

Abstract
Background aims
Heart failure therapy with human embryonic stem cell (hESC)-derived cardiomyocytes (hCM) has been limited by the low rate of spontaneous hCM differentiation. As others have shown that p38 mitogen-activated protein kinase (p38MAPK) directs neurogenesis from mouse embryonic stem cells, we investigated whether the p38MAPK inhibitor, SB203580, might influence hCM differentiation.
Methods
We treated differentiating hESC with SB203580 at specific time-points, and used flow cytometry, immunocytochemistry, quantitative real-time (RT)–polymerase chain reaction (PCR), teratoma formation and transmission electron microscopy to evaluate cardiomyocyte formation.
Results
We observed that the addition of inhibitor resulted in 2.1-fold enrichment of spontaneously beating human embryoid bodies (hEB) at 21 days of differentiation, and that 25% of treated cells expressed cardiac-specific α-myosin heavy chain. This effect was dependent on the stage of differentiation at which the inhibitor was introduced. Immunostaining and teratoma formation assays demonstrated that the inhibitor did not affect hESC pluripotency; however, treated hESC gave rise to hCM exhibiting increased expression of sarcomeric proteins, including cardiac troponin T, myosin light chain and α-myosin heavy chain. This was consistent with significantly increased numbers of myofibrillar bundles and the appearance of nascent Z-bodies at earlier time-points in treated hCM. Treated hEB also demonstrated a normal karyotype by array comparative genomic hybridization and viability in vivo following injection into mouse myocardium.
Conclusions
These studies demonstrate that p38MAPK inhibition accelerates directed hCM differentiation from hESC, and that this effect is developmental stage-specific. The use of this inhibitor should improve our ability to generate hESC-derived hCM for cell-based therapy.
Keywords: cardiomyocyte, cell fate, differentiation, human embryonic stem cell, p38 mitogen-activated protein kinase, teratoma
Introduction
Human cardiomyocytes (hCM) differentiated from various human embryonic stem cell (hESC) lines have been shown to exhibit properties similar to endogenous hCM, including cardiac-specific gene expression (1), sarcomere ultrastructure (2,3) and characteristic action potentials (4–6). Successful engraftment of isolated beating clusters into various models of myocardial disease has generated enthusiasm for hESC as a source of new hCM for myocardial cell therapy (1,5,7,8).
The differentiation of hESC into hCM, however, is a low-yield process, as most groups have relied on spontaneous differentiation in the presence of serum (9–11). For an effective therapy, the cardiac cells need to be generated in large numbers and with sufficient purity to avoid the formation of other tissues (11–13). Xu et al. (10) reported increased cardiogenesis with the addition of the de-methylation agent azacytidine, followed by Percoll gradient separation. Mummery et al. (14,15) subsequently showed an increased cardiomyocyte yield by co-culturing hESC with a mouse endoderm-like cell line with or without serum. Other groups have since reported increased cardiogenesis by the addition of various culture additives, such as activin A and bone morphogenetic proteins 2 and 4 (16,17).
Pathways regulating the activity of p38 mitogen-activated protein kinase (p38MAPK) have been implicated in regulating mammalian hCM proliferation (18) and neural differentiation (19). Recently, Graichen et al. (20) detected the expression of p38MAPK in hESC as well as differentiating hEB and suggested that its inhibition could induce cardiogenesis. They reported enhanced cardiogenesis from hESC grown in co-culture with visceral endoderm cells in the presence of a p38MAPK inhibitor (20).
In this study, we used SB203580, a small-molecule inhibitor of p38MAPK, to study the dynamics of cardiogenesis with p38MAPK inhibition. We report that inhibition of p38MAPK during the stage of hESC differentiation that coincides with ectoderm/mesoendoderm divergence results in directed, accelerated differentiation of hCM, and that the resulting hCM maintain properties, such as genomic stability and survival in vivo, that are essential for cell transplant therapy.
Methods
hESC culture and differentiation
All hESC procedures were approved by the Stem Cell Research Oversight Committee at the University of California (San Francisco, CA, USA). The H9 hESC line (WA09) expressing enhanced green fluorescent protein (GFP) under the control of the ubiquitin C promoter (21) was maintained on irradiated CF1 mouse embryonic fibroblasts (MEF), as described previously (22). All reagents were purchased from Invitrogen, Carlsbad, CA, USA, except where indicated. hESC were cultured between passages 35–90 in KSR medium [knock-out Dulbecco’s modified Eagle medium (DMEM) containing 20% knock-out serum replacer, 0.1 mM non-essential amino acids, 2 mM L-glutamine, 0.1 mM 2-mercaptoethanol and 12 ng/mL recombinant human basic fibroblast growth factor (bFGF) (R&D Systems, Minneapolis, MN, USA)]. MEF were cultured in DMEM containing 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA, SH 30071.03), 2 mM L-glutamine and 1× penicillin and streptomycin. hESC colonies were passaged every 3–4 days on fresh MEF using 1 mg/mL collagenase IV in KSR medium without bFGF and manual dissociation of hESC colonies.
hESC were differentiated by hEB formation as described previously (22). Proliferating hESC colonies were washed with calcium/magnesium-free phosphate-buffered saline (PBS) (PBS-cmf) and incubated with 1 mg/mL collagenase IV for 5 min at 37°C, washed with PBS-cmf, then resuspended in small clumps in differentiation medium [knock-out DMEM medium containing 20% FBS (Hyclone SH 30070.03), 0.1 mM non-essential amino acids, 2 mM L-glutamine and 0.1 mM 2-mercaptoethanol] and placed in low-attachment 6-well plates. The medium was replaced every other day. On day 7 following re-suspension, approximately 25–30 hEB/well were plated on 0.1% gelatin-coated, 12-well plates in the same medium.
