

Based upon the recent advances in genetic association studies, the age of genetically based personalized medicine may be finally arriving in the near future for many diseases. The field of hematopoietic cell transplantation (HCT) is well ahead of this curve. In the entire field of medicine, there is no better demonstration of the power of personalized medicine than the use of patient-donor histocompatibility antigen (HLA) matching to improve stem cell transplant outcomes. Nevertheless, even the HCT field will benefit significantly from the latest developments in genetic association studies, which has allowed expanded analyses of the human genome beyond the major histocompatibility complex (MHC).
Despite extensive efforts using HLA matching to improve transplant outcomes, the nonrelapse mortality (NRM) rate has remained relatively unchanged over the last decade. Recent data from our Center between 1990–2000 indicate that the cumulative incidence of NRM rate is 25% and 30% for matched sibling and unrelated transplants respectively undergoing a myeloablative transplant for a hematological malignancy (Figure 1). Although many of these deaths are related to graft versus host disease (GVHD), many are also attributable to other complications of HCT, including opportunistic infection and organ specific syndromes, some of which may reflect GVHD and related immune deficiency. Because there is evidence suggesting that the risk for some of these complications may be influenced by genetic loci outside of the MHC, many investigators have already begun to evaluate genetic variation in genes of candidate biologic pathways that play a role in the pathogenesis of these complication, trying to identify genetic variants that might serve as a biomarker for not only predicting the risk for developing these complicated outcomes, but also serve as potential future therapeutic targets. However, the results of this approach have been less than impressive. The majority of studies conducted thus far have yielded results which are largely still pending rigorous replication in independent validation studies. This is likely due to shortcomings related to issues specific to genetic association studies, such as appropriate selection of cases and controls, use of biologically relevant and well defined phenotypes, adequate sample size to achieve statistical power, incorporation of population stratification analysis, and most importantly, validation of the genetic association in an independent cohort [1,2].
Figure 1.
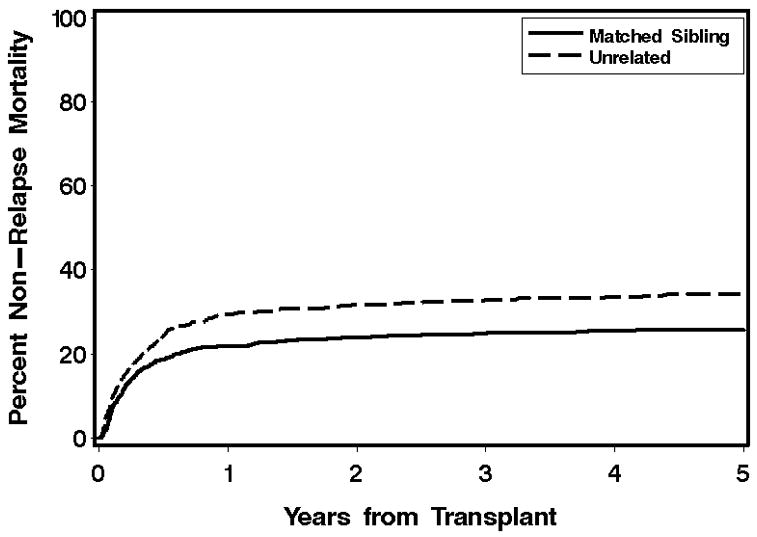
Cumulative incidence curve for nonrelapse mortality for all transplants between 1992 and 2005 conducted at the Fred Hutchinson Cancer Research Center.
