | 16.1 | 65.9 | 25.1 |
| +2 link (13mer) |  |
52.8 | 16.2 | 67.1 | 24.7 |
| +3 link (14mer) | 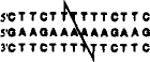 |
50.5 | 14.9 | 63.9 | 23.4 |
| +4 link (13mer) | 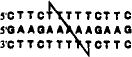 |
-- no crosslinking observed -- | |||
Conditions: 1.5 μM concentration each strand, 100 mM NaCl, 10 mM MgCl2, 10 mM Na·PIPES buffer.
Error limits for individual measurements are estimated at ±0.5 °C in Tm and ±5–10% in free energy.
First temperature is for triplex–duplex transition; second number is for duplex–single strands transition.
The positions of cross-linking groups are as indicated.
The intact incorporation (and successful deprotection) of the thiopropyne analog into the DNA was established in the following way: a deprotected DNA oligomer containing this residue (sequence 5′-dCTTCTTXTTTCTTC) was treated with N-ethylmaleimide to make a stable thiol adduct, and the oligomer was then enzymatically digested to nucleosides following the published method.33 HPLC analysis of the products showed only three significant peaks arising from the DNA: two of these were thymidine and deoxycytidine (confirmed by coinjection with authentic samples), and the third was the maleimide adduct of thiopropynyldeoxyuridine, also confirmed by coinjection with an authentic sample. The relative peak areas were consistent with the expected 9:4:1 ratio of nucleosides.
Further evidence for intact incorporation of the thiopropynyl derivative into DNA was obtained by 1H-NMR analysis of a short oligonucleotide having the sequence 5′-dT-X-T. The 500 MHz spectrum of the deprotected trinucleotide clearly showed the presence of a peak consistent with a propynyl –CH2– group having a chemical shift similar to that in the nucleoside.
Effect of Thiopropyne Modification on Helix Formation
To test whether the thiopropyne modification affects the ability of a strand to hybridize with its complement, we carried out a comparison of binding properties of modified and unmodified strands at pH 7.0. The sequence of the strands being tested was 5′-dCTTCTXTTTCTTC, where X is thiopropynyldeoxyuridine or unmodified thymidine. Thermal denaturation studies of these strands hybridized to the complement 5′-dGAAGAAAAAGAAG showed that they have virtually identical melting behavior. The Tm for the unmodified strand was 44.9 °C, with an estimated free energy of −11.2 kcal/mol; for the modified case the corresponding values were 45.6 °C and − 11.5 kcal/mol. Thus, the two analog strands bind with the same affinity, within experimental error, indicating that the thiopropyne modification does not measurably destabilize duplex formation even when it is near the center of the duplex sequence.
Cross-Linking of Triplex-Forming Strands
We aimed first to investigate whether two oligonucleotides, each containing one thiol group, could be crosslinked by formation of a triplex structure. The five pyrimidine sequences described above have sequence symmetry which allows them to bind a purine complement in 2:1 ratio, forming pyrimidine-purine-pyrimidine triple helices. The complexes are either 13 or 14 nucleotides in length and differ by the presence or absence of one T-A-T triad near the center. We anticipated the use of the purine complement as a template which would serve to bring two pyrimidine strands, and thus two thiols, in close proximity. Since the position of the thiol modification is varied systematically in these five sequences, we utilized the series to investigate whether the relative positioning of the two thiols would affect either crosslinking efficiency or binding efficiency of the resulting linked compounds. Use of these five compounds allowed testing of the 0, +1, +2, +3, and +4 geometries, where the number indicates the number of base steps moved in the 5′-direction on the opposite strand (see Table 1).
Cross-linking was carried out using crude oligomers just after deprotection. Attempts at gel purification of these oligomers as the free thiols gave compounds which subsequently could not be cross-linked, and so we avoided gel purification until after disulfides were formed. The cross-linking reactions were monitored by analytical denaturing gel electrophoresis; a successful reaction would be expected to give a band with mobility approximately equal to that of an oligomer twice the starting length. The reactions were carried out by mixing the pyrimidine and purine strands in 2:1 ratio (3 μM concentration for the pyrimidine strand) in a pH 5.0 buffer containing 10 mM Mg2+ and 100 mM Na+ and incubating at 23 °C for 6–8 h and then at 4 °C overnight. Analytical denaturing gels of the mixtures were used to determine extents of reaction.
Results show that three of the cases gave nearly complete crosslinking under these conditions (data not shown). The +1, +2, and +3 geometries showed efficient linking, with only a trace of monomer pyrimidine strand remaining. These are cases which modeling predicted to have the closest thiol–thiol distance. The 0 and +4 cases, by contrast, showed very little crosslinking, with only a trace of linked material visible on the gel. When carried out on 30 nmol scale, the products of the three successful reactions were isolated after preparative PAGE purification.
Binding Properties of Cross-Linked Pyrimidine Strands
The three cross-linked products were then examined for their ability to bind the complementary target sequence (either 5′-dGAAGAAAAAAGAAG or 5′-dGAAGAAAAAAGAAG). Thermal denaturation experiments were used to evaluate binding affinity and were carried out with 1:1 mixtures of linked oligomers and complements. For comparison we also examined unmodified 13mer and 14mer pyrimidine strands having the same sequences as the thiol-modified cases (see Table 1).
Results show that all three of the linked strands form strong, cooperative complexes with the complements. All three complexes are pH dependent, displaying tighter binding at pH 5.0 than at neutral pH, a result which is strongly indicative of the expected triple helical structure of the complexes. All three also show only a single, sharp melting transition having similar hyperchromicity to that of the unmodified triplexes (Figure 4). The latter, by contrast, show two clear transitions at neutral pH as a result of initial loss of the Hoogsteen strand followed by melting of the remaining duplex. Thus, the cross-links result in increased cooperativity for the triple helical complexes.
Figure 4.
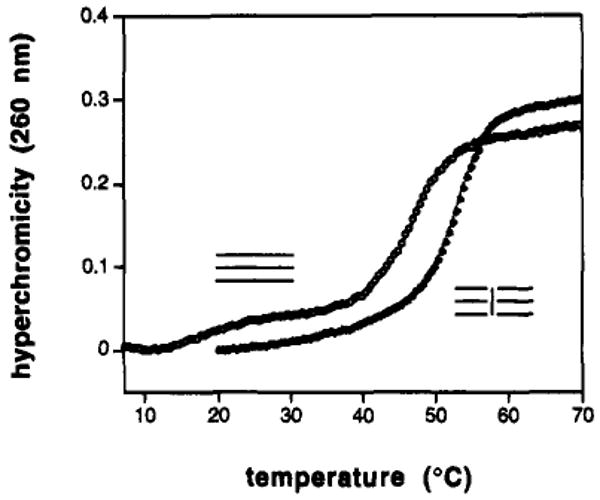
Effect of a central crosslink on triplex formation. Shown are thermal denaturation curves at pH 7.0 for a three-stranded triple helix 13 nucleotides in length and for the same sequence cross-linked in the +2 geometry (see Table 1 for sequences and conditions).
