

Abstract
Metastatic cancer is a major cause of morbidity and mortality. Current therapeutic options consist of chemotherapy, radiation, or targeted therapies. However, these therapies are often toxic, effective over a small range of cancer types, or result in drug resistance. Therefore, a more global, less toxic strategy for the management of metastatic cancer is required. Though most cancers display increased glucose metabolism, glutamine is also a major energy substrate for many cancers. We evaluated the anti-metastatic potential of 6-diazo-5-oxo-L-norleucine (DON), a glutamine analog, using the new VM mouse model of systemic metastasis. We found that primary tumor growth was approximately 20-fold less in DON treated mice than in untreated control mice. We also found that DON treatment inhibited metastasis to liver, lung, and kidney as detected by bioluminescence imaging and histology. Our findings provide proof of concept that metabolic therapies targeting glutamine metabolism can manage systemic metastatic cancer.
Keywords: metastasis, metabolic therapy, calorie restriction, VM mice, DON
Introduction
Tumor metastasis is the leading cause of morbidity and mortality in cancer patients 1, 2. Traditional chemotherapeutic agents such as cisplatin, though very effective in killing tumor cells, often result in extreme toxicities and drug resistance 3–5. In addition, targeted therapies such as epidermal growth factor receptor type 2 antibodies (Trastuzumab) are effective for only those cancers that over-express the particular antigen 6. Small molecule drugs are also available that target receptor tyrosine kinases involved in cell proliferation and survival 7. However, multiple pathways are often responsible for tumor cell survival and simultaneous inhibition of those pathways is usually necessary for global therapeutic effect 7. Alternative therapies are therefore needed that are less toxic and produce more global therapeutic effects.
Cancer cells frequently exhibit increased glycolysis and therapies that target glucose metabolism have been exploited in the past to include 2-deoxyglucose (2-DG) and calorie restriction (CR) 8–10. Calorie restriction is a powerful anti-angiogenic therapy and acts synergistically with 2-DG to reduce tumor growth in various experimental mouse and human brain cancer models 8–10. In addition, calorie restriction has been shown to target Akt signaling pathways involved in the anti-apoptotic and glycolytic phenotype of many tumors 11. However, targeting glucose alone may not be effective in managing those tumors that also rely heavily on glutamine for growth and survival 12–16. Currently, there are a number of drug targets of glutamine metabolism to include phenylbutyrate (PBA), and the glutamine analogs acivicin and 6-diazo-5-oxo-L-norleucine (DON) 17, 18. PBA has been used extensively in vitro and in human trials as a histone deacetylase inhibitor 19–22. In the body, PBA is metabolized to phenylacetate (PA), which covalently conjugates with glutamine 18. This glutamine-PA conjugate is then excreted, effectively reducing the amount of free glutamine in circulation 18. The glutamine analogs have also shown promising results in vitro and in murine models of cancer, as both inhibitors of nucleotide biosynthesis and inhibitors of glutaminolysis 23–26. However, limited success has been achieved with PBA, and high toxicities of the glutamine analogs limit their use for human studies 17, 25, 27, 28
The goal of this research was to examine the efficacy of glucose or glutamine targeting using the newly established pre-clinical VM-M3 mouse model of systemic metastatic cancer3. CR and other metabolic therapies have not been previously tested, to our knowledge, on natural models of systemic metastatic cancer 3. The VM-M3 tumor cells express the firefly luciferase gene, allowing for non-invasive detection of tumor growth and metastasis via bioluminescent imaging. This tumor arose spontaneously in the brain of a VM mouse and has multiple properties of glioblastoma multiforme to include systemic metastasis 29. While metastasis is not commonly seen in gliomas, GBM is highly metastatic once the tumor cells reach the blood stream 30–34. From a subcutaneous implantation site, the VM-M3 tumor recapitulates all the major hallmarks of metastasis, to include detachment from the primary tumor, intravasation into the blood stream, evasion of immune attack, extravasation at a distant capillary bed, and growth at distant sites 2, 3, 35, 36. In addition, this tumor has multiple properties of myeloid cells including macrophages/microglia, which are also seen in a number of human metastatic cancers to include lung, breast, colon, and skin 3, 36–41. A requirement for glutamine is a key metabolic hallmark for the growth of myeloid cells 42. We posited that metabolic therapies could have widespread inhibitory effects on tumor growth and metastasis.
In this study we found that the glutamine analog DON significantly reduced tumor growth and metastasis in the VM-M3 mouse model. In addition, survival was significantly enhanced in the DON treated group compared to the control group.
Materials and Methods
Tumor formation
The VM-M3 tumor arose spontaneously in the cerebrum of an adult male VM mouse as previously described 36. After a total of three i.c. passages, the tumors were grown subcutaneously (s.c.) and cell lines were prepared from the tumor as described previously 36.
Transduction of cell lines
The VM-M3 cell line was transduced with a lentivirus vector containing the firefly luciferase gene under control of the cytomegalovirus promoter (VM-M3/Fluc) as we previously described (gift from Miguel Sena-Esteves) 36.
Experimental Medias
DMEM powder (Sigma) was prepared as directed without the addition of glucose, glutamine or FBS and supplemented with 50 μg/ml penicillin-streptomycin (Sigma) and stored at 4°C. Using this minimal media as a base, all other experimental medias were prepared. Experimental medias include 25 mM glucose and 4 mM glutamine.
