

Abstract
Background
Cumulative evidence implicates the epidermal growth factor receptor (EGFR) as an important therapeutic target in head and neck squamous cell carcinomas (HNSCC). The basis for the lack of correlation between EGFR expression in the HNSCC tumor and clinical responses to EGFR inhibitors is incompletely understood. While a variety of mechanisms likely contribute to the effectiveness of EGFR blockade, this review focuses on the biological implications of known EGFR variations and the role of the immune system in mediating clinical responses to EGFR inhibitors.
Methods
A Medline review of articles published in the last 10 years (1999-Present) on EGFR in HNSCC was performed in combination with preliminary data from our laboratories.
Results
Studies published to date suggest no association between the expression of EGFR on HNSCC tumors and clinical responses to EGFR inhibitors. Several mechanisms have been proposed to mediate clinical response to EGFR inhibitors in HNSCC. Cumulative results from our laboratories support the role of several mechanisms, including cellular immune activation and mutated EGFR variants, in contributing to the discrepancy between level of EGFR expression and clinical response to EGFR inhibitors.
Conclusions
The efficacy of EGFR targeted therapies may be mediated, at least in part, by the immune system and the presence of the truncated EGFR variant, EGFRvIII, among other factors. Criteria to identify the subset of patients likely to be responsive to EGFR targeted therapies are needed.
Keywords: head and neck cancer, epidermal growth factor receptor, overexpression, EGFRvIII, mutations, polymorphisms, targeted therapy
Introduction
Despite recent advances in our understanding of the role of molecular and genetic abnormalities in the pathogenesis and clinical course of head and neck squamous cell carcinoma (HNSCC), this disease remains one of the most significant causes of morbidity and mortality among malignancies worldwide. These clinical findings have emphasized the urgency to develop effective therapeutic strategies for the treatment of this disease. The realization that the epidermal growth factor receptor (EGFR) is overexpressed and structurally altered in HNSCC and plays a role in its pathogenesis and clinical course has provided the rationale for the development and implementation of EGFR- targeted therapies. Alterations of EGFR structure and expression have been recently described and may contribute to the resistance of some cancers to current EGFR-targeted therapies (1). In this review we will first describe the characteristics of EGFR and its role in the pathogenesis and clinical course of the disease and then we will discuss the use of EGFR as a target of therapy for HNSCC.
EGFR in Oncogenesis
EGFR, a Type I receptor tyrosine kinase, is involved in a variety of cellular processes including survival and differentiation. The structure of EGFR includes an extracellular ligand-binding domain, a transmembrane segment, and an intracellular kinase domain with five autophosphorylation sites. EGFR is expressed in most epithelial tissues and EGFR knockout mice demonstrate a disorganized hair follicle phenotype, a fuzzy coat and early death (usually within 3 weeks of birth) (2). Dysregulation of EGFR has been widely implicated in epithelial oncogenesis [Table 1 (3-13)]. EGFR is expressed at higher levels in many epithelial malignancies including HNSCC than in the corresponding normal tissues (1). The association between EGFR expression levels in the primary tumor and decreased survival in cancers of the lung, pancreas, colon and head and neck has led to the development of FDA-approved EGFR targeting agents in these malignancies (14-16).
Table 1.
| Tumor Site | Citation |
|---|---|
| Bladder | Wang X et al. (2007) |
| Breast | Milanezi F et al. (2008) |
| Cervical | Fuchs I et al. (2007) |
| CNS | Voelzke WR et al. (2008) |
| Colorectal | Antonacopoulou et al. (2008) |
| Endometrial | Engelsen IB et al. (2008) |
| Esophageal | Wei Q et al. (2007) |
| Gastric | Kim MA et al. (2008) |
| Head + Neck | Sheikh A et al. (2008) |
| Lung | Tang X et al. (2008) |
| Ovarian | Lafky JM et al. (2008) |
Wild-type (wt) EGFR overexpression as a mechanism of oncogenesis is not unique to cancers of the head and neck. Overexpression of this receptor is associated with decreased survival in cancers of the bladder, cervix, esophagus, and ovary (17). A 2005 study by Pedersen et al demonstrated that, in addition to a simple increase in ligand-receptor interaction, wtEGFR overexpression also contributes to tumorigenesis in a ligand-independent manner (18). According to this study, malignant transformation and increased cellular motility may result from EGFR dimerization and constitutive signaling. As is seen in HNSCC, several potential mechanisms give rise to wtEGFR overexpression. We previously reported that transcriptional activation is likely the primary mechanism of EGFR overexpression in HNSCC (19). Results from multiple studies using FISH indicate that gene amplification occurs in 10-17% of HNSCC cases (20-22). EGFR gene amplification has been detected in breast cancers (23), glioblastoma multiforme (24), and non-small cell lung cancers (25), where increased EGFR levels are thought to result primarily, if not exclusively, from increased EGFR gene copy number.
In addition to alterations of the EGFR gene, mutations of the tumor suppressor p53, a common finding in HNSCC, have been shown to result in enhanced EGFR promoter activity (26). Transcriptional upregulation by mutant forms of p53 is thought to occur independently of the enhanced EGFR promoter activity caused by wild type p53. Although the significance of EGFR upregulation by wt p53 is not completely understood, p53 also upregulates transcription at the transforming growth factor alpha (TGF-α locus) (26).
Somatic mutations of the EGFR gene have been documented only rarely in patients with HNSCC, but have been documented in subsets of patients with non-small-cell lung cancer. Termed ‘activating mutations,’ these tyrosine kinase domain modifications are thought to be responsible for enhanced sensitivity to EGFR tyrosine kinase inhibitor (TKI) including gefitinib and erlotinib (27). Recent studies have identified several of these mutations that serve as favorable prognostic indicators and predictors of pharmacologic sensitivity in non-small-cell lung cancer (28).
