

Abstract
HIV-infected patients are at increased risk for development of pulmonary complications, including chronic obstructive pulmonary disease (COPD). Inflammation associated with sub-clinical infection has been postulated to promote COPD. Persistence of Pneumocystis (Pc) is associated with HIV and COPD, although a causal relationship has not been established. We used a simian/human immunodeficiency virus (SHIV) model of HIV infection to study pulmonary effects of Pc colonization. SHIV-infected/Pc-colonized monkeys developed progressive obstructive pulmonary disease characterized by increased emphysematous tissue and bronchial-associated lymphoid tissue. Elevated Th2 cytokines and pro-inflammatory mediators in bronchoalveolar lavage fluid coincided with Pc colonization and pulmonary function decline. These results support the concept that an infectious agent contributes to development of HIV-associated lung disease and suggests that Pc colonization may be a risk factor for the development of HIV-associated COPD. Furthermore, this model allows examination of early host responses important to disease progression thus identifying potential therapeutic targets for COPD.
Keywords: Pneumocystis, COPD, SHIV, AIDS, HIV
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is predicted to become the third leading cause of death worldwide by 2020[1]. COPD is characterized by development of irreversible airflow limitation and destruction of alveolar septa resulting in alveolar enlargement and airway obstruction. Although smoking is the primary risk factor for COPD, only 15–20% of smokers develop the disease suggesting other factors contribute to disease susceptibility.
COPD occurs earlier and more frequently in HIV-infected subjects compared to HIV-negative subjects[2, 3]. How these complications develop is not understood, but sub-clinical or latent infections might be involved[4, 5]. Evidence exists linking Pneumocystis jirovecii, a fungal opportunistic pathogen, to COPD development in HIV-negative smokers. Subjects with COPD tend to be colonized with Pneumocystis (Pc) more frequently than those with other chronic lung diseases, and Pc colonization is associated with severity of airflow obstruction[6, 7]. HIV-infected persons are also at risk for Pc colonization, with colonization prevalence up to 69%[8, 9]. Although these studies demonstrate association between Pc and COPD, a causal relationship has not been shown.
To examine whether persistent Pc colonization is a co-factor in HIV-related COPD pathogenesis, we developed a Pc colonization model using chimeric simian-human immunodeficiency virus (SHIV) in macaques. Excellent rationale exists for use of this model as studies have shown that Pc derived from humans and non-human primates to be phylogenetically most closely related[10, 11]. We performed longitudinal studies to determine association between Pc colonization and progression of airway obstruction and emphysema in the context of an AIDS model.
METHODS
Animals
Twelve adult, Chinese-origin, cynomolgus macaques (Macaca fasicularis) obtained from National Primate Centers or vendors approved by the Department of Laboratory Animal Research, University of Pittsburgh were individually housed and maintained in a BSL2+ primate facility at the University of Pittsburgh. Before purchase, all animals were screened and found negative for simian retroviral infections. Animal experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Clinical evaluations were conducted monthly or as needed[12].
Virus Infection
Monkeys were infected as described[13] with SHIV89.6P (gift of Dr. Opendra Narayan, University of Kansas), which induces CD4+ T cell lymphopenia and AIDS-like disease with wasting and opportunistic infections[13, 14]. Inoculations were repeated one month later to ensure infection in all animals. Viral loads were determined as described for blood and bronchoalveolar lavage samples[13].
Bronchoalveolar lavage (BAL)
Monkeys underwent BAL at baseline and at monthly intervals post-SHIV infection[12]. Unfractionated BAL fluid (BALF) aliquots were used for bacterial, fungal and viral culture (Antech Diagnostics, Pittsburgh, PA) and nested-PCR detection of Pc DNA[12]. The remainder was filtered through a 40-micron cell strainer after which cell counts were performed and supernatants were used for cytokine analysis and quantitation of SHIV[13] . 1 × 105 cells were removed and stained with modified Giemsa stain (Dade Behring, Newark, DE) and differential counts performed manually[15]. Recovered cells were prepared for flow cytometry as described[12].
Pc colonization of SHIV-infected macaques
To promote natural transmission of Pc, SHIV-infected macaques were continuously exposed by co-housing in the same room with 10–20 SIV- or SHIV-immunosuppressed macaques which served as a Pc source. None of the macaques (source or recipients) contracted fulminate Pneumocystis pneumonia (PcP) during the study. Determination of Pc colonization status was performed by detection of Pc DNA in BAL samples by nested PCR and by anti-Pc serology[12, 16]. Pc colonization was defined as a positive nested PCR of BAL fluid and >3 fold change in plasma anti-Pc KEX1 titers[16]. Additionally, BAL samples were stained for organisms by modified Giemsa and silver staining[12].
Peripheral blood collection
Peripheral blood was collected and processed as described[13]. T cells were analyzed as described[16].
Cytokine and chemokine analysis
Quantitative analysis of cytokines and chemokines in BALF was performed with Beadlyte Human Multi-Cytokine Flex Kit (Upstate, Temecula, CA) according to manufacturer’s instructions. Thirteen of the analytes shown in Table 3 were chosen based on cross-reactivity with non-human primate proteins[17]. IL-10 and IL-13 levels were analyzed using monkey-specific ELISA kits (BioSource, Camarillo, CA and Cell Sciences, Canton, MA respectively). Dilution effect of BALF samples was normalized based on plasma urea concentrations[18].
Table 3.
Serial analyses of BAL cytokines and chemokines in SHIV-infected monkeys.