Glucose and Glutamine Deprivation
Approximately 5 × 104 cells were seeded into two 24 well plates in complete DMEM. For imaging, 20 μl of a 300 μg/ml solution of D-luciferin (Promega) was added to the wells of one plate and the cells were imaged immediately on the Xenogen IVIS system for 3–5 minutes (Xenogen, Hopkington, MA) to record the bioluminescent signal from the cells. This reading is recorded as the 0 hr time point. After imaging, the cells in the remaining plate were allowed to settle for 6 hrs before being rinsed with minimal media and incubated in the experimental medias (25 mM glucose and 4 mM glutamine). Cells were also incubated in complete DMEM as a control. The cells were imaged again 24 hrs after the addition of the experimental medias. The data are represented as the percent of the initial cell number.
In vitro DON toxicity
Approximately 1 × 105 cells were seeded in 24-well plates and allowed to settle for 24 hrs. After 24 hrs the cells were imaged with the Xenogen IVIS system to obtain a 0 hr initial bioluminescent signal as described above. The wells were then rinsed with minimal media and incubated in complete DMEM media plus the various concentrations of 6-Diazo-5-oxo-L-norleucine (DON, Sigma lot # 1410096). After a 24 hr incubation, the cells were imaged for 3–5 minutes to record the bioluminescent signal as described above. Following imaging, fresh media containing drug was added and the cells were incubated for an additional 24 hrs. Data are represented as the percent of initial cell number relative to the 0 hour time point over 48 hrs.
Mice
Mice of the VM/Dk (VM) strain were obtained originally as gifts from H. Fraser (University of Edinburgh, Scotland). All Mice used in this study were housed and bred in the Boston College Animal Care Facility using husbandry conditions as previously described 43. All animal procedures were in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care Committee.
Subcutaneous Implants
VM mice were anaesthetized with Isoflurane (Halocarbon, River Edge, NJ), and the tumor was implanted by an s.c. injection of 0.1 ml of small tumor fragments suspended in 0.2 ml PBS by use of a 1 cc tuberculin syringe attached to an 18-gauge needle into the right flank as previously described 3. All mice recovered from their surgical procedure and were returned to their cages when they became fully active.
Dietary regimens, drug dosing, body weight, and food intake measurements
Adult male and female VM mice of approximately 60–90 days old were separated into individual housing 1–2 days before implantation. Individually housed mice were kept in plastic cages with filter tops containing Sani-Chip bedding (P.J. Murphy Forest Products Corp., Montville, NJ). Body weights and food intake measurements were recorded daily and food was provided ad libitum. We implanted tumor fragments on day zero. The mice were then separated into the various experimental groups and were matched for body weights 5–7 days post implantation. For the duration of the study, mice on calorie restriction received 40% of their normal food intake (60% restriction) at approximately 10 AM. The control mice continued to receive food ad libitum. All mice were weighed daily prior to food administration. For those mice that received 6-Diazo-5-oxo-l-norleucine (DON, Sigma lot # 1410096), a fresh stock was prepared on day 5 and diluted to an appropriate concentration in PBS. Drug was stored at 4°C for the duration of the study. Mice were dosed at approximately 10 AM daily. Some doses were skipped in the survival study if the mice appeared lethargic or if body weight loss exceeded 1.5 g from the previous day.
Imaging
The Xenogen IVIS system (Xenogen, Hopkington, MA) was used to record the bioluminescent signal from the labeled tumors as we recently described 36. Briefly, for in vivo imaging, mice recieved an intraperitoneal injection of d-Lucifierin (50 mg/kg, Promega) in PBS and Avertin (0.1 mL/10g). Imaging times ranged from 3 to 10 min, depending on the time point. For ex vivo imaging, tumors and organs were removed and were imaged separately in 300 μg/ml d-Luciferin in PBS for 3–10 min. The IVIS Lumina cooled CCD camera system was used for light acquisition. Data acquisition and analysis was performed with Living Image® software (Caliper LS).
Glucose Measurements
Mice were anesthetized with isoflurane and euthanized by exsanguination, involving collection of blood from the heart as previously described 10. The blood was centrifuged at 2,000 × g for 10 min, the blood supernatant was collected and was stored at −80°C before analysis. Serum or plasma glucose was measured in a spectrophotometer using an enzymatic assay (Stanbio Laboratories).
Histology
Liver samples were fixed in 10% neutral buffered formalin (Sigma) and embedded in paraffin. The samples were sectioned at 5 μm, were stained with haematoxylin and eosin (H & E) at the Harvard University Rodent Histopathology Core Facility (Boston, MA), and were examined by light microscopy as we previously described 8, 36. All histological sections were evaluated by a veterinary neuropathologist, (Roderick Bronson) at the Harvard University Rodent Histopathology Core Facility.
Results
The aim of this research was to determine the efficacy of glucose and glutamine targeting in reducing tumor growth and metastasis using the VM mouse model for systemic metastasis.
VM-M3 response to glucose and glutamine deprivation in vitro
In order to assess the metabolic requirements of the VM-M3 cell line, an in vitro bioluminescent- based cell viability assay was developed as described in the Materials and Methods to test the ability of the cells to survive under extreme energy stress in serum free media in the absence of either glucose or glutamine. As shown in Figure 1, the VM-M3 cell line was more dependent on glutamine (gln) than on glucose (gluc) for energy and survival. Glucose (gluc) alone was unable to support cell survival when grown in serum free medium.
Figure 1. Survival of VM-M3 cells in a media containing both glucose and glutamine.