EGFR Biology in HNSCC
EGFR ligands implicated in HNSCC include epidermal growth factor (EGF), TGF-α, and possibly amphiregulin. All three factors are involved to varying extents in normal cellular growth and differentiation, while TGF-α and amphiregulin are produced by HNSCC cells and have been implicated in dysregulation of these processes and subsequent transformation, EGF is primarily produced by salivary glands. The transformative effect of TGF-α is thought to arise as a consequence of its overexpression by mucosal cells, which typically occurs in conjunction with EGFR overexpression (29). Amphiregulin, a keratinocyte growth factor, has been recently identified as an additional contributor to carcinogenesis due to its apparent co-release with TGF-α and subsequent EGFR activation (30).
Ligand binding by EGFR monomers drives homodimerization or heterodimerization with the aforementioned ErbB family members, resulting in the initiation of downstream survival and proliferation signaling pathways. Among the best-studied pathways activated by EGFR ligand-binding are the Ras-Raf-MEK-Erk/MAPK and phosphotidylinositol-3-kinase (PI3K) pathways. These unique cascades converge with the upregulation of cyclin D1, a key mediator of mitosis and G1 to S-phase progression (Figure 1). EGFR overexpression, a ubiquitous phenomenon in HNSCC, leads to enhanced trafficking through this pathway and contributes to dysregulated growth.
Figure 1.
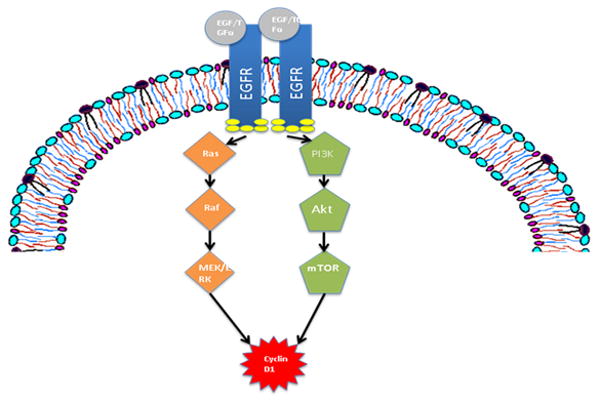
The epidermal growth factor receptor (EGFR) initiates pro-survival and proliferation signaling in the presence of ligand. Ras-Raf-MEK-Erk/MAPK and PI3K pathways converge with the upregulation of Cyclin D1, an important regulator of mitosis.
Studies in HNSCC have demonstrated an inverse relationship between EGFR expression and survival, regardless of treatment, prompting further investigation of the role of EGFR in clinical decision-making (17). The expression of EGFR in normal tissues, such as the skin, may account for the toxicities observed in the setting of EGFR-targeted therapies. Genetically and epigenetically altered forms of EGFR to date have primarily been described in other malignancies. These EGFR alterations appear to be restricted to cancer cells thereby enabling the design of more selective targeted therapeutics. EGFR variants detected in HNSCC include gene amplification of wild-type EGFR (wtEGFR), somatic tyrosine kinase domain mutants (E746_A750del, p.K745R, and p.G796S), and the phenotypically distinct EGFRvIII.
EGFR Polymorphisms in HNSCC
Genetic variations in EGFR may alter protein function, contribute to tumor formation, and possibly alter the therapeutic efficacy of EGFR inhibitors. Previous reports have implicated EGFR intron 1 CA repeat polymorphisms in altering basal transcription rates of EGFR (31, 32). Moreover, in a study involving more than 120 oral cancer cases and control subjects, individuals with fewer CA repeats were found to have increased risk for developing oral cancer (33). In regards to prognosis, the link between EGFR polymorphism and survival has been examined in only 78 HNSCC patients: both the R497K EGFR polymorphism (R521K in current nomenclature) and the CA repeat polymorphism were associated with clinical outcome (34). Similar results have been obtained in other cancers that exhibit EGFR overexpression (3, 35). Furthermore, the extent of EGFR polymorphism has been found to be higher in patients with lung cancer than in controls (36, 37). The EGFR intron CA repeat polymorphism has been associated with differential clinical responses to the EGFR TKI, gefitinib (38). In 2007, Bandres et al described the prognostic significance of four distinct EGFR alleles in patients with HNSCC (34). Two promotor-region polymorphisms, -216 G/T and -191C/A, were found to have no clinical or pathological significance. The R497K polymorphism of exon 13, however, was found to be associated with an increase in disease-specific mortality. This was particularly true of the homozygous Arg/Arg variant, which was present in 53% (40/76) of the study population. The intron 1 (CA)n repeat polymorphism was also found to be of significant prognostic value, as patients harboring 17 CA repeats demonstrated lower disease-specific mortality. These cumulative results demonstrate that genetic variants leading to reduced EGFR expression are associated with increased survival. An association between EGFR polymorphisms and response to EGFR inhibitors in patients with HNSCC has not been reported to date.
EGFR Mutations and Variations in HNSCC
Activating EGFR mutations, like those detected in lung cancer, occur rarely in HNSCC and likely do not mediate clinical responses to EGFR inhibition. Recent studies indicate that the incidence of EGFR mutations in HNSCC differs between ethnic groups, ranging from 0-4% in whites to 7% in Asians (39). Lee et al. (2005) were the first to report a tyrosine kinase domain mutation in HNSCC. The mutation, a somatic in-frame deletion termed E746_A750del, was detected in 3 of 41 Korean HNSCC patients (40). In 2006, Loeffler-Ragg, et al identified a somatic missense mutation (p.K745R) of the ATP binding cleft in one of 100 white patients (41). Another unique missense mutation of the tyrosine kinase domain (p.G796S) was identified in two of 127 white HNSCC patients in 2008 by Schwentner et al (39). At present, E746_A750del, p.K745R, and p.G796S are the only documented tyrosine kinase domain mutations in HNSCC. These mutations are clearly rare in HNSCC and their effect on disease progression and response to pharmacotherapy is unknown.
