

Abstract
The active site of myosin contains a group of highly conserved amino acid residues whose roles in nucleotide hydrolysis and energy transduction might appear to be obvious from the initial structural and kinetic analyses but become less clear on deeper investigation. One such residue is Ser236 (Dictyostelium discoideum myosin II numbering) which was proposed to be involved in a hydrogen transfer network during γ-phosphate hydrolysis of ATP, which would imply a critical function in ATP hydrolysis and motility. The S236A mutant protein shows a comparatively small decrease in hydrolytic activity and motility, and thus this residue does not appear to be essential. To understand better the contribution of Ser236 to the function of myosin, structural and kinetic studies have been performed on the S236A mutant protein. The structures of the D. discoideum motor domain (S1dC) S236A mutant protein in complex with magnesium pyrophosphate, MgAMPPNP, and MgADP·vanadate have been determined. In contrast to the previous structure of wild-type S1dC, the S236A·MgAMPPNP complex crystallized in the closed state. Furthermore, transient-state kinetics showed a 4-fold reduction of the nucleotide release step, suggesting that the mutation stabilizes a closed active site. The structures show that a water molecule approximately adopts the location of the missing hydroxyl of Ser236 in the magnesium pyrophosphate and MgAMPPNP structures. This study suggests that the S236A mutant myosin proceeds via a different structural mechanism than wild-type myosin, where the alternate mechanism is able to maintain near normal transient-state kinetic values.
The mechanism of myosin ATPase and its activation by actin have been studied extensively in order to understand the molecular basis of motility and other cellular events. To date, a similar kinetic mechanism has been found for all myosin classes, although the rates of particular steps and their coupling with actin vary widely with type, according to the specific function of the myosin (1−6). In this sequence, ATP binding, as well as Pi and ADP release, is at least a two-step process, which together with hydrolysis itself leads to a minimal seven-step scheme to describe the elementary pathway (eq 1).
 |
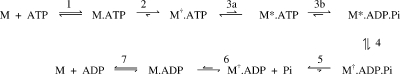
Intrinsic tryptophan fluorescence has been a key probe for distinguishing myosin conformers within the pathway. Many myosin isoforms show a fluorescence enhancement on binding ATP with a further enhancement associated with hydrolysis, as qualitatively represented by the * symbol. Wild-type Dictyostelium discoideum (Dd) myosin II, however, shows no enhancement on the initial ATP binding steps as it lacks tryptophan residues near the nucleotide binding pocket. Indeed, a small quench can be detected in some conditions (7). Mutant Dd constructs, in which tryptophan residues were introduced near the binding site to give a larger signal, demonstrated that ATP binding is at least two step (8), as with skeletal muscle myosin II (9−11). This binding process involves an initial weak encounter complex followed by a nearly irreversible trapping of the ATP, the latter step having a rate constant of >400 s−1. The large tryptophan fluorescence enhancement attributed to the hydrolysis step (20−160 s−1 for Dd myosin II (12,13)) was subsequently resolved into a rapid and reversible transition which changed the environment of a specific tryptophan (Trp501) and preceded ATP hydrolysis itself (14). The seven-step scheme was therefore expanded as shown in eq 2, in which the † symbol represents the small quench observed with wild-type Dd myosin II.
 |
These findings are consistent with structural studies which indicated that switch 2 must move from an “open” to “closed” position, in order that hydrolysis can occur. Switch 2 is located at one end of a helix, the “relay helix”, which transmits the information from the ATPase site to the C-terminal converter domain. The conserved Trp501 tryptophan is located in a loop at the other end of the relay helix and seems a nearly ideal reporter of events at the ATPase site, even though it is 50 Å away. Site-directed mutagenesis of Trp501 to another aromatic residue abolishes the nucleotide-induced fluorescence enhancement (7,15). This result indicates that the other three tryptophan residues in the myosin motor domain are insensitive to nucleotide binding and hydrolysis. Switch 2 closure and hence the relay helix tryptophan movement and fluorescence enhancement therefore precede the hydrolysis step itself.
There are now a large number of crystal structures available for myosins that define the major structural transitions that underlie the steps in the pathway and suggest the mechanisms involved in the conversion of chemical energy into directed movement (16−22). Currently, there are three primary conformations recognized for the myosin motor. Historically, the first of these was seen in chicken skeletal myosin subfragment 1, which most closely represents the “post-rigor” or prehydrolysis state in the kinetic cycle, where switch 2 is open (16). The second conformation is that of the pre-power stroke in which switch 2 is closed (18,23). The third conformation is presumed to represent the “rigor” state and is observed in the apo states (nucleotide free) of myosins V, VI, and II (19−22). These conformational states are defined by the extent of closure of the large cleft that splits the 50 kD region of the motor domain, the orientation of the lever arm, and the conformation of the active site residues.
In the “post-rigor” state the 50 kD cleft is open, but switch 1 in the nucleotide binding site is closed and favors high-affinity binding to ATP and low-affinity binding to actin. The transition to the pre-power stroke conformation is coupled with partial closure of the 50 kD cleft and switch 2 movement. This serves to orient the active site for nucleotide hydrolysis and prevent the loss of Pi. This transition is also accompanied by a large rotation of the light chain binding region that serves to prime the molecule for the power stroke. The final step in the conformational cycle is prompted by myosin rebinding to actin. The apo structures of myosins V, VI, and II, which are believed to mimic the actin-bound state for myosin, show that this transition is accompanied by closure of the 50 kD cleft and rotation of the lever arm to yield a power stroke. Importantly, actin binding is accompanied by the twisting of a large β-sheet that forms the backbone of the motor domain (24). This twisting motion changes the relationship between switch 1 and 2 and the P-loop such that Pi and ADP can no longer bind tightly (19−22). Thus, activation of the product release is coupled to translational movement of the actin filament relative to the myosin.
The above mechanism is based on attempts to correlate chemical states observed in solution with crystal structures. However, detailed kinetic studies have revealed that two or more myosin conformational states may be present at significant concentrations for a single nucleotide state (7,14). Crystallization necessarily will select a single conformational state. To date, ATPγS, ADP, PPi, and AMPPNP have yielded crystals of Dd S1dC1 in the open state (equivalent to the M†ATP and M†ADP states) (25,26), while ADP·AlF4 and ADP·VO4 have yielded the closed states, which serve as mimics of the Dd M*·ADP·Pi complex (eq 2) (18,23). On the other hand, ADP·BeFx complexes have been crystallized in both states (17,18). This is not surprising, given that the equilibrium between the open and closed states in solution is often within an order of magnitude of 1 and therefore little energy input is required to shift the dominant species. Low temperatures used for crystallization tend to favor the open state, which may account for AMPPNP generally crystallizing in this state, even though the closed state is present at significant concentration at 20 °C.
This study focuses on the function of Ser236 (Dd numbering) in the hydrolysis of ATP and maintenance of the metastable M*ADP·Pi state. Analysis of the active site of Dd myosin switch 2 open and closed structures shows there is no amino acid side chain that could be used as a catalytic base near the γ-phosphate. This suggests that the γ-phosphate acts as the fundamental general base in a reaction with a water molecule to produce hydrolysis. It was originally hypothesized that a highly conserved serine residue (Ser236) in the vicinity of the γ-phosphate may be involved with a proton relay mechanism to facilitate this proton rearrangement from the nucleophilically attacking water molecule to the γ-phosphate (18,23). However, mutational analysis in both smooth and Dd myosin II argued that this residue does not play a major role in nucleotide binding nor nucleotide hydrolysis, as the kinetics of these steps are only slowed by 2−5-fold (27,28). Furthermore, the S236A mutation in both Dd and smooth muscle myosin only causes a 7- and 2-fold decrease in motility, respectively, which further highlights the question of why this residue is conserved in all myosin classes. Classical molecular dynamics (MD) and combined quantum mechanical and molecular mechanical (QM/MM) calculations (29) have been utilized to calculate the rate-limiting activation barriers for the Ser236 assisted proton relay mechanism and for the direct proton transfer from water to the γ-phosphate. It was found that the Ser236 assisted mechanism had only a slightly lower rate-limiting barrier (29).