| Cytokine/ | Weeks After | SHIV/Pc+ | SHIV/Pc− | ||||
|---|---|---|---|---|---|---|---|
| Chemokine | SHIV Infection | Mean (pg/mL) | Std Dev | p (vs BL) | Mean (pg/mL) | Std Dev | p (vs BL) |
| IL-4 | BL | 71.25 | 30.7 | 70.21 | 28.9 | ||
| 4 | 78.7 | 30.5 | 0.549 | 96.07 | 39.7 | 0.310 | |
| 16 | 146.8 | 42.9 | 0.004 | 72.13 | 33.8 | 0.230 | |
| 35 | 134.3 | 40.6 | 0.026 | 94.74 | 61.2 | 0.390 | |
| IL-5 | BL | 65.72 | 26.2 | 64.3 | 31.5 | ||
| 4 | 68.44 | 25.4 | 0.774 | 77.83 | 31.4 | 0.642 | |
| 16 | 143.5 | 40 | 0.003 | 66.41 | 27.5 | 0.343 | |
| 35 | 153.6 | 48.5 | 0.007 | 83.51 | 48.8 | 0.221 | |
| IL-13 | BL | 11.8 | 4.4 | 12.6 | 6.1 | ||
| 4 | 11.0 | 7.3 | 0.797 | 18.3 | 4.8 | 0.148 | |
| 16 | 21.6 | 4.8 | 0.005 | 10.7 | 4.0 | 0.595 | |
| 35 | 15.0 | 6.4 | 0.247 | 8.5 | 1.0 | 0.213 | |
| IL-10 | BL | 583.5 | 303.6 | 573.3 | 442.6 | ||
| 4 | 774.4 | 357.2 | 0.797 | 718.5 | 283.7 | 0.507 | |
| 16 | 752.7 | 279.1 | 0.005 | 474.1 | 298.9 | 0.309 | |
| 35 | 442.8 | 155.6 | 0.247 | 384.8 | 410.8 | 0.652 | |
| IFN-γ | BL | 62.5 | 34.5 | 53.1 | 28.5 | ||
| 4 | 59.7 | 43.3 | 0.881 | 104.4 | 72.3 | 0.346 | |
| 16 | 179.2 | 71.8 | 0.005 | 53.9 | 45.6 | 0.528 | |
| 35 | 106.6 | 58.3 | 0.176 | 70.5 | 27.2 | 0.067 | |
| IL-12 (p40) | BL | 23.0 | 52.3 | 82.6 | 131.0 | ||
| 4 | 9.4 | 22.6 | 0.266 | 8.5 | 17.0 | 0.351 | |
| 16 | 305.3 | 388.8 | 0.058 | 78.2 | 81.6 | 0.367 | |
| 35 | 241.0 | 462.6 | 0.239 | 80.2 | 96.0 | 0.982 | |
| Lymphotoxin | BL | 117.0 | 43.5 | 124.0 | 68.1 | ||
| 4 | 128.1 | 61.5 | 0.579 | 137.9 | 39.8 | 0.782 | |
| 16 | 241.6 | 56.5 | 0.002 | 98.7 | 27.4 | 0.793 | |
| 35 | 248.9 | 93.9 | 0.019 | 152.3 | 84.2 | 0.241 | |
| TNF-α | BL | 85.9 | 45.7 | 36.5 | 31.1 | ||
| 4 | 109.2 | 51.9 | 0.309 | 78.4 | 36.4 | 0.204 | |
| 16 | 232.6 | 89.2 | 0.006 | 83.7 | 37.2 | 0.115 | |
| 35 | 153.2 | 61.4 | 0.079 | 66.4 | 30.7 | 0.013 | |
| IL-1β | BL | 81.3 | 31.2 | 83.2 | 43.9 | ||
| 4 | 93.0 | 49.5 | 0.476 | 100.2 | 17.9 | 0.508 | |
| 16 | 160.9 | 46.4 | 0.006 | 86.0 | 25.0 | 0.325 | |
| 35 | 192.0 | 81.4 | 0.018 | 120.7 | 76.2 | 0.177 | |
| IL-6 | BL | 113.7 | 43.7 | 114.5 | 54.1 | ||
| 4 | 120.8 | 58.5 | 0.776 | 144.6 | 28.9 | 0.499 | |
| 16 | 221 | 50.7 | 0.004 | 95.53 | 36.1 | 0.865 | |
| 35 | 196.4 | 69 | 0.04 | 137.4 | 71.4 | 0.296 | |
| IL-8 | BL | 161.1 | 83.4 | 185.4 | 141.0 | ||
| 4 | 251.3 | 258.2 | 0.327 | 174.2 | 45.7 | 0.893 | |
| 16 | 304.5 | 105.8 | 0.013 | 142.8 | 66.3 | 0.464 | |
| 35 | 385.8 | 248.4 | 0.059 | 400.2 | 416.2 | 0.224 | |
| GM-CSF | BL | 82.0 | 51.1 | 72.2 | 44.4 | ||
| 4 | 80.8 | 22.6 | 0.951 | 76.9 | 40.0 | 0.909 | |
| 16 | 198.1 | 73.0 | 0.007 | 66.8 | 26.9 | 0.584 | |
| 35 | 173.2 | 70.8 | 0.040 | 121.7 | 55.9 | 0.096 | |
| MIP-1α | BL | 148.8 | 172.3 | 0.0 | 0.0 | ||
| 4 | 109.1 | 144.0 | 0.541 | 155.9 | 192.8 | 0.204 | |
| 16 | 579.7 | 300.0 | 0.010 | 214.4 | 196.4 | 0.199 | |
| 35 | 307.1 | 219.1 | 0.181 | 1068 | 1931 | 0.350 | |
| MCP-1 | BL | 2006.0 | 1695.0 | 1951.0 | 2249.0 | ||
| 4 | 1549.0 | 1222.0 | 0.508 | 2736.0 | 2414.0 | 0.427 | |
| 16 | 3905.0 | 6292.0 | 0.325 | 725.4 | 184.7 | 0.607 | |
| 35 | 4947.0 | 5785.0 | 0.177 | 13150.0 | 22628.0 | 0.352 | |
| RANTES | BL | 91.2 | 86.8 | 176.4 | 136.2 | ||
| 4 | 55.5 | 32.2 | 0.318 | 70.8 | 25.5 | 0.248 | |
| 16 | 124.3 | 55.2 | 0.151 | 67.8 | 40.2 | 0.203 | |
| 35 | 438.4 | 471.9 | 0.060 | 122.5 | 36.1 | 0.528 | |
BL: baseline
Pc colonization was detected by 8 weeks post-SHIV infection.
p values, analyzed by paired t test, are for baseline measurements versus measurements for the indicated week post-SHIV infection in SHIV/Pc+ (n = 8) and SHIV/Pc− (n = 4) animals. Timepoints where significant changes in cytokine levels were detected are shaded light gray.
Pulmonary Function Testing
Pulmonary function tests (PFT) were performed at baseline and every other month after SHIV infection using whole body plethysmography and forced deflation technique. Monkeys were anesthetized with intravenous propofol and the oropharynx desensitized with 2% lidocaine followed by intubation. Endotracheal tube placement was verified by chest X-ray and monitored using a CO2 detector (Nellcor Pedi-cap, Boulder, CO). PFTs were performed using a Buxco whole body plethysmograph (Buxco Electronics, Inc., Sharon, CT), and BioSystems for Maneuvers Software (Buxco Electronics, Inc.) was used to collect data on flow rates and flow volumes. Tests were considered valid when three measurements for forced vital capacity were within 10% of each other.
For bronchodilator challenge, standard PFTs were performed, followed by administration of one pediatric dosette of nebulized albuterol (3 ml of 0.083% albuterol) (Nephron Pharmaceuticals Corp., Orlando, FL). Fifteen minutes after administration, PFTs were repeated and compared to baseline values.
Quantitative computed tomography (CT)
Conventional, non-contrast CT scans were performed on 10 of the 12 animals in a GE 9800 Highlight Advantage CT scanner (General Electric Medical Systems, Milwaukee, WI). Anesthetized, intubated animals were mechanically ventilated to 20 cm H2O to ensure scan to scan volume uniformity. Axial slices (1.25 mm) were acquired during end-inspiratory breath-hold. Calculation of densities used for determination of lung properties in Table 2 was performed for animals at baseline and repeated post-SHIV infection as described[19]. Briefly, mean CT scan attenuations of the lung were calculated and converted to density measurements in milligrams/milliliter which was then multiplied by lung volume to obtain lung mass approximation. Actual lung weights measured at necropsy correlated with lung weights calculated from endpoint scans by Pearson correlation analysis (p = 0.01). CT scan analysis was performed in a blinded manner using custom software (Emphylx: Department of Radiology/iCAPTURE Laboratory, University of British Columbia, Vancouver, BC, Canada) [20]. Small airway dimensions were calculated using the PV-Wave software package (Visual Numerics, Boulder, CO)[21].