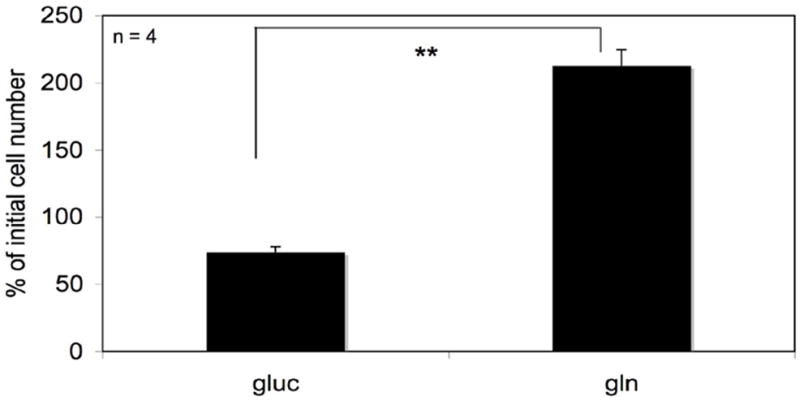
Approximately 5 × 104 VM-M3/Fluc cells were seeded in 24-well plates. Cells were immediately imaged on the Xenogen IVIS System in order to obtain a 0 hr baseline bioluminescent value. This value is proportional to the number of cells seeded. Cells were allowed to settle for 6 hrs before the addition of minimal DMEM media containing either 25 mM glucose (gluc) or 4 mM glutamine (gln). Cell viability was assessed 24 hrs later via bioluminescent imaging. Data are expressed as the mean percent increase in cell number relative to the 0 hr time point ± 95% C.I. of 4 independent samples per group. The asterisks indicate that the gln values differ significantly from the gluc values at p < 0.01
In vitro effect of DON on VM-M3 cells
As shown in Figure 2, DON was effective in inhibiting cell growth over 48 hours at both low (50μM) and high (250 μM) concentrations. Over the first 24 hours DON did not cause cell death but rather inhibited cell growth compared to the non-drug control as evidenced by continued ATP production and a lack of floating dead cells. A slight reduction in cell viability was observed over 48 hours. No morphological differences were observed between the control and the DON treated VM-M3 cells.
Figure 2. Effect of DON on VM-M3 cells in vitro.
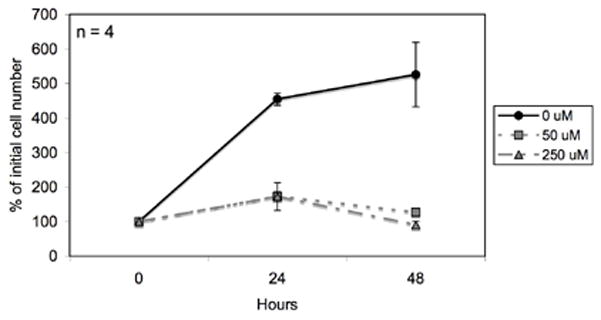
VM-M3/Fluc cells were seeded in DMEM in 24-well plates as described in the Materials and Methods and treated with DON (50 μM and 250 μM). Cells were imaged every 24 hrs using the Xenogen IVIS System. Data are expressed as the mean percent increase in cell number relative to the 0 hr time point ± 95% C.I. of 4 independent samples per group.
Effect of calorie restriction or DON on body weights and blood glucose
We next evaluated the effect of calorie restriction or the glutamine analog, DON, on body weights and blood glucose in mice implanted subcutaneously with the VM-M3 tumor. Calorically restricted mice received about 40% of their normal food intake (60% CR) as described in Materials and Methods (Figure 3A). DON was administered beginning on day 5 at a dose of 1 mg/kg/day. Calorie restriction significantly reduced the body weights of the mice over the course of the study (Figure 3B). The body weights of DON treated mice were similar during drug treatment compared to the control mice (Figure 3B). However, the body weights of DON treated mice declined over the last 3 days of the study. In addition, circulating glucose levels were significantly lower in the CR group compared to the control ad libitum (AL) fed group while blood glucose levels were similar between the DON group and control group (Figure 3C). As DON has been shown to inhibit glutaminase activity, circulating glutamine levels were not measured due to DON inhibition of the glutaminase enzyme reaction 44.
Figure 3. Experimental design and effect of calorie restriction or DON on body weights and blood glucose.
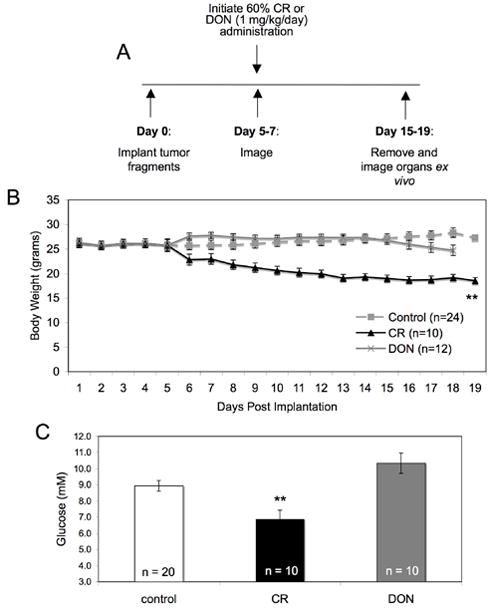
(A) The injection and treatment protocol as described in Materials and Methods. (B) Body weights were monitored daily. Prior to treatment, the body weights of all mice were averaged for a single value. (C) Mice were sacrificed 15–19 days post implantation and blood was collected for the analysis of glucose levels as described in the Materials and Methods. Values represent the mean ± SEM of 10–20 mice per group. The asterisks indicates that the CR values differ significantly from the AL control group at p < 0.01.
Effect of calorie restriction or DON on tumor growth and metastasis
As shown in Figure 4A, primary tumor growth was significantly lower in both the CR and DON group than in the AL group. In addition, primary tumor size was significantly lower in the DON group compared to the CR group. As seen in Figure 4B, the control mice had tumor metastasis to the liver, lung, kidney, and spleen, consistent with the behavior of this tumor 36. However, metastatic spread was not statistically different between the control and CR group for any of the organs (Figure 4B). On the other hand, the DON treated group had no detectable metastasis to the liver, lung, or kidney (Figure 4B). Interestingly, spleen metastasis in the DON group was similar to spleen metastasis in both the control and CR groups. In addition, we examined liver histology because it is an organ heavily infiltrated with tumor cells from the control group and is found in 100% of the control mice. As shown in Figure 5, histological analysis confirmed the lack of tumor cells in the liver of the DON treated mice in comparison to both the control AL non-treated group and the CR group.