Extensive investigation of EGFR structure and function has also lead to the identification of several structural variants, some of which have been observed in human malignancies. Wikstrand et al. described the most frequently detected genomic variant, termed EGFRvIII, in detail in 1995 (42). EGFRvIII, a 145kDa protein, which is expressed in 42% of HNSCC tumors (1), results from the deletion of amino acids 6-273 of the wtEGFR extracellular domain. Because the N-terminal signal peptide is spared, targeting and cell membrane insertion occur normally. The unique extracellular domain resulting from this deletion, with its novel glycine residue located at the junction of residues 5 and 274, causes a marked reduction in the binding affinity of monoclonal antibodies (mAbs) raised with wtEGFR. The transmembrane domain of EGFRvIII is thought to be identical to that of the wild-type protein, a hydrophilic sequence of 23 amino acids with a yet-unknown role in receptor function (43). Likewise, the mutant receptor's 542 amino acid intracellular domain is structurally identical to that of wtEGFR. EGFRvIII is unique, however, in its ability to initiate intracellular signaling in the absence of TGF-α via persistent phosphorylation of its protein kinase domain.
EGFRvIII is functionally distinct from its wild type counterpart. Chu et al. have demonstrated two important differences between survival signals initiated by wtEGFR and EGFRvIII: 1) Ligand-dependent wtEGFR signaling requires Her family receptor dimerization, while ligand-independent EGFRvIII signaling has no such requirement, and 2) wtEGFR signaling proceeds through the Ras-Raf-MEK-Erk/MAPK pathway in addition to the phosphotidylinositol-3-kinase (PI3K) pathway, while EGFRvIII signaling appears to proceed exclusively through the PI3K pathway (Figure 3) (44, 45). Following activation by EGFRvIII, the PI3K pathway initiates survival and anti-apoptotic signals that are not subject to the regulatory mechanisms that govern Ras-Raf-MEK-Erk/MAPK signaling (46). Constitutive survival and anti-apoptotic signaling thus appear to be central to the role of EGFRvIII in HNSCC.
Figure 3.
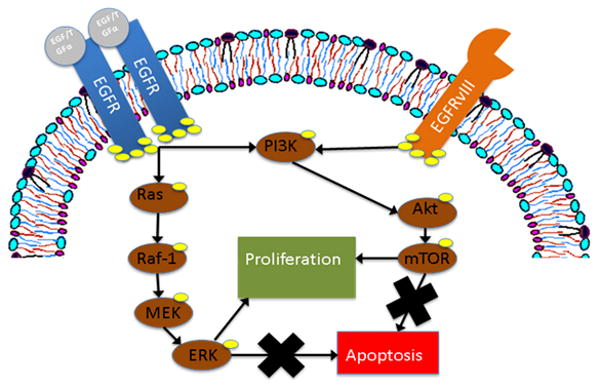
The mutant epidermal growth factor receptor, EGFRvIII, promotes cell survival and proliferation by preferential signaling through the phosphotidyinositol-3-kinase (PI3K) pathway. The wild-type receptor utilizes both the PI3K pathway and the Ras-Raf-MEK pathway. Downstream effectors of both pathways further promote cell survival via inhibition of apoptosis.
To date, expression of the 150 kDa EGFRvIII protein has yet to be documented in the absence of wild- type receptor (1). The phenotypic consequences of this co-expression are thought to include enhanced tumor growth and resistance to wtEGFR-targeted pharmacotherapy (1). Furthermore, recent data suggests that EGFRvIII expression enhances the ability of malignant cells to migrate and invade normal tissues, which are likely to be important events in tumor metastasis (47). Enhanced tumor growth may occur secondary to constitutive signaling through the p-Akt survival pathway. Drug resistance is presumably conferred by the specific alterations to the receptor's intra- and extra-cellular domains. The unique extracellular domain of EGFRvIII has recently become the focus of much research, as immunotherapy targeting this domain is already in development for cancers of the brain (48). This strategy is particularly promising as a therapy for cetuximab-refractory HNSCC which express EGFRvIII, since cetuximab binds with much less affinity to EGFRvIII than to wtEGFR.
EGFR Biology: Implications for Pharmacotherapy
As perhaps the most well-studied member of the ErbB receptor family, EGFR is known to contribute to both normal and neoplastic growth processes in humans. A variety of genomic alterations involving the EGFR gene, including amplifications and incomplete deletions, are now known to be tumorigenic. Current therapeutic strategies, including the only FDA approved pharmacotherapy for HNSCC (cetuximab), have been developed primarily for the treatment of tumors demonstrating wild type EGFR (wtEGFR) overexpression. This strategy of tumorigenesis is not unique to cancers of the head and neck, as wtEGFR overexpression has been documented in many epithelial malignancies. The ubiquity of wtEGFR overexpression in HNSCC, therefore, supports the use of EGFR-targeted therapies in this cancer.
mAbs recognizing determinants expressed on the EGFR extracellular ligand-binding domain represent more specific EGFR targeting approaches than TKIs, which may also block other kinases. However, these agents are unable to discriminate between wtEGFR expressed on normal and malignant tissues; the reactivity of mAbs with normal tissues is likely to account for the cutaneous toxicity observed in patients treated with EGFR-specific mAb-based immunotherapy. For example, systemic administration of toxin-linked antibodies targeted to the extracellular domain of wtEGFR is not possible due to the associated hepatotoxicity (49-51). In a phase III study Pryor et al reported increased toxicity in patients receiving cetuximab as an adjuvant to radiotherapy for HNSCC (52). Side effects included skin reactions and mucositis, reportedly resulting in decreased compliance with scheduled radiation therapy. Unlike mAbs, TKIs lack specificity for wtEGFR. Because these compounds are engineered to have high affinity for tyrosine kinase domains, cross-reactivity is possible with other members of the ErbB receptor family. Documented TKI toxicities include interstitial lung disease, rash, and diarrhea. Although not intrinsic to their design, TKIs have been found to demonstrate some specificity for malignant lung tissue as a consequence of increased affinity for mutated EGFR tyrosine kinase domains (27). Thus, in the case of pure EGFR overexpression, TKIs may provide less specificity than mAbs with a comparable toxicity profile.