Table 2.
Quantitative CT analysis of the lungs pre- and post-infection
| SHIV/Pc+‡ | SHIV/Pc− | |||
|---|---|---|---|---|
| Baseline* | Endpoint | Baseline | Endpoint | |
| Total Lung volume, ml | 352 ± 40 | 358 ± 49 | 372 ± 22 | 371 ± 34 |
| Airspace volume, ml | 300 ± 36 | 309 ± 44 | 321 ± 18 | 322 ± 28 |
| Tissue volume, ml | 52 ± 5 | 49 ± 5§ | 51 ± 6 | 49 ± 7 |
| Lung weight, g | 55 ± 5 | 52 ± 5§ | 54 ± 6 | 52 ± 7 |
| % Voxels > −910 HU† | 82 ± 6 | 74 ± 6§ | 68 ± 13 | 71 ± 12 |
| % Voxels ≤ −910 HU | 18 ± 6 | 26 ± 6§ | 32 ± 13 | 29 ± 12 |
Values (mean ± SEM) were calculated in monkeys before SHIV infection (baseline) and following SHIV infection in SHIV/Pc+ (n = 6) and SHIV/Pc− (n = 4) animals at the termination of the experiment (10–12 months post-SHIV infection).
HU: Hounsfield units
Different from baseline (p = 0.04 by paired t test analysis)
Two SHIV/Pc+ animals were not included in either the pre- or post-infection analyses because baseline scans were not performed. Both of these animals developed airway obstruction based on pulmonary function testing.
Lung tissue preparation and morphometry
Right lungs removed at necropsy were inflated to 25 cm H2O with 10% buffered formalin. Paraffin-embedded, serial mid-sagittal sections from each lobe were then stained. Modified Harris hematoxylin-stained (Sigma) tissue was used to estimate alveolar size by determination of mean chord lengths [22]. H&E-stained tissue sections were examined for the presence of bronchial-associated lymphoid tissue, defined by the presence of non-encapsulated lymphoid tissue within outer airway walls. Approximately 100 airways per monkey were examined.
Statistical analysis
Pulmonary function data analysis was performed using the R environment for statistical analysis and graphics in which mixed linear models were used to estimate and test the relationship among pulmonary function profiles (dependent variable), Pc colonization (independent variable), and time (independent variable). Differences in profiles over time were tested using restricted maximum likelihood. All other data were analyzed using Prism software, (GraphPad, La Jolla, CA) using paired or unpaired, two-tailed Student’s t test, where appropriate. A p value less than 0.05 was considered statistically significant.
RESULTS
Pc colonization of SHIV-infected macaques results in pulmonary obstruction
Twelve cynomolgus macaques were infected with SHIV89.6P [14] and exposed to Pc via co-housing with Pc-infected macaques[16]. Peripheral blood CD4+ T cell levels declined to ≤ 50% of baseline values by four weeks post-SHIV infection in all monkeys and remained depressed throughout the study (Fig. 1). Peak viremia ranged from 3.4 × 106 to 2.3 × 108 RNA copies/ml by week two post-infection (not shown). Serial bacterial and fungal cultures of BALF were negative throughout the study. Eight of 12 monkeys became colonized with Pc by nine weeks post-SHIV infection (SHIV/Pc+), as determined by nested-PCR of bronchoalveolar lavage fluid and Pc serology[16], while four remained Pc-negative throughout the study (SHIV/Pc−). After initial exposure to Pc, anti-Pc titers in the SHIV/Pc+ animals remained above baseline throughout the study (not shown). None of the monkeys tested positively by PCR at every time point which was most likely due to low level organism burden and sampling of different areas of the lung at each time point. Among these animals, two to nine time points were positive by PCR during the period studied. Modified Giemsa and silver staining were also performed on BAL samples but were not found to be positive for organisms at any time point. There was no significant difference in peak viral titers (mean peak viral titers (viral RNA copies/ml plasma): SHIV/Pc+: 4.42 × 107 ± 7.50 × 107, SHIV/Pc−: 3.19 × 107 ± 2.88 × 107; p = 0.76) or peripheral blood CD4+ T cells levels at any time post-SHIV infection between the groups (Fig. 1).
Figure 1.

Peripheral blood CD4+ T cell levels are not different for SHIV/Pc+ animals versus SHIV/Pc− animals. Peripheral blood mononuclear cells isolated from whole blood were stained with anti-CD4 antibody and analyzed by flow cytometry. Open circles represent SHIV/Pc+ animals and closed squares represent SHIV/Pc− animals. p = 0.79 by two-way repeated measures ANOVA for SHIV/Pc+ (n = 8) versus SHIV/Pc− group (n = 4).
To assess airway obstruction, pulmonary maneuvers using whole body plethysmography[23] were performed at baseline and every other month after SHIV infection. Peak expiratory flow, forced expiratory volume in 0.4 seconds and maximum mid-expiratory flow are the pulmonary function parameters chosen to evaluate obstructive disease. No significant differences in baseline physical characteristics and pulmonary function parameters between groups were observed (Table 1).
Table 1.
Baseline values for height, weight and pulmonary function parameters in SHIV/Pc− and SHIV/Pc+ animals.
| Parameter | Pc negative* | Pc positive | p value |
|---|---|---|---|
| Height, cm | 58.4 (53.3 – 61.0) | 61.0 (45.7 – 63.5) | 0.94 |
| Weight, kg | 4.7 (3.3 – 5.8) | 5.8 (3.8 – 7.6) | 0.19 |
| Pulmonary Function Parameters† | |||
| FEV0.1, ml | 39.1 (36.6 – 42.6) | 37.3 (32.9 – 44.8) | 0.66 |
| FEV0.2, ml | 89.4 (86.2 – 96.7) | 88.4 (77.3 – 101.2) | 0.80 |
| FEV0.4, ml | 187.2 (168.4–197.9) | 186.2 (163.6 – 204.4) | 0.97 |
| FVC, ml | 396.7 (223.8 – 454.6) | 415.9 (227.8 – 527.8) | 0.61 |
| FEV0.1/FVC, % | 10.4 (8.0 – 17.2) | 8.4 (7.2 – 19.7) | 0.74 |
| FEV0.2/FVC, % | 23.8 (19.0 – 38.6) | 19.9 (16.8 – 44.4) | 0.77 |
| FEV0.4/FVC, % | 49.3 (39.8 – 75.3) | 42.0 (35.3 – 85.4) | 0.78 |
| FEF25%, ml/s | 504.7 (483.6 – 527.5) | 497.3 (438.6 – 572.6) | 0.95 |
| FEF50%, ml/s | 453.9 (430.6 – 483.0) | 456.2 (404.1 – 542.7) | 0.72 |
| FEF75%, ml/s | 382.2 (349.8 – 430.6) | 391.7 (367.3 – 477.0) | 0.36 |
| FEF90%, ml/s | 230.8 (180.7 – 376.2) | 299.1 (170.8 – 370.5) | 0.53 |
| FEF25–75%, ml/s (MMEF) | 450.5 (423.3 – 480.6) | 450.6 (402.3 – 535.6) | 0.71 |
| PEF, ml/s | 517.7 (492.9 – 546.8) | 512.7 (447.1 – 576.2) | 0.89 |
Comparison of baseline values of animals that were subsequently infected with SHIV89.6P and were colonized with Pc (SHIV/Pc+, n = 8) or remained uncolonized (SHIV/Pc−, n = 4). No significant differences were observed in any of the parameters by unpaired t test.