Examination of melting transition temperatures and calculated free energies for the binding of the linked ligands indicates that they have considerably more favorable binding affinity relative to the three-stranded unmodified complexes or to simple duplexes alone (Table 1). At pH 5.0, where both unlinked and linked triplexes show cooperative melting behavior, we find that the cross-linking results in an increase in Tm of 14.5 to 19.1 °C relative to unmodified triplexes of the same sequence. At neutral pH the advantage is a smaller 2.6–6.6 °C as compared to simple duplexes; in free energy terms the advantage is found to be 3–5 kcal/mol (37 °C) at this pH.
Comparison of cross-linking geometries for the three successfully linked cases shows that there are small but significant differences. The three geometries examined encompass relative positions +1, +2, and +3. At pH 5.0 the Tm advantages for the linked triplexes relative to the unmodified analogs are 16.5°, 19.1°, and 14.5 °C, respectively. Thus, it would appear that the most stabilizing crosslink has the +2 relative geometry, at least for the sequences examined.
Previous studies have utilized a nucleotide loop as a means to linking of two triplex-forming binding domains.1,6,8 We wished to compare the relative efficacy of that approach as compared to the present disulfide links. To this end we synthesized the sequence 5′-dCTTCTTTTTTCTTCTTTTTCTTCTTTTTTCTTC, which contains the same 14-base binding domains as two of the above linked strands, but which links these domains with a pentanucleotide loop (underlined). Binding studies of this oligonucleotide under the same conditions show that it also forms a pH-dependent complex, but with affinity close to or slightly lower than that of the optimum disulfide-linked strands. At pH = 5.0 the Tm is 64.9 °C, as compared to 63.9 and 65.9 °C for the disulfide-linked strands which form the 14mer complexes. The optimized cross-linked ligand has a higher Tm of 67.1 °C even though it forms a shorter helix. Thus, the results show that a simple optimized disulfide link compares favorably to a loop composed of five nucleotides.
Synthesis of a Macrobicyclic Oligonucleotide
Since one of our long-term strategies in the design of DNA-binding ligands has been increasing the preorganization of the ligand, it seemed possible that an optimized disulfide link might be used in conjunction with nucleotide loops to considerably rigidify a triplex-forming ligand (Figures 1–3). Such a molecular strategy has evolved in naturally occurring cyclic oligopeptide antibiotics. For example, the Triostin antibiotics (Figure 1) are cyclic octapeptide-derived natural products which bind DNA sequence selectively,34 and they are crosslinked by disulfide bonds from two opposed cysteine residues.
Since the above studies of the effects of cross-link geometry indicated that a +2 relative linking orientation is likely the most favorable, we chose that geometry for incorporation into a circular DNA ligand. A precursor 36-base oligonucleotide (see Scheme 2) with a 3′-phosphate was synthesized and deprotected as described above. The complementary target sequence for the final product was 5′-dAAAGAGAGAGAAA.
Scheme 2.
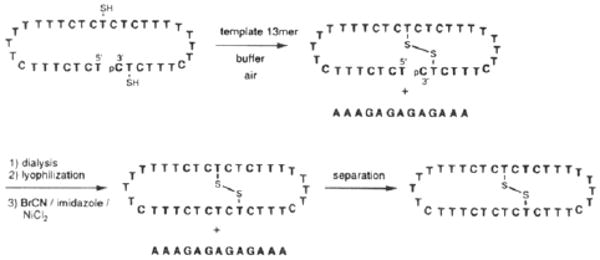
The strategy we developed for the synthesis of the macrobicyclic oligonucleotide is shown in Scheme 2. We have utilized BrCN-mediated esterifications as a means to cyclize oligonucleotides;1,6,17 however, it seemed possible that free thiols would not survive this reaction. Thus our strategy in this case involved initial intramolecular disulfide formation followed by the BrCN cyclization reaction. Both steps were carried out with crude oligonucleotide, and purification was done after both bonds were formed. A complementary template DNA strand was used to promote formation of both bonds, and the reactions were monitored by analytical denaturing gel electrophoresis.
Disulfide formation was carried out under the previous conditions. Results show (Figure 5) that the reaction goes nearly to completion, giving a new band with slower mobility than the starting material. The mobility change is consistent with ring formation, since cyclic oligonucleotides commonly travel more slowly than their linear precursors. The DNA was separated from buffer and salts by dialysis and lyophilization, and then it was subjected to our previously described cyclization conditions. After the reaction, gel analysis showed that a new product was formed with mobility slower yet than linear or crosslinked oligomers. After initial observation of products from these reactions, this same reaction sequence was carried out on a 15 nmol scale, and the final product was isolated by preparative gel electrophoresis. Four nanomoles (27%) of the product were isolated.
Figure 5.
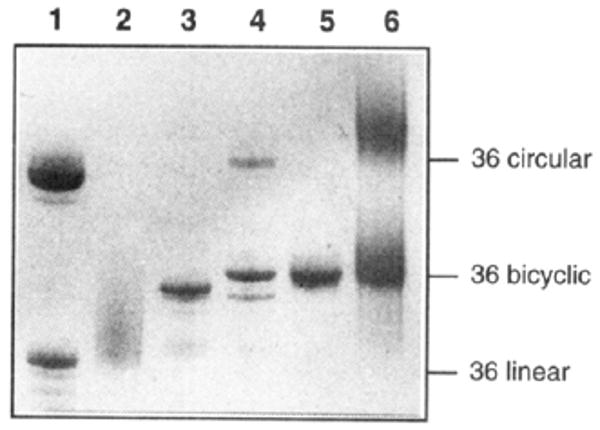
Photograph of stained 20% denaturing PAGE gel showing intermediates and products of synthesis of the bicyclic oligonucleotide. (lane 1) Mixture of unmodified circular (upper band) and linear (lower band) 36mers for size reference. (lane 2) Crude starting 36mer containing two thiol groups. (lane 3) Crude product after oxidation in the presence of 13mer template strand. (lane 4) Crude products after BrCN-mediated cyclization reaction. (lane 5) Purified bicyclic product. (lane 6) Treatment of bicyclic compound with dithiothreitol, showing mobility similar to that of unmodified cyclic 36mer.
Evidence as to the structure of this product comes from gel mobility relative to size markers and from experiments with nuclease enzymes and with dithiothreitol reduction (Figure 5). The presence of a disulfide bond in this product is clearly shown by treatment with dithiothreitol, a strong disulfide reducing agent. This treatment resulted in a product with binding properties very different than the starting compound (see below) and quite similar to those of a circular oligonucleotide lacking thiol groups. Moreover, the gel mobility after DTT treatment was retarded, and became similar to that of the unmodified circle of the same sequence, and this is considerably slower than the 36mer linear precursor to the compound. The circular nature of the DNA backbone was further established by treatment with T4 polynucleotide kinase and γ-32P-ATP; this enzyme did not label the bicyclic product, while a linear oligonucleotide also present in the reaction was successfully labeled (data not shown). This confirms the lack of a free 5′ end, as expected for the cyclized product.