In an effort to understand the role of Ser236, a kinetic analysis of the S236A mutant protein has been performed in conjunction with a structural analysis of the protein in the open and closed conformation. The S236A mutation favors the closed state more than the wild type and has permitted the structure of myosin-bound AMPPNP in the pre-power stroke in which switch 2 is closed to be determined. The structure was obtained in the presence of blebbistatin because this yielded better crystals; however, the same crystal form was obtained without this inhibitor. Blebbistatin is an inhibitor that favors the closed state (30,31). This allows a view of a γ-phosphoryl moiety in the closed state which provides insight into the hydrogen bonding that helps to maintain ATP and ADP·Pi in the closed state and suggests an alternative role for S236A in preventing hydrolysis until switch 2 has closed.
Experimental Procedures
Construction, Expression, and Purification of S236A
Escherichia coli strain TOP10 was used for all cloning. D. discoideum SldC myosin II consisting of Asp2−Asn759 followed by a sequence that encodes a non-native C-terminal peptide LPN was cloned into a modified pDXA-3C expression vector (32) that placed an N-terminal 6His tag prior to a tobacco etch virus (rTEV) protease cut site. The N-terminal sequence of the expressed protein was MHHHHHHGRHMSGSAENLYFQGGN. This introduces GGN in place of MD at the N-terminus of SldC after cleavage with rTEV. The S236A mutation was introduced using QuikChange site-directed mutagenesis with primers 5′-CCCGTAACAACAATGCATCTCGTITCGG-3′ and 5′-CCGAAACGAGATGCATTGTTGTTACGGG-3′, and the resulting construct was then sequence verified. D. discoideum ORF+ cells were transformed with the resulting expression vector using a Gene Pulser II (Bio-Rad) electroporator as follows. Cells were grown in HL5 medium to a density of (3−6) × 106 cells/mL, washed, and resuspended in ice-cold electroporation buffer (10 mM KH2PO4, 20 mM HEPES, 140 mM KCl, 10 mM NaCl, 2 mM MgCl2, pH 7.5) at 5 × l07 cells/mL. Electroporation (one pulse of 1.25 kV, 50 μF) was performed in a 0.4 cm cuvette after mixing 0.4 mL of cell suspension with 20 μL of 1 μg/mL plasmid. After electroporation, 0.6 mL of HL5 medium at room temperature was added, and 10 and 100 μL of the cell suspension was plated onto 100 mm Petri dishes containing 10 mL of HL5. After 24 h of incubation at 21 °C, the selection was started by replacing the medium with HL5 containing 20 μg/mL G418 (Invitrogen). Seven days posttransfection, large individual clones were picked into 24-well plates and expanded for expression tests (33). Clones that showed the highest level of recombinant protein expression were used for large-scale cell growth (1 L of HL5 medium containing 10 μg/mL G418 in a 2 L shaking flask seeded with (1−2) × 106 cells and shaken at 21 °C/200 rpm until stationary phase was reached).
The myosin motor domain was purified as before (34). Specifically, 50 g of cells was lysed in 200 mL of 50 mM Tris-HCl, pH 8.0, 2 mM EDTA, 0.2 mM EGTA, 20% ethylene glycol, 1 mM DTT, 5 μg/mL leupeptin, 1 mM PMSF, 0.1 mM TLCK, 15 μg/mL RNase A, 100 units of alkaline phosphatase, 1% Triton X-100, and one tablet of Complete EDTA-free protease inhibitor (Roche). This was followed by centrifugation at 200000g and homogenization of the pellet with 100 mL of HKM buffer (50 mM HEPES, pH 7.3, 30 mM potassium acetate, 10 mM MgSO4, 20% ethylene glycol, 150 mM NaCl, 7 mM β-mercaptoethanol, 5 μg/mL leupeptin, 1 mM PMSF, 0.1 mM TLCK, and a tablet of Complete EDTA-free protease inhibitor (Roche)). Another round of centrifugation and homogenization was performed with 12 mM ATP in the HKM buffer. The last centrifugation step produced the final supernatant that was applied to a Ni-NTA (Qiagen) column. The protein bound to the Ni-NTA column was washed first with high salt buffer (50 mM Hepes, pH 7.3, 300 mM potassium acetate, and 3 mM benzamidine) and then low salt buffer (50 mM Hepes, pH 7.3, 30 mM potassium acetate, and 3 mM benzamidine) followed by elution with a 0−500 mM imidazole gradient. Fractions containing the protein were pooled and dialyzed overnight against 10 mM Hepes, pH 7.4, 50 mM NaCl, 0.2 mM EDTA, 1 mM DTT, 0.2 mM NaN3, and complete EDTA-free protease inhibitor (Roche). Cleavage of the 6His tag was performed using rTEV protease (purified as described in ref (35)), and the solution was passed over Ni-NTA again to remove the rTEV protease and the 6His tag. Fractions containing S1dC were again pooled and dialyzed overnight in the above dialysis buffer. The protein was finally concentrated to 10 mg/mL in a Centriprep-50 microfiltration device, frozen as 30 μL droplets in liquid nitrogen, and stored at −80 °C.
Crystallization and Freezing
For the MgADP·VO4 and MgAMPPNP crystal forms, the S236A mutant protein of S1dC at 12 mg/mL was first mixed with blebbistatin to a final concentration of 0.5 mM. Blebbistatin was added to the crystallization because it improved the quality of the final crystals. Previous studies have shown that the active site is virtually identical in the presence and absence of blebbistatin (34). The protein solution was allowed to incubate at room temperature for 10 min in the dark before a 1/9th volume of 10× nucleotide analogue cocktail was added. The 10× MgADP·VO4 cocktail contained 10 mM MgCl2, 20 mM ADP, and 30 mM sodium vanadate. The 10× MgAMPPNP cocktail contained 20 mM MgCl2 and 20 mM AMPPNP. Both were allowed to incubate with the protein for 1 h on ice in the dark after mixing. The protein solutions were mixed with a 12% polyethylene glycol (PEG) 8K, 250 mM MgCl2, and pH 7.0 100 mM MOPS precipitant solution in a 1 to 1 μL ratio. The drops were streaked-seeded the following day using crushed crystals from an earlier crystallization experiment. Crystals continued to grow over the course of 2 weeks until they had attained their maximum dimensions of ∼700 × 200 × 80 μm. Crystals were cryoprotected prior to being frozen by being gradually transferred into a cryosolution of 16% PEG 8K, 300 mM MgCl2, 100 mM MOPS, pH 7.0, and 25% ethylene glycol solution. A synthetic mother liquor solution that matched the original well solution of 12% polyethylene glycol (PEG) 8K, 250 mM MgCl2, and pH 7.0 100 mM MOPS together with the ligand mixture was prepared and used to dilute the cryosolution in a mother liquor:cryoprotectant solution ratio 1:0, 3:1, 1:1, 1:3, and 0:1. The time of each soak was 7, 5, 5, 3, and 1 min, respectively. The crystals were then flash-frozen using a nitrogen cold stream. Crystals of the magnesium pyrophosphate complex (S236A·MgPPi) were grown by mixing a 10× trapping solution cocktail containing 20 mM each of MgCl2 and sodium pyrophosphate with the S236A mutant protein at 12 mg/mL to a final 1× concentration and incubated on ice for 1 h, in the absence of blebbistatin. This mixture was mixed 1:1 with a precipitant solution containing 12.5% methyl ether polyethylene glycol (MePEG) 5K, 117 mM ammonium acetate, and pH 8.0 100 mM Hepps. The drops were streaked-seeded the following day using crushed crystals from an earlier crystallization experiment. Crystals continued to grow over the course of 2−3 weeks. The cryoprotectant solution consisted of 16% MePEG 5K, 185 mM ammonium acetate, 25 mM NaCl, 100 mM Hepps, pH 8.0, 20% ethylene glycol, and 4% glycylglycine, together with 2 mM MgCl2 and 2 mM sodium pyrophosphate. The crystals were then flash-frozen using a nitrogen cold stream.