FEV0.1, FEV0.2, FEV0.4, forced expiratory volume in 0.1, 0.2 and 0.4 seconds respectively; FVC, forced vital capacity; FEF25%, FEF50%, FEF75%, FEF90%, forced expiratory flow through 25%, 50%, 75% and 90% of forced vital capacity respectively; FEF25–75% (MMEF), forced expiratory flow from 25% to 75% of forced vital capacity or maximum mid-expiratory flow; PEF, peak expiratory flow. Values are medians with ranges shown in parentheses.
Six of eight SHIV/Pc+ animals developed airway obstruction as determined by decreased pulmonary function. All parameters evaluated decreased significantly in these animals compared to SHIV/Pc− monkeys (Fig. 2a–c). Median change in peak expiratory flow from baseline to 10 months post-SHIV infection was −58.5 ml/sec and +2.5 ml/sec for SHIV/Pc+ and SHIV/Pc− animals, respectively (p=0.004). Median change from baseline forced expiratory volume in 0.4 seconds in SHIV/Pc+ animals was −16.0 ml versus +4.0 ml for SHIV/Pc− animals (p=0.001). For maximum mid-expiratory flow, median change from baseline for SHIV/Pc+ animals was −47.5 ml/sec versus +24.5 ml/sec for SHIV/Pc− monkeys. (p=0.001). Although the forced expiratory volume in 0.4 seconds to forced vital capacity ratio, another measure of COPD, declined in SHIV/Pc+ animals, the change was not significant by 10 months post-infection (Fig. 2d).
Figure 2.

Pneumocystis colonization results in progressive pulmonary function decline. Whole body plethysmography was used to evaluate serial measurements of: (a) Peak expiratory flow. p = 0.003 (b) Forced expiratory volume in 0.4 seconds. p = 0.003 (c) Maximum mid-expiratory flow. p = 0.002 (d) Forced expiratory volume in 0.4 seconds to forced vital capacity ratio. p = 0.32. For all graphs, SHIV/Pc+ animals are represented by dashed lines and SHIV/Pc− animals are represented by solid lines. Each p value is for the interaction between time and group (Pc-colonized (n = 8) versus non-colonized (n = 4)).
Since airflow limitation in COPD is poorly reversible in response to bronchodilator treatment, we tested the effect of administration of the bronchodilator, albuterol. No significant differences were observed post-treatment (not shown).
Pneumocystis colonization results in radiographic and pulmonary emphysema but not small airway thickening in SHIV-infected monkeys
Emphysema is associated with increased lung and airspace volumes, usually coupled with decreased lung weight. Quantitative computed tomography (CT) morphometry has been used to evaluate extent of emphysema in humans[19]. We applied this technique to evaluate baseline and post-infection lung CT scans by performing tissue density analysis based on a density mask cut-off of ≤ −910 Hounsfield units (HU), which is similarly used to identify emphysema in humans[19]. There was a significant increase compared to baseline values in lung percent at ≤ −910 HU in SHIV/Pc+ monkeys compared to SHIV/Pc− monkeys (Fig. 3a). Lobe by lobe comparison of percent change in ≤ −910 HU during the course of infection revealed significant increases in upper and middle lobes of SHIV/Pc+ monkeys but not in lower lobes. No significant changes were observed in individual lobes of SHIV/Pc− monkeys (Fig 3b).
Figure 3.


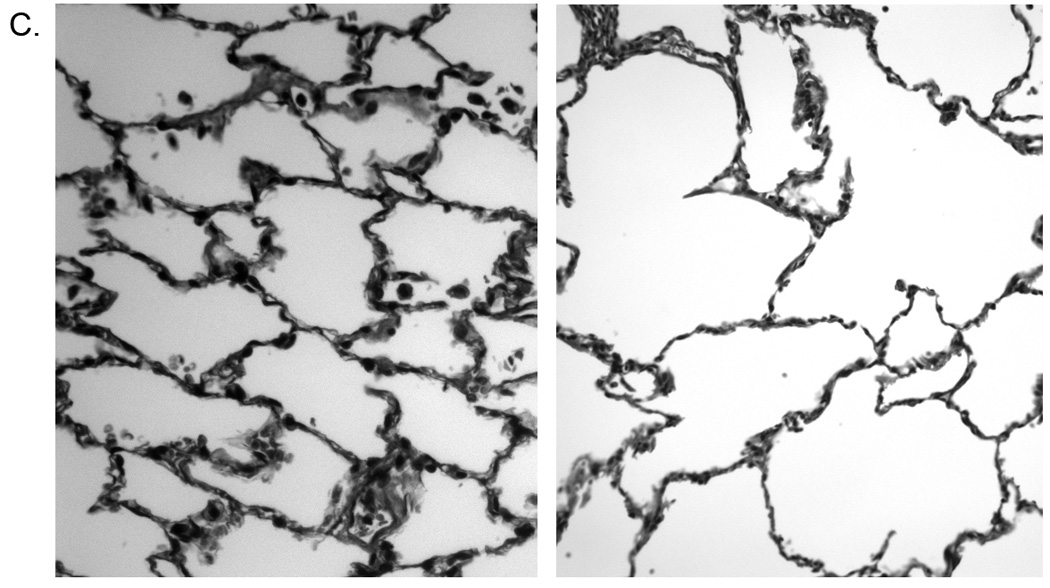

Pneumocystis colonization leads to an increase in the proportion of emphysematous tissue in the lungs. Quantitative computed tomography (CT) scans were performed at 20 cm H2O lung inflation pressure at baseline (BL) and post- SHIV infection. The cutoff mask of ≤ −910 Hounsfield units (HU) was used to assess amount of emphysematous lung tissue present at each scan. Boxes represent the range of values for the specified group with the median value represented by the line within the box. (a) Change in the proportion of emphysematous lung tissue for the animal groups; * p = 0.04 for SHIV/Pc− (n = 4) versus SHIV/Pc+ (n = 6†) animals by unpaired t test. (b) Comparison of the proportion of emphysematous lung tissue present at baseline and endpoint scan by lobe. For SHIV/Pc+ animals†: * p = 0.04 by paired t test, for the proportion of lung tissue that is emphysematous in both the upper and middle lobes for baseline versus endpoint scans; p = 0.78 by paired t test for proportion of lung tissue that is emphysematous in the lower lobe for baseline versus endpoint scans (n = 6). For SHIV/Pc− animals: p = 0.55, 0.80 and 0.11 by paired t test for proportion of lung tissue that is emphysematous in the upper, middle and lower lobes respectively for baseline versus endpoint scans (n = 4). (c) Representative hematoxylin-stained lung tissue sections from SHIV-infected monkeys; left: SHIV/Pc− and right: SHIV/Pc+. (d) Chord length analysis (mean ± SEM) of airspaces for animals exhibiting clinical type (≥ 12% decline in pulmonary function from baseline level) obstruction (COPD+, n = 5) versus non-obstructed animals (COPD−, n = 7), *p = 0.0001.
