

Abstract
High-risk human papillomaviruses (HPVs) contribute to cervical and other anogenital cancers, and they are also linked etiologically to a subset of head and neck squamous cell carcinomas (HNSCC). We previously established a model for HPV-associated HNSCC in which we treated transgenic mice expressing the papillomaviral oncoproteins with the chemical carcinogen 4-nitroquinoline-1-oxide (4-NQO). We found that the HPV-16 E7 oncoprotein was highly potent in causing HNSCC, and its dominance masked any potential oncogenic contribution of E6, a second papillomaviral oncoprotein commonly expressed in human cancers. In the current study, we shortened the duration of treatment with 4-NQO to reduce the incidence of cancers and discovered a striking synergy between E6 and E7 in causing HNSCC. Comparing the oncogenic properties of wild-type versus mutant E6 genes in this model for HNSCC uncovered a role for some but not other cellular targets of E6 previously shown to contribute to cervical cancer.
Keywords: High-risk human papillomaviruses, E6 oncoprotein, E7 oncoprotein, Head and neck squamous cell carcinoma
Introduction
High-risk human papillomaviruses (HPVs) are causative agents of nearly all cervical cancers and more recently have been implicated in the development of a subset of cancers of the head and neck. Approximately one fifth of all head and neck squamous cell carcinomas (HNSCCs) contain HPV DNA, and the vast majority of HPV-positive HNSCCs harbor HPV-16 DNA in particular (Gillison 2004). The papillomaviral E6 and E7 oncoproteins can bind to and stimulate the degradation of the tumor suppressors p53 (Huibregtse et al. 1991; Scheffner et al. 1993; Scheffner et al. 1990; Werness et al. 1990) and pRb (Boyer et al. 1996; Dyson et al. 1989), respectively, and although their oncogenic potentials are largely correlated with these interactions (Heck et al. 1992; Nguyen et al. 2002), their interference with the functions of other intracellular proteins likely plays important roles as well (Balsitis et al. 2006; Balsitis et al. 2005; Shai et al. 2007b; Song et al. 1998). The expression of E6 and E7 in HPV-positive HNSCC is correlated with an intact TP53 gene, reduced expression of pRb, and elevated levels of p16; in contrast, HPV-negative HNSCC generally retains an intact TP53 gene, displays normal expression of pRb, and displays an up-regulation of p16 (Balz et al. 2003; Hafkamp et al. 2003; Wiest et al. 2002). Furthermore, the expression of E6 and E7 are required for the maintenance of the transformed phenotype of cell lines derived from HPV-positive oropharyngeal cancers (Rampias et al. 2009). HPV-positive HNSCC often has a more basaloid morphology when compared with HPV-negative disease (Gillison et al. 2000) and individuals with HPV-positive HNSCC tend to be younger and less likely to be regular users of tobacco and alcohol than people with HPV-negative HNSCC (Lindel et al. 2001). In sum, these data suggest that HPVs in general and both viral oncogenes in particular play a causal role in the genesis of a subset of oropharyngeal cancers.
K14E6 (Song et al. 1999) and K14E7 (Herber et al. 1996) transgenic mice, in which expression of the individual HPV16 oncogenes are directed to the stratified squamous epithelium by the human keratin 14 (K14) promoter, were previously generated and characterized. In the murine cervix, HPV-16 E6 (Shai et al. 2007a) and E7 (Riley et al. 2003) each was found to be capable of cooperating with exogenous estrogen to induce cervical cancers, although E6 expressing mice required a longer period of treatment with exogenous estrogen to give rise to cancers. Additionally, the two oncogenes, when expressed together, led to increases in the incidence and size of cervical carcinomas compared to mice expressing either one oncogene (Brake et al. 2005; Riley et al. 2003). In the head and neck, E7 was likewise found to be the dominant oncogene, synergizing with the chemical carcinogen 4-nitroquinoline-1-oxide (4-NQO) to induce head and neck cancers in mice. In contrast, like-treated E6 transgenic mice failed to develop HNSCC (Strati et al. 2007). Consistent with the dominant oncogenic properties of E7 in the head and neck region, there was no significant difference in the severity or incidence of neoplastic disease arising in K14E7 transgenic versus K14E6/K14E7 bi-transgenic mice under the condition used in these prior studies (Strati & Lambert 2007; Strati et al. 2006).
HPV-16 E6 can interact with dozens of intracellular proteins, but its interference with the tumor suppressive role of p53 is by far its best-studied activity. E6 and the intracellular ubiquitin ligase E6-associated protein (E6-AP) can bind to p53 in a ternary complex and induce its proteasomal degradation (Huibregtse et al. 1991; Scheffner et al. 1993; Scheffner et al. 1990; Werness et al. 1990); however, whether this is the only mechanism by which E6 can cause the degradation of p53 currently is unclear (Massimi et al. 2008; Shai et al. 2007b). E6-AP binds to E6 using a conserved α-helical motif (Chen et al. 1998; Elston et al. 1998) which is shared by a variety of other binding partners of E6, including E6-binding protein (E6-BP) (Chen et al. 1995; Elston et al. 1998), paxillin (Chen et al. 1998; Vande Pol et al. 1998), tuberin (Elston et al. 1998; Lu et al. 2004), and interferon regulatory factor-3 (IRF-3) (Ronco et al. 1998). In addition to α-helical binding partners, E6 also can bind to several members of the post-synaptic density protein 95/Drosophila Discs large/zonula occludens-1 (PDZ)-domain family, which includes the Scribble (Scrib) (Massimi et al. 2008; Nakagawa et al. 2000) and Discs large (Dlg) (Gardiol et al. 1999; Kiyono et al. 1997) proteins; notably, both proteins are tumor suppressors in Drosophila (Bilder et al. 2000; Gateff 1978; Murphy 1974; Stewart et al. 1972). Lastly, E6 can interact with a host of other proteins involved in the regulation of apoptosis, genomic stability, epithelial differentiation, and transcription, many of which bind through alternative or unknown motifs (reviewed in (Tungteakkhun et al. 2008)).
To determine which families of binding partners of E6 are important in mediating its oncogenicity, we previously generated and used K14E6I128T (Nguyen et al. 2002) and K14E6Δ146-151 (Nguyen et al. 2003a; Nguyen et al. 2003b) transgenic mice, which express mutant forms of the E6 protein deficient in binding to α-helical (Liu et al. 1999) and PDZ-domain binding partners, respectively. K14E6I128T mice do not develop as many spontaneous epidermal carcinomas as K14E6 mice, nor do they develop as many epidermal papillomas and carcinomas as K14E6 mice in cooperation with topical treatment with initiating and promoting agents (Nguyen et al. 2002). In addition, studying K14E6I128T mice treated chronically with estrogen revealed that the interaction of E6 with α-helical binding partners contributes to the incidence and size of cervical cancers (Shai et al. 2007a). K14E6Δ146-151 mice do not develop significantly more spontaneous epidermal tumors than non-transgenic mice and display defects in the promotion of the development of papillomas in cooperation with topical treatment with chemical agents (Simonson et al. 2005). Unlike in K14E6I128T mice, however, deficiencies in the ability of E6Δ146-151 to contribute to cervical carcinogenesis appear only in the context of the co-expression of E7 (Shai et al. 2007a). These data indicate that both α-helical and PDZ-domain binding partners of E6 contribute to the genesis of neoplastic disease in the skin and in the cervix.