Figure 4. Effect of calorie restriction or DON on VM-M3 tumor growth and metastasis.
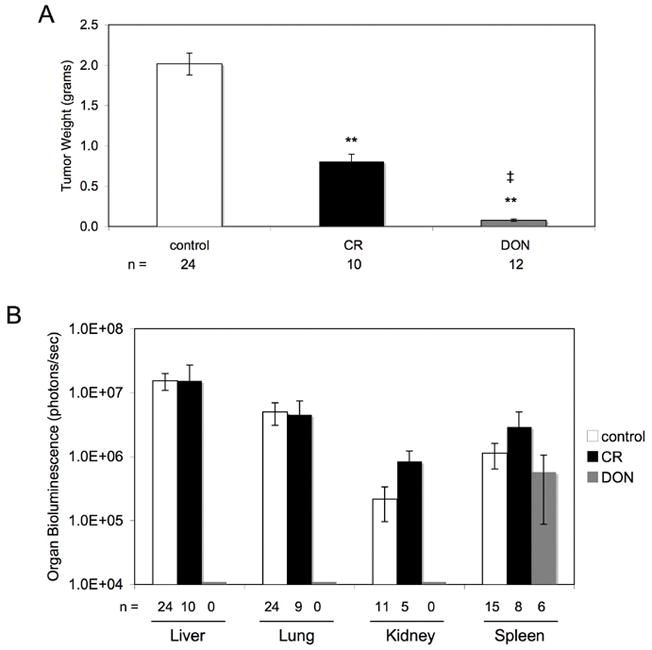
(A) Mice were implanted s.c. with the VM-M3/Fluc tumor as described in Materials and Methods. Mice were sacrificed 15–19 days post implantation and the tumors removed and weighed. The asterisks indicate that the CR or DON values differ significantly from the AL control group at p < 0.01. The ‡ indicates that the DON values differ significantly from the CR values at a p < 0.01. (B) At the time of sacrifice, the organs were removed and imaged ex vivo. Bioluminescence values were plotted on a log scale. All values represent the mean ± SEM of 6 to 24 mice per group. No detectable bioluminescence was found in the lung, liver or kidney of DON treated mice.
Figure 5. Influence of DON or CR on liver histology.
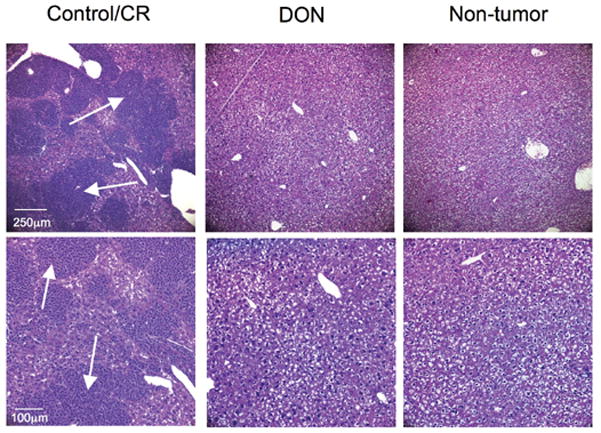
Removed livers were stained with haematoxylin and eosin (H & E) as described in Materials and Methods. Arrows indicate secondary tumor lesions in the control and CR group. Images are shown at 100X (top panel) and 200X (bottom panel).
Effect of calorie restriction or DON on mouse survival
As seen in Figure 6, all control mice reached morbidity 15–19 days post implantation. Mice in both the CR or DON group survived significantly longer than the control mice. However, the DON treated group reached morbidity due to drug toxicity, rather than from tumor metastasis, as indicated by loss of body weight, hind leg paralysis, and urinary blockages. The primary tumors in the DON-treated mice remained small and systemic metastasis detected with bioluminescent imaging was not apparent (data not shown).
Figure 6. Influence of DON or DON + CR on mouse survival.
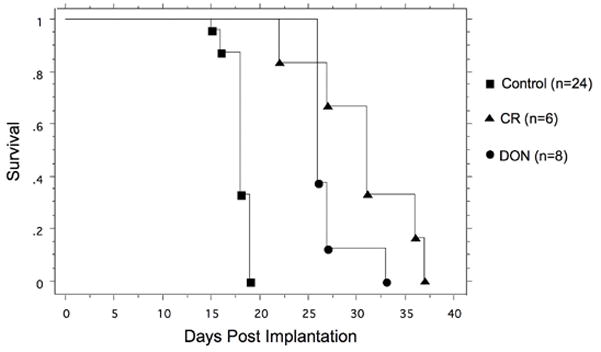
VM mice were implated s.c. with the VM-M3/Fluc tumor as described in Materials and Methods. All control mice reached morbidity 15–19 days post implantation. Both the CR and DON treated mice survived significantly longer than the controls. Survival in the CR mice was not significantly longer than the DON treated mice.