SB203580 (Calbiochem, Gibbstown, NJ, USA) was prepared in dimethylsulfoxide (DMSO) at 10 mM, and diluted in medium to the indicated final concentrations. SB203580 was removed by rinsing cultures with PBS-cmf, then resuspending hEB in differentiation medium.
Flow cytometry
Flow cytometric analysis was performed as described previously (22). hESC differentiated for 30 days in culture were harvested using 0.05% trypsin–ethylene diamine tetra acetic acid (EDTA) and fixed in 2% paraformaldehyde. Cells were permeabilized using perm/wash buffer (554723; BD Biosciences, San Jose, CA, USA), blocked with cold blocking buffer (PBS with 20% horse serum and 0.5 mM EDTA) and incubated on ice in blocking buffer with Alexa 647-conjugated anti-α myosin heavy chain (MHC) (1:50 dilution) or isotype control antibody (1:50 dilution). Anti-αMHC (αMHC ab15; Abcam, Cambridge, MA, USA) and isotype control antibodies were conjugated to Alexa Fluor 647 using an Alexa labeling kit (A-20186; Molecular Probes/Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. Following incubation with antibody, cells were washed with blocking buffer and analyzed on a FACSCalibur flow cytometer (BD Biosciences) using standard filter sets.
Immunocytochemistry
The lineage fate of differentiated hESC was determined by staining 30-day-old adherent differentiating hEB in culture plates. Cells were fixed with 2% paraformaldehyde for 20 min at 21°C, then permeabilized and blocked in 10% horse serum, 1% bovine serum albumin (BSA) and 0.1% Triton X-100 in PBS for 1 h at 21°C. hEB were incubated with primary antibody (1–5 μg/mL) in blocking buffer for 1 h at 21°C or overnight at 4°C. Primary antibodies used were mouse anti-human α-fetoprotein (Sigma-Aldrich, St. Louis, MO, USA, A8452), mouse anti-human nestin (R&D Systems MAB1259), mouse anti-human βIII tubulin (R&DSystemsMAB1195), mouse anti-humans mooth muscle actin (R&D Systems MAB1420) and mouse anti-human cardiac troponin T (cTnT; Lab Vision Corporation, Fremont, CA, USA, MS-295). Cells were washed with blocking buffer, incubated with a 1:200 dilution of phycoerythrin (PE)-conjugated donkey anti-mouse secondary antibody (Jackson Immuno Research Laboratories, West Grove, PA, USA) for 30 min at 21°C, washed with 1% Triton X-100 in PBS-cmf, and then analyzed by fluorescence microscopy using a Nikon Eclipse TE300 microscope.
Array comparative genomic hybridization
Genomic DNA (300–500 ng) from treated and untreated 30-day-old differentiated hEB was isolated using a QiaEasy DNA kit for tissue (Qiagen, Valencia, CA, USA). DNA was labeled with Cy3 and Cy5 using the BioPrime DNA labeling system (Invitrogen), hybridized to custom HumArray 3.2 human chromosome arrays, and analyzed for chromosomal composition in the Array Core at the UCSF Helen Diller Family Comprehensive Cancer Center according to published methods (http://cancer.ucsf.edu/array/protocols/index.php, 3 June, 2010) (23). Differentially labeled human male reference genomic DNA was run as a control. The custom HumArray 3.2 prepared in the Array Core contains 2464 bacterial artificial chromosome clones spotted in triplicate and distributed uniformly across the genome. Each clone contains at least one Sequence Tagged Site (STS) and is mapped to the human genome sequence. Clones containing unique sequences near the telomeres and genes known to be significant in cancer and medical genetics are included on these arrays.
Quantitative real-time–polymerase chain reaction
For analysis of hESC-derived hCM, beating areas from approximately 10 hEB were visualized with a Leica, Wetzlar, Germany, MZ6 microscope and manually excised using an 18-g needle. The collected tissue samples were treated with 0.05% trypsin–EDTA to generate a single-cell suspension prior to RNA isolation. RNA was isolated and cDNA synthesized from c. 50 000 hEB-derived cells or proliferating hESC using the TaqMan gene expression cells-to-CT kit (Ambion, Austin, TX, USA). cDNA was quantitated using a Nanodrop ND-1000 Spectrophotometer (Nanodrop Technologies/Thermo Scientific, Wilmington, DE, USA, ND software version 3.3.0). Linear pre-amplification of target sequences was accomplished using the Applied Biosystems PreAmp system. Relative expression was determined using a TaqMan assay (Applied Biosystems (ABI), Foster City, CA, USA) on an ABI 7300 real-time (RT)–polymerase chain reaction (PCR) system with the following commercially inventoried primer pairs (Applied Biosystems): α-fetoprotein (Hs00173490_m1), smooth muscle actin (Hs00242273_m1), βIII tubulin (Hs00964965_m1), GATA4 (Hs00171403_m1), Nkx2–5 (Hs00231763_m1), αMHC (Hs00411908_ m1), MLC4 (Hs00267321_m1), cTnT (Hs001 65960_m1) and GAPDH (4326317E). Cycle times to detection were normalized against a reference gene, GAPDH, and relative changes were calculated using ABI version 1.4 sequence detection software.
Transmission electron microscopy
Beating areas (c. 20–30) were excised from p38i-treated and untreated cultures at day 45 of differentiation and fixed in 2% glutaraldehyde, 1% paraformaldehyde in 0.1 M sodium cacodylate, pH 7.4. Tissue was post-fixed in 2% osmium tetroxide, block-stained in 2% aqueous uranyl acetate, dehydrated in acetone and embedded in LX-112 resin (Ladd Research Industries Inc, Williston, VT, USA). Ultrathin sections were contrast stained with 0.8% lead citrate. Samples were analyzed on a JEOL JEM-1230 electron microscope (JEOL USA, Inc. Portland, OR, USA) and photographed with a Gatan ultrascan USC1000 digital camera (Gatan, Inc., Pleasanton, CA, USA). At least 30 sections were analyzed for each condition.