The aim of the study described herein was to assess the contribution of E6 to HNSCC. In our previous studies assessing the roles of HPV-16 E6 and E7, we treated transgenic mice with the chemical carcinogen 4-NQO in their drinking water for 16 weeks and then maintained them for an additional eight weeks in the absence of the drug. Under those conditions, we were unable to discern an individual contribution of HPV-16 E6 or its possible synergy with HPV-16 E7 in HNSCC, possibly due to the combined oncogenic potencies of both E7 and 4-NQO. Therefore, we halved the duration of exposure to 4-NQO from 16 weeks to eight weeks, and we found that, although neither K14E6 nor K14E7 mice developed significantly more head and neck tumors compared to non-transgenic mice, similarly treated K14E6/K14E7 mice expressing both HPV-16 E6 and E7 developed a significantly higher incidence of HNSCC. Therefore, E6 can contribute to HNSCC, and it does so by synergizing with E7. By studying K14E6I128T/K14E7 and K14E6Δ146-151/K14E7 mice treated with 4-NQO, we found that the ability of E6 to synergize with E7 in causing head and neck cancer does not correlate with its ability to interact with its PDZ-domain binding partners and only partially correlates with its ability to interact with α-helical partners. We conclude that the ability of E6 to synergize with E7 in contributing to HNSCC likely involves its interactions with multiple families of its binding partners.
Results
HPV16 E6 is functionally expressed in head/neck epithelia in K14E6 transgenic mice
In K14E6 mice, the HPV16 E6 oncoprotein is directed in its expression from the human keratin 14 (K14) promoter, which should direct expression of E6 to all stratified squamous epithelia, including in head/neck tissues. Our prior studies with K14E7 mice clearly showed that HPV16 E7, when expressed from the same K14 promoter was expressed and functional as an oncoprotein in head/neck epithelia. In the current study in which we evaluated the role of E6 in HNSCC using K14E6 mice, we wanted to establish that E6 is expressed and functional in the head and neck epithelia. Specifically we asked whether in K14E6 mice HPV16 E6 can inactivate p53 in the epithelia of the tongue and esophagus. A functional readout for E6’s inactivation of p53 in vivo is its ability to abrogate normal DNA damage responses. We therefore irradiated nontransgenic and K14E6 transgenic mice with 12 Gy ionizing radiation, a dose sufficient to elicit growth arrest in the epithelium of the tongue and esophagus. 24 hours post irradiation, we scored for cell proliferation by counting the number of cells labeled with BrdU 1 hour prior to harvesting the tissue. As expected, the epithelia of the tongue (Fig 1A) and esophagus (Fig. 1B) in nontransgenic mice underwent growth arrest, as evidenced by a reduction in the frequency of BrdU-positive cells. In contrast, K14E6 mice were completely abrogated in this response in both the tongue (Fig. 1A) and esophagus (Fig. 1B); no decrease in the frequency of BrdU-positive cells was observed in the irradiated K14E6 mice. This data demonstrates that HPV16 E6 is expressed and functional in head/neck epithelia in K14E6 mice.
Fig. 1.
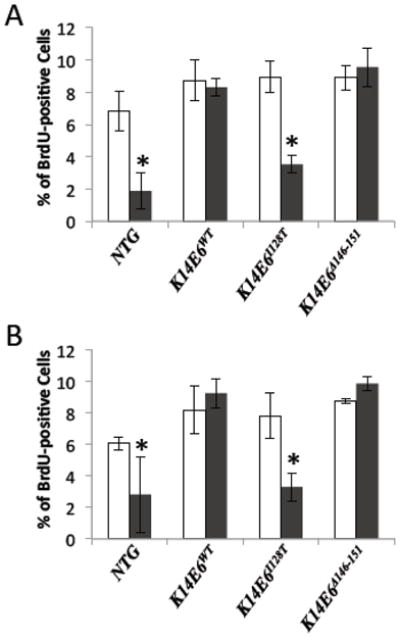
Response of head and neck epithelia to ionizing radiation. Shown in panels A and B are the percentage BrdU-positive cells present in tongue and esophagus epithelia, respectively, from mice that were (black bars) or were not (white bars) exposed to 12 Gy ionizing radiation (see methods for complete description of protocol used). For each condition and genotype, three mice were evaluated. In both tissues, significant reductions (p<0.05 based upon 2-sided Wilcoxon rank sum test) in Brdu-positive cells were observed only in the nontransgenic and the K14E6I128T transgenic mice (see *). In contrast there was the complete abrogation of growth arrest in the K14E6 and K14E6Δ146-151 mice, consistent with inactivation of p53.
We further wanted to confirm the properties of the mutant E6 proteins in the head and neck tissues of K14E6I128T and K14E6Δ146-151 transgenic mice, as these transgenic mice were used in this study to investigate the mechanism of action of E6 in HNSCC. E6I128T is predicted to be unable to inactivate p53; whereas E6Δ146-151 is known to retain this activity. Consistent with prior results in the skin of these transgenic mice, we observed that K14E6I128T mice were not abrogated in their response to ionizing radiation in the tongue (Fig. 1A) and esophagus (Fig. 1B); whereas K14E6Δ146-151 mice were abrogated in their response in these same tissues (Fig 1A and B).
HPV-16 E6 can contribute to HNSCC by synergizing with E7
Our laboratory previously investigated the roles of the papillomaviral oncogenes in HNSCC by treating adult K14E6, K14E7, and K14E6/K14E7 mice with 4-NQO for the first 16 weeks of a 24-week experimental period (Strati & Lambert 2007; Strati et al. 2006). Under this regimen of treatment, K14E6 mice did not display a significant increase in the incidence of head and neck tumors (23%) over that observed in similarly treated non-transgenic mice (16%). On the other hand, similarly treated K14E7 and K14E6/K14E7 mice developed tumors at high incidences (96% and 95%, respectively; p < 10−6 for both versus non-transgenic mice, two-sided Fisher’s exact tests). Based on these results, we concluded that E7 is the dominant papillomaviral oncogene in the head and neck, and its dominance masked any potential contribution of E6 to HNSCC.
To try to reveal a contribution of E6 to HNSCC, we halved the duration of treatment with 4-NQO from 16 weeks to eight weeks and subsequently held the mice in the absence of 4-NQO for the remainder of the aforementioned 24-week period. As with the previously used 16-week treatment with 4-NQO (Strati & Lambert 2007), the eight-week treatment did not lead to a significant increase in the severity of histopathological disease (Table 1); the incidences of tumors, cancers, or multiple tumors (Table 2); the multiplicity of tumors (Table 2); or the per-mouse average size of tumors (Fig. 2) in K14E6 mice when compared to non-transgenic mice.
Table 1.
Summary of overt disease in the head and neck.
| Genotype | nb | No Tumors | Histological Grade of Tumora |
||||
|---|---|---|---|---|---|---|---|
| Papilloma | Carcinoma |
||||||
| I | II | III | IV | ||||
| NTG | 34 | 32 | - | 1 | 1 | - | - |
| K14E6 | 20 | 18 | 1 | 1 | - | - | - |
| K14E7 | 22 | 17 | 4 | 1 | - | - | - |
| K14E6/K14E7c,d | 26 | 8 | 6 | 7 | 4 | - | 1 |
| K14E6I128T/K14E7c,d,e | 29 | 14 | 2 | 10 | 3 | - | - |
| K14E6Δ146-151/K14E7c,d,e | 27 | 13 | 5 | 6 | 1 | 2 | - |
Overt tumors were harvested and scored histopathologically as papillomas or graded (I – IV) as carcinomas, and mice then were assigned a diagnosis based on the most severe lesion observed. The number of mice assigned into each category is indicated.
Total number of mice examined for each genotype.
p < 0.001 vs. non-transgenic mice, two-sided Wilcoxon rank-sum test.
p < 0.02 vs. K14E7 mice, two-sided Wilcoxon rank-sum test.
p > 0.21 vs. K14E6/K14E7 mice, two-sided Wilcoxon rank-sum test.