Discussion
The goal of this study was to examine metabolic therapies using the newly established VM-M3 mouse model for metastatic cancer. We found that the VM-M3 tumor cell line was more dependent on glutamine than glucose for survival in vitro. In support of our in vitro data we showed that the glutamine analog, 6-diazo-5-oxo-L-norleucine (DON), had a significant inhibitory effect on VM-M3 growth in vitro and on tumor growth and metastasis in vivo. In contrast, calorie restriction, which lowers circulating glucose levels, did not significantly reduce metastasis. This in vivo finding supports the in vitro findings that the VM-M3 tumor cells depend more on glutamine than on glucose for survival. As long as glutamine is available, the VM-M3 cells can survive despite the targeting of glucose. We previously showed that CR is anti-angiogenic and anti-inflammatory against experimental brain tumors 8, 9. The anti-angiogenic and anti-inflammatory action of CR could be responsible for the reduction in primary tumor size observed in this study. However, evidence shows that circulating glutamine levels are increased in mice during calorie restriction 45. In addition, glutamine levels are normally higher than that of any other circulating amino acid 46–48. As glutamine promotes VM-M3 cell growth and survival, the failure of CR to target glutamine levels could be responsible for the failure of CR to inhibit VM-M3 tumor metastasis. Survival in CR mice was significantly longer than control mice in spite of the presence of systemic metastasis. This suggests that morbidity in the control mice may be due to a combination of factors to include primary tumor burden. As shown, the primary tumor size of mice on CR was approximately half the size of control mice. As previously shown, drugs, such as methotrexate, that inhibit metastasis but do not affect primary tumor size do not increase mouse survival 3. Therefore, at the time of control mouse morbidity, systemic metastasis may not be a major contributing factor.
Increasing evidence indicates that glutamine is a major energy substrate for cancer cells, to include glioma, HeLa, and prostate cancer cell lines 12–15. In addition, it is well known that glutamine is a major energy substrate for cells of myeloid origin 42. Interestingly, the VM-M3 cell line has a number of properties in common with cells of myeloid/mesenchymal lineage (macrophages and microglia) to include morphology, gene expression, lipid profile, and phagocytic capacity 36. We also found that the VM-M3 tumor cells require glutamine as a major energy substrate similar to cell of myeloid origin. Therefore, we suggest that the dependence of the VM-M3 cells on glutamine results from the myeloid origin of the cells.
We showed that DON administration in vitro inhibited cell growth, but did not significantly enhance cell death. DON administration in vitro therefore displayed a cytostatic effect over a wide range of concentrations, similar to other known cytostatic agents 3. Additionally, whole body bioluminescence was nearly unchanged over the course of DON treatment with the exception of spleen metastasis (data not shown). These findings suggest that DON inhibits cell growth, consistent with previous reports in human studies 28. There have also been reports of tumor regression, indicating that DON can be useful for early and late stage tumor management 17, 28.
Although DON inhibited VM-M3 metastasis to the liver, lung, and kidneys, DON treatment had no effect on metastasis to the spleen. The spleen is recognized as a reservoir for monocytes 49, and may represent a sanctuary for the myeloid-like metastatic cells. Interestingly, studies have shown increases in glutaminase activity in the spleens of tumor bearing mice 50. Glutaminase is the first enzyme involved in glutamine metabolism. This perhaps indicates that the spleen could support tumor growth due to an influx of glutamine originally intended to support immune function 51. Further studies are required to determine the factors involved in tumor cell metastasis to the spleen.
Because targeting glucose and glutamine individually increased mouse survival, we suggest that targeting both glucose and glutamine in vivo could potentially have a synergistic and less toxic effect 52. We previously found that CR administered together with low dose 2-deoxyglucose, an inhibitor of glucose metabolism, acted synergistically to reduce brain tumor growth 10. In addition, DON treatment, in combination with 2-deoxyglucose, had a greater inhibitory effect on myeloid leukaemia cells in vitro25. Previous studies suggest that glutamine inhibition also restricts glucose metabolism 53. This suggests that some tumor cells might become more susceptible to glycolysis inhibition following inhibition of glutamine metabolism. Additionally, diets low in glutamate may further inhibit tumor growth and metastasis when combined with glutamine antagonists 54. Further studies are required to evaluate the therapeutic efficacy of targeting both glutamine and glucose metabolism for the management of metastatic cancers.
Besides DON, phenylbutyrate (PBA), which reduces circulating glutamine levels, has been evaluated in human clinical trials 19–22, 27. PBA is metabolized to phenylacetate (PA) in humans, which then covalently conjugates with glutamine18, 55. This PA-glutamine conjugate is then excreted, effectively reducing circulating glutamine levels 18. Although current studies utilize PBA as a histone deacetylase inhibitor in vitro, part of its mechanism of action could be due to a reduction of circulating glutamine in vivo. We were unable to test this possibility in mice, as PBA is metabolized differently in mice than in humans. PA is conjugated to glycine rather than glutamine in mice 56. We therefore tested DON because of its previous use in mice 26. Because PBA has already been introduced in clinical trials and is well tolerated in humans 55, PBA can potentially be used in place of DON as a glutamine-targeting drug. Because PBA is well tolerated, the toxicities seen with DON treatment could be avoided. We suggest that new non-toxic inhibitors of glutamine metabolism could be broadly effective for managing systemic metastatic cancer.
In contrast to our data, previous studies showed that glutamine inhibited the formation of chemically induced squamous cell cancer 57. However, cancers of epithelial origin may have different requirements for glutamine than cells of myeloid origin. In fact, we have shown that the CT2A astrocytoma, which is of epithelial origin, is highly dependent on glucose and is responsive to CR 9. In addition, glutamine administration would be expected to enhance the activity of host immune cells such as macrophages. Macrophage function is highly dependent on glutamine 42. In contrast, some reports suggest that glutamine supplementation can inhibit tumor growth 58. Such inhibition was due to reductions in glutathione synthesis, which would render the tumor cells more susceptible to oxidative stress 58. However, our unpublished findings, using C13 glutamine, show that the VM-M3 cells actively synthesize glutathione. Hence, glutamine is a necessary nutrient for the VM-M3 metastatic cells and possibly for human metastatic cells with myeloid/mesenchymal properties.