Data Collection, Structure Determination, and Refinement
X-ray data for the S236A·MgADP·VO4 and S236A·MgAMPPNP complexes were collected at the Advanced Photon Source (APS) in Argonne, IL, using a MAR-165 detector at COM-CAT 32-ID. A total of 280 frames, each with an oscillation range of 0.5°, were collected at 150 mm. The data were integrated and scaled with the HKL2000 program package (36). The structures were determined by molecular replacement with the program Molrep (37) utilizing the structure of S1dC·MgADP·VO4 (PDB accession number 1VOM) (23) as the starting model. The structure was refined with Refmac5 in the CCP4 package, and water molecules were located and added using the model building program COOT (38,39). The conformations for AMPPNP and blebbistatin were obtained from coordinate sets 1MMN and 1YV3, respectively (26,34). The models were manually adjusted with the model building program COOT (38,39).
Data for S236A·MgPPi were collected at APS using a ADSC Q315 315 mm × 315 mm mosaic CCD detector at SBC-CAT 19-ID. A total of 220 frames, each with an oscillation range of 1°, were collected at 180 mm. Integration and solution of the structure were performed as above with the exception that the starting model was that of S1dC·MgPPi (PDB accession code 1MNE) (25). Data collection and refinement statistics are given in Table 1.
Table 1. Data Collection and Refinement Statistics.
| MgADP·VO4 | AMPPNP | MgPPi | |
|---|---|---|---|
| space group | C2221 | C2221 | P21212 |
| unit cell parameters | |||
| a, b, c (Å) | 88.4, 146.4, 153.7 | 88, 147, 153.4 | 104, 180.8, 53.9 |
| α = β = γ (deg) | 90 | 90 | 90 |
| wavelength (Å) | 0.97885 | 0.97885 | 1.00707 |
| resolution range (Å) | 50−2.0 | 50−1.84 | 30−2.0 |
| reflections: measured | 332841 | 503544 | 529646 |
| reflections: unique | 64281 | 84770 | 69055 |
| redundancy | 5.2 (4.0) | 6 (4.6) | 7.7 (7.8) |
| completeness (%) | 96.7 (90.1) | 98.7 (97.6) | 99.8 (98.6) |
| average I/σ | 18.6 (2.9) | 35.7 (5.3) | 17.6 (3.9) |
| Rsym (%) | 6.1 (23.7) | 5.1 (15.2) | 11.5 (40.6) |
| Rwork (%) | 19.3 | 19.9 | 18.9 |
| Rfree (%) | 23.4 | 22.7 | 23.6 |
| no. of protein atoms | 708 | 706 | 5976 |
| no. of water molecules | 352 | 445 | 736 |
| Wilson B-value (Å2) | 23.6 | 24 | 19.7 |
| average B factors (Å2) | |||
| S1dC | 26.3 | 26.8 | 20.9 |
| solvent | 28.5 | 32.3 | 30.2 |
| Ramachandran (%) | |||
| most favored | 92 | 92.3 | 92.9 |
| additionally allowed | 7.5 | 6.9 | 6.8 |
| generously allowed | 0.2 | 0.5 | 0.1 |
| disallowed | 0.3 | 0.3 | 0.1 |
| rms deviations | |||
| bond lengths (Å) | 0.013 | 0.011 | 0.013 |
| bond angles (deg) | 1.356 | 1.339 | 1.411 |
| chiral | 0.094 | 0.111 | 0.098 |
Kinetic Measurements
Transient tryptophan fluorescence measurements were recorded using an Applied Photophysics SX18MV stopped-flow instrument with 295 nm excitation and a WG320 cutoff filter in the emission channel (7). ATP-induced dissociation of actin complexes with S1dC was monitored by light scattering at 340 nm as described previously (13). Concentrations of components are stated for the reaction chamber (i.e., half the syringe concentrations). Slow time courses were also monitored by manual mixing using an SLM 8000 spectrofluorometer with the same excitation wavelength and emission filter. Reactions were performed in a buffer containing 40 mM NaCl, 20 mM TES or Hepes, and 2 mM MgCl2 at pH 7.5 and 20 °C, unless otherwise stated. Records were normally collected with a logarithmic time base to ensure similar weighting was given when fitting fast and slow phases in multiphasic reactions (40). Rate constants were obtained by fitting to single or double exponential functions using Kaleidagraph (Synergy Software) or the Applied Photophysics Pro-Data software. Standard errors in the fitted parameters were generally <1%, and standard deviations in parameter values between repeat measurements were <20%. Corrections were made to account for photobleaching, which was of the order of 1% decrease per minute in the stopped-flow apparatus.
To determine the extent of ATP hydrolysis following the binding step, 50 μL of 5 μM γ-32P-labeled ATP was mixed with an equal volume 20 μM S1dC construct, and the reaction was quenched after 10 s with 100 μL of 6% perchloric acid. The 32P-Pi was extracted using a phosphomolybdate-based procedure as described previously (7). The extent of ATP hydrolysis was corrected for the 32P-Pi detected in a control sample quenched at zero time.
Results and Discussion
Crystal Structures
Structures for the open and closed forms of S236A S1dC have been determined. Complexes exhibiting the closed form were obtained for both S236A·MgAMPPNP and S236A·MgADP·VO4, where the overall protein topologies are very similar to the previously determined closed forms of S1dC observed in MgADP·VO4 and MgADP·AlF4 complexes (18,23). The root mean square differences between the equivalent α-carbon atoms for S236A·MgAMPPNP and S236A·MgADP·VO4 and the reference data sets for S1dC·MgADP·VO4 (1YV3 and 1VOM) are 0.15 and 0.37 Å, respectively, for 645 structurally equivalent amino acid residues. As noted earlier, blebbistatin was utilized to improve the quality of the crystals of the S236A·MgAMPPNP complex. It is thus noteworthy that the S236A mutant protein complexed with blebbistatin and MgAMPPNP is exceedingly similar (rms 0.15 Å) to the equivalent wild-type protein complexed with vanadate in the presence of blebbistatin. The same crystal lattice was obtained for the S236A·MgAMPPNP complex in the absence of blebbistatin, where this crystal form is only commensurate with the closed form of myosin because of the 70° rotation of the converter domain. Likewise, the structure of S236A·MgPPi, which exhibits the open conformation, is very similar to that of the wild-type complex (1MNE(25)) where the root mean square difference is 0.35 Å for 704 structurally equivalent amino acid residues.
The structures for the S236A·AMPPNP, S236A·MgADP·VO4, and S236A·MgPPi complexes were determined to 1.84, 2.0, and 2.0 Å resolution, respectively (Table 1). It is noteworthy that Trp501 is ordered in all of the structures determined here. This is not unexpected for the two structures of the closed state (S236A·MgAMPPNP and S236A·MgADP·VO4) since this residue is typically ordered in this conformational state. It is unusual to observe Trp501 in the open state (S236A·MgPPi). Typically, residues 500−508 are disordered in the open conformation, whereas only residues 503−506 are disordered in the S236A·MgPPi complex. This further demonstrates that the contents of the active site can have subtle effects on the environment of this spectroscopically sensitive residue, even when the overall structure of the protein is exceedingly similar.