Table 2.
Incidence of tumors and carcinomas in the head and neck.
| Genotype | nb | Incidence of:a |
Average Multiplicity of Tumorsc | ||
|---|---|---|---|---|---|
| Tumors (%) | Carcinomas (%) | Multiple Tumors (%) | |||
| Non-transgenic | 34 | 2 (5.9) | 2 (5.9) | − (−) | 0.059 |
| K14E6 | 20 | 2 (10) | 1 (5) | − (−) | 0.10 |
| K14E7 | 22 | 5 (23) | 1 (4.6) | 1 (4.6) | 0.27 |
| K14E6/K14E7 | 26 | 18 (69)d,e | 12 (46)d,e | 8 (31)d,e | 1.0f,g |
| K14E6I128T/K14E7 | 29 | 15 (52)d,e,j | 13 (45)d,e,k | 2 (6.9)h | 0.59f,g,i |
| K14E6Δ146-151/K14E7 | 27 | 14 (52)d,e,j | 9 (33)d,e,k | 6 (22)d | 0.75f,g |
The number of mice of each genotype with tumors, cancers, or multiple tumors is listed, with the percentage indicated in parentheses.
Total number of mice examined for each genotype.
Total number of tumors divided by the total number of mice (n) for each genotype.
p < 0.008 vs. non-transgenic mice, two-sided Fisher’s exact test.
p < 0.05 vs. K14E7 mice, two-sided Fisher’s exact test.
p < 10−4 vs. non-transgenic mice, two-sided Wilcoxon rank-sum test.
p < 0.05 vs. K14E7 mice, two-sided Wilcoxon rank-sum test.
p < 0.03 vs. K14E6/K14E7 mice, two-sided Fisher’s exact test.
p < 0.05 vs. K14E6/K14E7 mice, two-sided Wilcoxon rank-sum test.
p > 0.25 vs. K14E6/K14E7 mice, two-sided Fisher’s exact test.
p > 0.40 vs. K14E6/K14E7 mice, two-sided Fisher’s exact test.
Fig. 2.

Total size per mouse of head and neck tumors in mice treated with 4-NQO. Each dot represents the cumulative measured size of tumors in one mouse. The average total size of tumors per mouse is listed in parentheses under the genotypes. *p < 10−4 versus non-transgenic mice and †p < 0.03 versus K14E7 mice, two-sided Wilcoxon rank-sum tests. There were no significant differences between K14E6mutant/K14E7 and K14E6/K14E7 mice (p > 0.20, two-sided Wilcoxon rank-sum tests).
When we examined K14E7 mice treated with 4-NQO for eight weeks, we observed a sharp reduction in the oncogenic phenotypes previously observed when they were treated for 16 weeks (Strati & Lambert 2007). Compared with non-transgenic mice, we observed only marginal increases in K14E7 mice in the severity of head and neck neoplastic disease (p ≈ 0.087, two-sided Wilcoxon rank-sum test; Table 1), the incidence of tumors (23% versus 5.9%, p ≈ 0.099, two-sided Fisher’s exact test; Table 2), the multiplicity of tumors (0.27 versus 0.059, p ≈ 0.061, two-sided Wilcoxon rank-sum test; Table 2), and the average total size of tumors per mouse (0.59 mm versus 0.18 mm, p ≈ 0.075, two-sided Wilcoxon rank-sum test; Fig. 2). Furthermore, K14E7 mice treated for eight weeks did not display an increase in the incidence of cancer or of multiple tumors over that observed in non-transgenic mice treated similarly (Table 2). Therefore, shortening the 16-week treatment with 4-NQO to eight weeks strongly reduces the ability of E7 alone to induce head and neck tumors.
Our interest then turned to the incidence and severity of neoplastic disease in K14E6/K14E7 bi-transgenic mice. In stark contrast to what we observed in K14E6 and K14E7 singly transgenic mice, halving the duration of treatment of bi-transgenic mice with 4-NQO still led to a significant increase over what was seen in non-transgenic mice with regard to the severity of histopathological disease (p < 10−5, two-sided Wilcoxon rank-sum test; Table 1); the incidences of tumors, cancers, and multiple tumors (69% versus 5.9%, p < 10−6; 46% versus 5.9%, p < 10−3; and 31% versus 0%, p < 10−3; respectively, two-sided Fisher’s exact tests; Table 2); the multiplicity of tumors (1.0 versus 0.059, p < 10−6, two-sided Wilcoxon rank-sum test; Table 2); and the average size of tumors per mouse (3.1 mm versus 0.18 mm, p < 0.003, two-sided Wilcoxon rank-sum test; Fig. 2). Moreover, all of these measures of disease were increased in K14E6/K14E7 mice compared with K14E7 mice (p < 0.05, two-sided Wilcoxon rank-sum or Fisher’s exact tests; Tables 1 and 2; Fig. 2). These comparisons demonstrate that after the eight-week treatment with 4-NQO, E6 can synergize with E7 to drive the development of HNSCC.
Eliminating the interaction between E6 and its PDZ-domain binding partners does not affect its ability to synergize with E7 in HNSCC
We next investigated which of the families of binding partners of E6 were contributing to its ability to synergize with E7 to drive HNSCC. To do this, we used two previously generated lines of transgenic mice that harbor mutant versions of the HPV-16 E6 gene. K14E6Δ146-151 transgenic mice express a truncated form of E6 incapable of binding to or degrading proteins that contain PDZ domains (Nguyen et al. 2003a), such as Dlg (Gardiol et al. 1999; Kiyono et al. 1997) and Scrib (Massimi et al. 2008; Nakagawa & Huibregtse 2000), both of which are considered to be tumor suppressors in Drosophila (Bilder et al. 2000). K14E6I128T transgenic mice express an E6 protein that has a severely reduced ability to bind to the α-helical partners (Nguyen et al. 2002), including E6-AP (Liu et al. 1999); notably, a ternary complex of E6 and E6-AP can interact with and induce the degradation of p53 (Huibregtse et al. 1991; Scheffner et al. 1993; Scheffner et al. 1990; Werness et al. 1990). By comparing the severity, incidence, and multiplicity of disease between K14E6/K14E7 and these K14E6mutant/K14E7 mice, we were able to determine whether the α-helical or PDZ-domain binding partners of E6 are important in contributing to its synergistic role in HNSCC.
We first compared K14E6Δ146-151/K14E7 mice to K14E6/K14E7 mice to assess whether there were defects in the ability of E6Δ146-151 to synergize oncogenically with E7 compared with wild-type E6. We found no difference in the severity of disease between the two cohorts treated for eight weeks with 4-NQO (Table 1). In addition, the incidences of tumors, cancers, and multiple tumors, as well as the multiplicity of tumors, were statistically indistinguishable between the two groups of mice (Table 2). Lastly, the per-mouse average size of tumors in K14E6/K14E7 and K14E6Δ146-151/K14E7 mice was not significantly different (Fig. 2). The similarities between the phenotypes observed in K14E6Δ146-151/K14E7 mice and K14E6/K14E7 mice suggest that the interactions between E6 and its PDZ-domain binding partners are not critical for its synergy with E7 in inducing head and neck cancer in this model.