Our research group recently published three studies evaluating genes in candidate pathways that are involved in the development of infections and airways injury after allogeneic HCT. While these are not the only studies available, they are representative examples of genetic association studies using a two-phase discovery validation approach. In the first study, we comprehensively screened innate immunity genes to discover and verify that single nucleotide polymorphism (SNP) haplotypes across the highly polymorphic gene bactericidal permeability increasing (BPI) gene influences the risk of developing rapid airflow decline after allogeneic HCT [3]. We subsequently confirmed that BPI, an anti-inflammatory lipopolysaccaride binding protein, is produced by small airway epithelial cells [4], further supporting its potential role in the development of bronchiolitis obliterans syndrome, a form of GVHD in the lungs [3]. In a second study, we discovered that a SNP haplotype containing a putative functional SNP in the promoter region of the lipopolysaccharide binding protein (LBP) gene is associated with a two-fold increase in risk of developing Gram-negative bacteremia after allogeneic HCT [5]. We then confirmed this association in a prospective cohort, where we demonstrated that the homozygous rare genotype is associated with a nearly three-fold increase in circulating LBP levels, which likely plays a significant role in the down regulation of the innate immune response to Gram-negative bacteria, potentially resulting in unchecked bacterial replication [5]. Finally, we recently conducted a study that comprehensively evaluate toll-like receptors (TLR) 2, 4, 6, and 9 that discovered and validated the finding that a SNP haplotype in TLR4 that contain previously known functional SNPs is associated with a two to five-fold increase in risk of developing invasive aspergillus infection [6].
Despite these and other successes, the candidate gene approach remains extremely limited in scope [7]. Furthermore, the quality of genetic association studies continues to improve. The genome wide genetic association studies published in 2007 and 2008 clearly indicate that the field of genetic epidemiology has entered a new era. These studies examined complex phenotypes such as diabetes [8,9], rheumatoid arthritis [10,11], and multiple sclerosis [12] by scanning the genomes of tens of thousands of individuals using high throughput genotyping arrays containing over 500,000 single nucleotide polymorphisms (SNPs). Recently, we also embarked upon a genome wide analysis of infectious and organ specific complications of allogeneic HCT. Although the stem cell transplant population is much smaller than those used for other more common diseases, our population benefits from phenotypes that occur with higher frequency and specificity, both of which can significantly decrease the false discovery rate and increase the likelihood that we will discover a genetic relationship that is biologically plausible. From 3,177 patients undergoing allogeneic HCT at our Center between 1992 and 2005 for whom cryopreserved peripheral blood mononuclear cells (PBMC) and/or B-lymphoblastoid cell lines (B-LCL) were available, we randomly selected a discovery cohort consisting of 1,560 donor-patient pairs (total of 3,120 specimens), and reserved the remainder patient/donor pairs as the validation cohort. Each of these specimens was genotyped using the Affymetrix GeneChip® Genome-Wide Human Single Nucleotide Polymorphism (SNP) Array 5.0, which contains over 500,000 SNPs placed evenly across the genome (methods will be explained in detail later in this proposal). Population stratification was accounted for using a principle components approach to identify a subset of SNPs that capture genetic differences between ethnic populations.
Our analyses thus far have focused on a number of infectious and organ specific endpoints. Some initial results from our analysis of Gram-negative bacteremia and bronchiolitis obliterans syndrome are promising. To develop the Gram-negative bacteremia phenotype, an a priori list of GN bacterial organisms was assembled from a review of our laboratory database [5]. Patients were considered a “case” if they had one or more positive blood cultures with one of these organisms prior to discharge from our Center. A time to event allelic and genotypic analysis was performed for each SNP to calculate hazard ratios and 95% confidence intervals for all allele types and genotypes respectively. Death was treated as a competing event. Pretransplant neutropenia, acute GVHD, and time to engraftment, defined as three consecutive days with a neutrophil count >500 cells/ul, were included as covariates in the genetic model. Overall, there were 303 cases of GN bacteremia in the discovery cohort. After eliminating SNPs that were identified to be in Hardy-Weinberg disequilibrium at p<0.001 or had a genotype call rate of <80%, 17 SNP genotypes in the patient were significantly associated with the GN bacteremia phenotype, with p-values ranging from 1 × 10−6.1 to 1 × 10−8.1 (Figure 2 and Table 1). Using a similar approach for bronchiolitis obliterans syndrome, which was defined according to recent recommendations from the National Institutes of Health [13], we have identified four patient and three donor genetic loci that are associated with a higher risk of developing BOS, with p-values ranging from 1 × 10−6.0 to 1 × 10−8.1.