Teratomas
All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of California. Teratomas were generated as previously described (22,24). Treated and untreated hEB were differentiated for 7 days in suspension, then 5 × 105 hEB-derived cells were mixed with an equal volume of 1 mg/mL Phaseolus vulgaris lectin (PHA-P L1668; Sigma), pelleted and incubated in growth medium overnight at 37°C, 5% CO2 in a 0.4-μm MILLICELL (Millipore, Billerica, MA, USA). At least two cell pellets were grafted under each kidney capsule of 8-week-old male CB17 SCID-Beige mice (three mice per condition) using standard techniques (24,25). The resulting teratomas were analyzed 10 weeks after grafting. Teratomas were fixed in 10% buffered formalin, embedded in paraffin, and 5-μm sections stained with hematoxylin and eosin (H&E) to identify tissue structures.
Tracking of SB203580-treated hESC in mouse hearts
hESC treated at differentiation days 4–6 with a 10-μm final concentration of SB203580 were differentiated for 30 days and hCM enriched by Percoll gradient using established methods (10). hCM were dissociated into single cells using Accutase (Sigma A6964) and 106 cells were delivered into the hearts of 12-week-old CB17 SCID-Beige mice (Taconic, Hudson, NY, USA) using closed-chest ultrasound-guided injection, as published previously (26). Hearts were harvested at 60 days post-injection, fixed in 10% buffered formalin and embedded in paraffin. Sections (5 μm) were stained with H&E for tissue analysis, and with anti-GFP antibody (Invitrogen A11122) to determine retention of injected hESC-derived hCM. Antigen retrieval was performed by incubating the sections with trypsin for 5 min at 37°C followed by incubations with anti-GFP (1:100), rabbit-on-rodent horseradish peroxidase (HRP) (1:200; Biocare Medical RMR622) and 3,3′-diaminodbenzidine (DAB) chromogen substrate (Biocare Medical, Concord, CA, USA, BDB2004).
Statistical analysis
Percentages of beating hEB were determined as a ratio of hEB exhibiting spontaneous contractile activity to total number of hEB plated. At least 120 hEB were plated for each group in each experiment, with five replicates performed for each experiment. Multiple synchronously beating areas within one hEB were counted as one beating hEB. For comparison among groups [i.e. dose and interval of inhibitor treatment, cytometric analysis of αMHC expression and quantitative RT–PCR (qPCR) analysis of relative expression], analysis of variance (ANOVA) with Fisher’s post-hoc test was used. Where ANOVA indicated significant differences among groups, multiple comparisons were made using Student’s t-test with Bonferroni correction. A P-value less than 0.05 was considered significant.
Results
Inhibition of p38MAPK directs differentiation of hESC-derived hCM in a stage- and dose-specific manner
To determine whether inhibition of p38MAPK alters the dynamics of hESC differentiation, we exposed hEB to the p38MAPK inhibitor, SB203580, for sequential time periods during in vitro differentiation (Figure 1A) and counted the number of beating hEB on day 21, based on earlier reports that contractile activity peaks by this time-point (9). We observed a significant increase in hCM formation, as evidenced by an increase in the percentage of hEB with spontaneous contractile activity, with exposure to the inhibitor during days 2–6 of differentiation; a maximal effect was observed with exposure specifically from days 4 to 6 (26 ± 4 versus 12 ± 1.5; P < 0.0001). Addition of the compound at later time-points had no significant effect. To establish the optimal concentration of SB203580 for directed differentiation of hEB into hCM, we generated a dose–response curve for exposure from days 4 to 6, which established an optimal concentration of 5–10 μM (Figure 1B). As hEB may contain non-uniform cell numbers, we quantitated the percentage of hEB-derived cells expressing αMHC by flow cytometry (Figure 2). This demonstrated that inhibition of p38MAPK between days 4 and 6 resulted in c. 2.1-fold increase in the number of αMHC-expressing hCM (26.3 ± 4.6 versus 12.3 ± 3.9; P < 0.01).
Figure 1.

Inhibition of p38MAPK directs differentiation of hEB into hCM in a developmental stage- and dose-specific manner. (A) hEB were incubated in the presence of 10 μM SB203580 in culture during the indicated days. Treatment during days 2–6 resulted in an increase in the number of beating hCM at day 21, with the maximal effect seen (c. 2.3-fold) with treatment during days 4–6. Data shown are mean percentage ± SEM (n = 5; refers to separate experiments in all figure legends). *P < 0.01, **P < 0.0001. (B) hEB were cultured in the presence of 5, 10 or 20 μM SB203580 during days 4–6 days of differentiation. Treatment with 5–10 μM SB203580 resulted in an increase in the number of beating hCM at day 21, with the maximal effect seen (c. 2.3-fold) with 5 μM. Data shown are mean percentage ± SEM (n = 5). *P < 0.01.
Figure 2.

Inhibition of p38MAPK results in a 2.1-fold increase in total number of αMHC+ hCM. Single-cell suspensions of SB203580-treated (p38i) and untreated (Ctl) hEB differentiated for 30 days were analyzed by flow cytometry for the expression of αMHC. Isotype control antibody was used to set the positive gate (left). SB203580-treated samples demonstrated a 2.1-fold increase (26.3% versus 12.3%; P < 0.01) in the number of αMHC+ hCM compared with untreated cells. Representative data are shown (n = 3). FSC, forward light scatter.