We also compared K14E6Δ146-151/K14E7 mice to K14E7 mice to determine whether E6Δ146-151 still was capable of increasing the severity or incidence of disease observed in the presence of E7 alone. The severity of disease (p < 0.02, two-sided Wilcoxon rank-sum test; Table 1), incidences of tumors and cancers (52% versus 23%, p < 0.05 and 33% versus 4.6%, p < 0.02, respectively, two-sided Fisher’s exact test; Table 2), multiplicity of tumors (0.75 versus 0.27, p < 0.03, two-sided Wilcoxon rank-sum test; Table 2), and average total size of tumors per mouse (4.3 mm versus 0.59 mm, p < 0.008, two-sided Wilcoxon rank-sum test; Fig. 2) were increased in K14E6Δ146-151/K14E7 mice when compared with K14E7 mice, indicating that E6Δ146-151 still can synergize with E7 to increase most of the parameters of head and neck neoplastic disease. Interestingly, however, the incidence of multiple tumors in K14E6Δ146-151/K14E7 mice was only marginally increased when compared to K14E7 mice (22% versus 4.6%, p ≈ 0.11, two-sided Fisher’s exact test; Table 2), indicating that K14E6Δ146-151 may have very subtle defects in its oncogenic potential when compared with wild-type E6. Overall, though, these data indicate that E6Δ146-151 still can synergize with E7 to contribute to HNSCC.
Strongly reducing the interaction of E6 with its α-helical binding partners reduces its ability to synergize with E7 in HNSCC
In order to determine the importance of the interaction of E6 with α-helical binding partners in its ability to synergize with E7, we next examined the oral cavities and esophagi of K14E6I128T/K14E7 mice treated with 4-NQO for eight weeks and compared them with those of similarly treated K14E6/K14E7 mice. As with K14E6Δ146-151/K14E7 mice, K14E6I128T/K14E7 mice displayed no differences when compared to K14E6/K14E7 mice with respect to the severity of disease (Table 1), the incidence of tumors or cancers (Table 2), or the per-mouse average size of tumors (Fig. 2). However, we noted a significant reduction in the incidence of multiple tumors (6.9% versus 31%, p < 0.04, two-sided Fisher’s exact test; Table 2) and the multiplicity of tumors (0.59 versus 1.0, p < 0.05, two-sided Wilcoxon rank-sum test; Table 2) in K14E6I128T/K14E7 versus K14E6/K14E7 mice, suggesting that the interaction of E6 with α-helical binding partners contributes subtly to its ability to synergize with E7.
When we compared K14E6I128T/K14E7 mice to K14E7 mice, we found that E6I128T still was able to increase the severity of disease (p < 0.01, two-sided Wilcoxon rank-sum test; Table 1), the incidences of tumors and cancers (52% versus 23%, p < 0.05 and 45% versus 4.6%, p < 0.002, respectively, two-sided Fisher’s exact tests; Table 2), and the average total size of tumors per mouse (2.1 mm versus 0.59 mm, p < 0.03, two-sided Wilcoxon rank-sum test; Fig. 2) over what we observed in K14E7 singly transgenic mice. The incidence of multiple tumors in K14E6I128T/K14E7 mice, however, was not significantly altered compared to K14E7 mice (Table 2). Lastly, even though the average multiplicity of tumors in K14E6I128T/K14E7 mice was reduced compared to K14E6/K14E7 mice, it was still higher than what was observed in K14E7 mice (0.59 versus 0.27, p < 0.05, two-sided Wilcoxon rank-sum test; Table 2). Thus, interfering with the binding of E6 to its α-helical partners strongly reduces its ability to synergize with E7 to induce multiple head and neck tumors; however, E6I128T still can contribute subtly in combination with E7 to increase the multiplicity of tumors. Overall, E6I128T is partially defective in its ability to synergize with E7 in the development HNSCC.
Discussion
Using our mouse model, we unmasked the contribution of HPV-16 E6 to HNSCC and found that it can synergize with E7 to drive the development of head and neck tumors. We used transgenic mice encoding mutated versions of E6 to show that its interactions with PDZ-domain binding partners are not critical for this synergy, whereas its interactions with α-helical partners partially contribute to it. Our data indicate that the mechanisms by which E6 synergizes with E7 in the head and neck differ from those observed in the cervix, where the interactions of E6 with both PDZ-domain and α-helical binding partners contribute to the growth of cervical cancer (Shai et al. 2007a).
HPV-16 E6 can synergize with E7 to contribute to the development of HNSCC
Previously in the head and neck, we were unable to determine whether the expression of E6 is important to carcinogenesis because the 16-week treatment with 4-NQO that we had used led to similar incidences of tumors, cancers, and multiple cancers in K14E7 and K14E6/K14E7 mice (Strati & Lambert 2007). By reducing the duration of treatment with 4-NQO from 16 weeks to eight weeks, we were able to reduce the severity of disease and incidences of tumors and cancers in K14E7 mice sufficiently to reveal significant differences in several parameters of neoplastic head and neck disease when we compared them to similarly treated K14E6/K14E7 mice (Tables 1 and 2; Fig. 2). Therefore, E6 can synergize with E7 to drive the genesis of head and neck tumors, although based on these and previous (Strati & Lambert 2007) results it is a weaker oncogene in the head and neck than E7.
In the murine cervix, as in the head and neck, E7 is a more potent oncogene than E6, as K14E7 mice develop cervical cancers within six months of chronic treatment with estrogen (Riley et al. 2003) whereas K14E6 mice require nine months of continuous treatment (Shai et al. 2007a). In the skin, a small proportion of K14E6 mice develop spontaneous carcinomas late in life, but K14E7 mice develop only benign papillomas; furthermore, E6 but not E7 can induce the progression of chemically induced epidermal papillomas to carcinomas, indicating that E6 is the more potent of the two oncogenes in the skin (Song et al. 2000). The reasons that E6 is more oncogenically potent in the epidermis than at mucosal sites are not understood, but they may involve the differential expression of target proteins of the viral oncogenes in different tissues or a differential importance of the pathways with which the oncogenes interfere in controlling proliferation in different types of stratified squamous epithelia.
Binding to PDZ-domain partners is not critical to the ability of E6 to synergize with E7 in HNSCC
The incidence of multiple tumors in K14E6Δ146-151/K14E7 (22%) was intermediate between that of K14E7 (4.6%) and K14E6/K14E7 (31%) mice (Table 2). Therefore, E6Δ146-151 does have a subtle defect in contributing to the growth of multiple head and neck tumors when compared with wild-type E6, but the defect was not strong enough to lead to a statistically significant reduction in the incidence of multiple tumors in K14E6Δ146-151/K14E7 mice compared to K14E6/K14E7 mice. In the context of other parameters of tumorigenicity, there was effectively no difference in the ability of E6Δ146-151 versus wild-type E6 to synergize with E7 (Tables 1 and 2; Fig. 2).
We were surprised to see so little an effect of eliminating the interaction of E6 with its PDZ-domain binding partners, because the PDZ-binding motif is found exclusively in the E6 proteins of high-risk HPVs and therefore is thought to be critical to their ability to cause cancers. Two PDZ-domain binding partners of E6, Dlg and Scrib, are tumor suppressors in Drosophila (Bilder et al. 2000; Gateff 1978; Murphy 1974; Stewart et al. 1972), and these and many other members of the PDZ-domain family targeted by E6 regulate several processes important to malignancy, including cellular polarity, adhesion, and proliferation (reviewed in (Thomas et al. 2008)). Although the E6 protein of rhesus papillomavirus type 1 (RhPV-1), which is the only other type of papillomavirus besides HPV that is a causative agent of cervical cancers in its host, lacks a PDZ-binding motif, the RhPV-1 E7 protein contains one (Tomaic et al. 2009). Furthermore, K14E6Δ146-151 mice display obvious defects in the growth of spontaneous and chemically induced epidermal papillomas when compared with K14E6 mice (Simonson et al. 2005), and K14E6Δ146-151/K14E7 mice develop fewer and smaller cervical carcinomas than K14E6/K14E7 mice when treated chronically with estrogen (Shai et al. 2007a), showing that the PDZ-binding motif of E6 is important for its oncogenicity in the skin and cervix. While our study is the first to examine the role of the interaction between E6 and the PDZ-domain proteins in head and neck neoplasia in vivo, it was shown recently that E6Δ146-151 is defective in promoting the anchorage-independent growth and immortalization of primary human tonsillar epithelial cells (HTECs) (Spanos et al. 2008). These cells are particularly relevant in that tonsillar cancers are predominantly HPV-positive. Thus, previous studies have suggested an important oncogenic role for the binding of PDZ-domain proteins by E6.