†Two SHIV/Pc+ animals were not included in either the pre- or post-infection analyses because baseline scans were not performed. Both of these animals developed airway obstruction based on pulmonary function testing.
Consistent with an increase in percentage of emphysematous tissue, total tissue volume and lung weight were significantly decreased from baseline in SHIV/Pc+ monkeys (Table 2), but not in SHIV/Pc− monkeys. No significant changes in small airway wall dimensions, including thickness, were observed for either group (not shown).
Airspace enlargement was also evaluated in lung tissue sections by determination of mean chord length, the average distance between opposing walls of a single alveolus. Figure 3c shows representative lung tissue sections from both groups. In support of our radiologic findings, mean chord length was significantly larger in obstructed versus non-obstructed monkeys (Fig. 3d).
Pneumocystis colonization results in increased bronchial-associated lymphoid tissue in SHIV-infected monkeys
As an indicator of inflammation due to increased pathogen burden, lung tissue was examined for presence of bronchial-associated lymphoid tissue. SHIV/Pc+ monkeys had significantly higher bronchial-associated lymphoid tissue frequency compared to SHIV/Pc− monkeys, indicating persistent pulmonary inflammation in these animals (Fig. 4a, b).
Figure 4.
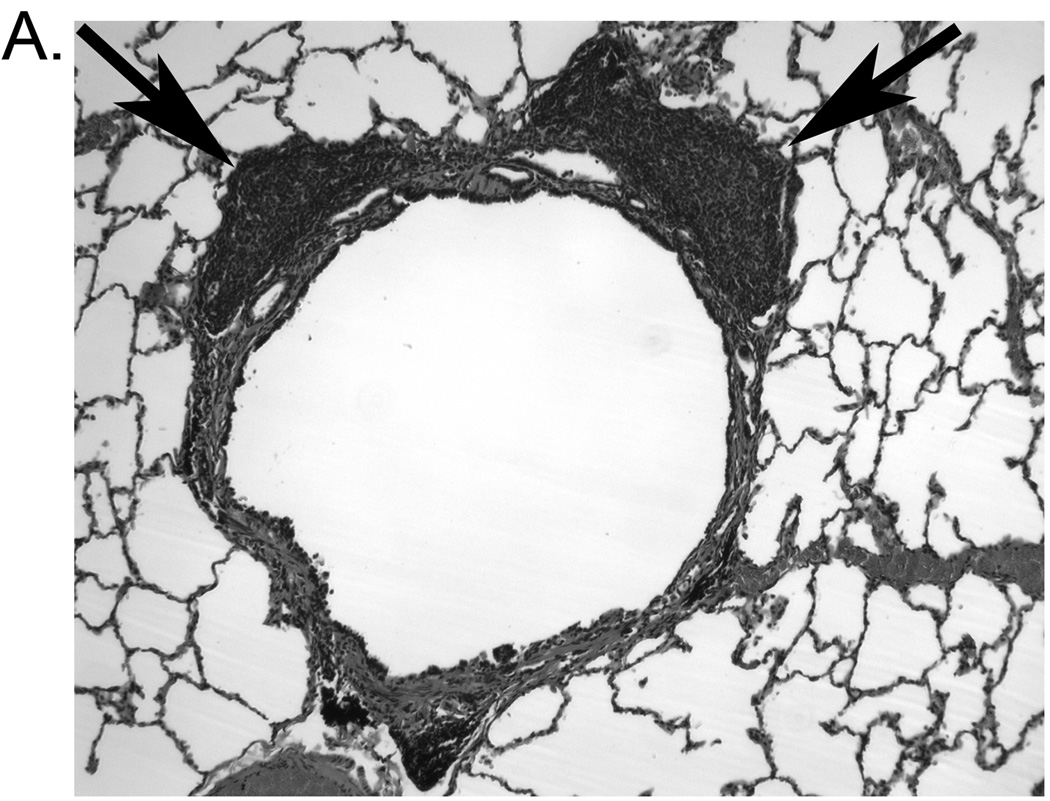

Pneumocystis colonization results in increased bronchial-associated lymphoid tissue formation. (a) Representative hematoxylin and eosin-stained lung tissue section from a SHIV/Pc+ animal showing an airway associated with lymphoid follicles (indicated by arrows). (b) Analysis of percent of airways with bronchial-associated lymphoid tissue in SHIV/Pc- versus SHIV/Pc+ monkeys. For SHIV/Pc− (n = 4) and SHIV/Pc+ (n = 8) animals, an average of 114 ± 43 and 103 ± 17 airways per animal were evaluated respectively, p = 0.04 by unpaired t test.
Pneumocystis colonization induces inflammatory and Th2-associated cytokines in the bronchoalveolar lavage fluid of SHIV-infected monkeys
Because COPD and PcP have been associated with vigorous inflammatory responses[24, 25], we evaluated inflammation indicators in serial BALF samples following SHIV infection and Pc colonization. Interestingly, there were no significant changes in absolute number or percentage of T cells, macrophages, neutrophils, or CD4+/CD8+ T cell ratios, in BALF of infected monkeys regardless of Pc status (up to 12 months post-SHIV infection) (not shown). Serial cytokine and chemokine analysis of BALF revealed changes from baseline in SHIV/Pc+, but not SHIV/Pc−, monkeys (Table 3). Assays were performed at baseline, four weeks post-SHIV infection (after significant CD4+ T cell decline, but prior to detectable Pc colonization), and weeks 16 and 35 (after detection of persistent Pc colonization). Increases at weeks 16 and 35 in interleukin (IL)-4, IL-5, IL-6, granulocyte macrophage-colony stimulating factor (GM-CSF) and lymphotoxin-α and transient increases in IL-8, IL-13, interferon (IFN)-γ, macrophage inflammatory protein (MIP)-1α and tumor necrosis factor (TNF)-α were observed in SHIV/Pc+ monkeys. Conversely, SHIV/Pc− animals did not exhibit increases in cytokine levels, except for TNF-α at 35 weeks. These results demonstrate that SHIV infection alone had little effect on induction of inflammatory mediators in alveolar spaces, while Pc colonization induced a pro-inflammatory and Th2-skewed cytokine response, which was coincident with declining pulmonary function (Fig. 1).