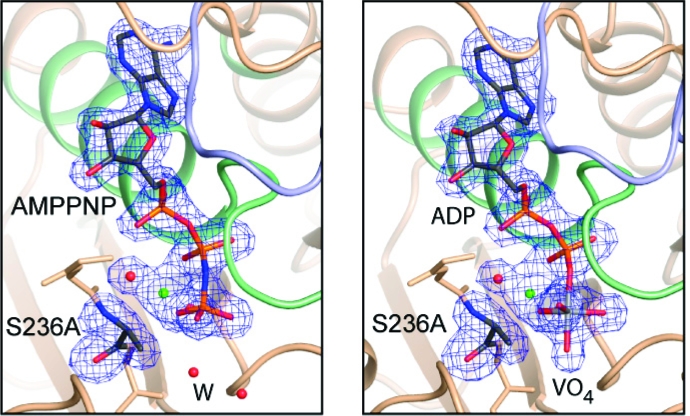
In all complexes, the electron density for the ligands in the active sites is unequivocal, as is the electron density surrounding the S236A mutation (Figure 1 and Supporting Information Figure 1). The mutation itself does not introduce any significant changes in the positions of the amino acid residues in either the open or closed forms (Figures 2 and 3). Indeed, the only obvious changes are in the position of the water molecules associated with the γ-phosphate binding pocket.
Figure 1.

Stereoview of the electron density for S236A·MgAMPPNP in the closed conformation at 1.84 Å resolution. The electron density map, contoured at 3σ, was calculated from coefficients of the form Fo − Fc where the MgAMPPNP, Ala236, and the associated water molecules were omitted from the phase calculation and refinement. Mg2+ is shown as a green sphere. Figures 1−5 were prepared with the program Pymol (42).
Figure 2.

Stereoviews the γ-phosphoryl binding site in the S236A·MgPPi complex and a comparison with wild-type S1dC·MgATP. Both of these complexes represent the open state of myosin. (A) shows the coordination of the water molecule that fills the cavity left by the removal of the γ-hydroxyl group of Ser236. The new water molecule is coordinated by one of the nonbridging oxygen atoms of the terminal phosphoryl group of magnesium pyrophosphate, the carbonyl oxygen of Ser237, and the guanadinium Nη of Arg238 (distances in Å). The water molecule is within van der Waals distance of the β-carbon of Ala236. (B) shows the superposition of the S236A·MgPPi complex with the wild-type S1dC·MgATP complex (PDB accession code 1FMW(41)). The S236A·MgPPi complex is depicted in color whereas the S1dC·MgATP complex is shaded in gray. This panel reveals the very close similarity between the structures of these complexes and demonstrates that the S236A mutation does not introduce a large perturbation in the overall structure of the myosin motor domain. The new water molecule in the S236A·MgPPi complex is labeled “W” for clarity. As can be seen, a water molecule could not adopt this position in the wild-type structure since it would clash with the serine hydroxyl (black dashed arrows). Introduction of the new water molecule displaces the primary waters observed in the wild-type S1dC·MgATP complex (red dashed arrows). The structures described in this figure and elsewhere were superimposed with the program uw_align (43). The coordinates for the wild-type S1dC·MgATP complex were obtained from PDB file 1FMW(41).
Figure 3.

Stereo comparison of the S236A·MgADP·VO4 and wild-type S1dC·MgADP·VO4 complexes. The S236A mutant protein is depicted in colors whereas the wild-type protein is colored in gray. This figure shows that the protein in both complexes adopts an essentially identical conformation in the closed state. Likewise, the two water molecules associated with the γ-phosphoryl binding pocket occupy very similar locations (arrows). Furthermore, the locations of the magnesium ion and its associated waters are the same within experimental error. This leaves a small cavity in the S236A mutant protein active site associated with the removal of the serine γ-hydroxyl moiety. The coordinates for the wild-type S1dC·MgADP·VO4 complex were obtained from PDB file 1VOM(23).
It is widely accepted that the open conformation of myosin, as exemplified by the S1dC·MgPPi or S1dC·MgATP complex (25,41), is not competent to hydrolyze ATP because the water molecule responsible for hydrolysis is not in an appropriate location for nucleophilic in-line attack on the γ-phosphoryl moiety. Furthermore, the water molecules that are in the active site are hydrogen bonded in such a way that the putative locations of lone pairs of electrons that would be expected to attack the γ-phosphorus atom are coordinated to other components of the active site.
Examination of the structure of the S236A·MgPPi complex shows that removal of the hydroxyl on Ser236 allows, relative to the wild-type structure, the intrusion of a water molecule that hydrogen bonds between one of the nonbridging oxygen atoms of the terminal phosphoryl group, the carbonyl oxygen of Ser237, and the guanadinium Nη of Arg238 (Figure 2A). The water molecule is within van der Waals distance (3.5 Å) of the β-carbon of Ala236. This prompts a rearrangement of two of the primary water molecules in the structure of S1dC·MgATP (Figure 2B).
The water structure of the S236A·MgADP·VO4 complex is identical to that of wild-type S1dC·MgADP·VO4 (Figure 3). The absence of the Ser236 hydroxyl in the S236A complex introduces a hole that is not filled in the closed conformation. This is not surprising from a structural point of view, since the γ-phosphate binding pocket is quite small and only contains two water auxiliary molecules outside of the axial water molecule coordinated by the vanadate anion. (There are two other water molecules associated with the magnesium ion.) All of the active site water molecules in the vanadate complexes exhibit a complete four-coordinate hydrogen-bonding network and as such would be difficult to displace. As a consequence, removal of the hydroxyl from Ser236 leaves a hole, rather than prompting a rearrangement of the contents of the active site.
Comparison of the water structure in S236A·MgAMPPNP relative to that seen in S236A·MgADP·VO4 shows that the γ-phosphate binding pocket contains three water molecules (outside of the two water molecules associated with the magnesium ion). Two of these occupy essentially identical locations to the auxiliary waters seen in both S236A·MgADP·VO4 and wild-type S1dC·MgADP·VO4 (Figure 4A). The third occupies essentially the same location as the water molecule that occupies the hole left by removal of the serine hydroxyl in the S236A·MgPPi open complex (Figure 4B). This water molecule in the S236A·MgAMPPNP complex forms a four-coordinate hydrogen-bonding network with one of the nonbridging oxygen atoms of the γ-phosphoryl group, the carbonyl oxygen of Ser237, the guanadinium Nη of Arg238, and one of the conserved auxiliary water molecules. The only difference in the coordination of this water molecule, relative to that seen in the S236A·MgPPi complex, is the additional hydrogen bond to the conserved water molecule that is absent in the open conformation.
Figure 4.

Stereo comparison of (A) S236A·MgAMPPNP with S236A·MgADP·VO4 and (B) S236A·MgAMPPNP with S236A·MgPPi. The bonds in the S236A·MgAMPPNP are depicted in color, whereas the other complexes are shown in gray. In (A) the water structures for the two closed complexes of the S236A mutant are compared. These mimic the ATP and transition state for the closed conformation. The hydrogen-bonding network is shown for the water molecules in the S236A·MgAMPPNP. For clarity the hydrogen bond distances have been omitted; however, all of the distances lie within the range of 2.6−2.9 Å. The arrow indicates that the new water molecule (W) would only have to move 1.5 Å to adopt an axial position relative to the γ-phosphoryl moiety. Regardless of whether this is the nucleophile, this water molecule must move in order to form the transition state for hydrolysis. (B) shows the superposition of the open and closed states of the S236A mutant protein as represented by the MgPPi and MgAMPPNP complexes. This reveals that the new water molecule that takes the place of the serine hydroxyl adopts the same location in the open and closed complexes.