Inhibition of p38MAPK leads to elevated levels of cardiac-specific gene expression
To correlate the increase in numbers of hESC-derived hCM with p38MAPK inhibition to gene expression, we compared cardiac-specific gene expression by qPCR between total hEB at day 4 prior to treatment and at 14 days of differentiation relative to undifferentiated hESC (Figure 3A). This analysis showed that levels of αMHC and cTnT were increased in treated hEB compared with spontaneously differentiated hEB (αMHC, 3.6-fold increase, P < 0.01; cTnT, 1.3-fold increase, P < 0.03). These data suggested an increase in cardiac-specific gene expression in total hEB cultures exposed to the p38MAPK inhibitor. To establish whether cardiac-specific gene expression was specifically up-regulated in hEB with spontaneous contractile activity, we manually dissected beating areas from the surrounding hEB tissue at 30 days of differentiation, and analyzed for expression of early cardiac transcription factors, GATA4 and Nkx2-5, and sarcomeric proteins, cTnT, αMHC and myosin light chain 4 (MLC4). Expression of these proteins was very low in non-beating areas, with no detectable difference between treated and untreated samples (Figure 3B). As expected, the expression of GATA4, Nkx2-5, cTnT, αMHC and MLC4 in beating areas was substantially increased compared with non-beating areas (Figure 3B), with no detectable expression of the early germ layer markers Pax6 (ectoderm), Pdx1 (endoderm) and Brachyury (mesoderm) (data not shown). Remarkably, beating areas from cultures that had been treated with p38MAPK inhibitor demonstrated substantially increased expression of these transcripts, compared with beating areas isolated from untreated cultures (GATA4, 3.1-fold increase, P < 0.001; Nkx2-5, 7.7-fold increase, P < 0.001; cTnT, 6.7-fold increase, P < 0.001; αMHC, 5.2-fold increase, P < 0.001; MLC4, 1.9-fold increase, P < 0.03).
Figure 3.

Inhibition of p38MAPK results in up-regulation of cardiac-specific gene expression during early stages of differentiation. (A) hEB from SB203580-treated (p38i) and untreated (Ctl) cultures collected prior to treatment at days 4 and 14 were assayed for cardiac-specific gene expression by qPCR. Expression was normalized to GAPDH and calculated relative to proliferating, undifferentiated hESC. Increased expression of αMHC and cTnT were observed in treated hEB compared with untreated cultures. Data shown are mean percentage ± SEM (n = 3). *P < 0.03; **P < 0.01. (B) Beating (B) and non-beating (NB) hEB from SB203580-treated (p38i) and untreated (Ctl) cultures collected at day 30 were assayed for cardiac-specific gene expression by qPCR. Expression was normalized to GAPDH and calculated relative to untreated, non-beating hEB. All five cardiac-specific transcripts were highly expressed in beating areas compared with non-beating areas irrespective of SB203580 treatment. For each transcript, however, SB203580 treatment resulted in significantly increased expression in beating hEB compared with untreated beating hEB. Data shown are mean percentage ± SEM (n = 3). *P < 0.03; ***P < 0.001.
Inhibition of p38MAPK accelerates sarcomere development in hESC-derived hCM
While the increase in expression of GATA4, Nkx2-5, cTnT, αMHC and MLC4 in beating areas of cultures treated with p38MAPK inhibitor may in part be the result of an increase in the number of hESC-derived hCM, we also hypothesized that this could be the result of accelerated differentiation. This was supported by our observation that, at day 14 of differentiation, treated hEB already showed an increase in cTnT and αMHC expression compared with spontaneously differentiating control hEB (Figure 3A). To examine the rate of hCM development at the ultrastructural level, we examined hEB at day 45 of differentiation by transmission electron microscopy (TEM) for evidence of sarcomere formation (Figure 4). Compared with the scattered areas of myofibril bundles seen among hEB from untreated cultures, hEB that had been treated with the p38MAPK inhibitor showed extensive regions of myofibril formation, with nascent Z-bodies and numerous mitochondria, consistent with developing sarcomeres.
Figure 4.
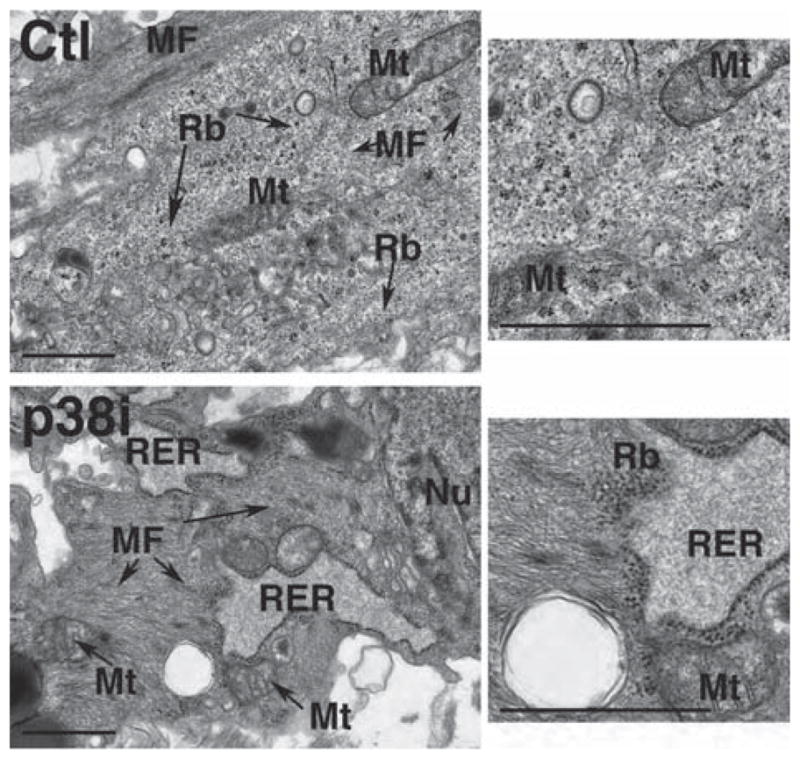
Inhibition of p38MAPK accelerates sarcomere development in hESC-derived hCM. Ultrastructural analysis of hESC-derived hCM at day 45 of differentiation showed scattered myofibril bundles (MF) in untreated samples, compared with extensive myofibrillar organization with nascent Z-bodies (arrows) surrounded by prominent rough endoplasmic reticulum (RER) and ribosomes (Rb), as evidence of active protein synthesis, in SB203580-treated samples. Representative transmission electron micrographs are shown. Mt, mitochondrion; Nu, nucleus. Bar 1 μm.