Figure 2.

Genome wide allelic and genotypic association in with Gram negative bacteremia for the patient genome. Solid line indicates significance threshold of 1 × 10−6. Circles indicate loci that meet or exceed this significance threshold.
Table 1.
Summary of significant associations between patient genome and Gram negative bacteremia phenotype.
| Chromosome | rs# | Position | Alleles | MAF | HWE | % genotype call | Allelic or genotypic association | −log10(p)* | Hazard ratio* |
|---|---|---|---|---|---|---|---|---|---|
| 3 | rs2712964 | 111590926 | A/G | 0.034 | 0.812 | 100.0 | Allelic | 6.4 | 2.541 |
| 3 | rs2669100 | 111616022 | A/G | 0.036 | 0.833 | 99.9 | Allelic | 6.5 | 2.544 |
| 4 | rs16879412 | 27082225 | A/C | 0.005 | 0.992 | 100.0 | Allelic | 6.4 | 4.469 |
| 4 | rs7686272 | 38985157 | A/T | 0.006 | 0.644 | 99.9 | Allelic | 7.9 | 5.051 |
| 4 | rs2062230 | 38996761 | C/T | 0.01 | 0.379 | 100.0 | Allelic | 7.7 | 2.561 |
| 4 | rs16856486 | 43471432 | C/T | 0.005 | 0.803 | 100.0 | Genotypic | 6.1 | 4.569 |
| 7 | rs42716 | 28422139 | A/G | 0.086 | 0.953 | 99.9 | Genotypic | 6.3 | 4.923 |
| 8 | rs7816574 | 140875208 | A/G | 0.078 | 0.015 | 100.0 | Genotypic | 6.6 | 5.077 |
| 12 | rs2654662 | 59308693 | A/T | 0.17 | 0.215 | 98.0 | Genotypic | 6.2 | 3.522 |
| 12 | rs273812 | 59342417 | C/T | 0.169 | 0.177 | 99.9 | Genotypic | 6.3 | 3.499 |
| 12 | rs273782 | 59353980 | A/T | 0.169 | 0.092 | 99.1 | Genotypic | 6.5 | 3.632 |
| 13 | rs7331598 | 76752154 | C/T | 0.029 | 0.848 | 99.0 | Allelic | 6.5 | 2.351 |
| 15 | rs16945332 | 89543178 | A/G | 0.062 | 0.949 | 100.0 | Allelic | 6.3 | 1.981 |
| 16 | rs10500532 | 64526117 | C/T | 0.009 | 0.567 | 100.0 | Allelic | 6.7 | 4.402 |
| 16 | rs7188110 | 79148871 | C/T | 0.013 | 0.201 | 100.0 | Allelic | 7.7 | 3.26 |
| 16 | rs7187446 | 79148886 | A/G | 0.013 | 0.201 | 100.0 | Allelic | 8.1 | 3.852 |
| 16 | rs16956238 | 80617787 | A/C | 0.005 | 0.622 | 100.0 | Genotypic | 6.4 | 4.736 |
MAF=minor allele frequency; HWE=p-value for Hardy Weinberg equilibrium test
−log(10)p’s and hazard ratios refer to the minor allele for allelic models and the homozygous rare genotype for the genotype model.
Despite these promising genetic leads, it should be recognized that this approach is not without its own challenges. For instance, when over half a million analyses are conducted at the same time, the multiple comparison penalty paid restricts the focus of attention to the loci that meet the most stringent threshold for significance. While this conservative approach will reduce false positive discoveries, it also means that promising associations that do not quite meet the significance threshold will not receive the critical attention they deserve. Another major limitation of the agnostic genome wide approach is that these discoveries rarely lead to a candidate gene or pathway whose biologic role is immediately obvious. In fact, most genome wide studies thus far have resulted in associations with loci in gene deserts, leaving scientists at the beginning of the long and arduous task of elucidating the mechanisms that account for the observed associations. Finally, we should recognize that while the era genomics is technologically attractive, environmental influences such as clinical factors still play a major role in the development of many of these phenotypes. Our task is to integrate these genetic discoveries with the critical clinical factors to develop a biologically plausible hypothesis that will one day help us improve the outcome of stem cell transplantation.