The γ-phosphoryl group of the S236A·MgAMPPNP closed complex provides insight into the structural transitions that occur between the open and closed conformations of the myosin motor domain. In particular, it shows how the γ-phosphoryl moiety is coordinated in the active site in the closed state. As can be seen, the location of the γ-phosphoryl group relative to the P-loop and upper 50 kD section of the motor domain is very similar in the S236A·MgAMPPNP closed complex and in the S1dC·MgATP open complexes. This is consistent with the general understanding that the position of ATP does not change in the active site in order to facilitate hydrolysis. Rather, it is repositioning of switch 2 which supports movement of a water molecule into the axial location relative to the γ-phosphorus and its bond to the bridging oxygen to ADP.
Transient-State Kinetics
Shimada et al. reported a less that 2-fold reduction in the basal steady-state Dd Mg·ATPase rate on introduction of the S236A mutation (28). Li et al. found a 4-fold reduction for the equivalent mutation in smooth muscle myosin (27). We readdressed this measurement using single turnover analysis, as this approach is less sensitive to uncertainties in the active protein concentration. Using intrinsic tryptophan fluorescence, single ATP turnovers of the Dd S1dC wild-type and S236A mutant revealed a 3.4-fold reduction in the rate-limiting product release step, from 0.021 to 0.0058 s−1 (Figure 5). In a separate experiment, acid quenching the reaction after 10 s, when the ATP binding phase was nearly complete, followed by Pi analysis, showed 37% of the ATP was hydrolyzed to the M*·ADP·Pi state for the S236A mutant, compared with 40−80% typically obtained for wild-type preparations (7). As an independent check of the ATP turnover rate, a manual assay was set up in which 100 μM ATP was mixed with the either 4 μM wild-type or S236A construct, and after 100 s, when the tryptophan fluorescence level reflected the steady state, 5 mM PPi was added as a competitive ligand (Supporting Information Figure S2A). The decay in tryptophan fluorescence followed a single exponential with rate constant 0.028 s−1 for the wild-type construct and 0.0068 s−1 for S236A. These values are similar to the rate constants determined from single turnover analysis in a stopped-flow instrument, as described above, and reflect the decay of the M*ADP·Pi complex in both cases. These measurements confirm the previous findings (26,27) that Ser236 is not essential for near-normal catalytic activity but that the basal ATPase of the S236A construct is lower.
Figure 5.

Single turnover of 1 μM ATP by 2 μM S1dC as monitored by tryptophan fluorescence: (A) wild-type; (B) S236A mutant. The reaction was measured in 40 mM NaCl, 10 mM Hepes, and 2 mM MgCl2 at pH 7.5 and 20 °C. The signals were normalized such that the fluorescence of the apo state was equal to 1. Superposed fits to a single exponential yield rate constants of 0.02 and 0.0058 s−1, respectively.
ATP binding and hydrolysis yielded a 40% enhancement in tryptophan fluorescence for both wt and S236A Dd S1dC (Supporting Information Figure S2B), although the mutant showed a greater proportion (30%, cf. 15% total amplitude) of a concentration-independent slow phase (0.5 s−1, cf. 2 s−1, respectively). The rate constant of the fast phase increased with increasing [ATP] and reached a maximum of 125 s−1 for wild type (Kapp = 208 μM) and 97 s−1 for S236A (Kapp = 192 μM). The values for the observed rate constants at saturating ATP are higher than those we reported previously for wild-type Dd M759 preparations (17 s−1(13)) and the Trp501+ construct (30 s−1(7)) but are within the range reported for Dd constructs with differing C-terminal truncations (12). ADP binding to both preparations was accompanied by a small quench in fluorescence (around 2%) that was not sufficient for detailed kinetic characterization but indicated that ADP bound with an apparent kon in the region of 106 M−1 s−1. This is in line with previous studies on the mutant Dd Trp501+, containing a single tryptophan residue, where the signal to background was much larger (7). ADP displacement with ATP yielded a dissociation rate constant of 0.28 s−1 for S236A compared with 1.6 s−1 for wild type. Overall, these studies indicate that S236A shows near wild-type behavior, but with a tendency to release nucleotides around 4 times slower than the wild type, suggesting stabilization of a closed state.
As with ATP, AMPPNP binding to wild type and S236A also gave a biphasic fluorescence enhancement, but the second slower phase was more marked in both cases (Figure 6). The overall fluorescence enhancement was 17.6 ± 1.7% (SD n = 5) for wt and 31.6 ± 1.9% (SD n = 5) for S236A. For S236A, the fast phase increased linearly with [AMPPNP] to yield an apparent second-order rate constant of 4.8 (±1.0 SE) × 104 M−1 s−1 and exceeded a value of 12 s−1 (kobs at 500 μM AMPPNP). The second slow phase showed little concentration dependence and had a maximum rate constant of 0.29 s−1. For the wild-type preparation, the corresponding rate constants for the fast and slow phases were 2 and 3 times larger, respectively (Figure 6). The dissociation rate constant for the M*·AMPPNP complex was too small to be measured by stopped flow, where photobleaching and drift due to back-diffusion over >1000 s made analysis difficult. Displacement by manual addition of 4 mM PPi to wild-type M*·AMPPNP (and using an excitation slit width of 0.5 nm to minimize photobleaching) yielded a rate constant of about 0.001 s−1. For the S236A mutant, the reaction was more difficult to analyze because PPi also gave an enhancement in tryptophan fluorescence (see below), and the overall amplitude of the signal on exchanging AMPPNP for PPi (8%) was 4 times smaller than the change for wild type. However, the observed rate constant for displacement appeared comparable to that for wild type. Overall, we conclude that the interactions of AMPPNP with wild-type S1dC and the S236A construct are similar, but the larger fluorescence enhancement with S236A suggests that a larger fraction exists in the closed state at equilibrium.
Figure 6.
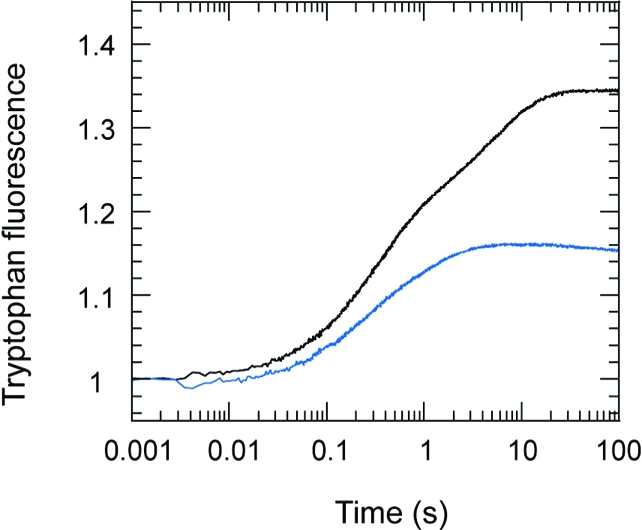
Enhancement on tryptophan fluorescence on binding 60 μM AMPPNP to 2 μM S1dC: wt (blue trace); S236A mutant (black trace). Conditions were as in Figure 5. Fitting a biexponential to the data between 0.1 and 10 s yielded rate constants of 3.4 s−1 (amplitude = 0.17) and 0.24 s−1 (amplitude = 0.14) for the S236A construct. For the wild-type construct values were 5.4 s−1 (amplitude = 0.085) and 0.92 s−1 (amplitude = 0.081). The fluorescence signal was normalized to 1 for the apo state, so that the total fluorescence enhancement in these records was 31% for the S236A construct and 16.6% for the wild type.