In conclusion, we found that glutamine is a major energy metabolite for the metastatic VM-M3 cells and suggest that targeting glutamine could be effective for managing systemic metastatic cancer in humans. We suggest that glutamine-targeting drugs could be more therapeutic and possibly less toxic than other current therapies for cancer metastasis. As glucose and glutamine are the primary energy metabolites of most malignant cancers, therapeutic synergy can be expected if these metabolites are targeted simultaneously.
Acknowledgments
This work was supported from NIH grants [NS-055195; CA-102135] and from the Boston College Research expense fund. The authors would like to thank Ivan Urits and Roderick Bronson for technical assistance and evaluation of histological sections. The authors would also like to thank Purna Mukherjee for editorial assistance.
List of Abbreviations
- CR
calorie restriction
- DON
6-diazo-5-oxo-L-norleucine
- gln
glutamine
- gluc
glucose
- PA
phenylacetate
- PBA
phenybutyrate
References
- 1.Mehlen P, Puisieux A. Metastasis: a question of life or death. Nature reviews. 2006;6:449–58. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- 2.Bacac M, Stamenkovic I. Metastatic cancer cell. Annual review of pathology. 2008;3:221–47. doi: 10.1146/annurev.pathmechdis.3.121806.151523. [DOI] [PubMed] [Google Scholar]
- 3.Huysentruyt LC, Shelton LM, Seyfried TN. Influence of methotrexate and cisplatin on tumor progression and survival in the VM mouse model of systemic metastatic cancer. International journal of cancer. 2010;126:65–72. doi: 10.1002/ijc.24649. [DOI] [PubMed] [Google Scholar]
- 4.Sanborn RE. Cisplatin versus carboplatin in NSCLC: is there one “best” answer? Current treatment options in oncology. 2008;9:326–42. doi: 10.1007/s11864-009-0085-5. [DOI] [PubMed] [Google Scholar]
- 5.McWhinney SR, Goldberg RM, McLeod HL. Platinum neurotoxicity pharmacogenetics. Molecular cancer therapeutics. 2009;8:10–6. doi: 10.1158/1535-7163.MCT-08-0840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muss HB. Targeted therapy for metastatic breast cancer. The New England journal of medicine. 2006;355:2783–5. doi: 10.1056/NEJMe068260. [DOI] [PubMed] [Google Scholar]
- 7.Patel PH, Chaganti RS, Motzer RJ. Targeted therapy for metastatic renal cell carcinoma. British journal of cancer. 2006;94:614–9. doi: 10.1038/sj.bjc.6602978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mukherjee P, El-Abbadi MM, Kasperzyk JL, Ranes MK, Seyfried TN. Dietary restriction reduces angiogenesis and growth in an orthotopic mouse brain tumour model. Br J Cancer. 2002;86:1615–21. doi: 10.1038/sj.bjc.6600298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mukherjee P, Abate LE, Seyfried TN. Antiangiogenic and proapoptotic effects of dietary restriction on experimental mouse and human brain tumors. Clin Cancer Res. 2004;10:5622–9. doi: 10.1158/1078-0432.CCR-04-0308. [DOI] [PubMed] [Google Scholar]
- 10.Marsh J, Mukherjee P, Seyfried TN. Drug/diet synergy for managing malignant astrocytoma in mice: 2-deoxy-D-glucose and the restricted ketogenic diet. Nutrition & metabolism. 2008;5:33–7. doi: 10.1186/1743-7075-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marsh J, Mukherjee P, Seyfried TN. Akt-dependent proapoptotic effects of caloric restriction on late-stage management of a PTEN/TSC2-deficient mouse astrocytoma. Proc Amer Assoc Cancer Res. 2008;14:7751–62. doi: 10.1158/1078-0432.CCR-08-0213. [DOI] [PubMed] [Google Scholar]
- 12.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19345–50. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. The Journal of biological chemistry. 1979;254:2669–76. [PubMed] [Google Scholar]
- 14.Yang C, Sudderth J, Dang T, Bachoo RG, McDonald JG, Deberardinis RJ. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or akt signaling. Cancer research. 2009;69:7986–93. doi: 10.1158/0008-5472.CAN-09-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matheson BK, Adams JL, Zou J, Patel R, Franklin RB. Effect of metabolic inhibitors on ATP and citrate content in PC3 prostate cancer cells. The Prostate. 2007;67:1211–8. doi: 10.1002/pros.20617. [DOI] [PubMed] [Google Scholar]
- 16.Yuneva M. Finding an “Achilles’ heel” of cancer: the role of glucose and glutamine metabolism in the survival of transformed cells. Cell cycle (Georgetown, Tex. 2008;7:2083–9. doi: 10.4161/cc.7.14.6256. [DOI] [PubMed] [Google Scholar]
- 17.Livingston RB, Venditti JM, Cooney DA, Carter SK. Glutamine antagonists in chemotherapy. Advances in pharmacology and chemotherapy. 1970;8:57–120. doi: 10.1016/s1054-3589(08)60594-3. [DOI] [PubMed] [Google Scholar]
- 18.Darmaun D, Welch S, Rini A, Sager BK, Altomare A, Haymond MW. Phenylbutyrate-induced glutamine depletion in humans: effect on leucine metabolism. The American journal of physiology. 1998;274:E801–7. doi: 10.1152/ajpendo.1998.274.5.E801. [DOI] [PubMed] [Google Scholar]