DISCUSSION
The results presented here support the concept that Pc colonization contributes to the development of COPD in a non-human primate model of HIV infection. SHIV-infected monkeys that became naturally colonized with Pc developed progressive pulmonary obstruction that was unresponsive to bronchodilator treatment. Additionally, Pc colonization correlated with anatomic evidence of emphysema, increased bronchial-associated lymphoid tissue frequency, and increased levels of pro-inflammatory mediators and Th2-type cytokines in BALF. In contrast, SHIV infection alone did not exert such effects. These data support the hypothesis that in HIV-associated COPD, persistent Pc carriage, common among HIV+ subjects[8, 9], induces lung inflammation, possibly promoting tissue damage and COPD development.
COPD is a complex disorder resulting from a combination of genetic and environmental factors associated with persistent lung inflammation[25]. While cigarette smoking is the main risk factor, other factors likely influence disease progression, as only ~25% of smokers develop COPD[26]. HIV infection is also associated with increased COPD risk, particularly in smokers. Diaz et al. reported 37% of HIV- infected smokers had emphysema by pulmonary function or chest CT scan, in contrast to no demonstrable emphysema in HIV-negative controls[2]. Crothers et al. showed that HIV+ subjects are more likely to have a COPD diagnosis compared with HIV-negative controls, and that HIV infection was an independent predictor of COPD[3]. Reports of high Pc colonization frequency in HIV-infected subjects and HIV-negative COPD patients [7, 27], suggests persistent Pc carriage may promote pulmonary function decline and COPD development. The primate model of HIV infection described here supports these clinical findings and enables longitudinal characterization of factors associated with development of COPD pathogenesis.
Simian immunodeficiency virus (SIV) and SHIV have been used extensively in rhesus and cynomolgus macaques as models of HIV [28] . As in HIV infection, PcP is common in SIV− and SHIV-infected macaques and susceptibility correlates with peripheral blood CD4+ T cell decline[15, 29]. In contrast to SIV, SHIV produces rapid decline in blood CD4+ T cells, facilitating long-term studies of persistent infection. We previously described SIV infection/Pc colonization in macaques using both intrabronchial inoculation and airborne transmission of Pc[12, 16]. Airborne transmission is more likely representative of natural Pc transmission, and eliminates potential transient inflammatory responses associated with intrabronchial inoculations[12], allowing examination of inflammatory responses associated with persistent colonization.
In serial pulmonary function studies, we found significant obstruction in six of eight Pc-colonized monkeys, but not in monkeys infected with SHIV alone. These data suggest that viral infection is insufficient to induce emphysema, but SHIV-induced immunosuppression may increase Pc carriage susceptibility, which may result in obstructive changes. Interestingly, one SHIV/Pc+ monkey without measurable pulmonary function decline showed evidence of emphysema based on increased lung volume and percentage of lung tissue ≤ −910 HU (not shown). This suggests that SHIV infection and Pc colonization may result in emphysema without airflow obstruction, a COPD phenotype described in humans[30]. No significant changes between baseline and endpoint small airway wall dimensions were observed in either group, suggesting the observed pulmonary obstruction was an emphysema-dominant phenotype with minimal small airway involvement[31].
Several studies have examined inflammatory responses in COPD patients, with conflicting results likely due to disease heterogeneity, variability in disease severity, and lung region sampling differences[32, 33]. Neutrophils have been implicated in COPD pathogenesis. Severe COPD patients have neutrophilic infiltration of airway walls, and increased neutrophil counts in BALF and sputum samples that correlate with disease severity[34, 35]. In contrast, mild emphysema is not commonly associated with BAL neutrophilia[36]. Similarly, studies have shown T cell infiltration in small airway walls and alveolar spaces of COPD patients with general increases in CD8+ T cell proportions though their role in COPD-associated inflammatory damage is unknown. CD4+ and CD8+ T cells can elaborate pro-inflammatory cytokines that may contribute to lung damage. Th1-skewed cytokine production has been reported in COPD patients in several studies[37], although a mixed or Th2-dominant response has also been reported[38].
We detected inflammatory changes in airspaces only after Pc colonization was evident, with little evidence of inflammation due to persistent SHIV infection. Increases in IL-4, IL-5, and IL-13 with minimal increases in IFN-γ and no detectable IL-12 in SHIV/Pc+ monkeys suggested a Th2-skewed response. Although Th2 cytokines are more commonly associated with asthma[39], these results support reports of increased IL-4 in emphysematous human lung tissue[40]. Additionally, Ma et al. demonstrated that in mice genetically predisposed toward a Th1 response, over-expression of IL-4 resulted in emphysematous pulmonary destruction and reduced protease inhibitor levels in the lung[41].
Increased IL-13 observed in Pc-colonized monkeys is interesting in light of reports that emphysema was associated with IL-13 expression in a transgenic mouse model[42], and in murine models of Nippostrongylus brasiliensis[43] and persistent viral infection[44]. Although its role in alveolar destruction progression is unclear, increased IL-13 in transgenic mice correlated with increased matrix metalloprotease and cathepsin production in lung tissue[22], which may promote lung tissue degradation in emphysema[45].
IL-1β, IL-6 and GM-CSF, pro-inflammatory cytokines associated with macrophage activation, were also increased in BALF of SHIV/Pc+ monkeys. These results are consistent with reports of increased levels of these cytokines in pulmonary and plasma samples from COPD patients[46] and in animal models of emphysema[47], indicating a key role for macrophage activation in the early process of lung damage in this model.
Contrary to studies reporting inflammatory cellular infiltration associated with human and animal COPD, we found no significant changes in absolute numbers or proportions of T cells or neutrophils in BALF of either monkey group, even after significant pulmonary function decline was evident. This may be due to the fact that in human studies, patients have had clinical disease for years whereas the primate model is capturing early events in disease progression. Unlike our previous study showing infiltration of CD8+ T cells and neutrophils in intrabronchially infected macaques[12], the Pc doses associated with natural colonization reported here were likely much lower. It is likely that as Pc burden increases, a CD8+ T cell-and neutrophil-dominant response may develop and amplify inflammation-mediated pulmonary damage.
Innate inflammatory responses initiated by alveolar macrophages or other cells such as mast cells, NK or NKT cells may be activated early in response to Pc colonization thus elaborating pro-inflammatory cytokines prior to detectable activation of adaptive responses and subsequent cellular infiltration. This hypothesis is consistent with studies suggesting macrophages and NKT cells are key effectors in murine models demonstrating IL-13-mediated emphysematous destruction[43]. Additionally, a role for mast cells in human COPD has been suggested[48], possibly via IL-4 upregulation[49]. We further postulate that persistent Pc colonization is associated with an adaptive immune response, as indicated by increased frequency of bronchial-associated lymphoid tissue in Pc-colonized monkeys with COPD. These results are consistent with the finding of an increased frequency of bronchial-associated lymphoid tissue in COPD patients, and support the concept that persistent infection and host immune response is associated with COPD development[4, 35].