Inhibition of p38MAPK does not alter genomic fidelity in hESC-derived hCM
The genomic stability of hESC has been shown to be sensitive to culture conditions (27,28). To determine whether p38MAPK inhibition affected genomic stability, we analyzed treated hEB by array comparative genomic hybridization (aCGH). This is a sensitive method for detecting chromosomal abnormalities at the level of individual regions of the genome, allowing for the detection of single copy gains and losses compared with diploid even in the presence of normal cell contamination. This analysis demonstrated that treatment with SB203580 did not affect the chromosomal content of hESC-derived hEB at day 30 of differentiation (Figure 5).
Figure 5.

Inhibition of p38MAPK does not alter genomic fidelity in hESC-derived hCM. aCGH was carried out on DNA isolated from SB203580-treated (p38i) and untreated (Ctl) hEB on day 30 of differentiation after exposure to SB203580 during days 4–6. There was no evidence of single or multiple copy deletion or duplication in the treated sample (bottom) compared with a reference sample (top). Male DNA was used as a reference, so that the second X-chromosome from female hEB could be seen (arrows).
Inhibition of p38MAPK does not affect hESC pluripotency
As p38MAPK inhibition directed the accelerated differentiation of hEB toward hCM, we wanted to know whether this inhibition affected the pluripotency of treated hESC. To accomplish this, we evaluated treated hEB differentiated for 30 days by immunostaining with antibodies against markers of the three embryonic germ layers: nestin and βIII tubulin (ectoderm), smooth muscle actin and cTnT (mesoderm) and α-fetoprotein (endoderm) (Figure 6A). We observed evidence of differentiation into all three embryonic germ layers in hEB that had been treated with SB203580, suggesting that while p38MAPK inhibition may direct differentiation toward hCM, it does not completely disrupt differentiation of other tissues derived from mesoderm, or from ectoderm or endoderm.
Figure 6.

p38MAPK inhibitor-treated hESC differentiate into all three germ layers in vitro. (A) Immunofluorescence staining of SB203580-treated (p38i) and untreated (Ctl) hEB on day 30 of differentiation demonstrated expression of markers of all three embryonic germ layers, namely nestin and βIII tubulin (ectoderm), α-fetoprotein (AFP; endoderm) and smooth muscle actin (SMA) and cTnT (mesoderm). Bar 10 μm. (B) hEB from SB203580-treated (p38i) and untreated (Ctl) cultures collected prior to treatment, at day 4, and at day 14 were assayed for germ layer-specific gene expression by qPCR. Expression was normalized to GAPDH and calculated relative to proliferating, undifferentiated hESC. There was no significant difference between expression levels of βIII tubulin, SMA or α-fetoprotein in treated and untreated cultures. Data shown are mean percentage ± SEM (n = 3).
To determine whether p38MAPK inhibition influenced the expression of early genes associated with embryonic germ layer differentiation, we performed qPCR analysis of α-fetoprotein, smooth muscle actin and βIII tubulin in day-14 cultures. There were no significant differences between treated and untreated hEB, and each showed an appropriate increase in expression of all three proteins with differentiation (Figure 6B).
p38MAPK inhibitor-treated hESC differentiate into all three germ layers in vivo
While qPCR results suggested that treated hESC maintained their ability to differentiate into all three germ layers, we wanted to determine whether this was true in vivo using a teratoma formation assay. Analysis of teratoma tissues showed no qualitative difference in the distribution of ectoderm, mesoderm or endoderm from the p38MAPK inhibitor-treated versus untreated differentiated hESC (Figure 7), suggesting that although p38MAPK inhibition directs hESC differentiation toward hCM, differentiation toward the three embryonic germ layers is not disrupted.
Figure 7.