EGFR-Targeted Therapy in HNSCC
The overexpression of EGFR in nearly all HNSCCs has led to the development of pharmacotherapy directed against this cell-surface receptor (38). Current strategies include mAbs and TKIs, both of which have demonstrated efficacy against HNSCC lines in preclinical models. Immunotherapies, which include the clinically-tested agent cetuximab (Erbitux®, Bristol-Myers Squibb - New York, NY), are administered parenterally and are highly specific for EGFR. TKIs, often referred to as small molecule inhibitors, are less specific but may offer more convenient oral dosing.
TKIs antagonize normal EGFR signaling by occupying the receptor's intracellular ATP-binding site, thereby inhibiting autophosphorylation (Figure 2). Current investigation of TKIs in head and neck cancer is limited to phase I and phase II trials. The best-studied TKIs (erlotinib and gefitinib) demonstrate modest increases in overall survival and progression-free survival in the treatment of HNSCC (53). In a phase II study in patients with recurrent or metastatic HNSCC, erlotinib treatment as a single agent was associated with an objective response rate of 4.3% with a median progression-free survival of 9.6 weeks (53). The combination of erlotinib plus cisplatin in recurrent or metastatic HNSCC was associated with a median progression-free survival of 3.3 months (54). These cumulative results demonstrate that erlotinib has antitumor activity in HNSCC comparable to standard chemotherapy regimens. Another clinical EGFR TKI, gefitinib, was tested in a phase III trial compared with methotrexate for recurrent HNSCC. In this study, neither 250 mg nor 500 mg dosing of daily gefitinib resulted in improved survival compared with intravenous methotrexate. The convenient oral dosing of TKIs makes them an attractive treatment option for HNSCC, however, the lack of positive phase III data limits their incorporation into a standard treatment approach.
Figure 2.
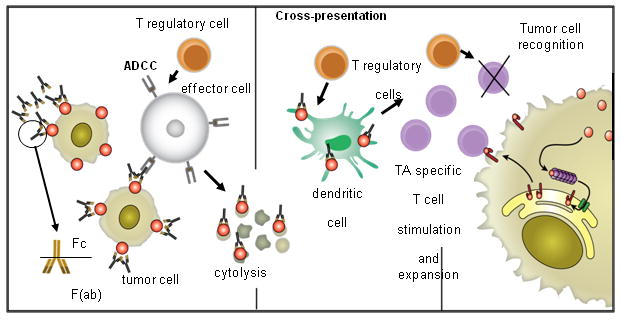
A proposed model of cellular cascades triggered by activation of NK cells with cetuximab-coated HNSCC targets. Cetuximab-mediated NK cell-dependent tumor cell lysis results in the generation of EGFR-cetuximab immune complexes which are taken up by DC, processed and presented to TA-specific T cells. CTL recognize and eliminate tumor cells. T regulatory cells may down-regulate NK activity, DC functions and/or CTL activity, leading to tumor immune escape. In addition, defects in the antigen processing machinery component expression contribute to tumor escape from CTL recognition.
There are currently two FDA-approved mAbs targeting EGFR. Cetuximab is a chimeric IgG1 mAb and panitumumab (Vectibix®, Amgen - Thousand Oaks, CA) is a fully human IgG2a mAb. mAbs recognizing determinants expressed on the extracellular domain of EGFR, such as cetuximab or panitumimab, antagonize normal ligand-receptor interactions and therefore disrupt downstream signaling. In addition mAbs mediate NK cell-dependent lysis of HNSCC cells (Figure 2). Extensive clinical testing of cetuximab has shown this agent to be particularly useful as an adjuvant to primary radiotherapy, with improvements in overall survival, progression-free survival, and duration of locoregional control versus radiation alone in the treatment of HNSCC (55). In the definitive phase III trial that led to FDA-approval of this agent, cetuximab combined with radiation was shown to improve locoregional control (from 14.9 to 24.4 months) and overall survival (from 29.3 to 49.0 months) in patients with locoregionally advanced HNSCC. More recently, cetuximab plus platinum-based chemotherapy was tested in recurrent or metastatic HNSCC where the addition of cetuximab prolonged the median progression-free survival from 3.3 to 5.6 months (56).
The experience acquired from the treatment of a large number of patients in multicenter trials has shown that the treatment is effective in approximately 20% of the patients. At present, the mechanism(s) underlying the clinical response to EGFR-specific mAb-based immunotherapy are poorly understood. Cell-mediated cytotoxicity of target cells triggered by EGFR-specific mAbs appears to play a role in the clinical outcome of colorectal carcinoma patients (57). Our results in vitro are consistent with a similar mechanism in HNSCC patients, a finding which is currently under intense study in clinical trial cohorts (58). The variables influencing the extent of lysis of HNSCC cells by NK cells and EGFR-specific mAbs have been characterized, and have been shown to include the level of EGFR expression, the amount of mAb, and the genotype of the Fcγ receptor (FcγR) which mediates the interactions of NK cells with the mAbs bound to target cells, i.e. FcγR IIIa (59). Interestingly, our data also suggest that NK cell-dependent lysis mediated by cetuximab is not altered in the setting of co-expression of EGFR vIII mutant on HNSCC cells. The discrepancy between the role of the level of EGFR expression in in vitro lysis and in the clinical outcome of patients treated with cetuximab raises the possibility that other mechanisms play a role in vivo. Among them is the possibility that the lysis of HNSCC cells by NK cells and the EGFR-specific mAb cetuximab triggers a series of events which lead to the generation of cytotoxic T lymphocytes (CTL) recognizing tumor antigens expressed on the HNSCC cells. Because the mAbs are most effective when administered in combination with radiotherapy and/or chemotherapy, these factors may also influence the efficacy and mechanism(s) underlying the clinical outcomes observed.