This study establishes a novel model for HIV-associated COPD and provides evidence supporting a role for Pc colonization in obstructive disease development. These results support the paradigm that infectious agents, directly or indirectly, can promote COPD pathogenesis[4, 43]. A detrimental inflammatory response may be amplified by continuous or repeated colonization by various pulmonary pathogens leading to disease progression, as has been clinically shown[50]. As in human COPD pathogenesis, it is likely that COPD development in SHIV-infected macaques is multifactorial and that genetic and environmental factors contribute to susceptibility. This study supports the concept that Pc colonization contributes to COPD pathogenesis in SHIV-infected macaques, but does not exclude a role for other factors. The non-human primate model allows for serial examination of various parameters associated with development of obstructive changes and should help define host responses that promote tissue destruction at early disease stages. Additionally, these results identify Pc as a potentially treatable risk factor for COPD development in HIV-infected and non-infected individuals.
ACKNOWLEDGEMENTS
We thank Drs. Anita Trichel and Chris Janssen for clinical veterinary care and Nicole Banichar and Tim Sturgeon for technical assistance.
Footnotes
This work was supported by the NIH, National Heart, Lung, Blood Institute (KAN).
The authors declare no conflict of interest regarding this work.
AUTHORS CONTRIBUTIONS
K.A.N., T.W.S., and A.M. designed and supervised the research, analyzed the data and wrote the paper. T.W.S, H.M.K, S.P., J.K., S.E.G, J.M.M., and X.S. performed experiments; H.O.C., J.C.H., F.C.S., P,T. and R.M.R assisted in data analysis; T.W.S., A.M. and T.R., performed statistical analyses and all other authors provided technical advice, discussed results and assisted in manuscript preparation.
References
- 1.Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet. 1997;349:1498–1504. doi: 10.1016/S0140-6736(96)07492-2. [DOI] [PubMed] [Google Scholar]
- 2.Diaz PT, King MA, Pacht ER, et al. Increased susceptibility to pulmonary emphysema among HIV-seropositive smokers. Ann Intern Med. 2000;132:369–372. doi: 10.7326/0003-4819-132-5-200003070-00006. [DOI] [PubMed] [Google Scholar]
- 3.Crothers K, Butt AA, Gibert CL, Rodriguez-Barradas MC, Crystal S, Justice AC. Increased COPD among HIV-positive compared to HIV-negative veterans. Chest. 2006;130:1326–1333. doi: 10.1378/chest.130.5.1326. [DOI] [PubMed] [Google Scholar]
- 4.Hogg JC. Role of latent viral infections in chronic obstructive pulmonary disease and asthma. Am J Respir Crit Care Med. 2001;164:S71–S75. doi: 10.1164/ajrccm.164.supplement_2.2106063. [DOI] [PubMed] [Google Scholar]
- 5.Morris A, Sciurba FC, Norris KA. Pneumocystis: a novel pathogen in chronic obstructive pulmonary disease? Copd. 2008;5:43–51. doi: 10.1080/1541255070181756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calderon EJ, Rivero L, Respaldiza N, et al. Systemic inflammation in patients with chronic obstructive pulmonary disease who are colonized with Pneumocystis jiroveci. Clin Infect Dis. 2007;45:e17–e19. doi: 10.1086/518989. [DOI] [PubMed] [Google Scholar]
- 7.Morris A, Sciurba FC, Lebedeva IP, et al. Association of chronic obstructive pulmonary disease severity and Pneumocystis colonization. Am J Respir Crit Care Med. 2004;170:408–413. doi: 10.1164/rccm.200401-094OC. [DOI] [PubMed] [Google Scholar]
- 8.Huang L, Crothers K, Morris A, et al. Pneumocystis colonization in HIV-infected patients. J Eukaryot Microbiol. 2003;50 Suppl:616–617. doi: 10.1111/j.1550-7408.2003.tb00651.x. [DOI] [PubMed] [Google Scholar]
- 9.Morris A, Kingsley LA, Groner G, Lebedeva IP, Beard CB, Norris KA. Prevalence and clinical predictors of Pneumocystis colonization among HIV-infected men. Aids. 2004;18:793–798. doi: 10.1097/00002030-200403260-00011. [DOI] [PubMed] [Google Scholar]
- 10.Norris KA, Wildschutte H, Franko J, Board KF. Genetic variation at the mitochondrial largesubunit rRNA locus of Pneumocystis isolates from simian immunodeficiency virus-infected rhesus macaques. Clin Diagn Lab Immunol. 2003;10:1037–1042. doi: 10.1128/CDLI.10.6.1037-1042.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guillot J, Demanche C, Norris K, et al. Phylogenetic relationships among Pneumocystis from Asian macaques inferred from mitochondrial rRNA sequences. Mol Phylogenet Evol. 2004;31:988–996. doi: 10.1016/j.ympev.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 12.Board KF, Patil S, Lebedeva I, et al. Experimental Pneumocystis carinii pneumonia in simian immunodeficiency virus-infected rhesus macaques. J Infect Dis. 2003;187:576–588. doi: 10.1086/373997. [DOI] [PubMed] [Google Scholar]
- 13.Pawar SN, Mattila JT, Sturgeon TJ, et al. Comparison of the effects of pathogenic simian human immunodeficiency virus strains SHIV-89.6P and SHIV-KU2 in cynomolgus macaques. AIDS Res Hum Retroviruses. 2008;24:643–654. doi: 10.1089/aid.2007.0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reimann KA, Li JT, Veazey R, et al. A chimeric simian/human immunodeficiency virus expressing a primary patient human immunodeficiency virus type 1 isolate env causes an AIDS-like disease after in vivo passage in rhesus monkeys. J Virol. 1996;70:6922–6928. doi: 10.1128/jvi.70.10.6922-6928.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Croix DA, Board K, Capuano S, 3rd, et al. Alterations in T lymphocyte profiles of bronchoalveolar lavage fluid from SIV- and Pneumocystis carinii-coinfected rhesus macaques. AIDS Res Hum Retroviruses. 2002;18:391–401. doi: 10.1089/088922202753519179. [DOI] [PubMed] [Google Scholar]
- 16.Kling HM, Shipley TW, Patil S, Morris A, Norris KA. Pneumocystis colonization in immunocompetent and simian immunodeficiency virus-infected cynomolgus macaques. J Infect Dis. 2009;199:89–96. doi: 10.1086/595297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giavedoni LD. Simultaneous detection of multiple cytokines and chemokines from nonhuman primates using luminex technology. J Immunol Methods. 2005;301:89–101. doi: 10.1016/j.jim.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 18.Rennard SI, Basset G, Lecossier D, et al. Estimation of volume of epithelial lining fluid recovered by lavage using urea as marker of dilution. J Appl Physiol. 1986;60:532–538. doi: 10.1152/jappl.1986.60.2.532. [DOI] [PubMed] [Google Scholar]
- 19.Coxson HO, Rogers RM, Whittall KP, et al. A quantification of the lung surface area in emphysema using computed tomography. Am J Respir Crit Care Med. 1999;159:851–856. doi: 10.1164/ajrccm.159.3.9805067. [DOI] [PubMed] [Google Scholar]