The wild-type and S236A mutant protein differed most markedly in their binding of PPi (Figure 7). At 20 °C, wild-type S1dC gave a 2.5% quench on binding PPi with a single exponential profile and yielded an apparent second-order association rate constant of 3.7 × 104 M−1 s−1. On the other hand, S236A showed a 22% enhancement with at least two phases. On reducing the temperature to 5 °C, the fluorescence of the apo S236A preparation increased by 14%, in line with the typical temperature dependence of tryptophan (14), but the fluorescence enhancement induced by PPi was reduced to 7%. Furthermore, an additional small quench phase was observed between the two enhancement phases. These results cannot be interpreted in terms of interconversion between a single open and single closed state, as modeled previously (7). However, the temperature dependence is in line with the earlier finding that low temperature favors an open state with low or no fluorescence enhancement (14). The higher fluorescence enhancement amplitudes for PPi and AMPPNP binding to S236A compared with wild-type S1dC indicate that the mutation favors the closed state. This likely accounts for the ability to crystallize the mutant protein in the closed state with MgAMPPNP at low temperatures. As noted earlier, blebbistatin is not required to obtain these crystals, though it does improve their quality. Thus, the enhanced propensity for the closed state reflects an intrinsic property of the mutant protein.
Figure 7.
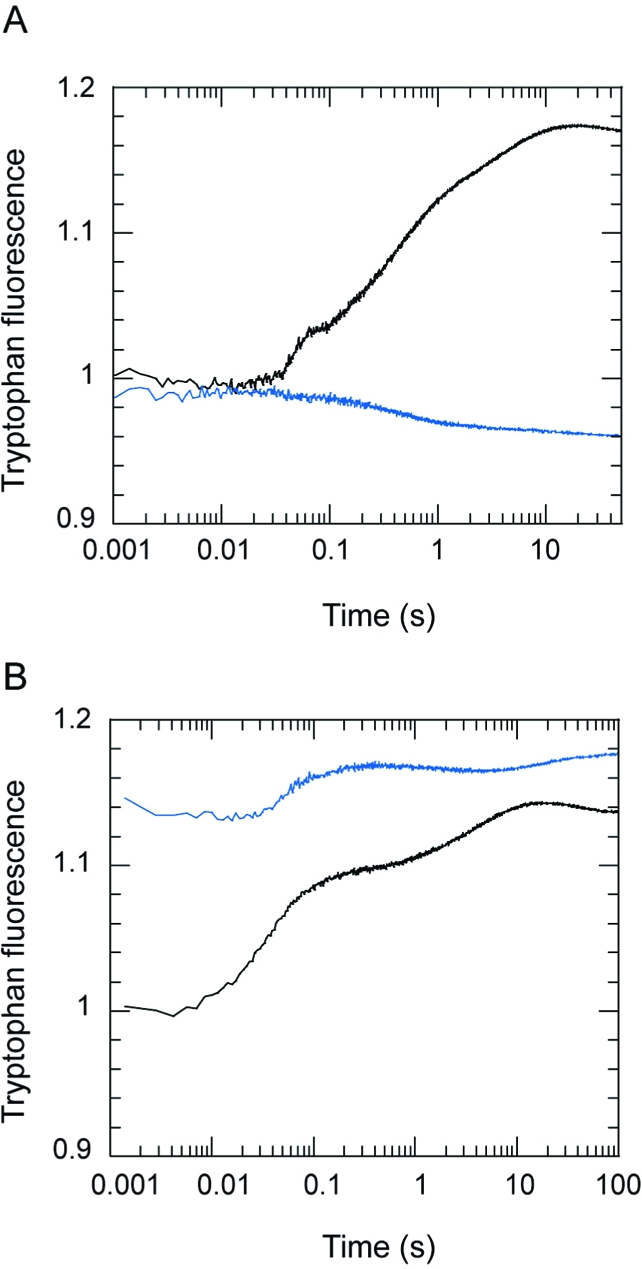
(A) Stopped-flow records showing the change in tryptophan fluorescence on binding 50 μM PPi to 2 μM S1dC (wild-type showing quench in fluorescence (blue trace) and S236A (black trace) showing an enhancement). (B) The fluorescence enhancement on binding 500 μM PPi to 2 μM S236A at 20 °C (black) or 5 °C (blue). The records are normalized to the fluorescence of the apo state at 20 °C.
The S236A construct formed a complex with actin whose ATP-induced dissociation was monitored using light scattering. The apparent second-order rate constant for ATP binding to acto-S236A at low [ATP] was smaller (0.3 μM−1 s−1) than for the wild-type construct (1.1 μM−1 s−1), but the observed rate constant at 1 mM ATP was similar (170 s−1). However, while the wild-type construct was close to saturation at 1 mM ATP, the S236A construct may show a higher maximum rate constant (∼400 s−1) at saturating [ATP]. Changes in ionic strength at higher [ATP] make experimental determination difficult. Nevertheless, at the typical [ATP] used for actin-activated steady-state ATPase and in vitro motility assays (27), the rate constant for the actin dissociation step would be comparable. Previous studies (27) indicated a 4-fold reduction in the Vmax of the steady-state actin-activated ATPase and a 7-fold reduction in the velocity of actin filament sliding for the S236A mutation. It therefore appears the S236A mutant is compromised in steps following actin dissociation.
Role of Ser236
The structure of the S236A·MgADP·VO4 complex might suggest that the closed state in this mutant protein would be less stable than the open state due to the creation of a cavity in the γ-phosphoryl binding pocket. This hypothesis is not supported by the kinetic studies which show that the S236A mutation favors the closed state by a factor of about 4-fold. The greater stability of the closed state for the S236A mutant protein relative to the open state is also supported by the observation that it can be crystallized in the closed state in the presence of AMPPNP. There are two possible explanations for this apparent dichotomy. First, the modifications in the water structure lead to a stabilization of the closed state. Second, the mutation changes the energy of the open state in a manner that favors the closed state. This latter viewpoint recognizes that the stability of the closed state is reflective of the difference in energy between the open and closed states. Mutations can influence either or both states.
These two explanations are not mutually exclusive. Careful examination of the environment of Ser236 in the wild-type structures shows that this residue participates in one or two hydrogen-bonding interactions in the open state for Dd S1dC. Likewise, in the structure of the rigor-like state seen in myosin V (1OE9(19)) the equivalent residue is partially coordinated. In contrast, in the closed state Ser236 is approximately tetrahedrally coordinated by the substrate, protein, and water molecules in the active site. This again would suggest that the S236A mutant protein would have a less stable closed state, because these interactions would be removed. Comparison of the three structures for the S236A mutant shows that the water molecule that takes the place of the γ-hydroxyl group of Ser236 is also highly coordinated in both of the closed state structures, whereas it is only bicoordinate in the open state. This provides an explanation for why the mutation might not be destabilizing relative to the wild-type structure, but it does not explain why the mutation leads to a net stabilization of the closed state. One possible solution lies in the putative location of the hydrolytic water molecule in the wild-type enzyme.