Binding Affinity of the Bicyclic Ligand
The macrobicyclic ligand was then examined for its ability to bind a complementary DNA strand. For comparison we synthesized a circular oligonucleotide having the same sequence as the bicyclic one but with no modified nucleotides (Table 2). In addition, we tested a linear 13-base oligodeoxynucleotide which is complementary to the same target in simple Watson–Crick fashion. As before, binding was evaluated by thermal denaturation experiments in buffers containing 100 mM Na+ and 10 mM Mg2+.
Table 2.
Thermal and Thermodynamic Stabilities of Complexes of Cross-Linked and Unlinked Pyrimidine Oligonucleotide Strands Binding the DNA Sequence 5′-dAAAGAGAGAGAAAa,b,d
| ligand | pH = 5.0 | pH = 7.0 | pH = 8.0 | |||
|---|---|---|---|---|---|---|
| Tm (°C) | -ΔG°37 (kcal/mol) | Tm (°C) | ΔG°37 (kcal/mol) | Tm (°C) | -ΔG°37 (kcal/mol) | |
| TTTCTCTCTCTTT (Watson-Crick complement) | 46.4 | 12.1 | 43.8 | 10.6 | 43.5 | 10.4 |
| TTTCTCTCTCTTT TTTCTCTCTCTTT (two separate strands) | 56.7 | -- | 25.3, 45.1c | -- | 11.9, 44.8c | -- |
| 73.8 | 28.4 | 54.7 | 17.5 | 45.7 | 12.3 | |
| 67.9 | 26.1 | 57.5 | 17.1 | 49.3 | 13.4 | |
| 83.3 | 40.8 | 64.3 | 25.2 | 57.6 | 18.6 | |
Conditions: 1.5 μM concentration each strand, 100 mM NaCl, 10 mM MgCl2, 10 mM Na·PIPES buffer.
Error limits for individual measurements are estimated at ±0.5 °C in Tm and ±5–10% in free energy.
First temperature is for triplex–duplex transition; second number is for duplex– single strands transition.
The positions of thiopropyne cross-linking groups are as indicated.
Examples of melting profiles for some of these complexes are shown in Figure 6, and Tm and free energy data are presented in Table 2. Examination of the melting plots for three triplexes–one termolecular, one with a circular ligand, and one with the bicyclic ligand–shows that there are large differences in the three cases despite the fact that the triple helices all have the same sequence. The three-stranded complex melts in noncooperative fashion and at considerably lower temperatures than the two complexes involving cyclic ligands. In contrast to this, the two bimolecular complexes melt with sharp single transitions.
Figure 6.
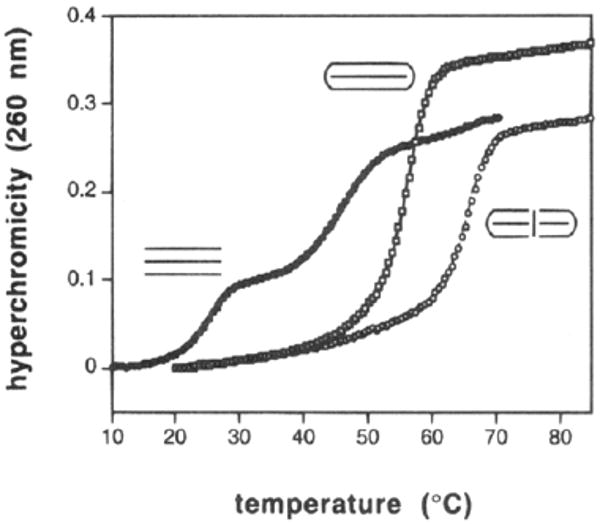
Comparison of binding behavior, as determined by thermal denaturation at pH 7.0, for three triple helical complexes having the same sequence but different linkage geometries as shown. See Table 2 for sequences and conditions.
The binding data show (Table 2) that the bicyclic oligonucleotide binds its complement with extremely high affinity. At neutral pH it binds the complement with a Tm of 64.3 °C and a free energy estimated at −25 kcal/mol. This is nearly 10 °C and 8 kcal/mol more favorable than binding by the unmodified circular oligomer, and it is 20 °C and 15 kcal/mol more favorable than binding by a standard Watson–Crick complement. At pH 5.0 the affinity of the bicyclic ligand increases further, with a Tm rising to 83.3 °C. Although such triplexes are usually weaker at pH values above neutral,35 we find that at pH 8.0 the bicyclic compound still binds with a large (∼8 kcal) advantage over a simple Watson–Crick complement. All the complexes in Table 2 show considerable pH dependence, consistent with the expected Hoogsteen-type binding in a pyr·pur·pyr triple helical structure.36
The effect of the disulfide cross-link is also shown by comparison of the bicyclic ligand with its reduced dithiol form (Table 2). Treatment of the linked compound with 65 mM dithiothreitol followed by melting in the presence of 2.6 mM of the reducing agent gives a binding affinity considerably lower than the intact bicyclic ligand and not far from that of the unmodified circle. Thus, the results show that it is clearly the presence of this single disulfide bond which imparts the unusually favorable binding properties to the bicyclic compound.
Sequence Selectivity
Since the presence of the extra crosslink undoubtedly adds a significant degree of conformational rigidity to the bicyclic oligodeoxynucleotide, it seemed possible that not only binding affinity but also binding selectivity might be affected. To measure sequence selectivity of ligands we synthesized three variants of the target sequence shown in Table 2 which had a single mismatched base at a central position. The sequence of the mismatched oligomers is 5′-dAAAGAGAXAGAAA (where X = C, T, A); this pairs the mismatch position with cytidine residues in the ligands. We define sequence selectivity as the difference in free energy of binding the correctly matched target versus that for the mismatched targets,1 as determined by thermal denaturation experiments. We compared the selectivities of the bicyclic compound, an unmodified circle having the same sequence (see Table 2 for sequences), and a Watson–Crick complement both at pH 5.0 and 7.0.
Figure 7 compares the selectivities of these three oligonucleotide ligands at neutral pH. Results show that selectivity of the unmodified circle is considerably higher than that of the Watson–Crick complement, as we have previously observed.1,37 Interestingly, the data show that the bicyclic ligand has even higher sequence selectivity than the unmodified circular compound. At pH 7.0 the selectivity of the linear complement against these single mismatches is 4.9–5.6 kcal/mol. The circular compound has a selectivity of 8.6–9.2 kcal/mol against the same mismatches, and the bicyclic compound, 10.0 to 11.8 kcal/mol. Results at pH 5.0 show the same trends (data not shown), but the selectivities of the cyclic ligands are yet higher than those at neutral pH, an effect noted previously for circular oligonucleotides.37
Figure 7.