There are few molecules that are exclusively expressed on HNSCC cells compared with normal cells. Since EGFRvIII is expressed exclusively by tumor cells, targeting of EGFRvIII may offer the opportunity of tissue specificity in the treatment of HNSCC. The conjugation of cytotoxic compounds to a mAb specific for the extracellular domain of EGFRvIII has proven efficacious in early animal studies of glioblastoma multiforme (48). This immunotoxin, MR1-1 [MR1-1(dsFv)-PE38KDEL] has been shown to have high specificity, potency and lack of an identified mechanism for resistance in preclinical testing. A phase I trial is presently underway in glioma patients at Duke University using this approach. The anti-tumor effect of these compounds appears not to be exclusively cytotoxic, but rather a combination of direct cytotoxicity due to receptor/toxin endocytosis and an immunotoxin-provoked immune response to malignant tissue (48). Similar success in the use of immunotoxins against EGFRvIII-expressing HNSCC likely depends on several prerequisite conditions: 1) EGFRvIII signaling is the predominant mechanism of resistance to anti-wtEGFR therapy in HNSCC, 2) EGFRvIII expression in HNSCC is comparable to that of CNS tumors, and 3) a similar immunotoxin-induced tumor antigen-specific immune response is observed in non-CNS tissues. In addition to immunotoxins, EGFRvIII-specific mAbs and irreversible EGFR/Her2 kinase inhibitors have shown activity in preclinical cancer models as well as early phase trials of patients whose tumors express EGFRvIII (60, 61).
Side Effects of EGFR Inhibitors in HNSCC
To date, the most common side effect of EGFR inhibitors in HNSCC (including TKI or mAbs) is a cutaneous acneiform rash that when severe, can require dose-reduction or cessation of therapy in addition to specific therapeutic measures to mitigate this toxicity. In the southeastern United States, the presence of IgE specific for galactose-alpha-1,3-galactose has been reported to result in a high incidence of anaphylaxis (infusion reactions) in HNSCC patients treated with cetuximab in this region (62). Infusion reactions (IgE-dependent or –independent) may be prevented by premedication with antihistamines, acetaminophen and/or corticosteroids. EGFR TKIs have been reported to cause gastrointestinal distress in addition to a cutaneous rash.
Clinical Resistance to EGFR Inhibitors in HNSCC
To date, EGFR expression levels in the tumor have not been consistently correlated with response to EGFR inhibitors, either mAb or TKI. Theoretically, resistance can either be present at the time of initial treatment (de novo) or acquired during therapy. For example primary resistance to EGFR TKI in lung cancer patients has been reportedly due to amplification of MET whereas the development of new mutations has been implicated in the setting of acquired resistance (63, 64). The phase III trial of cetuximab that served as the basis for FDA approval in HNSCC demonstrated that despite an effect on the primary tumor, cetuximab did not abrogate recurrence or metastasis. Analysis of primary and recurrent tumors in the setting of EGFR targeted therapies is needed to determine the molecular basis of clinical resistance in HNSCC.
Other potential factors that may contribute to resistance to EGFR inhibitors include: 1) the concentrations of oral TKIs given to patients may not be high enough to block the EGFR kinase activity of tumors, which needs to be balanced with the relative lack of specificity of these agents at higher doses; 2) HNSCC cell lines are quite heterogeneous in their response to EGFR kinase inhibitors in the absence of mutations of the kinase domain implicating other mechanisms that mediate sensitivity; 3) lack of understanding of EGFR “oncogene addiction” in HNSCC despite ubiquitous overexpression of this tyrosine kinase in HNSCC tumors; and 4) conflicting reports to date regarding the sensitivity of EGFRvIII-expressing cells and tumors to EGFR TKI.
Summary and Future Directions
The EGFR is a 170kDa cell-surface protein involved in a variety of cellular processes, including DNA synthesis, proliferation, migration, and adhesion. EGFR variations are thought to contribute to the development of cancers of the head and neck via dysregulation of these processes. Current pharmacotherapeutic agents for HNSCC include the FDA-approved cetuximab, a mAb, and the TKIs gefitinib and erlotinib. Of the known EGFR variations, wtEGFR overexpression and the mutant EGFRvIII are the best studied. The aforementioned agents have demonstrated efficacy against tumors overexpressing wtEGFR, however, EGFRvIII is thought to be an important contributor to the development of cetuximab-resistant and metastatic tumors. Unlike those identified in non-small-cell lung cancer, somatic mutations of the EGFR tyrosine kinase domain are not thought to enhance tumorigenicity nor pharmacosensitivity. The future development of therapeutic agents in HNSCC is likely to include further refinement of TKIs inhibitors and immunotherapeutic targeting of epitopes that are unique to malignant tissues (i.e. EGFRvIII). Selective targeting of altered forms of EGFR may prove particularly useful in the treatment of refractory disease.
Figure 4.
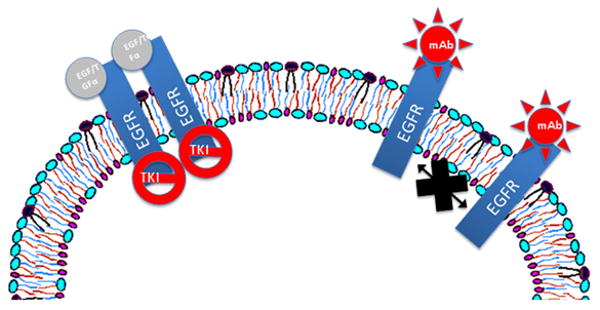
Current EGFR-targeted therapies for HNSCC include mAbs like cetuximab and TKIs like gefitinib and erlotinib. mAbs prevent EGFR ligand-binding, while TKIs inhibit receptor autophosphorylation. Both mechanisms prevent the initiation of survival and proliferation signaling.