- 19.Dyer ES, Paulsen MT, Markwart SM, Goh M, Livant DL, Ljungman M. Phenylbutyrate inhibits the invasive properties of prostate and breast cancer cell lines in the sea urchin embryo basement membrane invasion assay. International journal of cancer. 2002;101:496–9. doi: 10.1002/ijc.10609. [DOI] [PubMed] [Google Scholar]
- 20.Li XN, Parikh S, Shu Q, Jung HL, Chow CW, Perlaky L, Leung HC, Su J, Blaney S, Lau CC. Phenylbutyrate and phenylacetate induce differentiation and inhibit proliferation of human medulloblastoma cells. Clin Cancer Res. 2004;10:1150–9. doi: 10.1158/1078-0432.ccr-0747-3. [DOI] [PubMed] [Google Scholar]
- 21.Engelhard HH, Homer RJ, Duncan HA, Rozental J. Inhibitory effects of phenylbutyrate on the proliferation, morphology, migration and invasiveness of malignant glioma cells. Journal of neuro-oncology. 1998;37:97–108. doi: 10.1023/a:1005865125588. [DOI] [PubMed] [Google Scholar]
- 22.Lopez CA, Feng FY, Herman JM, Nyati MK, Lawrence TS, Ljungman M. Phenylbutyrate sensitizes human glioblastoma cells lacking wild-type p53 function to ionizing radiation. International journal of radiation oncology, biology, physics. 2007;69:214–20. doi: 10.1016/j.ijrobp.2007.04.069. [DOI] [PubMed] [Google Scholar]
- 23.Rabinovitz M, Olson ME, Greenberg DM. Effect of glutamine analogs on amino acid incorporation into protein of some normal and neoplastic cells in vitro. Cancer research. 1959;19:388–92. [PubMed] [Google Scholar]
- 24.Lyons SD, Sant ME, Christopherson RI. Cytotoxic mechanisms of glutamine antagonists in mouse L1210 leukemia. The Journal of biological chemistry. 1990;265:11377–81. [PubMed] [Google Scholar]
- 25.Griffiths M, Keast D, Patrick G, Crawford M, Palmer TN. The role of glutamine and glucose analogues in metabolic inhibition of human myeloid leukaemia in vitro. The International journal of biochemistry. 1993;25:1749–55. doi: 10.1016/0020-711x(88)90303-5. [DOI] [PubMed] [Google Scholar]
- 26.Ovejera AA, Houchens DP, Catane R, Sheridan MA, Muggia FM. Efficacy of 6-diazo-5-oxo-L-norleucine and N-[N-gamma-glutamyl-6-diazo-5-oxo-norleucinyl]-6-diazo-5-oxo-norleucine against experimental tumors in conventional and nude mice. Cancer research. 1979;39:3220–4. [PubMed] [Google Scholar]
- 27.Phuphanich S, Baker SD, Grossman SA, Carson KA, Gilbert MR, Fisher JD, Carducci MA. Oral sodium phenylbutyrate in patients with recurrent malignant gliomas: a dose escalation and pharmacologic study. Neuro-oncology. 2005;7:177–82. doi: 10.1215/S1152851704000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magill GB, Myers WP, Reilly HC, Putnam RC, Magill JW, Sykes MP, Escher GC, Karnofsky DA, Burchenal JH. Pharmacological and initial therapeutic observations on 6-diazo-5-oxo-1-norleucine (DON) in human neoplastic disease. Cancer. 1957;10:1138–50. doi: 10.1002/1097-0142(195711/12)10:6<1138::aid-cncr2820100608>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 29.Ng WH, Yeo TT, Kaye AH. Spinal and extracranial metastatic dissemination of malignant glioma. J Clin Neurosci. 2005;12:379–82. doi: 10.1016/j.jocn.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 30.Youness E, Barlogie B, Ahearn M, Trujillo JM. Tumor cell phagocytosis. Its occurrence in a patient with medulloblastoma. Arch Pathol Lab Med. 1980;104:651–3. [PubMed] [Google Scholar]
- 31.Kauffman HM, Cherikh WS, McBride MA, Cheng Y, Hanto DW. Deceased donors with a past history of malignancy: an organ procurement and transplantation network/united network for organ sharing update. Transplantation. 2007;84:272–4. doi: 10.1097/01.tp.0000267919.93425.fb. [DOI] [PubMed] [Google Scholar]
- 32.Rubinstein LJ. Atlas of Tumor Pathology. Washington, D.C: Armed Forces Institute of Pathology; 1972. Tumors of the Central Nervous Systemed. [Google Scholar]
- 33.Taha M, Ahmad A, Wharton S, Jellinek D. Extra-cranial metastasis of glioblastoma multiforme presenting as acute parotitis. Br J Neurosurg. 2005;19:348–51. doi: 10.1080/02688690500305506. [DOI] [PubMed] [Google Scholar]
- 34.Hoffman HJ, Duffner PK. Extraneural metastases of central nervous system tumors. Cancer. 1985;56:1778–82. doi: 10.1002/1097-0142(19851001)56:7+<1778::aid-cncr2820561309>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 35.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nature reviews. 2002;2:563–72. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 36.Huysentruyt LC, Mukherjee P, Banerjee D, Shelton LM, Seyfried TN. Metastatic cancer cells with macrophage properties: evidence from a new murine tumor model. International journal of cancer. 2008;123:73–84. doi: 10.1002/ijc.23492. [DOI] [PubMed] [Google Scholar]
- 37.Spivak JL. Phagocytic tumour cells. Scandinavian journal of haematology. 1973;11:253–6. doi: 10.1111/j.1600-0609.1973.tb00126.x. [DOI] [PubMed] [Google Scholar]
- 38.Abodief WT, Dey P, Al-Hattab O. Cell cannibalism in ductal carcinoma of breast. Cytopathology. 2006;17:304–5. doi: 10.1111/j.1365-2303.2006.00326.x. [DOI] [PubMed] [Google Scholar]
