

Abstract
Interleukin-2 receptor (IL-2R) signaling regulates tolerance and immunity. Here, we review recent work concerning the structure, signaling, and function of the IL-2R, emphasizing the contribution of IL-2 for T cell-dependent activity in vivo. IL-2R signaling influences two discrete aspects of immune responses by CD8+ T cells, terminal differentiation of effector cells in primary responses, and aspects of memory recall responses. IL-2 also delivers essential signals for thymic development of regulatory T (Treg) cells and later to promote their homeostasis and function. Each of these outcomes on T effector and Treg cells requires distinct amounts of IL-2R signaling, with low IL-2R signaling sufficient for many key aspects of Treg cells. Thus, tolerance is readily maintained and favored with limited IL-2.
Introduction
Adaptive immune responses depend on antigen activation of B and T lymphocytes into antibody-producing plasma and T effector cells. Responses to antigen are in turn limited in part by the capacity of conventional T cells to also develop into suppressive induced T regulatory (iTreg) cells through upregulation of the transcription factor Foxp3. The high diversity of the adaptive immune response also places a never-ending risk of responding to autoantigens. Thymic-derived natural Treg (nTreg) cells represent a major mechanism to keep potential autoreactive T lymphocytes in check.
The development of mature immunocompetent lymphoid cells from less-committed precursors, their subsequent antigen-driven immune responses, and the suppression of these and unwanted autoreactive responses are highly dependent and regulated by cytokines (including interleukin-2 [IL-2], IL-4, IL-7, IL-9, IL-15, and IL-21) that utilize receptors in the common γ-chain (γc) family (Rochman et al., 2009). IL-2 is no exception from this paradigm because it is essential for thymic development of nTreg cells and critically regulates several key aspects of mature peripheral Treg and antigen-activated conventional T cells. Because of its potent T cell growth factor activity in vitro, IL-2 has been extensively studied in part because this activity offered a potential means to directly boost immunity, e.g., in cancer and AIDS-HIV patients, or a target to antagonize unwanted responses, e.g., transplantation rejection and autoimmune diseases. Although in vitro studies with IL-2 provided a strong rationale for these studies, the function of IL-2 in vivo is clearly much more complex as first illustrated in IL-2-deficient mice, where a rapid lethal autoimmune syndrome, not lack of immunity, was observed (Sadlack et al., 1993, 1995). Similar observations were later made when the gene encoding IL-2Rα (Il2ra) and IL-2Rβ (Il2rb) were individually ablated (Suzuki et al., 1995; Willerford et al., 1995). We now know that the reason for this lethal autoimmunity is because of failed thymic development of nTreg cells and their impaired peripheral homeostasis (Malek, 2008). Indeed, a singular transfer of Treg cells into newborn IL-2Rβ-deficient mice results in life-long protection from this potent autoimmunity (Malek et al., 2002), providing the first direct evidence that the lack of mature Treg cells is the primary driver of this lethal disease. Moreover, by correcting the Treg cell defect, the role of IL-2 in T cell immunity can now be studied in model systems where all conventional T cells remain IL-2R-deficient but without the severe complications of widespread autoimmune disease associated with IL-2- or IL-2R-deficient mice. Such studies leave a clear picture that antigen-driven T cell expansion and contraction does not markedly depend on IL-2. Rather, aspects of T memory development (Bachmann et al., 2007; Williams et al., 2006) and terminal differentiation of T effector cells (Kalia et al., 2010; Pipkin et al., 2010) require IL-2R signaling.
The past few years have resulted in rethinking the contribution of IL-2 within the immune system to one that is a pivotal player in regulation of both tolerance and immunity. Much past work has established the structure, functional, and signaling consequences of the IL-2-IL-2R interaction. This review will primarily focus on our current understanding of the immunobiology of IL-2-IL-2R in regulation of T cell-mediated immunity in vivo but first touch on recent developments in IL-2R structure and signaling.
IL-2-IL-2R Binding
IL-2 is a 4-bundle α-helical protein of 15 kDa predominately produced by antigen-activated T cells that binds to a high-affinity receptor consisting of three subunits, IL-2Rα (CD25), IL-2Rβ (CD122), and γc (CD132), that are readily found on Treg cells and recently antigen-activated T lymphocytes. The three IL-2R subunits are not preformed as stable heterotrimers. The crystal structure of IL-2 bound to the IL-2R provides molecular support for a model where IL-2 drives assembly of the high-affinity trimeric receptor (Stauber et al., 2006; Wang et al., 2005). IL-2 is first captured by IL-2Rα through a large hydrophobic binding surface surrounded by a polar periphery that results in a relatively weak interaction (Kd 10−8 M) with rapid on-off binding kinetics. The IL-2Rα-IL-2 binary complex leads to a very small conformational change in IL-2 that promotes association with IL-2Rβ through a distinct polar interaction between IL-2 and IL-2Rβ. Notably, the extracellular domain IL-2Rα does not interact with IL-2Rβ, but rather, the binary complex of IL-2Rα-IL-2 appears to present in cis IL-2 to IL-2Rβ. The ternary IL-2Rα-IL-2Rβ-IL-2 complex then recruits γc through a weak interaction with IL-2 and a stronger interaction with IL-2Rβ to produce a stable quaternary high-affinity IL-2R (Kd 10−11 M).