Acknowledgments
This work was supported by NIH grants P50 CA097190 and R01 CA77308
References
- 1.Sok JC, Coppelli FM, Thomas SM, et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin Cancer Res. 2006;12(17):5064–73. doi: 10.1158/1078-0432.CCR-06-0913. [DOI] [PubMed] [Google Scholar]
- 2.Hansen LA, Alexander N, Hogan ME, et al. Genetically null mice reveal a central role for epidermal growth factor receptor in the differentiation of the hair follicle and normal hair development. Am J Pathol. 1997;150(6):1959–75. [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X, Zhang S, MacLennan GT, et al. Epidermal growth factor receptor protein expression and gene amplification in small cell carcinoma of the urinary bladder. Clin Cancer Res. 2007;13(3):953–7. doi: 10.1158/1078-0432.CCR-06-2167. [DOI] [PubMed] [Google Scholar]
- 4.Milanezi F, Carvalho S, Schmitt FC. EGFR/HER2 in breast cancer: a biological approach for molecular diagnosis and therapy. Expert Rev Mol Diagn. 2008;8(4):417–34. doi: 10.1586/14737159.8.4.417. [DOI] [PubMed] [Google Scholar]
- 5.Fuchs I, Vorsteher N, Buhler H, et al. The prognostic significance of human epidermal growth factor receptor correlations in squamous cell cervical carcinoma. Anticancer Res. 2007;27(2):959–63. [PubMed] [Google Scholar]
- 6.Voelzke WR, Petty WJ, Lesser GJ. Targeting the epidermal growth factor receptor in high-grade astrocytomas. Curr Treat Options Oncol. 2008;9(1):23–31. doi: 10.1007/s11864-008-0053-5. [DOI] [PubMed] [Google Scholar]
- 7.Antonacopoulou AG, Tsamandas AC, Petsas T, et al. EGFR, HER-2 and COX-2 levels in colorectal cancer. Histopathology. 2008;53(6):698–706. doi: 10.1111/j.1365-2559.2008.03165.x. [DOI] [PubMed] [Google Scholar]
- 8.Engelsen IB, Stefansson IM, Beroukhim R, et al. HER-2/neu expression is associated with high tumor cell proliferation and aggressive phenotype in a population based patient series of endometrial carcinomas. Int J Oncol. 2008;32(2):307–16. [PubMed] [Google Scholar]
- 9.Wei Q, Chen L, Sheng L, Nordgren H, Wester K, Carlsson J. EGFR, HER2 and HER3 expression in esophageal primary tumours and corresponding metastases. Int J Oncol. 2007;31(3):493–9. [PubMed] [Google Scholar]
- 10.Kim MA, Lee HS, Lee HE, Jeon YK, Yang HK, Kim WH. EGFR in gastric carcinomas: prognostic significance of protein overexpression and high gene copy number. Histopathology. 2008;52(6):738–46. doi: 10.1111/j.1365-2559.2008.03021.x. [DOI] [PubMed] [Google Scholar]
- 11.Sheikh MS, Carrier F, Johnson AC, Ogdon SE, Fornace AJ., Jr Identification of an additional p53-responsive site in the human epidermal growth factor receptor gene promotor. Oncogene. 1997;15(9):1095–101. doi: 10.1038/sj.onc.1201264. [DOI] [PubMed] [Google Scholar]
- 12.Tang X, Varella-Garcia M, Xavier AC, et al. Epidermal growth factor receptor abnormalities in the pathogenesis and progression of lung adenocarcinomas. Cancer Prev Res (Phila Pa) 2008;1(3):192–200. doi: 10.1158/1940-6207.CAPR-08-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lafky JM, Wilken JA, Baron AT, Maihle NJ. Clinical implications of the ErbB/epidermal growth factor (EGF) receptor family and its ligands in ovarian cancer. Biochim Biophys Acta. 2008;1785(2):232–65. doi: 10.1016/j.bbcan.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Morgan S, Grandis JR. ErbB receptors in the biology and pathology of the aerodigestive tract. Exp Cell Res. 2009;315(4):572–82. doi: 10.1016/j.yexcr.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galizia G, Lieto E, Ferraraccio F, et al. Prognostic significance of epidermal growth factor receptor expression in colon cancer patients undergoing curative surgery. Ann Surg Oncol. 2006;13(6):823–35. doi: 10.1245/ASO.2006.05.052. [DOI] [PubMed] [Google Scholar]
- 16.Ueda S, Ogata S, Tsuda H, et al. The correlation between cytoplasmic overexpression of epidermal growth factor receptor and tumor aggressiveness: poor prognosis in patients with pancreatic ductal adenocarcinoma. Pancreas. 2004;29(1):e1–8. doi: 10.1097/00006676-200407000-00061. [DOI] [PubMed] [Google Scholar]
- 17.Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37 4:S9–15. doi: 10.1016/s0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- 18.Pedersen MW, Pedersen N, Damstrup L, et al. Analysis of the epidermal growth factor receptor specific transcriptome: effect of receptor expression level and an activating mutation. J Cell Biochem. 2005;96(2):412–27. doi: 10.1002/jcb.20554. [DOI] [PubMed] [Google Scholar]
- 19.Rubin Grandis J, Zeng Q, Tweardy DJ. Retinoic acid normalizes the increased gene transcription rate of TGF-alpha and EGFR in head and neck cancer cell lines. Nat Med. 1996;2(2):237–40. doi: 10.1038/nm0296-237. [DOI] [PubMed] [Google Scholar]
- 20.Temam S, Kawaguchi H, El-Naggar AK, et al. Epidermal growth factor receptor copy number alterations correlate with poor clinical outcome in patients with head and neck squamous cancer. J Clin Oncol. 2007;25(16):2164–70. doi: 10.1200/JCO.2006.06.6605. [DOI] [PubMed] [Google Scholar]