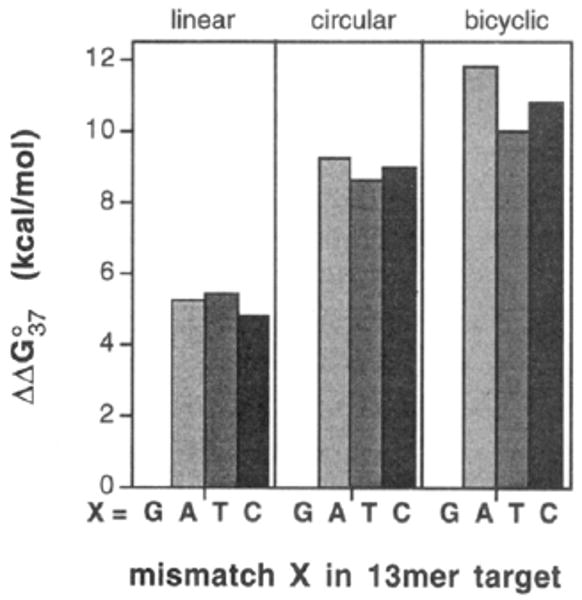
Comparison of sequence selectivities for three different types of DNA-binding oligonucleotides. Shown are the free energy differences (pH 7.0, 37 °C) for binding the complement 5′-dAAAGAGAGAGAAA relative to mismatched targets having the sequence 5′dAAAGAGAXAGAAA (X = A, T, C).
Discussion
The thiopropyne modification for deoxyuridine is a convenient and advantageous molecular strategy for introduction of thiols into DNA. The synthesis of the phosphoramidite derivative is relatively short and proceeds with high yields. The phosphoramidite can be incorporated into oligodeoxynucleotides in high yield using standard automated DNA synthesis procedures. In addition, the benzoyl group protecting the thiol and the rest of the groups protecting the oligonucleotide can be removed simultaneously by a convenient fast-deprotection scheme.32 Finally, this thiopropyne-containing derivative is unusual in that it can pair normally with adenine and without destabilization of DNA helices.
This specific thiol-carrying nucleoside makes possible the formation of stable cross-links in triple helical DNAs. The linking of two triplex-forming DNA strands in the middle of the helix is a novel strategy for improving binding properties of oligonucleotides. It is clear that such linking, like that previously reported at the ends of helices,2,3,6 is significantly stabilizing to the resulting complex. Unlike previous approaches, however, this new strategy requires no additional nucleotides beyond those directly involved in binding. By comparison, we have shown that a standard nucleotide loop which links two triplex-forming domains is optimally 4–5 nucleotides in length.6 The new cross-Unking approach thus implies a shorter, higher-yield synthesis of such ligands, and the presence of a single cross-link results in binding affinities at least as high as those with a nucleotide loop.
The incorporation of nucleotide loops at both ends of the triple helix and a disulfide bond across the center allows the construction of a novel macrobicyclic oligonucleotide. It is clear that the bicyclic structure brings about large improvements in binding properties. We have previously shown that circular triplex-forming oligonucleotides bind complementary targets with association constants which are several orders of magnitude higher than standard Watson–Crick complements.2,13,17 Significantly, we find that the addition of the central disulfide crosslink to such a ligand increases affinity even further. Indeed, at pH 7.0 we find that this bicyclic ligand binds its complement with an estimated free energy of −25 kcal/mol (37 °C), for a 14 kcal advantage over Watson–Crick binding. This corresponds to a Kassoc of ∼1017 M−1, which is three orders of magnitude greater than the biotin–streptavidin complex,38 an archetypal very strong noncovalent complex. At pH 5 we estimate for the bicyclic ligand an even more favorable free energy of complexation of an impressive −40 kcal/mol. We know of no DNA-binding ligand which has been documented to have such a binding advantage over standard Watson–Crick binding at conditions near physiological ionic strength. Since it would appear that the same noncovalent bonds are formed in both circular and bicyclic ligands, it seems likely that the source of increased affinity in the latter case is the greater conformational rigidity which the additional bridge must impart to the ligand.
Not only is binding affinity greatly increased for this bicyclic ligand but also sequence selectivity is significantly increased as well. The combination of these two properties is important, since increasing affinity without increasing selectivity can result in formation of tight nonspecific complexes with undesired activities. We find that the bicyclic ligand displays 10–12 kcal/mol of selectivity (37 °C) against a single mismatch out of 13 bases of target sequence; this corresponds to difference in binding constant of 7–8 orders of magnitude. This is much greater than the 3–4 orders of magnitude in selectivity seen for a standard Watson–Crick complement. We are aware of no other natural or synthetic DNA-binding ligands which show sequence selectivity this high. We have recently shown that the increased selectivity of circular oligonucleotides relative to Watson–Crick complements is due in large part to the favorable protonation of cytosines in the Hoogsteen strand.37 Since the bicyclic compound likely undergoes protonation in similar fashion, we surmise that the still higher selectivity arises primarily from the increased rigidity of the compound.
Work is underway to explore further applications of this new thiol-carrying nucleotide derivative and the properties of resulting disulfide bridges in DNA. We anticipate that the linking of triplex-forming strands using this new strategy may result in the construction of further novel nucleic acid-binding ligands and that the resulting improvements in binding affinity and selectivity will be generally useful in specific targeting of nucleic acid sequences.
Experimental Section
General Procedures
1H, 13C, and 31P NMR spectra were recorded on General Electric Company QE 300 NMR instruments at 300.2, 75.6, and 121.7 MHz, respectively. Chemical shifts are reported in δ (ppm) relative to solvents as internal lock standards and to H3PO4 as the external standard; coupling constants (J) are reported in hertz. Mass spectral analyses were performed at the University of California-Riverside Mass Spectrometry Facility. All organic solvents for carrying out reactions were purified and dried by standard procedures. Imidazole was made anhydrous by azeotropic removal of moisture with dry benzene in a Dean-Stark water separator, followed by drying under high vacuum for several hours. Propargyl alcohol was redistilled under reduced pressure prior to use. 4,4′-Dimethoxytrityl chloride was dried over P2O5 in a vacuum desiccator for 24 h prior to use. Other reagents, chemicals, and solvents were used as obtained from Aldrich, Sigma, Lancaster, Fisher Scientific, or J. T. Baker Inc. Reagents, chemicals and solid supports for solid-phase synthesis of DNA were purchased from Applied Biosystems (Foster City, CA). Ac-dC-CE phosphoramidite and Ac-dC-lcaa-CPG were obtained from Glen Research (Sterling, VA). Column chromatography was carried out with Baker silica gel (40 μm) under a positive pressure of air. For compounds containing the 4,4′-dimethoxytrityl protecting group, silica columns were pretreated with a 1% solution (v/v) of triethylamine in the appropriate eluant. Melting points were determined using a Thomas Hoover capillary apparatus and are uncorrected.