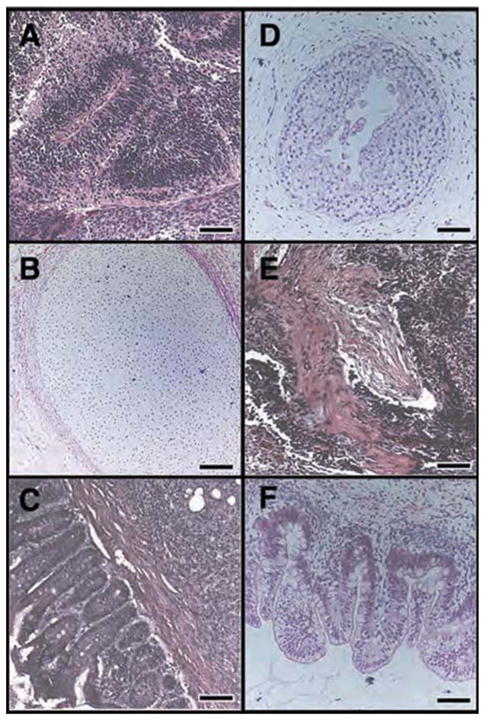
p38MAPK inhibitor-treated hESC differentiate into all three germ layers in vivo. Cultures of SB203580-treated hESC or untreated hESC were used to form teratomas by renal capsule grafting. Teratomas were sectioned and stained with H&E to identify embryonic tissues. (A–C) As expected, untreated hESC gave rise to tissues derived from all three embryonic germ layers, ectoderm (A), mesoderm (B) and endoderm (C). (D–F) SB203580-treated hESC also gave rise to tissues derived from embryonic ectoderm (D), mesoderm (E) and endoderm (F). (A) Nascent neural tube structure. (B) Cartilage surrounded by capsule of condensed mesenchyme. (C) Glandular intestinal structure surrounded by smooth muscle. (D) Primitive squamous epithelium. (E) Skeletal muscle surrounded by dense mesenchyme. (F) Glandular intestinal structure. Bar 100 μm (except C); Bar 200 μm (C).
p38MAPK inhibitor-treated hCM remain viable in vivo
To determine the viability of p38MAPK inhibitor-exposed hCM in vivo, we differentiated GFP-expressing, p38MAPK inhibitor-treated hESC for 30 days and purified hCM by Percoll gradient separation (10). We injected 106 hCM into the hearts of CB17 SCID-Beige mice and evaluated cell survival 60 days after injection. The injected hESC-derived hCM were retained in the mouse hearts, as demonstrated by the presence of GFP-expressing cells 60 days post-injection (Figure 8A–D). In addition, there was no evidence of local teratoma formation (Figure 8E, F).
Figure 8.
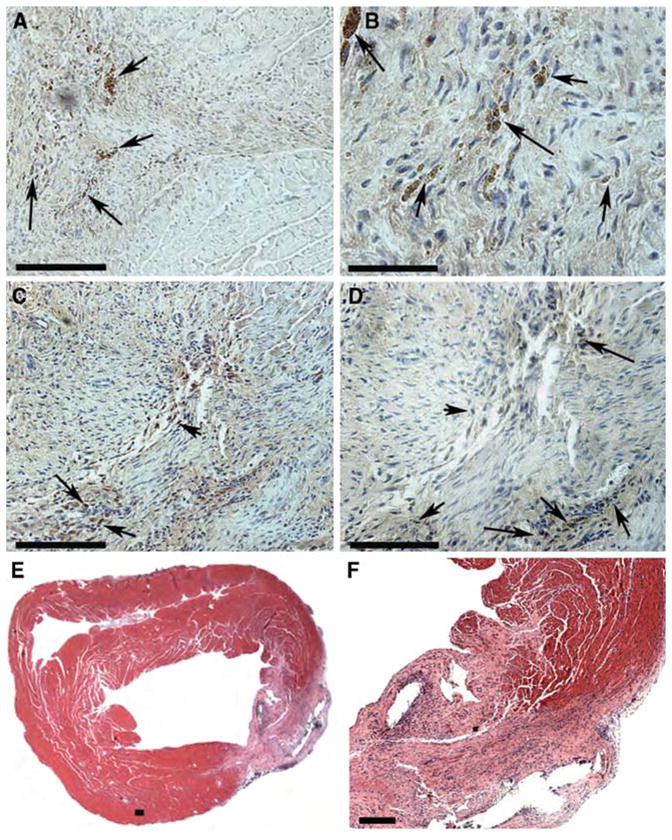
p38MAPK inhibition does not affect survival or teratoma formation in vivo. (A–D) Sections of mouse hearts injected with SB203580-treated hESC-derived hCM were stained with anti-GFP antibody to detect retention of viable hCM 60 days after injection. Sections were counterstained with DAPI (blue) to detect nuclei. (A, B) and (C, D) are from two representative hearts (n = 5). Clusters of GFP-expressing hESC-derived hCM (brown) were detected (arrows) in all injected hearts. Bar 100 μm (A,C); 50 μm (B,D). (E, F) Sections of mouse heart injected with SB203580-treated hESC-derived hCM were stained with H&E to survey for teratoma tissue. “(E, F) 4X and 20X magnification of injection site (lower right) in representative heart. There was no evidence of teratoma formation in any of the injected hearts (n = 5). Bar 100 μm.
Discussion
In the current study, we have shown that treatment of hESC with an inhibitor of p38MAPK (a) accelerated the directed differentiation of hESC into hCM in a developmental stage-specific manner, (b) did not affect hESC pluripotency, (c) did not induce genomic instability and (d) maintained viability of transplanted hCM in mouse myocardium.
During the course of our experiments, another group reported increased cardiogenesis after p38MAPK inhibition in endoderm co-cultures using hESC lines (20). Despite seemingly similar results, our study differs from their work in several important ways. Foremost, they differentiated hESC using serum-free medium conditioned by visceral endoderm-like mouse cells, whereas we used serum induction, a frequently employed method for spontaneous hESC differentiation (2,9,17). Second, they studied HES2, HES3, HES3-GFP hESC lines, while we carried out our studies using another extensively characterized (H9) hESC line. The increase in cardiogenesis observed irrespective of differentiation protocol across three hESC lines [our work and (20)] suggests that the effects of p38MAPK inhibition are not hESC line-specific. Third, the previously reported study examined cardiogenesis in a dose-dependent manner only, while we evaluated the effect of SB203580 in a developmental stage-specific manner. We show that the combination of timing and dose has an additive effect on cardiogenic enrichment. Our results emphasize the need for identifying the appropriate time-points during differentiation in culture to achieve a maximal effect. Each study adds significant details to the primary observation of p38MAPK inhibition as promoting hESC cardiogenesis, and the similar results using different hESC lines and different culture conditions (feeder-free serum replacement versus visceral endoderm co-culture) suggest that p38MAPK activity is important during hESC differentiation, and its manipulation may be useful in directing the differentiation of other mesoderm-derived tissues as well.