- 21.Koynova DK, Tsenova VS, Jankova RS, Gurov PB, Toncheva DI. Tissue microarray analysis of EGFR and HER2 oncogene copy number alterations in squamous cell carcinoma of the larynx. J Cancer Res Clin Oncol. 2005;131(3):199–203. doi: 10.1007/s00432-004-0627-y. [DOI] [PubMed] [Google Scholar]
- 22.Freier K, Joos S, Flechtenmacher C, et al. Tissue microarray analysis reveals site-specific prevalence of oncogene amplifications in head and neck squamous cell carcinoma. Cancer Res. 2003;63(6):1179–82. [PubMed] [Google Scholar]
- 23.Bhargava R, Gerald WL, Li AR, et al. EGFR gene amplification in breast cancer: correlation with epidermal growth factor receptor mRNA and protein expression and HER-2 status and absence of EGFR-activating mutations. Mod Pathol. 2005;18(8):1027–33. doi: 10.1038/modpathol.3800438. [DOI] [PubMed] [Google Scholar]
- 24.Wong AJ, Bigner SH, Bigner DD, Kinzler KW, Hamilton SR, Vogelstein B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc Natl Acad Sci U S A. 1987;84(19):6899–903. doi: 10.1073/pnas.84.19.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki S, Dobashi Y, Sakurai H, Nishikawa K, Hanawa M, Ooi A. Protein overexpression and gene amplification of epidermal growth factor receptor in nonsmall cell lung carcinomas. An immunohistochemical and fluorescence in situ hybridization study. Cancer. 2005;103(6):1265–73. doi: 10.1002/cncr.20909. [DOI] [PubMed] [Google Scholar]
- 26.Ludes-Meyers JH, Subler MA, Shivakumar CV, et al. Transcriptional activation of the human epidermal growth factor receptor promoter by human p53. Mol Cell Biol. 1996;16(11):6009–19. doi: 10.1128/mcb.16.11.6009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 28.Riely GJ. The use of first-generation tyrosine kinase inhibitors in patients with NSCLC and somatic EGFR mutations. Lung Cancer. 2008;60 2:S19–22. doi: 10.1016/S0169-5002(08)70101-6. [DOI] [PubMed] [Google Scholar]
- 29.Grandis JR, Tweardy DJ. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993;53(15):3579–84. [PubMed] [Google Scholar]
- 30.Zhang Q, Thomas SM, Xi S, et al. SRC family kinases mediate epidermal growth factor receptor ligand cleavage, proliferation, and invasion of head and neck cancer cells. Cancer Res. 2004;64(17):6166–73. doi: 10.1158/0008-5472.CAN-04-0504. [DOI] [PubMed] [Google Scholar]
- 31.Gebhardt F, Burger H, Brandt B. Modulation of EGFR gene transcription by a polymorphic repetitive sequence--a link between genetics and epigenetics. Int J Biol Markers. 2000;15(1):105–10. doi: 10.1177/172460080001500120. [DOI] [PubMed] [Google Scholar]
- 32.Etienne-Grimaldi MC, Pereira S, Magne N, et al. Analysis of the dinucleotide repeat polymorphism in the epidermal growth factor receptor (EGFR) gene in head and neck cancer patients. Ann Oncol. 2005;16(6):934–41. doi: 10.1093/annonc/mdi189. [DOI] [PubMed] [Google Scholar]
- 33.Kang D, Gridley G, Huang WY, et al. Microsatellite polymorphisms in the epidermal growth factor receptor (EGFR) gene and the transforming growth factor-alpha (TGFA) gene and risk of oral cancer in Puerto Rico. Pharmacogenet Genomics. 2005;15(5):343–7. doi: 10.1097/01213011-200505000-00010. [DOI] [PubMed] [Google Scholar]
- 34.Bandres E, Barricarte R, Cantero C, et al. Epidermal growth factor receptor (EGFR) polymorphisms and survival in head and neck cancer patients. Oral Oncol. 2007;43(7):713–9. doi: 10.1016/j.oraloncology.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 35.Dubey S, Stephenson P, Levy DE, et al. EGFR dinucleotide repeat polymorphism as a prognostic indicator in non-small cell lung cancer. J Thorac Oncol. 2006;1(5):406–12. [PubMed] [Google Scholar]
- 36.Zhang W, Stabile LP, Keohavong P, et al. Mutation and polymorphism in the EGFR-TK domain associated with lung cancer. J Thorac Oncol. 2006;1(7):635–47. [PubMed] [Google Scholar]
- 37.Choi JE, Park SH, Kim KM, et al. Polymorphisms in the epidermal growth factor receptor gene and the risk of primary lung cancer: a case-control study. BMC Cancer. 2007;7:199. doi: 10.1186/1471-2407-7-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim S, Grandis JR, Rinaldo A, Takes RP, Ferlito A. Emerging perspectives in epidermal growth factor receptor targeting in head and neck cancer. Head Neck. 2008;30(5):667–74. doi: 10.1002/hed.20859. [DOI] [PubMed] [Google Scholar]
- 39.Schwentner I, Witsch-Baumgartner M, Sprinzl GM, et al. Identification of the rare EGFR mutation p.G796S as somatic and germline mutation in white patients with squamous cell carcinoma of the head and neck. Head Neck. 2008;30(8):1040–4. doi: 10.1002/hed.20831. [DOI] [PubMed] [Google Scholar]
- 40.Lee JW, Soung YH, Kim SY, et al. Somatic mutations of EGFR gene in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2005;11(8):2879–82. doi: 10.1158/1078-0432.CCR-04-2029. [DOI] [PubMed] [Google Scholar]
- 41.Loeffler-Ragg J, Witsch-Baumgartner M, Tzankov A, et al. Low incidence of mutations in EGFR kinase domain in Caucasian patients with head and neck squamous cell carcinoma. Eur J Cancer. 2006;42(1):109–11. doi: 10.1016/j.ejca.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 42.Wikstrand CJ, Hale LP, Batra SK, et al. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res. 1995;55(14):3140–8. [PubMed] [Google Scholar]