5-Iodo-2′-deoxyuridine 3′,5′-Di-tert-butyldimethylsilyl Ether24
To a cooled solution of tert-butyldimethylsilyl chloride (2.26 g, 15.0 mmol) in 10 mL of anhydrous DMF were added anhydrous imidazole (1.7 g, 25.0 mmol) in one portion and after stirring for 10 min (+)-5-iodo-2′-deoxyuridine (1.77 g, 5.0 mmol). The resulting mixture was stirred at 23 °C for 2 h. This was poured into 25 mL of ice water with vigorous stirring, and the product was extracted with diethyl ether (3 × 30 mL). The combined extract was washed with water (2 × 15 mL) and brine (15 mL). After drying with anhydrous Na2SO4, ether was removed to obtain a thick oil which was purified by silica column chromatography eluting with EtOAc:hexanes (40:60, v/v). The pure silyl ether was obtained as a white foam: yield 2.6 g, 90%; 1H NMR (CDC13) δ 8.35 (1H, b.s., NH), 8.12 (1H, s, 6-H), 6.3 (1H, dd, J = 9.0 and 6.0, 1′-H), 4.43–4.41 (1H, m, 3′-H), 4.02–4.0 (1H, m, 4′-H), 3.92 (1H, dd, J = 12.0 and 3.0, 5′-H), 3.78 (1H, dd, J = 12.0 and 3.0, 5″-H), 2.4-2.28 (1H, m, 2′-Hα), 2.08–1.96 (1H, m, 2′-Hβ), 0.97 (9H, s), 0.92 (9H, s), 0.18 (3H, s), 0.17 (3H, s), 0.11 (3H, s) and 0.1 (3H, s).
5-(3-Hydroxypropyn-1-yl)-2′-deoxyuridine 3′,5′-Di-tert-butyldimethylsilyl Ether24
To a solution of the silyl ether (2.26 g, 3.88 mmol), redistilled propargyl alcohol (655 mg, 11.68 mmol), and dry triethylamine (790 mg, 7.8 mmol) in 20 mL of dry and deaerated DMF were added tetrakis(triphenylphosphine)palladium(0) (450 mg, 0.39 mmol) and copper(I) iodide (148.5 mg, 0.78 mmol) under nitrogen. The resulting mixture was stirred at 23 °C for 2 h and quenched with 30 mL of cold water. Product was isolated by extraction with EtOAc (3 × 30 mL) followed by washing (water, 2 × 15 mL; brine, 15 mL), drying (Na2SO4), and removal of solvent by rotary evaporator. Purification by silica column chromatography (EtOAc:hexanes, 70:30, v/v) produced the hydroxypropynyl nucleoside: yield 1.32 g, 67%; 1H NMR (DMSO-d6) δ 11.64 (1H, s, exch. with D2O), NH), 7.89 (1H, s, 6-H), 6.08 (1H, pseudo t, J = 6.6, 1′-H), 5.25 (1H, t, J = 6.0, exch. with D2O, OH), 4.4–4.34 (1H, m, 3′-H), 4.19 (2H, d, J = 6.0, CH2), 3.82–3.66 (3H, m, 4′,5′,5″-H), 2.3–2.08 (2H, m, 2′,2″-H)), 0.88 (9H, s), 0.85 (9H, s), 0.08 (6H, s) and 0.06 (6H, s).
5-(3-Benzoylthiopropyn-1-yl)-2′-deoxyuridine 3′,5′-di-tert-Butyldimethylsilyl Ether
A solution of the hydroxypropynyl nucleoside (810 mg, 1.59 mmol) and triethylamine (415 mg, 4.1 mmol) in 5 mL of anhydrous CH2Cl2 was cooled to −55 °C under nitrogen and held at that temperature for 10 min. Methanesulfonyl chloride (218 mg, 1.9 mmol) was introduced as a solution in 200 μL of CH2Cl2 by means of a syringe. After stirring at −50 °C for 40 min, thiobenzoic acid (90%, 265 mg, 1.74 mmol in 200 μL of CH2C12) was added to the mixture, and it was allowed to warm to 23 °C during 3 h. The mixture was then diluted with 50 mL of EtOAc and washed with water (2 × 15 mL) and brine (15 mL). Drying with anhydrous Na2SO4 followed by removal of solvent under reduced pressure yielded a thick oil, which was purified by silica chromatography using EtOAc:hexanes (40:60, v/v) as the eluant to obtain the thiobenzoate as a pale yellow foamy solid: yield 775 mg, 77%; mp 145–147 °C; 1H NMR (CDCl3) δ 8.16 (1H, b.s., NH), 7.99 (1H, s, 6-H), 7.94 (2H, d, J = 8.4), 7.59 (1H, t, J = 8.4), 7.46 (2H, t, J = 8.4), 6.28 (1H, pseudo t, J = 6.3, 1′-H), 4.42–4.38 (1H, m, 3′-H), 4.08 (2H, s, CH2), 4.0–3.96 (1H, m, 4′-H), 3.92–3.72 (2H, ill-resolved ‘d’ of an AB ‘q’ 5′,5″-H), 2.38–2.26 (1H, m, 2′-Hα), 2.08–1.98 (1H, m, 2′-Hβ), 0.9 (9H, s). 0.88 (9H, s), 0.12–0.06 (12H, four singlets); 13C NMR (CDCl3) δ 190.8, 161.8, 149.6, 143.2, 136.5, 134.1, 129.1, 127.8, 100.2, 89.2, 88.8, 86.3, 74.7, 72, 8, 63.4, 42.5, 26.4, 26.2, 19.0, 18.9, 18.4, −4.2, −4.4, −4.9 and −5.1; HRFAB/NBA/PEG exact mass, calcd for C31H46N2O6SSi2 + H+ 631.2693, found 631.2670.
5-(3-Benzoylthiopropyn-1-yl)-2′-deoxyuridine
The thiobenzoate silyl ether (740 mg, 1.17 mmol) was treated with 1 M n-Bu4NF/2M pyridinium hydrogen fluoride in dry pyridine (2.8 mL) for 20 h at 23 °C. After removal of pyridine under reduced pressure, the crude residue was adsorbed on 6 g of silica gel for column chromatography, and product was eluted with EtOAc containing 3% of methanol (v/v). The pure deprotected nucleoside was obtained as a white foam: yield 440 mg, 93%; mp 50–65 °C; 1H NMR (methanol-d4) δ 8.29 (1H, s, 6-H), 7.96 (2H, d, J = 8.4), 7.65 (1H, t, J = 8.4), 7.52 (2H, t, J = 8.4), 6.23 (1H, pseudo t, J = 6.3, 1′-H), 4.42-4.36 (1H, m, 3′-H), 4.09 (2H, s, CH2), 3.94–3.88 (1H, m, 4′-H), 3.81 (1H, dd, J = 12.0 and 3.6, 5′-H), 3.72 (1H, dd, J = 12.0 and 5.0, 5″-H) and 2.32–2.16 (2H, m, 2′,2″-H); 13C NMR (methanol-d4) δ 190.0, 162.7, 149.4, 143.7, 136.0, 133.3, 128.3, 126.5, 98.4, 87.7, 87.4, 85.3, 73.4, 70.3, 60.9, 39.9 and 17.4; HRFAB/NBA/PEG exact mass, calcd for C19H18N2O6S + H+ 403.0964, found 403.0968.