At this time, no structure is available for MgATP in the closed state. Consequently, the location of the true hydrolytic water molecule, prior to its movement into the axial position, is not known. As noted earlier, the new water molecule observed in the S236A·MgAMPPNP complex cannot occupy the same location as the true hydrolytic water in the wild-type protein because its location is too close to the γ-hydroxyl of Ser236 (2.1 Å). Indeed, it seems likely that Ser236 in the wild-type protein serves to localize a water molecule in the true axial position. It was proposed many years ago that the conformation of the closed state stabilized a water molecule in the axial position so that one of its lone pairs of electrons would be collinear with the P−O bridging bond that would be broken (23). Ser236 was proposed to play a key role in the process. Energetically, this would be an unstable situation for a water molecule since in this position it can only form three hydrogen bonds. Nevertheless, myosin spends a considerable amount of time with ATP in its active site when it is in the closed state, since this state contains an equilibrium mixture of ATP and ADP·Pi. This means that for a substantial fraction of the time there will be a thermodynamically unstable water in the active site. This would destabilize the closed state in the wild-type enzyme. In contrast, in the S236A mutant the water molecule appears to adopt a stable location prior to the transition state (as modeled by ADP·VO4), which would then favor the closed state. This provides a satisfactory answer to the modest increase in the stability of the closed state for the S236A mutant protein.
The fact that the S236A mutant hydrolyzes ATP does not mean that Ser236 is unimportant in the hydrolytic mechanism of wild-type myosin, because the S236A mutant protein probably proceeds via a different mechanism than the wild type. The mutation introduces a new water molecule into the active site which probably creates an alternative route by which ATP is hydrolyzed. In the case of the mutant protein, the new water molecule either must function as the nucleophile or must move away to allow another water molecule to enter the γ-phosphate binding pocket. It seems likely that the water molecule that takes the place of the serine hydroxyl serves as the nucleophile, since it is observed in both the open and closed conformations and is only located ∼1.5 Å from the axial water molecule of the vanadate complex (Figure 4A). It is already hydrogen-bonded to a terminal oxygen of the γ-phosphoryl group, which means that a four-center proton transfer from the attacking water to the γ-phosphate would be more favorable from proximity arguments. It also means that from entropic considerations fewer changes are necessary to bring the water into position for nucleophilic attack, so that even if the four-center proton transfer is unfavorable, the proximity of the water will counterbalance this cost. The new water molecule is functionally different from the serine γ-hydroxyl group and hence demands a different structural mechanism since there is no longer an adjacent hydroxyl group to facilitate orientation of the nucleophile or proton transfer.
This study explains why the S236A mutation retains function, but it still does not account for the conservation of serine in this position in the myosin superfamily. Interestingly, there is a conserved serine in the kinesin superfamily of molecular motors that lies in approximately the same location as Ser236, but its role in the ATPase cycle is not well characterized. In contrast, the corresponding amino acid in the G-proteins, which have a very similar P-loop and analogous switch 1 and 2, is a proline residue. This establishes a precedent that a hydroxyl group coordinated directly to the γ-phosphoryl moiety is not necessary for hydrolysis, which is consistent with these studies of the S236A mutation. Furthermore, this comparison emphasizes that, although motor or signaling proteins might utilize a similar mechanism for nucleotide hydrolysis, the biological function is tightly coupled to the kinetics of product release (myosin and kinesin) and activation (G-proteins) which differ among proteins that exhibit a strikingly similar nucleotide binding motif. Clearly, an alanine residue in myosin would have been sampled during evolution since it is only one base removed from serine in the genetic code. So, obviously, there must be a functional advantage to retaining a serine. The myosin superfamily of proteins exhibits a wide range of rate constants that encompass the differences in enzymatic function observed between the wild-type and S236A mutant protein. Thus, the reason for the conservation of Ser236 still remains unclear, though the explanation for its retention of function after replacement with alanine is now better understood.
Acknowledgments
We thank Robert Smith and Kirsten Dennison for assistance in purification and cloning, Jane Dickens and Kenji Imamura for assistance in some kinetic measurements, and Dr. John Allingham for helpful discussions. We also thank Laura Walker for assistance in crystal growth.
Supporting Information Available
Stereoviews of the electron density for (a) MgADP·VO4 and (b) MgPPi in the S236A mutant protein complexes both at 2.0 Å resolution (Figure S1) and determination of the ATP turnover rate constant by displacement with PPi and the dependence of the observed rate constant for tyrptophan enhancement on [ATP] (Figure S2). This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by funds from the NIH to I.R. (GM086351). Stopped-flow instrumentation and upgrades were funded by Wellcome Trust grants to C.R.B. Use of the SBC ID19 and ComCat ID32 beamlines at the Argonne National Laboratory Advanced Photon Source was supported by the U.S. Department of Energy, Office of Energy Research, under Contract No. W-31-109-ENG-38.
X-ray coordinates for the complexes between the S236A mutant of S1dC complexed with MgADP·VO4, MgAMPPNP, and MgPPi have been deposited in the Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (Protein Data Bank entries 3MYH, 3MYK, and 3MYL, respectively).
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: S1dC, myosin motor domain; APS, Advanced Photon Source, Argonne National Laboratory; PEG, polyethylene glycol; PPi, pyrophosphate.
Supplementary Material
References
- Geeves M. A.; Holmes K. C. (1999) Structural mechanism of muscle contraction. Annu. Rev. Biochem. 68, 687–728. [DOI] [PubMed] [Google Scholar]
- Howard J. (2001) Mechanics of motor proteins and the cytoskeleton, Sinauer Associates, Sunderland, MA. [Google Scholar]
- Wakelin S.; Conibear P. B.; Woolley R. J.; Floyd D. N.; Bagshaw C. R.; Kovacs M.; Malnasi-Csizmadia A. (2002) Engineering Dictyostelium discoideum myosin II for the introduction of site-specific fluorescence probes. J. Muscle Res. Cell Motil. 23, 673–683. [DOI] [PubMed] [Google Scholar]
- De La Cruz E. M.; Ostap E. M. (2004) Relating biochemistry and function in the myosin superfamily. Curr. Opin. Cell Biol. 16, 61–67. [DOI] [PubMed] [Google Scholar]
- Zeng W.; Conibear P. B.; Dickens J. L.; Cowie R. A.; Wakelin S.; Malnasi-Csizmadia A.; Bagshaw C. R. (2004) Dynamics of actomyosin interactions in relation to the cross-bridge cycle. Philos. Trans. R. Soc. London, Ser. B 359, 1843–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mezgueldi M., and Bagshaw C. R. (2008) The myosin family: biochemical and kinetic properties, in Myosins: a superfamily of molecular motors (Coluccio L. M., Ed.) pp 55−93, Springer, Berlin. [Google Scholar]