There are several possible reasons that may explain why our results contrast so sharply with previous studies. We have shown previously that HPV-16 E6 does not induce the degradation of Dlg or Scrib in the murine epidermis (Simonson et al., unpublished results), but it does reduce the levels of Scrib in the murine lens (Nguyen et al. 2003b). It is possible, therefore, that E6 does not induce the degradation of some or all of the PDZ-domain proteins to which it binds in murine head and neck epithelial cells. This might allow them to retain some function even if they are bound by E6, although degradation is not necessarily the only way in which E6 could interfere with the functions of its PDZ-domain binding partners. It is also conceivable that PDZ-domain proteins play less important tumor suppressive roles in the head and neck epithelium than they do in the epidermis and cervical epithelium, so their inactivation by E6 may not be critical to the genesis of head and neck tumors. E6 proteins from different high-risk types of HPVs have been shown to bind with different affinities to various PDZ-domain proteins—HPV-18 E6 binds to Dlg and membrane-associated guanylate kinase with inverted domain structure (MAGI)-1 more strongly than HPV-16 E6 (Pim et al. 2000; Thomas et al. 2001), for example—and the proteins bound most strongly by HPV-16 E6, which include Scrib (Thomas et al. 2005), may be the less important for the maintenance of head and neck epithelial cell polarity and adhesion. In addition, there could be some functional compensation (Kocher et al. 2003; Misawa et al. 2001) for the loss of PDZ-domain proteins in the oral cavity by proteins either not bound or bound only weakly by HPV-16 E6, although it is important to note that such compensation has not been shown to occur among the PDZ-domain proteins bound by E6. While the aforementioned experiments performed in HTECs (Spanos et al. 2008) showed the importance of the PDZ-domain binding motif of E6 in immortalizing oral keratinocytes in tissue culture, they relied on the transduction of HTECs with HPV-16 E6 and E7 and may not be predictive of the behavior of cancers in vivo. Furthermore, it is unlikely that anchorage-independent growth and immortalization correlate perfectly with malignancy. On the other hand, our carcinogenesis study was carried out in mice, which may differ in their sensitivity to PDZ protein function at least in the head/neck region. Furthermore our mouse model relies upon use of a chemical carcinogen, 4NQO, which could mask a role of certain activities of E6. Characterizing the levels of expression of PDZ-domain proteins in the presence and absence of E6 in the oral epithelium, investigating the effects on the oral epithelium of the loss of their individual or combined expression, and comparing results obtained with HPV-16 E6 with future studies examining the E6 proteins from other high-risk types of HPVs will be important in addressing further the mechanism of action of E6 in HNSCC. Such experiments could help to elucidate the reasons behind the surprisingly subtle defects in the oncogenicity of HPV-16 E6Δ146-151 in the head and neck.
The binding of HPV-16 E6 to α-helical partners weakly contributes to its ability to synergize with E7 in HNSCC
In contrast to PDZ-domain binding partners of E6, the interaction with α-helical binding partners by E6 was critical for its ability to induce multiple tumors with E7 (Table 2). In addition, the average multiplicity of tumors in K14E6I128T/K14E7 mice (0.59 per mouse) was intermediate between the multiplicities observed in K14E7 (0.27) and K14E6/K14E7 (1.0) transgenic mice (Table 2), indicating that E6I128T has defects in synergizing with E7 to increase the multiplicity of head and neck tumors. In contrast, in the cervix, the interactions of E6 with α-helical partners contribute to the incidence and size of tumors in the absence of E7 but only to the size of tumors in the presence of E7 (Shai et al. 2007a). Comparing our results with these previous conclusions from the study in the cervix suggests that in different tissues, the binding of E6 to its families of partners may contribute to carcinogenesis differently. However, we cannot exclude the possibility that the two different carcinogens used in this study and in the study investigating cervical cancer—4-NQO and estrogen, respectively—differentially affect the importance of the interactions of E6 with its binding partners to tumorigenesis. Regardless, E6128T was still able to synergize with E7 in driving most aspects of neoplastic disease in the head and neck.
The best-studied interaction affected by the E6I128T mutation is the one between E6 and E6-AP, which together can bind to p53 and induce its degradation (Huibregtse et al. 1991; Scheffner et al. 1993; Scheffner et al. 1990; Werness et al. 1990). E6I128T binds to E6-AP and induces the degradation of p53 with less than 5% the ability of wild-type E6 (Liu et al. 1999), and its reduced ability to degrade p53 is probably the most obvious mechanism by which this mutation in E6 could lead to a decrease in the both the incidence of multiple tumors and the multiplicity of tumors (Table 2). Importantly, E6I128T does retain some binding to E6-AP (Liu et al. 1999), so it remains possible that residual degradation of p53 by an E6I128T/E6-AP complex may be sufficient to synergize with E7 in head and neck tumorigenesis; this may be one reason that we did not observe a more drastic reduction in neoplastic phenotypes when comparing K14E6I128T/K14E7 mice to K14E6/K14E7 mice. It has been shown previously that knocking out p53 contributes to both epidermal (Kemp et al. 1993) and cervical (Shai et al. 2008) carcinogenesis, but in neither tissue is eliminating p53 sufficient to recapitulate the full oncogenic potential of E6. Because the E6I128T mutation does not ablate selectively the degradation of p53 (Liu et al. 1999), further studies involving p53−/− mice or mice in which p53 is deleted conditionally likely are necessary to assess the importance of the degradation of p53 to the oncogenicity of E6 in the head and neck.
In addition to E6-AP, E6I128T is also deficient in binding to E6-BP (Liu et al. 1999), which is a calcium-binding protein that may be involved in the differentiation of epithelial cells (Chen et al. 1995), although its role and the effect of its binding to E6 are not understood (Sherman et al. 2002). Many other proteins—minichromosome maintenance protein 7 (MCM7) (Kuhne et al. 1998; Kukimoto et al. 1998), a licensing factor for the replication of DNA; IRF-3 (Ronco et al. 1998), a transcription factor that induces the expression of interferons; paxillin (Chen et al. 1998; Tong et al. 1997), which is associated with focal adhesion proteins and is thought to play a role in the organization of actin; tuberin (Elston et al. 1998), a putative tumor suppressor; and transcriptional regulator interacting with the plant homeodomain-bromodomain 1 (TRIP-Br1) (Gupta et al. 2003), a transcriptional co-activator and regulator of the cell cycle— are predicted to associate with E6 using the same α-helical motif shared by E6-AP and E6-BP. Whether the E6I128T mutant is defective for binding to any of them has not been investigated specifically. Furthermore, several binding partners of E6 other than p53, including the proto-oncogene N-Myc (Gross-Mesilaty et al. 1998) and the 91-kDa splice variant of nuclear transcription factor, X-box binding 1 (NFX1-91) repressor of telomerase (Gewin et al. 2004), and possibly the proto-oncogene c-Myc (Gross-Mesilaty et al. 1998; Veldman et al. 2003) and the O6-methylguanine-DNA methyltransferase (MGMT) (Srivenugopal et al. 2002), may bind to E6 only in the presence of E6-AP, so the E6I128T mutation probably affects the binding of these proteins indirectly. Potentially, the interaction of E6 with a variety of α-helical partners and with proteins that bind only to the E6/E6-AP complex likely contributes to some aspects of its ability to synergize with E7 in HNSCC, although based on our data, other binding partners also play critical roles in this synergy.