5-(3-Benzoylthiopropyn-1-yl)-2′-deoxy-5′-O-(4,4′-dimethoxytrityl)uridine
A mixture of the deprotected nucleoside (430 mg, 1.07 mmol), 4,4′-dimethoxytrityl chloride (90%, 655 mg, 1.74 mmol) and N,N-diisopropylethylamine (368 mg, 2.85 mmol) in anhydrous CH2Cl2 (10 mL), was heated under gentle reflux in a nitrogen atmosphere for 1 h. It was then cooled, diluted with EtOAc (60 mL), washed with water (15 mL) and brine (15 mL), and dried (Na2SO4). Removal of solvent afforded a thick liquid which was purified by silica column chromatography using 2% methanol in EtOAc as the eluant. The tritylated nucleoside was obtained as a white foam: yield 420 mg, 56%; 1H NMR (CDCl3) δ 8.23 (1H, b.s., NH), 8.1 (1H, s, 6-H), 7.81 (2H, d, J = 8.4), 7.55 (1H, t, J = 8.4), 7.44–7.16 (11H, m), 6.83 (4H, d, J = 9.0), 6.3 (1H, pseudo t, J = 6.6, 1′-H), 4.6–4.54 (1H, m, 3′-H), 4.12– 4.08 (1H, m, 4′-H), 3.82–3.68 (8H, m, containing a singlet at δ 3.74, CH2 and 2 × OCH3), 3.42–3.36 (2H, m, 5′-5″-H), 2.6–2.48 (1H, m, 2′-Hα) and 2.38–2.22 (1H, m, 2′-Hβ); 13C NMR (CDC13) δ 189.9, 161.4, 158.3, 149.1, 144.2, 142.6, 136.0, 135.3, 135.1, 133.2, 129.7, 128.3, 127.7, 127.6, 126.9, 126.6, 115.0, 113.0, 99.6, 88.3, 86.6, 86.4, 85.6, 73.5, 72.0, 63.2, 54.9, 41.2 and 18.2; HRFAB/NBA/PPG exact mass, calcd for C40H36O8S + H+ 705.2270, found 705.2263.
5-(3-Benzoylthiopropyn-1-yl)-2′-deoxy-5′-O-(4,4′-dimethoxytrityl)uridine 3′-O-(2-cyanoethyl N,N-diisopropylphosphoramidite)
To a solution of the triylated nucleoside (390 mg, 0.55 mmol) in 10 mL of anhydrous CH2Cl2 containing N,N-diisopropylethylamine (363 mg, 2.8 mmol) was added 2-cyanoethyl N,N-diisopropylchlcirophosphoramidite (340 mg, 1.44 mmol) under nitrogen, and the resulting mixture was stirred at 23 °C for 1 h. It was diluted with EtOAc (60 mL), washed with water (15 mL) and brine (15 mL), and dried with anhydrous Na2SO4. Evaporation of the solvent under reduced pressure furnished a crude oil which was purified by silica chromatography (eluant: 4% methanol in EtOAc) to obtain a white foam of pure phosphoramidite as two diastereoisomers: Yield 470 mg, 94%; 1H NMR (CDCl3, higher Rf) δ 8.19 (1H, s, 6-H), 7.83 (2H, d, J = 9.0), 7.58 (1H, t, J = 9.0)), 7.5–7.2 (11H, m), 6.87 (4H, d, J = 9.0), 6.3 (1H, pseudo t, J = 6.3, 1′-H), 4.72–4.6 (1H, m, 3′-H), 4.28–4.2 (1H, m, 4′-H), 3.78 (6H, s, 2 × OCH3), 3.76–3.52 (6H, m, CH2, OCH2 and 2 × NCH), 3.5–3.32 (2H, ill-resolved AB ‘q’, 5′,5″-H), 2.65–2.52 (1H, m,2′-Hα), 2.47 (2H, t, J = 6.0, CH2CN), 2.42–2.3 (1H, m, 2′-Hβ) and 1.19 (12H, d, J = 6.0, 2 × CH(CH3)2); 1H NMR (CDCl3, lower Rf) δ 8.15 (1H, s, 6-H), 7.84 (2H, d, J = 8.4), 7.58 (1H, distorted ‘t’, J = 8.4), 7.5–5.2 (11H, m), 6.86 (4H, d, J = 9.0), 6.32 (1H, pseudo t, J = 6.3, 1′-H), 4.72–4.6 (1H, m, 3′-H), 4.22–4.18 (1H, m, 4′-H), 3.92–3.5 (12H, m, containing a singlet at δ 3.78, 2 × OCH3, OCH2, 2 × NCH, CH2), 3.46–3.3 (2H, ill-resolved AB ‘q’, 5′,5″-H), 2.7– 2.6 (3H, m, CH2CN and 2′-Hα), 2.41–2.28 (1H, m, 2′-Hβ), 1.19 (6H, d, J = 6.0, CH(CH3)2) and 1.1 (6H, d, J = 6.6, CH(CH3)2); 13C NMR (CDCl3) δ 190.6, 162.0, 159.1, 149.8, 149.7, 144.9, 143.3, 136.0, 135.9, 135.8, 134.0, 130.5, 129.1, 128.5, 127.7, 127.4, 118.0, 117.8, 113.8, 100.4, 100.3, 88.8, 87.6, 86.6, 86.3, 86.2, 74.3, 74.2, 74.0, 73.7, 66.3, 63.7, 63.5, 59.0, 58.9, 58.7, 58.6, 55.7, 43.9, 43.8, 43.7, 43.6, 41.2, 41.1, 25.1, 25.0, 20.9, 20.8, 20.7, 20.6, 19.0 and 15.7; 31P NMR (CDCl3) δ 149.70 and 149.27; HRFAB/NBA/PPG exact mass, calcd for C49H53N4O9PS + H+ 905.3349, found 905.3375.
Alkylation of 5-(3-Thiopropyn-1-yl)-2′-deoxyuridine with N-Ethvlmaleimide
To a solution of 5-(3-benzoylthiopropyn-l-yl)-2′-deoxyuridine (10 mg, 0.025 mmol) and N-ethylmaleimide (15.6 mg, 0.125 mmol) in 0.3 mL of methanol was added 25 μL of 29.5% NH4OH, and the mixture was stirred in a closed vial at 23 °C for 4 h. After removal of methanol and NH4OH in vacuo, the residue was purified by silica chromatography (eluant, 5% methanol in EtOAc) to obtain the 2-alkylthio-N-ethylsuccinimide as a mixture of two diastereoisomers: yield 5.5 mg, 52%; 1H NMR (methanol-d4) δ 8.31 and 8.3 (1H, two singlets, 6-H), 6.24 (1H, pseudo t, J = 6.3, 1′-H), 4.45– 4.38 (1H, m, 3′-H), 4.24–4.16 (1H, m, SCHC=O), 3.94–3.88 (1H, m, 4′-H), 3.83 (1H, dd, J = 12.0 and 3.6, 5′-H), 3.74 (1H, dd, J = 12.0 and 5.0, 5″-H), 3.55 (2H, two overlapping quartets, J = 7.0, NCH2), 3.4–3.2 (3H, m, partially obscured by solvent peaks, CHC=O and CH2S), 2.75–2.62 (1H, m, CHC=O), 2.38–2.2 (2H, m, 2′,2″-H), and 1.13 (3H, two overlapping triplets, J = 7.0, CH3).