- 39.Fais S. Cannibalism: a way to feed on metastatic tumors. Cancer Lett. 2007;258:155–64. doi: 10.1016/j.canlet.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 40.Lugini L, Matarrese P, Tinari A, Lozupone F, Federici C, Iessi E, Gentile M, Luciani F, Parmiani G, Rivoltini L, Malorni W, Fais S. Cannibalism of live lymphocytes by human metastatic but not primary melanoma cells. Cancer research. 2006;66:3629–38. doi: 10.1158/0008-5472.CAN-05-3204. [DOI] [PubMed] [Google Scholar]
- 41.Moonda A, Fatteh S. Metastatic colorectal carcinoma: an unusual presentation. Journal of cutaneous pathology. 2009;36:64–6. doi: 10.1111/j.1600-0560.2008.01007.x. [DOI] [PubMed] [Google Scholar]
- 42.Newsholme P, Costa Rosa LF, Newsholme EA, Curi R. The importance of fuel metabolism to macrophage function. Cell biochemistry and function. 1996;14:1–10. doi: 10.1002/cbf.644. [DOI] [PubMed] [Google Scholar]
- 43.Ranes MK, El-Abbadi M, Manfredi MG, Mukherjee P, Platt FM, Seyfried TN. N -butyldeoxynojirimycin reduces growth and ganglioside content of experimental mouse brain tumours. Br J Cancer. 2001;84:1107–14. doi: 10.1054/bjoc.2000.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shapiro RA, Clark VM, Curthoys NP. Inactivation of rat renal phosphate-dependent glutaminase with 6-diazo-5-oxo-L-norleucine. Evidence for interaction at the glutamine binding site. The Journal of biological chemistry. 1979;254:2835–8. [PubMed] [Google Scholar]
- 45.Selman C, Kerrison ND, Cooray A, Piper MD, Lingard SJ, Barton RH, Schuster EF, Blanc E, Gems D, Nicholson JK, Thornton JM, Partridge L, et al. Coordinated multitissue transcriptional and plasma metabonomic profiles following acute caloric restriction in mice. Physiological genomics. 2006;27:187–200. doi: 10.1152/physiolgenomics.00084.2006. [DOI] [PubMed] [Google Scholar]
- 46.Bode BP. Recent molecular advances in mammalian glutamine transport. The Journal of nutrition. 2001;131:2475S–85S. doi: 10.1093/jn/131.9.2475S. discussion 86S–7S. [DOI] [PubMed] [Google Scholar]
- 47.Newsholme P, Procopio J, Lima MM, Pithon-Curi TC, Curi R. Glutamine and glutamate--their central role in cell metabolism and function. Cell biochemistry and function. 2003;21:1–9. doi: 10.1002/cbf.1003. [DOI] [PubMed] [Google Scholar]
- 48.Darmaun D, Matthews DE, Bier DM. Glutamine and glutamate kinetics in humans. The American journal of physiology. 1986;251:E117–26. doi: 10.1152/ajpendo.1986.251.1.E117. [DOI] [PubMed] [Google Scholar]
- 49.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, et al. Science (New York, NY. Vol. 325. 2009. Identification of splenic reservoir monocytes and their deployment to inflammatory sites; pp. 612–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aledo JC, Segura JA, Barbero LG, Marquez J. Early differential expression of two glutaminase mRNAs in mouse spleen after tumor implantation. Cancer letters. 1998;133:95–9. doi: 10.1016/s0304-3835(98)00214-6. [DOI] [PubMed] [Google Scholar]
- 51.Medina MA. Glutamine and cancer. The Journal of nutrition. 2001;131:2539S–42S. doi: 10.1093/jn/131.9.2539S. discussion 50S–1S. [DOI] [PubMed] [Google Scholar]
- 52.Seyfried TNa, Shelton LM. Cancer as a Metabolic Disease. Journal of Nutrition and Metabolism. 2010:7. doi: 10.1186/1743-7075-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaadige MR, Looper RE, Kamalanaadhan S, Ayer DE. Glutamine-dependent anapleurosis dictates glucose uptake and cell growth by regulating MondoA transcriptional activity. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14878–83. doi: 10.1073/pnas.0901221106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshioka K, Takehara H, Okada A, Komi N. Glutamine antagonist with diet deficient in glutamine and aspartate reduce tumor growth. The Tokushima journal of experimental medicine. 1992;39:69–76. [PubMed] [Google Scholar]
- 55.Thibault A, Cooper MR, Figg WD, Venzon DJ, Sartor AO, Tompkins AC, Weinberger MS, Headlee DJ, McCall NA, Samid D, et al. A phase I and pharmacokinetic study of intravenous phenylacetate in patients with cancer. Cancer research. 1994;54:1690–4. [PubMed] [Google Scholar]
- 56.James MO, Smith RL, Williams RT, Reidenberg M. The conjugation of phenylacetic acid in man, sub-human primates and some non-primate species. Proceedings of the Royal Society of London Series B, Containing papers of a Biological character. 1972;182:25–35. doi: 10.1098/rspb.1972.0064. [DOI] [PubMed] [Google Scholar]
- 57.Lim V, Korourian S, Todorova VK, Kaufmann Y, Klimberg VS. Glutamine prevents DMBA-induced squamous cell cancer. Oral oncology. 2009;45:148–55. doi: 10.1016/j.oraloncology.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 58.Carretero J, Obrador E, Pellicer JA, Pascual A, Estrela JM. Mitochondrial glutathione depletion by glutamine in growing tumor cells. Free radical biology & medicine. 2000;29:913–23. doi: 10.1016/s0891-5849(00)00392-0. [DOI] [PubMed] [Google Scholar]