Others have observed that p38MAPK controls an early switch between commitment to mesoderm versus ectoderm during mouse embryonic stem cell (mESC) differentiation (19,29). There appear to be two waves of p38MAPK activity, one peaking at an early time-point between days 2 and 5 of mESC differentiation, and one at a later time-point between days 12 and 16. The early activity appears to control the decision between mesoderm and ectoderm (29), which is consistent with our finding that inhibition of p38MAPK at an early time-point (days 4–6) shifts differentiation toward cardiogenesis.
The early up-regulation of cardiac-specific genes in p38MAPK-inhibited hEB prompted our investigation into the ultrastructural maturity of treated versus untreated hCM. Surprisingly, we observed more mature sarcomere organization in the treated hEB. Previous studies have shown a progressive increase in the amount and organization of sarcomere structures coinciding with the developmental stage of hEB (3) or differentiation conditions (30). While we observed more extensive myofibrillar arrangements and evidence of nascent sarcomere formation earlier in treated cells than in untreated cells, we did not see characteristics of fully mature hCM in any of our samples. This variability in ultrastructural maturity between cultures, and even compared with fetal hearts, has been noted by others and thought to be because of differences between individual culture systems (3). This variability emphasizes the need to specify the differences between hESC differentiation protocols, the use of various culture additives, and the role of different culture systems, such as MEF and visceral endoderm-like cells (30).
In summary, we show that inhibition of p38MAPK during differentiation of hESC in co-culture with MEF results in accelerated differentiation of hCM and a 2.1-fold increase in hCM yield. We demonstrate that this enrichment is dependent on inhibiting p38MAPK at the time of ectoderm–mesoendoderm discrimination. We show that p38MAPK inhibition does not abolish pluripotency, as treated hESC remain competent at forming tissues from all three embryonic germ layers, and that the resulting hCM are genetically stable, can be purified by established methods (Percoll gradient) and are viable upon in vivo transplantation into mouse myocardium. These studies highlight further the utility of this method for producing hCM for therapeutic purposes.
Acknowledgments
This work was supported by a Comprehensive Research Grant from the California Institute for Regenerative Medicine (RC1-00104), a Public Health Service Grant (HL085377) from NHLBI, and a gift from the Pollin Foundation to HSB; and funds from the UCSF Cardiac Stem Cell Foundation to YY. We thank Frank King for technical advice and Angela Feraco for assistance with statistical analysis.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Laflamme MA, Gold J, Xu C, Hassanipour M, Rosler E, Police S, et al. Formation of human myocardium in the rat heart from human embryonic stem cells. Am J Pathol. 2005;167:663–71. doi: 10.1016/S0002-9440(10)62041-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Norstrom A, Akesson K, Hardarson T, Hamberger L, Bjorquist P, Sartipy P. Molecular and pharmacological properties of human embryonic stem cell-derived cardiomyocytes. Exp Biol Med (Maywood) 2006;231:1753–62. doi: 10.1177/153537020623101113. [DOI] [PubMed] [Google Scholar]
- 3.Snir M, Kehat I, Gepstein A, Coleman R, Itskovitz-Eldor J, Livne E, et al. Assessment of the ultrastructural and proliferative properties of human embryonic stem cell-derived cardiomyocytes. Am J Physiol Heart Circ Physiol. 2003;285:H2355–63. doi: 10.1152/ajpheart.00020.2003. [DOI] [PubMed] [Google Scholar]
- 4.Binah O, Dolnikov K, Sadan O, Shilkrut M, Zeevi-Levin N, Amit M, et al. Functional and developmental properties of human embryonic stem cell-derived cardiomyocytes. J Electrocardiol. 2007;40:S192–6. doi: 10.1016/j.jelectrocard.2007.05.035. [DOI] [PubMed] [Google Scholar]
- 5.Caspi O, Huber I, Kehat I, Habib M, Arbel G, Gepstein A, et al. Transplantation of human embryonic stem cell-derived cardiomyocytes improves myocardial performance in infarcted rat hearts. J Am Coll Cardiol. 2007;50:1884–93. doi: 10.1016/j.jacc.2007.07.054. [DOI] [PubMed] [Google Scholar]
- 6.Kehat I, Gepstein A, Spira A, Itskovitz-Eldor J, Gepstein L. High-resolution electrophysiological assessment of human embryonic stem cell-derived cardiomyocytes: a novel in vitro model for the study of conduction. Circ Res. 2002;91:659–61. doi: 10.1161/01.res.0000039084.30342.9b. [DOI] [PubMed] [Google Scholar]
- 7.Leor J, Gerecht S, Cohen S, Miller L, Holbova R, Ziskind A, et al. Human embryonic stem cell transplantation to repair the infarcted myocardium. Heart. 2007;93:1278–84. doi: 10.1136/hrt.2006.093161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xue T, Cho HC, Akar FG, Tsang SY, Jones SP, Marban E, et al. Functional integration of electrically active cardiac derivatives from genetically engineered human embryonic stem cells with quiescent recipient ventricular cardiomyocytes: insights into the development of cell-based pacemakers. Circulation. 2005;111:11–20. doi: 10.1161/01.CIR.0000151313.18547.A2. [DOI] [PubMed] [Google Scholar]