Oligodeoxynucleotides
DNA oligonucleotides were synthesized by automated methods on an ABI 392 DNA synthesizer using β-cyanoethyl phosphoramidite chemistry on 0.2 or 1.0 μmol scales. The synthetic thiopropynyl nucleoside phosphoramidite coupled as 0.11 M solution in dry CH3CN with >97% efficiency. In the synthesis of the thiol-derivatized oligomers, Ac-dC-CE phosphoramidite and Ac-dC-lcaa-CPG were used instead of the standard Bz-dC-CE phosphoramidite and Bz-dC-lcaa-CPG. All unmodified oligonucleotides were cleaved and deprotected with 29.5% NH4OH (55 °C, 12 h), purified by preparative gel electrophoresis on 20% polyacrylamide under denaturing conditions, isolated by the crush-soak and dialysis method, and quantitated by UV absorbance at 260 nm. Molar extinction coefficients were calculated by the nearest neighbor method. Thiol-modified oligomers were first treated on the solid support with dry triethylamine for 2 h at 23 °C and then were cleaved form solid support and deprotected with the UltraFAST system32 (AMA, 50:50, v/v mixture of 29.5% NH4OH and 40% MeNH2 in water) containing 330 mM dithiothreitol (DTT) at 23 °C for 90 min. These oligomers were isolated by dialysis (water, 4 × 2.0 L, 12 h) and lyophilization, quantitated by UV absorbance at 260 nm, and were submitted to disulfide cross-linking reactions without further purification. Molar extinction coefficients for oligonucleotides containing the nonnatural residue were obtained in the following way: The extinction coefficient for 5-(3-thiopropyn-l-yl)-2′-deoxyuridine was taken as that of 5-(l-propynyl)-2′-deoxy-uridine (Glen Research, molar ∈260 = 3.2 × 103). For an unmodified oligomer (i.e., containing a T in place of the thiol-modifier X), the sum of the individual molar extinction coefficients of all the bases was compared to its molar extinction coefficient obtained by the nearest neighbor method. Then for a given oligomer of the same sequence (but containing an X in place of a T), the corresponding sum of the molar extinction coefficients of the individual bases was scaled downward with this ratio to obtain its molar extinction coefficient. For HPLC analysis of enzymatically digested oligomers,33 thiol-containing DNA was first treated with N-ethylmaleimide following the published procedure.39 HPLC was performed on a reverse-phase Hypersil ODS C-18 5U column (250 mm × 4.6 mm) using a linear gradient of acetonitrile (5–15% in the first 20 min and 15–90% in the next 20 min) in 0.1 M triethylammonium acetate, pH = 6.7 at a flow rate of 1.0 mL/min.
General Procedure for the Preparation of Disulfide-Cross-Linked Oligonucleotides
To a solution of thiol-derivatized oligonucleotide (30.0 nmol) and template strand (15.0 nmol) in 3.0 mL of water was added 1.4× PIPES buffer (pH = 5.04, 7.0 mL) containing 143.0 mM NaCl and 14.3 mM MgCl2. The resulting mixture was kept exposed to air at 23 °C for 6–8 h and at 0–4 °C overnight. The solution was then dialyzed against water (4 × 2.0 L) for 16 h and lyophilized in a speed-vac. The dried oligomers were purified by gel electrophoresis on 20% polyacrylamide under denaturing conditions followed by crush-soak and dialysis method and were quantitated by UV absorbance at 260 nm. Molar extinction coefficients of the cross-linked oligomers were calculated as the sum of the molar extinction coefficients of the two corresponding uncross-linked strands. Yields of the purified oligomers were in the range of 5.0–9.0 nmol (33–60%).
Preparation of the Bicyclic Oligonucleotide
Thiol-modified precircle-3′-phosphate (15.0 nmol) was mixed with template strand 5′-dAAAGAGAGAGAAA (15.0 nmol) in 3.0 mL of water and to this was added 1.4× PIPES buffer (pH = 5.04, 7.0 mL) containing 143.0 mM NaCl and 14.3 mM MgCl2. This solution was treated as described in the preparation of cross-linked oligonucleotides above. The dried oligomer, without further purification and in presence of the template, was treated with 300 μL of a circularizing solution containing 200 mM imidazole·HCl (pH = 7.0), 100 mM NiCl2, and 125 mM BrCN at 23 °C for 30 h. Salts were removed by dialysis at 23 °C against water (4 × 2.0 L; 12 h), and the crude oligomer-template mixture (after lyophilization) was submitted to purification by gel electrophoresis on 20% polyacrylamide under denaturing conditions. Pure product was isolated from gel by crush-soak and dialysis method: yield 4.0 nmol (27%). The molar extinction coefficient of this bicyclic oligomer was approximated to be the same as that of the corresponding unmodified circular oligomer having the same sequence.
Thermal Denaturation Studies
Solutions for the thermal denaturation studies contained a one-to-one ratio of a given pyrimidine oligomer and complementary purine target oligomer (1.5 μM each). Also present were 100 mM NaCl and 10 mM MgCl2. Solutions were buffered with 10 mM Na·PIPES (l,4-piperazine-bis(ethanesulfonate), Sigma) at the pH values indicated. The buffer pH is that of a 1.4× stock solution at 25 °C containing the buffer and salts. After the solutions were prepared they were heated to 90 °C and allowed to cool slowly to room temperature prior to the melting experiments.
The melting studies were carried out in Teflon-stoppered 1 cm pathlength quartz cells under nitrogen atmosphere on a Varian Cary 1 UV–vis spectrophotometer equipped with thermoprogrammer. Absorbance (260 nm) was monitored, while temperature was raised from 10 °C at a rate of 0.5 °C/min; a slower heating rate with this apparatus does not affect the results. In all bimolecular cases the complexes displayed sharp, apparently two-state transitions, with all-or-none melting from bound complex to free oligomers. Melting temperatures (Tm) were determined by computer fit of the first derivative of absorbance with respect to 1/T. Uncertainty in Tm is estimated at ±0.5 °C based on repetitions of experiments. Free energy values were estimated by nonlinear least squares fitting of the denaturation data, using a two-state model with linear sloping baselines.40 Precision in individual free energy measurements is estimated at ±5–10% based on repetitions of experiments. For complexes with higher Tm values the free energy is likely to be less accurate than for those with lower Tm values because of the longer extrapolation made from the melting temperature.
Figure 2.
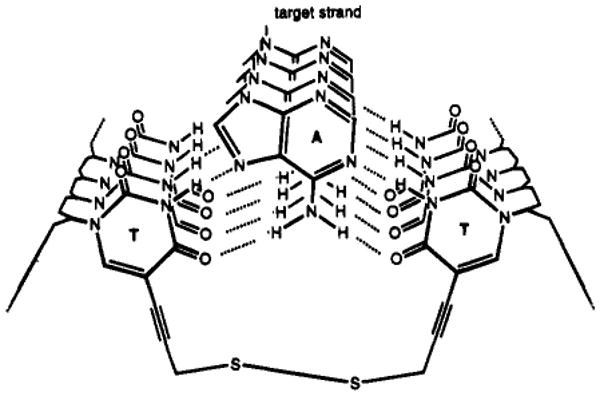
Illustration of a disulfide link formed between two thiopropynyluracils spanning across a triple helix.