The interaction of HPV-16 E6 with multiple binding partners likely plays a role in its ability to synergize with E7 in the head and neck
HPV-16 interacts with over two dozen intracellular proteins (reviewed in (Tungteakkhun & Duerksen-Hughes 2008)), and many of them do not bind to E6 through any identified or well-characterized motifs. It is possible that the interaction of E6 with binding partners outside of the α-helical and PDZ-domain families are important to head and neck tumorigenesis. The interaction of E6 with Bak (Thomas et al. 1999), Fas-associated death domain protein (FADD) (Filippova et al. 2004), procaspase-8 (Filippova et al. 2007), and tumor necrosis factor receptor 1 (TNF R1) (Filippova et al. 2002), all of which play a role in apoptosis, are some of the most obvious candidates, although the binding of E6 to several other proteins involved in genomic stability, epithelial differentiation, and transcriptional regulation also probably contributes to its oncogenicity in the head and neck. Until detailed mutational analysis of E6 is completed and the regions of the oncoprotein that are important to binding to individual intracellular proteins are identified, dissecting the specific contributions of any one of these interactions to tumorigenesis will remain difficult, if not impossible.
In addition, it remains possible that eliminating the interaction of E6 with both the α-helical and PDZ-domain families simultaneously would result in a more dramatic effect than what we observed when the interaction with either family was abolished individually (Tables 1 and 2; Fig. 1). Perhaps the two families of binding partners play overlapping roles in the head and neck, or maybe there is some functional compensation between the α-helical and PDZ-domain partners of E6 when only one of the two families is inactivated by the viral oncoprotein. Interfering with the interaction of E6 with both families simultaneously, perhaps by using an E6I128T/Δ146-151 double-mutant, would permit an initial investigation of these possibilities, although one important consideration would be whether mutating more than one region of E6 would affect the structure of the oncoprotein in a way that renders it functionally deficient in unexpected manners.
Materials and methods
Transgenic Mice
K14E6 (Song et al. 1999), K14E6I128T (Nguyen et al. 2002), K14E6Δ146-151 (Nguyen et al. 2003a), and K14E7 (Herber et al. 1996) transgenic mice have been described previously. All mice were maintained on the FVB/n inbred genetic background, and all mice were housed in the McArdle Laboratory Animal Care Unit, approved by the Association for Assessment of Laboratory Animal Care, at the University of Wisconsin Medical School. All protocols for animal work were approved by the University of Wisconsin Medical School Institutional Animal Care and Use Committee.
Irradiation studies
Groups of three 12–14 week old mice of each genotype were or were not exposed to 12Gy radiation from a 137Cs source. 23 hours post exposure, mice were injected intraperitoneally with BrdU (300 ul of a 12.5mg/ml solution). 1 hour later mice were sacrificed, tongue and esophagus formallin fixed, paraffin embedded, and sectioned. Histological sections were deparaffinized in Xylenes, rehydrated in a series of alcohols, boiled in 10mM citrate buffer for 17 mins to unmask antigens, blocked in 10% horse serum in PBS for 1 hour, then incubated overnight at 4°C with primary antibody specific for BrdU (Ab-2; Calbiochem, San Diego, CA) diluted1:100 in block. A universally biotinylated secondary antibody was applied for 30 minutes (Vectastain universal secondary), washed in PBS, and incubated in ABC (Vectastain, Vector labs, Burlingame, CA) reagent for 30 minutes. Sections were developed with DAB reagent for appropriate time, counterstained with hemotoxylin, dehydrated in a series of alcohols and cover slipped. The percentage of BrdU-positive cells were counted in the tongue and esophagus epithelia of each mouse (ten 400X microscope frames per mouse).
Treatment with 4-NQO
Six- to eight-week-old mice were treated for eight weeks with 10 μg/mL 4-NQO (Sigma-Aldrich Corporation, St. Louis, MO) in their drinking water, diluted from a stock solution of 0.2% 4-NQO (w/v) in propylene glycol that was protected from light and stored at 4°C. The mice then were returned to a normal supply of water for an additional 16 weeks.
Harvest and Analysis of Tongues, Esophagi, and Overt Tumors of the Head and Neck
After completion of the treatment regimen with 4-NQO, mice were sacrificed. At the time of sacrifice, overt tumors in the oral cavity and esophagus were scored, measured across their largest dimension with a ruler, and harvested; once the overt tumors were harvested, the entire remaining tongue and esophagus also were harvested. Specimens were fixed overnight at 4°C in 10% buffered formalin (v/v) and then embedded in paraffin. Embedded tumors were cut into 5-μm-thick sections, and 3 to 10 sections per tumor then were stained with hematoxylin and eosin (H&E) and scored as papillomas or carcinomas. Carcinomas were graded from I – IV based on morphology and the degree of keratinization. For statistical comparisons of the severity of disease between genotypes, mice were given a numerical value based on the assigned grade of tumors. The values assigned were: 0 for no overt tumors, 1 for papilloma, 2 for grade I carcinoma, 3 for grade II carcinoma, 4 for grade III carcinoma, and 5 for grade IV carcinoma.
Statistical Analyses
Specific comparisons done and statistical tests used are cited within the text and in the captions of figures and tables.