- Malnasi-Csizmadia A.; Woolley R. J.; Bagshaw C. R. (2000) Resolution of conformational states of Dictyostelium myosin II motor domain using tryptophan (W501) mutants: implications for the open-closed transition identified by crystallography. Biochemistry 39, 16135–16146. [DOI] [PubMed] [Google Scholar]
- Kovacs M.; Malnasi-Csizmadia A.; Woolley R. J.; Bagshaw C. R. (2002) Analysis of nucleotide binding to Dictyostelium myosin II motor domains containing a single tryptophan near the active site. J. Biol. Chem. 277, 28459–28467. [DOI] [PubMed] [Google Scholar]
- Bagshaw C. R.; Eccleston J. F.; Eckstein F.; Goody R. S.; Gutfreund H.; Trentham D. R. (1974) The magnesium ion-dependent adenosine triphosphatase of myosin. Two-step processes of adenosine triphosphate association and adenosine diphosphate dissociation. Biochem. J. 141, 351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K. A.; Taylor E. W. (1978) Intermediate states of subfragment 1 and actosubfragment 1 ATPase: reevaluation of the mechanism. Biochemistry 17, 3432–3442. [DOI] [PubMed] [Google Scholar]
- Millar N. C.; Geeves M. A. (1988) Protein fluorescence changes associated with ATP and adenosine 5′-[gamma-thio]triphosphate binding to skeletal muscle myosin subfragment 1 and actomyosin subfragment 1. Biochem. J. 249, 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurzawa S. E.; Manstein D. J.; Geeves M. A. (1997) Dictyostelium discoideum myosin II: characterization of functional myosin motor fragments. Biochemistry 36, 317–323. [DOI] [PubMed] [Google Scholar]
- Kuhlman P. A.; Bagshaw C. R. (1998) ATPase kinetics of the Dictyostelium discoideum myosin II motor domain. J. Muscle Res. Cell Motil. 19, 491–504. [DOI] [PubMed] [Google Scholar]
- Malnasi-Csizmadia A.; Pearson D. S.; Kovacs M.; Woolley R. J.; Geeves M. A.; Bagshaw C. R. (2001) Kinetic resolution of a conformational transition and the ATP hydrolysis step using relaxation methods with a Dictyostelium myosin II mutant containing a single tryptophan residue. Biochemistry 40, 12727–12737. [DOI] [PubMed] [Google Scholar]
- Batra R.; Manstein D. J. (1999) Functional characterisation of Dictyostelium myosin II with conserved tryptophanyl residue 501 mutated to tyrosine. Biol. Chem. 380, 1017–1023. [DOI] [PubMed] [Google Scholar]
- Rayment I.; Rypniewski W. R.; Schmidt-Bäse K.; Smith R.; Tomchick D. R.; Benning M. M.; Winkelmann D. A.; Wesenberg G.; Holden H. M. (1993) Three-dimensional structure of myosin subfragment-1: a molecular motor. Science 261, 50–58. [DOI] [PubMed] [Google Scholar]
- Dominguez R.; Freyzon Y.; Trybus K. M.; Cohen C. (1998) Crystal structure of a vertebrate smooth muscle myosin motor domain and its complex with the essential light chain: visualization of the pre-power stroke state. Cell 94, 559–571. [DOI] [PubMed] [Google Scholar]
- Fisher A. J.; Smith C. A.; Thoden J. B.; Smith R.; Sutoh K.; Holden H. M.; Rayment I. (1995) X-ray structures of the myosin motor domain of Dictyostelium discoideum complexed with MgADP·BeFx and MgADP·AlF4. Biochemistry 34, 8960–8972. [DOI] [PubMed] [Google Scholar]
- Coureux P. D.; Wells A. L.; Menetrey J.; Yengo C. M.; Morris C. A.; Sweeney H. L.; Houdusse A. (2003) A structural state of the myosin V motor without bound nucleotide. Nature 425, 419–423. [DOI] [PubMed] [Google Scholar]
- Coureux P. D.; Sweeney H. L.; Houdusse A. (2004) Three myosin V structures delineate essential features of chemo-mechanical transduction. EMBO J. 23, 4527–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menetrey J.; Bahloul A.; Wells A. L.; Yengo C. M.; Morris C. A.; Sweeney H. L.; Houdusse A. (2005) The structure of the myosin VI motor reveals the mechanism of directionality reversal. Nature 435, 779–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Gourinath S.; Kovacs M.; Nyitray L.; Reutzel R.; Himmel D. M.; O’Neall-Hennessey E.; Reshetnikova L.; Szent-Gyorgyi A. G.; Brown J. H.; Cohen C. (2007) Rigor-like structures from muscle myosins reveal key mechanical elements in the transduction pathways of this allosteric motor. Structure 15, 553–564. [DOI] [PubMed] [Google Scholar]
- Smith C. A.; Rayment I. (1996) X-ray structure of the magnesium(II)·ADP·vanadate complex of the Dictyostelium discoideum myosin motor domain to 1.9 Å resolution. Biochemistry 35, 5404–5417. [DOI] [PubMed] [Google Scholar]
- Holmes K. C.; Schroder R. R.; Sweeney H. L.; Houdusse A. (2004) The structure of the rigor complex and its implications for the power stroke. Philos. Trans. R. Soc. London, Ser. B 359, 1819–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. A.; Rayment I. (1995) X-ray structure of the magnesium(II)-pyrophosphate complex of the truncated head of Dictyostelium discoideum myosin to 2.7 Å resolution. Biochemistry 34, 8973–8981. [DOI] [PubMed] [Google Scholar]
- Gulick A. M.; Bauer C. B.; Thoden J. B.; Rayment I. (1997) X-ray structures of the MgADP, MgATPgammaS, and MgAMPPNP complexes of the Dictyostelium discoideum myosin motor domain. Biochemistry 36, 11619–11628. [DOI] [PubMed] [Google Scholar]
- Li X. D.; Rhodes T. E.; Ikebe R.; Kambara T.; White H. D.; Ikebe M. (1998) Effects of mutations in the gamma-phosphate binding site of myosin on its motor function. J. Biol. Chem. 273, 27404–27411. [DOI] [PubMed] [Google Scholar]
- Shimada T.; Sasaki N.; Ohkura R.; Sutoh K. (1997) Alanine scanning mutagenesis of the switch I region in the ATPase site of Dictyostelium discoideum myosin II. Biochemistry 36, 14037–14043. [DOI] [PubMed] [Google Scholar]
- Li G. H.; Cui Q. (2004) Mechanochemical coupling in myosin: a theoretical analysis with molecular dynamics and combined QM/MM reaction path calculations. J. Phys. Chem. B 108, 3342–3357. [Google Scholar]
- Kovacs M.; Toth J.; Hetenyi C.; Malnasi-Csizmadia A.; Sellers J. R. (2004) Mechanism of blebbistatin inhibition of myosin II. J. Biol. Chem. 279, 35557–35563. [DOI] [PubMed] [Google Scholar]
- Xu S.; White H. D.; Offer G. W.; Yu L. C. (2009) Stabilization of helical order in the thick filaments by blebbistatin: further evidence of coexisting multiple conformations of myosin. Biophys. J. 96, 3673–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manstein D. J.; Schuster H. P.; Morandini P.; Hunt D. M. (1995) Cloning vectors for the production of proteins in Dictyostelium discoideum. Gene 162, 129–134. [DOI] [PubMed] [Google Scholar]
- Manstein D. J.; Hunt D. M. (1995) Overexpression of myosin motor domains in Dictyostelium: screening of transformants and purification of the affinity tagged protein. J. Muscle Res. Cell Motil. 16, 325–332. [DOI] [PubMed] [Google Scholar]
- Allingham J. S.; Smith R.; Rayment I. (2005) The structural basis of blebbistatin inhibition and specificity for myosin II. Nat. Struct. Mol. Biol. 12, 378–379. [DOI] [PubMed] [Google Scholar]
- Blommel P. G.; Fox B. G. (2007) A combined approach to improving large-scale production of tobacco etch virus protease. Protein Expression Purif. 55, 53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. (2000) An approach to multi-copy search in molecular replacement. Acta Crystallogr., Sect. D: Biol. Crystallogr. 56, 1622–1624. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Walmsley A. R.; Bagshaw C. R. (1989) Logarithmic timebase for stopped-flow data acquisition and analysis. Anal. Biochem. 176, 313–318. [DOI] [PubMed] [Google Scholar]
- Bauer C. B.; Holden H. M.; Thoden J. B.; Smith R.; Rayment I. (2000) X-ray structures of the apo and MgATP-bound states of Dictyostelium discoideum myosin motor domain. J. Biol. Chem. 275, 38494–38499. [DOI] [PubMed] [Google Scholar]
- DeLano W. L. (2002) The PyMOL Molecular Graphics System.
- Cohen G. H. (1997) ALIGN: a program to superimpose protein coordinates, accounting for insertions and deletions. J. Appl. Crystallogr. 30, 1160–1161. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.