- 20.Perez At, Coxson HO, Hogg JC, Gibson K, Thompson PF, Rogers RM. Use of CT morphometry to detect changes in lung weight and gas volume. Chest. 2005;128:2471–2477. doi: 10.1378/chest.128.4.2471. [DOI] [PubMed] [Google Scholar]
- 21.Nakano Y, Wong JC, de Jong PA, et al. The prediction of small airway dimensions using computed tomography. Am J Respir Crit Care Med. 2005;171:142–146. doi: 10.1164/rccm.200407-874OC. [DOI] [PubMed] [Google Scholar]
- 22.Zheng T, Zhu Z, Wang Z, et al. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest. 2000;106:1081–1093. doi: 10.1172/JCI10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Proskocil BJ, Sekhon HS, Clark JA, et al. Vitamin C prevents the effects of prenatal nicotine on pulmonary function in newborn monkeys. Am J Respir Crit Care Med. 2005;171:1032–1039. doi: 10.1164/rccm.200408-1029OC. [DOI] [PubMed] [Google Scholar]
- 24.Gigliotti F, Wright TW. Immunopathogenesis of Pneumocystis carinii pneumonia. Expert Rev Mol Med. 2005;7:1–16. doi: 10.1017/S1462399405010203. [DOI] [PubMed] [Google Scholar]
- 25.Sarir H, Henricks PA, van Houwelingen AH, Nijkamp FP, Folkerts G. Cells, mediators and Toll-like receptors in COPD. Eur J Pharmacol. 2008;585:346–353. doi: 10.1016/j.ejphar.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 26.Lokke A, Lange P, Scharling H, Fabricius P, Vestbo J. Developing COPD: a 25 year follow up study of the general population. Thorax. 2006;61:935–939. doi: 10.1136/thx.2006.062802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Probst M, Ries H, Schmidt-Wieland T, Serr A. Detection of Pneumocystis carinii DNA in patients with chronic lung diseases. Eur J Clin Microbiol Infect Dis. 2000;19:644–645. doi: 10.1007/s100960000329. [DOI] [PubMed] [Google Scholar]
- 28.Ambrose Z, KewalRamani VN, Bieniasz PD, Hatziioannou T. HIV/AIDS: in search of an animal model. Trends Biotechnol. 2007;25:333–337. doi: 10.1016/j.tibtech.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 29.Joag SV, Li Z, Foresman L, et al. Characterization of the pathogenic KU-SHIV model of acquired immunodeficiency syndrome in macaques. AIDS Res Hum Retroviruses. 1997;13:635–645. doi: 10.1089/aid.1997.13.635. [DOI] [PubMed] [Google Scholar]
- 30.Gelb AF, Schein M, Kuei J, et al. Limited contribution of emphysema in advanced chronic obstructive pulmonary disease. Am Rev Respir Dis. 1993;147:1157–1161. doi: 10.1164/ajrccm/147.5.1157. [DOI] [PubMed] [Google Scholar]
- 31.Kim WD, Ling SH, Coxson HO, et al. The association between small airway obstruction and emphysema phenotypes in COPD. Chest. 2007;131:1372–1378. doi: 10.1378/chest.06-2194. [DOI] [PubMed] [Google Scholar]
- 32.Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J. 2008;31:1334–1356. doi: 10.1183/09031936.00018908. [DOI] [PubMed] [Google Scholar]
- 33.Kim V, Rogers TJ, Criner GJ. New concepts in the pathobiology of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2008;5:478–485. doi: 10.1513/pats.200802-014ET. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stanescu D, Sanna A, Veriter C, et al. Airways obstruction, chronic expectoration, and rapid decline of FEV1 in smokers are associated with increased levels of sputum neutrophils. Thorax. 1996;51:267–271. doi: 10.1136/thx.51.3.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–2653. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- 36.Betsuyaku T, Nishimura M, Takeyabu K, et al. Neutrophil granule proteins in bronchoalveolar lavage fluid from subjects with subclinical emphysema. Am J Respir Crit Care Med. 1999;159:1985–1991. doi: 10.1164/ajrccm.159.6.9809043. [DOI] [PubMed] [Google Scholar]
- 37.Curtis JL, Freeman CM, Hogg JC. The immunopathogenesis of chronic obstructive pulmonary disease: insights from recent research. Proc Am Thorac Soc. 2007;4:512–521. doi: 10.1513/pats.200701-002FM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barcelo B, Pons J, Fuster A, et al. Intracellular cytokine profile of T lymphocytes in patients with chronic obstructive pulmonary disease. Clin Exp Immunol. 2006;145:474–479. doi: 10.1111/j.1365-2249.2006.03167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson DS, Hamid Q, Ying S, et al. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 40.Zhu J, Majumdar S, Qiu Y, et al. Interleukin-4 and interleukin-5 gene expression and inflammation in the mucus-secreting glands and subepithelial tissue of smokers with chronic bronchitis. Lack of relationship with CD8(+) cells. Am J Respir Crit Care Med. 2001;164:2220–2228. doi: 10.1164/ajrccm.164.12.2009060. [DOI] [PubMed] [Google Scholar]
- 41.Ma B, Blackburn MR, Lee CG, et al. Adenosine metabolism and murine strain-specific IL-4-induced inflammation, emphysema, and fibrosis. J Clin Invest. 2006;116:1274–1283. doi: 10.1172/JCI26372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoshino T, Kato S, Oka N, et al. Pulmonary inflammation and emphysema: role of the cytokines IL-18 and IL-13. Am J Respir Crit Care Med. 2007;176:49–62. doi: 10.1164/rccm.200603-316OC. [DOI] [PubMed] [Google Scholar]
- 43.Marsland BJ, Kurrer M, Reissmann R, Harris NL, Kopf M. Nippostrongylus brasiliensis infection leads to the development of emphysema associated with the induction of alternatively activated macrophages. Eur J Immunol. 2008;38:479–488. doi: 10.1002/eji.200737827. [DOI] [PubMed] [Google Scholar]
- 44.Kim EY, Battaile JT, Patel AC, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med. 2008;14:633–640. doi: 10.1038/nm1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Churg A, Wright JL. Proteases and emphysema. Curr Opin Pulm Med. 2005;11:153–159. doi: 10.1097/01.mcp.0000149592.51761.e3. [DOI] [PubMed] [Google Scholar]
- 46.Gan WQ, Man SF, Senthilselvan A, Sin DD. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax. 2004;59:574–580. doi: 10.1136/thx.2003.019588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol. 2005;32:311–318. doi: 10.1165/rcmb.2004-0309OC. [DOI] [PubMed] [Google Scholar]
- 48.Gizycki MJ, Hattotuwa KL, Barnes N, Jeffery PK. Effects of fluticasone propionate on inflammatory cells in COPD: an ultrastructural examination of endobronchial biopsy tissue. Thorax. 2002;57:799–803. doi: 10.1136/thorax.57.9.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barczyk A, Pierzchala W, Kon OM, Cosio B, Adcock IM, Barnes PJ. Cytokine production by bronchoalveolar lavage T lymphocytes in chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2006;117:1484–1492. doi: 10.1016/j.jaci.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 50.Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med. 2008;359:2355–2365. doi: 10.1056/NEJMra0800353. [DOI] [PubMed] [Google Scholar]