Acknowledgments
We thank the University of Wisconsin Carbone Cancer Center Experimental Pathology facility for their support and technical assistance and Dr. Norman Drinkwater for consultations regarding statistical analyses. This study was supported by grants from the NIH (DE017315 and CA098428) to PFL.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balsitis S, Dick F, Dyson N, et al. Critical roles for non-pRb targets of human papillomavirus type 16 E7 in cervical carcinogenesis. Cancer Res. 2006;66 (19):9393–400. doi: 10.1158/0008-5472.CAN-06-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsitis S, Dick F, Lee D, et al. Examination of the pRb-dependent and pRb-independent functions of E7 in vivo. J Virol. 2005;79 (17):11392–402. doi: 10.1128/JVI.79.17.11392-11402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balz V, Scheckenbach K, Gotte K, et al. Is the p53 inactivation frequency in squamous cell carcinomas of the head and neck underestimated? Analysis of p53 exons 2–11 and human papillomavirus 16/18 E6 transcripts in 123 unselected tumor specimens. Cancer Res. 2003;63 (6):1188–91. [PubMed] [Google Scholar]
- Bilder D, Li M, Perrimon N. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science. 2000;289 (5476):113–6. doi: 10.1126/science.289.5476.113. [DOI] [PubMed] [Google Scholar]
- Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996;56 (20):4620–4. [PubMed] [Google Scholar]
- Brake T, Lambert PF. Estrogen contributes to the onset, persistence, and malignant progression of cervical cancer in a human papillomavirus-transgenic mouse model. Proc Natl Acad Sci U S A. 2005;102 (7):2490–5. doi: 10.1073/pnas.0409883102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JJ, Hong Y, Rustamzadeh E, et al. Identification of an alpha helical motif sufficient for association with papillomavirus E6. J Biol Chem. 1998;273 (22):13537–44. doi: 10.1074/jbc.273.22.13537. [DOI] [PubMed] [Google Scholar]
- Chen JJ, Reid CE, Band V, et al. Interaction of papillomavirus E6 oncoproteins with a putative calcium-binding protein. Science. 1995;269 (5223):529–31. doi: 10.1126/science.7624774. [DOI] [PubMed] [Google Scholar]
- Dyson N, Howley PM, Munger K, et al. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243 (4893):934–7. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- Elston RC, Napthine S, Doorbar J. The identification of a conserved binding motif within human papillomavirus type 16 E6 binding peptides, E6AP and E6BP. J Gen Virol. 1998;79 (Pt 2):371–4. doi: 10.1099/0022-1317-79-2-371. [DOI] [PubMed] [Google Scholar]
- Filippova M, Johnson MM, Bautista M, et al. The large and small isoforms of human papillomavirus type 16 E6 bind to and differentially affect procaspase 8 stability and activity. J Virol. 2007;81 (8):4116–29. doi: 10.1128/JVI.01924-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippova M, Parkhurst L, Duerksen-Hughes PJ. The human papillomavirus 16 E6 protein binds to Fas-associated death domain and protects cells from Fas-triggered apoptosis. J Biol Chem. 2004;279 (24):25729–44. doi: 10.1074/jbc.M401172200. [DOI] [PubMed] [Google Scholar]
- Filippova M, Song H, Connolly JL, et al. The human papillomavirus 16 E6 protein binds to tumor necrosis factor (TNF) R1 and protects cells from TNF-induced apoptosis. J Biol Chem. 2002;277 (24):21730–9. doi: 10.1074/jbc.M200113200. [DOI] [PubMed] [Google Scholar]
- Gardiol D, Kuhne C, Glaunsinger B, et al. Oncogenic human papillomavirus E6 proteins target the discs large tumour suppressor for proteasome-mediated degradation. Oncogene. 1999;18 (40):5487–96. doi: 10.1038/sj.onc.1202920. [DOI] [PubMed] [Google Scholar]
- Gateff E. Malignant neoplasms of genetic origin in Drosophila melanogaster. Science. 1978;200 (4349):1448–59. doi: 10.1126/science.96525. [DOI] [PubMed] [Google Scholar]
- Gewin L, Myers H, Kiyono T, et al. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes Dev. 2004;18 (18):2269–82. doi: 10.1101/gad.1214704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillison ML. Human papillomavirus-associated head and neck cancer is a distinct epidemiologic, clinical, and molecular entity. Semin Oncol. 2004;31 (6):744–54. doi: 10.1053/j.seminoncol.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Gillison ML, Koch WM, Capone RB, et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 2000;92 (9):709–20. doi: 10.1093/jnci/92.9.709. [DOI] [PubMed] [Google Scholar]
- Gross-Mesilaty S, Reinstein E, Bercovich B, et al. Basal and human papillomavirus E6 oncoprotein-induced degradation of Myc proteins by the ubiquitin pathway. Proc Natl Acad Sci U S A. 1998;95 (14):8058–63. doi: 10.1073/pnas.95.14.8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Takhar PP, Degenkolbe R, et al. The human papillomavirus type 11 and 16 E6 proteins modulate the cell-cycle regulator and transcription cofactor TRIP-Br1. Virology. 2003;317 (1):155–64. doi: 10.1016/j.virol.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Hafkamp HC, Speel EJ, Haesevoets A, et al. A subset of head and neck squamous cell carcinomas exhibits integration of HPV 16/18 DNA and overexpression of p16INK4A and p53 in the absence of mutations in p53 exons 5–8. Int J Cancer. 2003;107 (3):394–400. doi: 10.1002/ijc.11389. [DOI] [PubMed] [Google Scholar]
- Heck DV, Yee CL, Howley PM, et al. Efficiency of binding the retinoblastoma protein correlates with the transforming capacity of the E7 oncoproteins of the human papillomaviruses. Proc Natl Acad Sci U S A. 1992;89 (10):4442–6. doi: 10.1073/pnas.89.10.4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herber R, Liem A, Pitot H, et al. Squamous epithelial hyperplasia and carcinoma in mice transgenic for the human papillomavirus type 16 E7 oncogene. J Virol. 1996;70 (3):1873–81. doi: 10.1128/jvi.70.3.1873-1881.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huibregtse JM, Scheffner M, Howley PM. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. Embo J. 1991;10 (13):4129–35. doi: 10.1002/j.1460-2075.1991.tb04990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp CJ, Donehower LA, Bradley A, et al. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell. 1993;74 (5):813–22. doi: 10.1016/0092-8674(93)90461-x. [DOI] [PubMed] [Google Scholar]
- Kiyono T, Hiraiwa A, Fujita M, et al. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci U S A. 1997;94 (21):11612–6. doi: 10.1073/pnas.94.21.11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocher O, Pal R, Roberts M, et al. Targeted disruption of the PDZK1 gene by homologous recombination. Mol Cell Biol. 2003;23 (4):1175–80. doi: 10.1128/MCB.23.4.1175-1180.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhne C, Banks L. E3-ubiquitin ligase/E6-AP links multicopy maintenance protein 7 to the ubiquitination pathway by a novel motif, the L2G box. J Biol Chem. 1998;273 (51):34302–9. doi: 10.1074/jbc.273.51.34302. [DOI] [PubMed] [Google Scholar]
- Kukimoto I, Aihara S, Yoshiike K, et al. Human papillomavirus oncoprotein E6 binds to the C-terminal region of human minichromosome maintenance 7 protein. Biochem Biophys Res Commun. 1998;249 (1):258–62. doi: 10.1006/bbrc.1998.9066. [DOI] [PubMed] [Google Scholar]
- Lindel K, Beer KT, Laissue J, et al. Human papillomavirus positive squamous cell carcinoma of the oropharynx: a radiosensitive subgroup of head and neck carcinoma. Cancer. 2001;92 (4):805–13. doi: 10.1002/1097-0142(20010815)92:4<805::aid-cncr1386>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Liu Y, Chen JJ, Gao Q, et al. Multiple functions of human papillomavirus type 16 E6 contribute to the immortalization of mammary epithelial cells. J Virol. 1999;73 (9):7297–307. doi: 10.1128/jvi.73.9.7297-7307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Hu X, Li Y, et al. Human papillomavirus 16 E6 oncoprotein interferences with insulin signaling pathway by binding to tuberin. J Biol Chem. 2004;279 (34):35664–70. doi: 10.1074/jbc.M403385200. [DOI] [PubMed] [Google Scholar]
- Massimi P, Shai A, Lambert P, et al. HPV E6 degradation of p53 and PDZ containing substrates in an E6AP null background. Oncogene. 2008;27 (12):1800–4. doi: 10.1038/sj.onc.1210810. [DOI] [PubMed] [Google Scholar]
- Misawa H, Kawasaki Y, Mellor J, et al. Contrasting localizations of MALS/LIN-7 PDZ proteins in brain and molecular compensation in knockout mice. J Biol Chem. 2001;276 (12):9264–72. doi: 10.1074/jbc.M009334200. [DOI] [PubMed] [Google Scholar]