Acknowledgments
We thank the National Institutes of Health (GM46625) and the Army Research Office for support. E.T.K. acknowledges a Dreyfus Foundation Teacher-Scholar Award and an Alfred P. Sloan Foundation Fellowship. We also thank Kevin Ryan for assistance with labeling experiments.
Footnotes
Abstract published in Advance ACS Abstracts, October 1, 1995.
References
- 1.Kool ET. J Am Chem Soc. 1991;113:6265–6266. [Google Scholar]
- 2.Prakash G, Kool ET. J Chem Soc, Chem Commun. 1991:1161–1162. 646. doi: 10.1039/C39910001161. 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giovannangeli C, Montenay-Garestier T, Rougée M, Chassignol M, Thuong NT, Hélène C. J Am Chem Soc. 1991;113:7775–7776. [Google Scholar]
- 4.Nielsen PE, Egholm M, Berg RH, Buchardt O. Science. 1991;254:1497–1500. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]
- 5.Rumney S, Kool ET. Angew Chem. 1992;104:1686–1689. [Google Scholar]; Angew Chem, Int Ed Engl. 1992;31:1617–1619. doi: 10.1002/anie.199216171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prakash G, Kool ET. J Am Chem Soc. 1992;114:3523–3528. doi: 10.1021/ja00035a056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salunkhe M, Wu T, Letsinger RL. J Am Chem Soc. 1992;114:8768–8772. [Google Scholar]
- 8.D'Souza DJ, Kool ET. J Biomol Struct Dyn. 1992;10:141–152. doi: 10.1080/07391102.1992.10508634. [DOI] [PubMed] [Google Scholar]
- 9.Giovannangeli C, Thuong NT, Hé1ène C. Proc Natl Acad Sci USA. 1993;90:10013–10017. doi: 10.1073/pnas.90.21.10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gryaznov SM, Lloyd DH. Nucleic Acids Res. 1993;21:5909–5915. doi: 10.1093/nar/21.25.5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hudson RHE, Damha MJ. Nucleic Acids Symp Ser. 1993;29:97–99. [PubMed] [Google Scholar]
- 12.D'Souza DJ, Kool ET. Bioorg Med Chem Lett. 1994;4:965–970. doi: 10.1016/S0960-894X(01)80664-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang S, Kool ET. J Am Chem Soc. 1994;116:8857–8858. doi: 10.1021/ja00098a075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kandimal ER, Agrawal S. Gene. 1994;149:115–121. doi: 10.1016/0378-1119(94)90419-7. [DOI] [PubMed] [Google Scholar]
- 15.Noll DM, O'Rear JL, Cushman CD, Miller PS. Nucleosides & Nucleotides. 1994;13:997–1005. [Google Scholar]
- 16.Trapane TL, Christopherson MS, Roby CD, Ts'o POP, Wang D. J Am Chem Soc. 1994;116:8412–8413. [Google Scholar]
- 17.Wang S, Kool ET. Nucleic Acids Res. 1994;22:2326–2333. doi: 10.1093/nar/22.12.2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reynolds MA, Arnold LJ, Almazan MT, Beck TA, Hogrefe RI, Metzler MD, Stoughto SR, Tseng BY, Trapane TL, Ts'o POP. Proc Natl Acad Sci USA. 1994;91:2433–2437. doi: 10.1073/pnas.91.26.12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bandaru R, Hashimoto H, Switzer C. J Org Chem. 1995;60:786–788. [Google Scholar]
- 20.Wang S, Kool ET. Nucleic Acids Res. 1995;23:1157–1164. doi: 10.1093/nar/23.7.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferentz AE, Verdine GL. J Am Chem Soc. 1991;113:4000. [Google Scholar]
- 22.Glick GD, Osborne SE, Knitt DS, Marino JP. J Am Chem Soc. 1992;114:5447–5448. [Google Scholar]
- 23.Ferentz AE, Keating TA, Verdine GL. J Am Chem Soc. 1993;115:9006–9014. [Google Scholar]
- 24.Goodwin JT, Glick GD. Tetrahedron Lett. 1993;34:5549–5552. [Google Scholar]
- 25.Goodwin JT, Glick GD. Tetrahedron Lett. 1994;35:1647–1650. [Google Scholar]
- 26.Goodwin JT, Osborne SE, Swanson PC, Glick GD. Tetrahedron Lett. 1994;35:4527–4530. [Google Scholar]
- 27.Gao H, Yang M, Cook AF. Nucleic Acids Res. 1995;23:285–292. doi: 10.1093/nar/23.2.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sági J, Szemzö A, Ébinger K, Szabolcs A, Sági G, Ruff E, Ötvös L. Tetrahedron Lett. 1993;34:2191–2194. [Google Scholar]
- 29.Froehler BC, Wadwani S, Terhorst TJ, Gerrard SR. Tetrahedron Lett. 1992;33:5307–5310. [Google Scholar]
- 30.Robins MJ, Barr PJ. J Org Chem. 1983;48:1854–1862. [Google Scholar]
- 31.Hobbs FW., Jr J Org Chem. 1989;54:3420. [Google Scholar]
- 32.Reddy MP, Hanna NB, Farooqui F. Tetrahedron Lett. 1994;35:4311–4314. [Google Scholar]
- 33.Evaluating and Isolating Synthetic Oligonucleotides. Applied Biosystems Inc.; 1992. Appendix 1. [Google Scholar]
- 34.Wang AHJ, Ughetto G, Quigley GJ, Hakoshima T, van der Marel GA, van Boom JH, Rich A. Science. 1984;225:1115–1121. doi: 10.1126/science.6474168. [DOI] [PubMed] [Google Scholar]
- 35.Lee JS, Woodsworth ML, Latimer LJP, Morgan AR. Nucleic Acids Res. 1984;12:6603–6614. doi: 10.1093/nar/12.16.6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moser HE, Dervan PB. Science. 1987;238:645–650. doi: 10.1126/science.3118463. [DOI] [PubMed] [Google Scholar]
- 37.Wang S, Friedman A, Kool ET. Biochemistry. 1995;34:9774–9784. doi: 10.1021/bi00030a015. [DOI] [PubMed] [Google Scholar]
- 38.Green NM. Methods Enzymol. 1990;184:51–67. doi: 10.1016/0076-6879(90)84259-j. [DOI] [PubMed] [Google Scholar]
- 39.Swanson PC, Glick GD. Bioorg Med Chem Lett. 1993;3:2117–2118. [Google Scholar]
- 40.Petersheim M, Turner DH. Biochemistry. 1983;22:256–263. doi: 10.1021/bi00271a004. [DOI] [PubMed] [Google Scholar]