- 9.Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A, et al. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. 2001;108:407–14. doi: 10.1172/JCI12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu C, Police S, Rao N, Carpenter MK. Characterization and enrichment of cardiomyocytes derived from human embryonic stem cells. Circ Res. 2002;91:501–8. doi: 10.1161/01.res.0000035254.80718.91. [DOI] [PubMed] [Google Scholar]
- 11.Wong SSY, Bernstein HS. Cardiac regeneration using human embryonic stem cells: producing cells for future therapy. Regen Med. 2010 doi: 10.2217/rme.10.52. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel S, Tsang J, Harbers GM, Healy KE, Li S. Regulation of endothelial cell function by GRGDSP peptide grafted on interpenetrating polymers. J Biomed Mater Res A. 2007;83:423–33. doi: 10.1002/jbm.a.31320. [DOI] [PubMed] [Google Scholar]
- 13.Ramos GA, Hare JM. Cardiac cell-based therapy: cell types and mechanisms of actions. Cell Transplant. 2007;16:951–61. doi: 10.3727/096368907783338208. [DOI] [PubMed] [Google Scholar]
- 14.Mummery C, van der Heyden MA, de Boer TP, Passier R, Ward D, van den Brink S, et al. Cardiomyocytes from human and mouse embryonic stem cells. Methods Mol Med. 2007;140:249–72. doi: 10.1007/978-1-59745-443-8_14. [DOI] [PubMed] [Google Scholar]
- 15.Mummery C, Wardvan Oostwaard D, Doevendans P, Spijker R, van den Brink S, Hassink R, et al. Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation. 2003;107:2733–40. doi: 10.1161/01.CIR.0000068356.38592.68. [DOI] [PubMed] [Google Scholar]
- 16.Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25:1015–24. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 17.Pal R, Khanna A. Similar patterns in cardiac differentiation of human embryonic stem cell lines, BG01V and Reli-CellhES1, under low serum concentrations supplemented with bone morphogenetic protein-2. Differentiation. 2007;75:112–22. doi: 10.1111/j.1432-0436.2006.00123.x. [DOI] [PubMed] [Google Scholar]
- 18.Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, et al. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005;19:1175–87. doi: 10.1101/gad.1306705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aouadi M, Bost F, Caron L, Laurent K, Le Marchand Brustel Y, Binetruy B. p38 mitogen-activated protein kinase activity commits embryonic stem cells to either neurogenesis or cardiomyogenesis. Stem Cells. 2006;24:1399–406. doi: 10.1634/stemcells.2005-0398. [DOI] [PubMed] [Google Scholar]
- 20.Graichen R, Xu X, Braam SR, Balakrishnan T, Norfiza S, Sieh S, et al. Enhanced cardiomyogenesis of human embryonic stem cells by a small molecular inhibitor of p38 MAPK. Differentiation. 2008;76:357–70. doi: 10.1111/j.1432-0436.2007.00236.x. [DOI] [PubMed] [Google Scholar]
- 21.Nicholas CR, Gaur M, Wang S, Pera RA, Leavitt AD. A method for single-cell sorting and expansion of genetically modified human embryonic stem cells. Stem Cells Dev. 2007;16:109–17. doi: 10.1089/scd.2006.0059. [DOI] [PubMed] [Google Scholar]
- 22.King FW, Ritner C, Liszewski W, Kwan HC, Pedersen A, Leavitt AD, et al. Subpopulations of human embryonic stem cells with distinct tissue-specific fates can be selected from pluripotent cultures. Stem Cells Dev. 2009;18:1441–50. doi: 10.1089/scd.2009.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pinkel D, Albertson DG. Comparative genomic hybridization. Annu Rev Genomics Hum Genet. 2005;6:331–54. doi: 10.1146/annurev.genom.6.080604.162140. [DOI] [PubMed] [Google Scholar]
- 24.Ritner C, Bernstein HS. Fate mapping of human embryonic stem cells by teratoma formation. J Vis Exp. 2010 doi: 10.3791/2036. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cunha GR. Epitheliomesenchymal interactions in primordial gland structures which become responsive to androgenic stimulation. Anat Rec. 1972;172:179–95. doi: 10.1002/ar.1091720206. [DOI] [PubMed] [Google Scholar]
- 26.Springer ML, Sievers RE, Viswanathan MN, Yee MS, Foster E, Grossman W, et al. Closed-chest cell injections into mouse myocardium guided by high-resolution echocardiography. Am J Physiol Heart Circ Physiol. 2005;289:H1307–14. doi: 10.1152/ajpheart.00164.2005. [DOI] [PubMed] [Google Scholar]
- 27.Draper JS, Smith K, Gokhale P, Moore HD, Maltby E, Johnson J, et al. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat Biotechnol. 2004;22:53–4. doi: 10.1038/nbt922. [DOI] [PubMed] [Google Scholar]
- 28.Mitalipova MM, Rao RR, Hoyer DM, Johnson JA, Meisner LF, Jones KL, et al. Preserving the genetic integrity of human embryonic stem cells. Nat Biotechnol. 2005;23:19–20. doi: 10.1038/nbt0105-19. [DOI] [PubMed] [Google Scholar]
- 29.Binetruy B, Heasley L, Bost F, Caron L, Aouadi M. Concise review. Regulation of embryonic stem cell lineage commitment by mitogen-activated protein kinases. Stem Cells. 2007;25:1090–5. doi: 10.1634/stemcells.2006-0612. [DOI] [PubMed] [Google Scholar]
- 30.Bin Z, Sheng LG, Gang ZC, Hong J, Jun C, Bo Y, et al. Efficient cardiomyocyte differentiation of embryonic stem cells by bone morphogenetic protein-2 combined with visceral endoderm-like cells. Cell Biol Int. 2006;30:769–76. doi: 10.1016/j.cellbi.2006.05.011. [DOI] [PubMed] [Google Scholar]