As we wait for these discoveries to be validated, we have already learned three valuable lessons regarding the future of genetic research in HCT. First, while candidate gene approaches can succeed, they are less likely to identify genetic variants that will have a clinically meaningful effect when the entire genome is considered. However, this does not mean that candidate gene approaches should be abandoned. Rather, we believe that the candidate gene approach should take advantage of the flood of genetic information available from genome scans, such that each candidate gene can be more comprehensively evaluated. Second, these results demonstrate that an agnostic genome wide approach toward transplant outcomes can reveal highly statistically significant associations. While these discoveries still require rigorous validation, it is inevitable that we have identified leads that will ultimately lead us down a pathway that have never been previously considered. Importantly, this unbiased genome wide approach will likely identify targets that are not only clinically meaningful as potential predictive markers, but will also serve as high yield therapeutic targets. Finally, as we analyze genome wide data, we are repeatedly reminded of the importance of collaboration. Success in genome wide studies requires expertise in multiple areas of research (i.e. clinical and genetic epidemiology, statistical genetics, bioinformatics, molecular biology, immunobiology, and multiple clinical subspecialties), and is highly dependent upon cohort size and replication. As a community of scientists and patients, we are much smaller than those associated with more common diseases. In order for us to fully capitalize upon the coming technological advances, we will need to pool our resources and work together collaboratively in this exciting new era of genome sciences.
Acknowledgments
HL087690, HL088201
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 2.Botstein D, Risch N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease (Review) Nat Genet. 2003;33 (Suppl):228–237. doi: 10.1038/ng1090. [DOI] [PubMed] [Google Scholar]
- 3.Chien JW, Zhao LP, Hansen JA, Fan WH, Parimon T, Clark JG. Genetic variation in bactericidal/permeability-increasing protein influences the risk of developing rapid airflow decline after hematopoietic cell transplantation. Blood. 2006;107:2200–2207. doi: 10.1182/blood-2005-06-2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elsbach P, Weiss J. The bactericidal/permeability-increasing protein (BPI), a potent element in host-defense against gram-negative bacteria and lipopolysaccharide (Review) Immunobiology. 1993;187:417–429. doi: 10.1016/S0171-2985(11)80354-2. [DOI] [PubMed] [Google Scholar]
- 5.Chien JW, Boeckh MJ, Hansen JA, Clark JG. Lipopolysaccharide binding protein promoter variants influence the risk for gram-negative bacteremia and mortality after allogeneic hematopoietic cell transplantation. Blood. 2008;111:2462–2469. doi: 10.1182/blood-2007-09-101709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bochud P-Y, Chien JW, Marr KA, et al. Toll-like receptor 4 polymorphisms and aspergillosis in stem-cell transplantation. N Engl J Med. 2008;359:1766–1777. doi: 10.1056/NEJMoa0802629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kruglyak L, Nickerson DA. Variation is the spice of life. Nat Genet. 2001;27:234–236. doi: 10.1038/85776. [DOI] [PubMed] [Google Scholar]
- 8.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hakonarson H, Grant SF, Bradfield JP, et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature. 2007;448:591–594. doi: 10.1038/nature06010. [DOI] [PubMed] [Google Scholar]
- 10.Plenge RM, Cotsapas C, Davies L, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007;39:1477–1482. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomson W, Barton A, Ke X, et al. Rheumatoid arthritis association at 6q23. Nat Genet. 2007;39:1431–1433. doi: 10.1038/ng.2007.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas G, Jacobs KB, Yeager M, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008;40:310–315. doi: 10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- 13.Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant. 2005;11:945–956. doi: 10.1016/j.bbmt.2005.09.004. [DOI] [PubMed] [Google Scholar]