- Murphy C. Cell death and autonomous gene action in lethals affecting imaginal discs in Drosophila melanogaster. Dev Biol. 1974;39 (1):23–36. doi: 10.1016/s0012-1606(74)80005-9. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Huibregtse JM. Human scribble (Vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6AP ubiquitin-protein ligase. Mol Cell Biol. 2000;20 (21):8244–53. doi: 10.1128/mcb.20.21.8244-8253.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M, Song S, Liem A, et al. A mutant of human papillomavirus type 16 E6 deficient in binding alpha-helix partners displays reduced oncogenic potential in vivo. J Virol. 2002;76 (24):13039–48. doi: 10.1128/JVI.76.24.13039-13048.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen ML, Nguyen MM, Lee D, et al. The PDZ ligand domain of the human papillomavirus type 16 E6 protein is required for E6’s induction of epithelial hyperplasia in vivo. J Virol. 2003a;77 (12):6957–64. doi: 10.1128/JVI.77.12.6957-6964.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MM, Nguyen ML, Caruana G, et al. Requirement of PDZ-containing proteins for cell cycle regulation and differentiation in the mouse lens epithelium. Mol Cell Biol. 2003b;23 (24):8970–81. doi: 10.1128/MCB.23.24.8970-8981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pim D, Thomas M, Javier R, et al. HPV E6 targeted degradation of the discs large protein: evidence for the involvement of a novel ubiquitin ligase. Oncogene. 2000;19 (6):719–25. doi: 10.1038/sj.onc.1203374. [DOI] [PubMed] [Google Scholar]
- Rampias T, Sasaki C, Weinberger P, et al. E6 and E7 Gene Silencing and Transformed Phenotype of Human Papillomavirus 16-Positive Oropharyngeal Cancer Cells. J Natl Cancer Inst. 2009 doi: 10.1093/jnci/djp017. [DOI] [PubMed] [Google Scholar]
- Riley RR, Duensing S, Brake T, et al. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003;63 (16):4862–71. [PubMed] [Google Scholar]
- Ronco LV, Karpova AY, Vidal M, et al. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998;12 (13):2061–72. doi: 10.1101/gad.12.13.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M, Huibregtse JM, Vierstra RD, et al. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75 (3):495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, et al. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63 (6):1129–36. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- Shai A, Brake T, Somoza C, et al. The human papillomavirus E6 oncogene dysregulates the cell cycle and contributes to cervical carcinogenesis through two independent activities. Cancer Res. 2007a;67 (4):1626–35. doi: 10.1158/0008-5472.CAN-06-3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shai A, Nguyen ML, Wagstaff J, et al. HPV16 E6 confers p53-dependent and p53-independent phenotypes in the epidermis of mice deficient for E6AP. Oncogene. 2007b;26 (23):3321–8. doi: 10.1038/sj.onc.1210130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shai A, Pitot HC, Lambert PF. p53 Loss synergizes with estrogen and papillomaviral oncogenes to induce cervical and breast cancers. Cancer Res. 2008;68 (8):2622–31. doi: 10.1158/0008-5472.CAN-07-5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman L, Itzhaki H, Jackman A, et al. Inhibition of serum- and calcium-induced terminal differentiation of human keratinocytes by HPV 16 E6: study of the association with p53 degradation, inhibition of p53 transactivation, and binding to E6BP. Virology. 2002;292 (2):309–20. doi: 10.1006/viro.2001.1263. [DOI] [PubMed] [Google Scholar]
- Simonson SJ, Difilippantonio MJ, Lambert PF. Two distinct activities contribute to human papillomavirus 16 E6’s oncogenic potential. Cancer Res. 2005;65 (18):8266–73. doi: 10.1158/0008-5472.CAN-05-1651. [DOI] [PubMed] [Google Scholar]
- Song S, Gulliver GA, Lambert PF. Human papillomavirus type 16 E6 and E7 oncogenes abrogate radiation-induced DNA damage responses in vivo through p53-dependent and p53-independent pathways. Proc Natl Acad Sci U S A. 1998;95 (5):2290–5. doi: 10.1073/pnas.95.5.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Liem A, Miller JA, et al. Human papillomavirus types 16 E6 and E7 contribute differently to carcinogenesis. Virology. 2000;267 (2):141–50. doi: 10.1006/viro.1999.0106. [DOI] [PubMed] [Google Scholar]
- Song S, Pitot HC, Lambert PF. The human papillomavirus type 16 E6 gene alone is sufficient to induce carcinomas in transgenic animals. J Virol. 1999;73 (7):5887–93. doi: 10.1128/jvi.73.7.5887-5893.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanos WC, Geiger J, Anderson ME, et al. Deletion of the PDZ motif of HPV16 E6 preventing immortalization and anchorage-independent growth in human tonsil epithelial cells. Head Neck. 2008;30 (2):139–47. doi: 10.1002/hed.20673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivenugopal KS, Ali-Osman F. The DNA repair protein, O(6)-methylguanine-DNA methyltransferase is a proteolytic target for the E6 human papillomavirus oncoprotein. Oncogene. 2002;21 (38):5940–5. doi: 10.1038/sj.onc.1205762. [DOI] [PubMed] [Google Scholar]
- Stewart M, Murphy C, Fristrom JW. The recovery and preliminary characterization of X chromosome mutants affecting imaginal discs of Drosophila melanogaster. Dev Biol. 1972;27 (1):71–83. doi: 10.1016/0012-1606(72)90113-3. [DOI] [PubMed] [Google Scholar]
- Strati K, Lambert PF. Role of Rb-dependent and Rb-independent functions of papillomavirus E7 oncogene in head and neck cancer. Cancer Res. 2007;67 (24):11585–93. doi: 10.1158/0008-5472.CAN-07-3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strati K, Pitot HC, Lambert PF. Identification of biomarkers that distinguish human papillomavirus (HPV)-positive versus HPV-negative head and neck cancers in a mouse model. Proc Natl Acad Sci U S A. 2006;103 (38):14152–7. doi: 10.1073/pnas.0606698103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, Banks L. Human papillomavirus (HPV) E6 interactions with Bak are conserved amongst E6 proteins from high and low risk HPV types. J Gen Virol. 1999;80 ( Pt 6):1513–7. doi: 10.1099/0022-1317-80-6-1513. [DOI] [PubMed] [Google Scholar]
- Thomas M, Glaunsinger B, Pim D, et al. HPV E6 and MAGUK protein interactions: determination of the molecular basis for specific protein recognition and degradation. Oncogene. 2001;20 (39):5431–9. doi: 10.1038/sj.onc.1204719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, Massimi P, Navarro C, et al. The hScrib/Dlg apico-basal control complex is differentially targeted by HPV-16 and HPV-18 E6 proteins. Oncogene. 2005;24 (41):6222–30. doi: 10.1038/sj.onc.1208757. [DOI] [PubMed] [Google Scholar]
- Thomas M, Narayan N, Pim D, et al. Human papillomaviruses, cervical cancer and cell polarity. Oncogene. 2008;27 (55):7018–30. doi: 10.1038/onc.2008.351. [DOI] [PubMed] [Google Scholar]
- Tomaic V, Gardiol D, Massimi P, et al. Human and primate tumour viruses use PDZ binding as an evolutionarily conserved mechanism of targeting cell polarity regulators. Oncogene. 2009;28 (1):1–8. doi: 10.1038/onc.2008.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Howley PM. The bovine papillomavirus E6 oncoprotein interacts with paxillin and disrupts the actin cytoskeleton. Proc Natl Acad Sci U S A. 1997;94 (9):4412–7. doi: 10.1073/pnas.94.9.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tungteakkhun SS, Duerksen-Hughes PJ. Cellular binding partners of the human papillomavirus E6 protein. Arch Virol. 2008;153 (3):397–408. doi: 10.1007/s00705-007-0022-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Pol SB, Brown MC, Turner CE. Association of Bovine Papillomavirus Type 1 E6 oncoprotein with the focal adhesion protein paxillin through a conserved protein interaction motif. Oncogene. 1998;16 (1):43–52. doi: 10.1038/sj.onc.1201504. [DOI] [PubMed] [Google Scholar]
- Veldman T, Liu X, Yuan H, et al. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc Natl Acad Sci U S A. 2003;100 (14):8211–6. doi: 10.1073/pnas.1435900100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248 (4951):76–9. doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- Wiest T, Schwarz E, Enders C, et al. Involvement of intact HPV16 E6/E7 gene expression in head and neck cancers with unaltered p53 status and perturbed pRb cell cycle control. Oncogene. 2002;21 (10):1510–7. doi: 10.1038/sj.onc.1205214. [DOI] [PubMed] [Google Scholar]