- 43.Pedersen MW, Meltorn M, Damstrup L, Poulsen HS. The type III epidermal growth factor receptor mutation. Biological significance and potential target for anti-cancer therapy. Ann Oncol. 2001;12(6):745–60. doi: 10.1023/a:1011177318162. [DOI] [PubMed] [Google Scholar]
- 44.Chu CT, Everiss KD, Wikstrand CJ, Batra SK, Kung HJ, Bigner DD. Receptor dimerization is not a factor in the signalling activity of a transforming variant epidermal growth factor receptor (EGFRvIII) Biochem J. 1997;324(Pt 3):855–61. doi: 10.1042/bj3240855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moscatello DK, Montgomery RB, Sundareshan P, McDanel H, Wong MY, Wong AJ. Transformational and altered signal transduction by a naturally occurring mutant EGF receptor. Oncogene. 1996;13(1):85–96. [PubMed] [Google Scholar]
- 46.Moscatello DK, Holgado-Madruga M, Emlet DR, Montgomery RB, Wong AJ. Constitutive activation of phosphatidylinositol 3-kinase by a naturally occurring mutant epidermal growth factor receptor. J Biol Chem. 1998;273(1):200–6. doi: 10.1074/jbc.273.1.200. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki S, Thomas SM, Sen M, et al. Epidermal growth factor variant III mediates head and neck cancer cell motility and invasion via STAT3 activation. Submitted, Oncogene. 2009 doi: 10.1038/onc.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ochiai H, Archer GE, Herndon JE, 2nd, et al. EGFRvIII-targeted immunotoxin induces antitumor immunity that is inhibited in the absence of CD4+ and CD8+ T cells. Cancer Immunol Immunother. 2008;57(1):115–21. doi: 10.1007/s00262-007-0363-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pavlovskis OR, Voelker FA, Shackelford AH. Pseudomonas aeruginosa exotoxin in mice: histopathology and serum enzyme changes. J Infect Dis. 1976;133(3):253–9. doi: 10.1093/infdis/133.3.253. [DOI] [PubMed] [Google Scholar]
- 50.Schumann J, Angermuller S, Bang R, Lohoff M, Tiegs G. Acute hepatotoxicity of Pseudomonas aeruginosa exotoxin A in mice depends on T cells and TNF. J Immunol. 1998;161(10):5745–54. [PubMed] [Google Scholar]
- 51.Morgan AC, Jr, Manger R, Pearson JW, et al. Immunoconjugates of Pseudomonas exotoxin A: evaluation in mice, monkeys, and man. Cancer Detect Prev. 1991;15(2):137–43. [PubMed] [Google Scholar]
- 52.Pryor DI, Porceddu SV, Burmeister BH, et al. Enhanced toxicity with concurrent cetuximab and radiotherapy in head and neck cancer. Radiother Oncol. 2009;90(2):172–6. doi: 10.1016/j.radonc.2008.09.018. [DOI] [PubMed] [Google Scholar]
- 53.Soulieres D, Senzer NN, Vokes EE, Hidalgo M, Agarwala SS, Siu LL. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J Clin Oncol. 2004;22(1):77–85. doi: 10.1200/JCO.2004.06.075. [DOI] [PubMed] [Google Scholar]
- 54.Siu LL, Soulieres D, Chen EX, et al. Phase I/II trial of erlotinib and cisplatin in patients with recurrent or metastatic squamous cell carcinoma of the head and neck: a Princess Margaret Hospital phase II consortium and National Cancer Institute of Canada Clinical Trials Group Study. J Clin Oncol. 2007;25(16):2178–83. doi: 10.1200/JCO.2006.07.6547. [DOI] [PubMed] [Google Scholar]
- 55.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354(6):567–78. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 56.Vermorken JB, Mesia R, Rivera F, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359(11):1116–27. doi: 10.1056/NEJMoa0802656. [DOI] [PubMed] [Google Scholar]
- 57.Levy EM, Sycz G, Arriaga JM, et al. Cetuximab-mediated cellular cytotoxicity is inhibited by HLA-E membrane expression in colon cancer cells. Innate Immun. 2009;15(2):91–100. doi: 10.1177/1753425908101404. [DOI] [PubMed] [Google Scholar]
- 58.Bibeau F, Lopez-Crapez E, Di Fiore F, et al. Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol. 2009;27(7):1122–9. doi: 10.1200/JCO.2008.18.0463. [DOI] [PubMed] [Google Scholar]
- 59.Lopez-Albaitero A, Lee SC, Morgan S, et al. Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol Immunother. 2009;58(11):1853–64. doi: 10.1007/s00262-009-0697-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scott AM, Lee FT, Tebbutt N, et al. A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc Natl Acad Sci U S A. 2007;104(10):4071–6. doi: 10.1073/pnas.0611693104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ji H, Zhao X, Yuza Y, et al. Epidermal growth factor receptor variant III mutations in lung tumorigenesis and sensitivity to tyrosine kinase inhibitors. Proc Natl Acad Sci U S A. 2006;103(20):7817–22. doi: 10.1073/pnas.0510284103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chung CH, Mirakhur B, Chan E, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med. 2008;358(11):1109–17. doi: 10.1056/NEJMoa074943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cappuzzo F, Janne PA, Skokan M, et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol. 2009;20(2):298–304. doi: 10.1093/annonc/mdn635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Balak MN, Gong Y, Riely GJ, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12(21):6494–501. doi: 10.1158/1078-0432.CCR-06-1570. [DOI] [PubMed] [Google Scholar]