The binding characteristics of the IL-2-IL-2Rα have important implications for the function of this cytokine receptor in vivo. Even though the structural topography of the IL-2-IL-2Rα complex is permissive for transpresentation, i.e., IL-2-IL-2Rα on one cell presents IL-2 to IL-2Rβ and γc on another cell, the very rapid dissociation of IL-2 from IL-2Rα does not favor this mode of receptor binding. Thus, for IL-2-dependent responses, both the production of IL-2 and expression of the IL-2R must temporally occur within the same microenvironments. Consistent with this idea, IL-2 is directionally secreted within the immunological synapse for use by IL-2R-bearing cells (Huse et al., 2006; Sabatos et al., 2008). Moreover, as production of IL-2 and IL-2Rα are highly dependent upon TCR stimulation, IL-2-dependent responses by Treg and T effector cells in vivo are placed under tight physiologic and antigen-dependent control.
The direct use of IL-2 as an agonist to bind the IL-2R and modulate immune responses therapeutically has been problematic due to its short half-life (15–30 min) and high toxicity. Anti-IL-2-IL-2 complexes have recently been shown to also exert potent IL-2 agonist activity in vivo at much lower doses of IL-2, minimizing toxicity associated with high dose IL-2 immunotherapy (Boyman et al., 2006). The mechanism of action of these anti-IL-2 complexes is not fully characterized but is partially explained by substantially increasing IL-2 half-life (Létourneau et al., 2010; Phelan et al., 2008). IL-2 complexed to the S4B6 and JES-6.1 mAbs show preferential activation of cells that express only IL-2Rβ and γc versus the high-affinity IL-2R, expressing the α, β, and γc subunits, respectively. The quaternary structure of IL-2-IL-2R readily provides a model to explain the agonist properties of anti-IL-2-IL-2 complexes. The S4B6 mAb likely binds to an epitope on IL-2 that recapitulates the way IL-2Rα binds to IL-2. As such, S4B6-IL-2 complexes could readily display IL-2 to IL-2Rβ and γc in a manner analogous to the IL-2Rα-IL-2 binary complex. Consistent with this view, S4B6-IL-2 complexes are protected from interacting with IL-2Rα. In contrast, JES-6.1 likely binds to a distinct surface of IL-2, leaving exposed those regions of IL-2 that engage IL-2Rα, IL-2Rβ, and γc. Of note, the agonist activity of only this latter complex depends on FcR-mediated transpresentation to the high-affinity IL-2R (Létourneau et al., 2010).
IL-2R Signaling
The formation of the high-affinity quaternary IL-2-IL-2R complex leads to signal transduction through the tyrosine kinases Jak1 and Jak3, which are associated with IL-2Rβ and γc, respectively (Nelson and Willerford, 1998). Three tyrosine residues within the cytoplasmic tail of IL-2Rβ are phosphorylated to promote recruitment of the adaptor Shc (Y338 human; Y341 mouse), leading to activation of the MAPK and PI-3K kinase pathways, and predominately the Stat5 transcription factor (Y392 and Y510 human; Y398 and Y505 mouse), resulting in Stat5-dependent gene regulation (Gaffen, 2001). The quaternary IL-2-IL-2R complex is rapidly internalized, where IL-2, IL-2Rβ, and γc are rapidly degraded, but IL-2Rα is recycled to the cell surface (Hémar et al., 1995; Yu and Malek, 2001). Thus, those functional activities that require sustained IL-2R signaling require a continued source of IL-2 to engage IL-2Rα and form additional IL-2-IL-2R signaling complexes.
The above model gives the impression that activation of MAPK and PI3K through Shc versus Stat5 are modular and independent of each other. However, the factors controlling these receptor associated components and subsequent signaling are clearly much more complex and not fully appreciated. Several recent studies demonstrate that the IL-2R is not modular and solely based on Shc and Stat5 recruitment to these tyrosine residues. First, truncated IL-2Rβ, which is only permissive for recruitment of Shc, does not substantially activate downstream mediators of Akt unless some Stat5 activation is provided, presumably due to a requirement for induction of a Stat5-dependent gene (Lockyer et al., 2007). Second, full IL-2-dependent activation of the PI3K pathway or Stat5 does not occur when the respective key tyrosine residues are mutated to phenylalanine (Yu et al., 2009). Perhaps even more surprising, IL-2-dependent tyrosine phosphorylation of Stat5 readily occurs, albeit to an initial reduced amount that was not sustained, in T cells expressing IL-2Rβ in which all three critical tyrosine residues are mutated to phenylalanine (Yu et al., 2009). This finding suggests that these tyrosine residues may function to stabilize an IL-2R signaling complex for sustained activation.
IL-2R subunits are not randomly associated with the cell membrane but are selectively found in lipid rafts (Cho et al., 2010; Vámosi et al., 2004). Such colocalization facilitates IL-2R-dependent oligomerization and signaling. More recently, the Wiskot-Aldrich syndrome (WAS) interacting protein, WIP, has been implicated in regulating IL-2R-dependent Stat5 activation and IL-2-dependent proliferation in T cells (Le Bras et al., 2009). WIP functions in cytoskeleton organization. In antigen-activated T cells, WIP associates with the WAS protein such that it stabilizes the resulting immunological synapse. Thus, a role for WIP in IL-2R signaling may be to recruit IL-2R subunits into the immune synapse to receive IL-2 and/or it may provide a more ordered membrane structure required for the assembly of signaling molecules associated with the cytoplasmic tails of IL-2Rβ and γc.
IL-2 dependent signal transduction in T cells is quantitatively distinctive. Although CD4+ and CD8+ T effector and CD4+ Treg cells similarly activate Stat5 after IL-2-IL-2R binding, each of these cell populations distinctively activates the S6 kinase, a downstream target of the PI3K-Akt and mTOR kinase pathway (Yu et al., 2009). Intracellular staining with a mAb to phosphoryated S6 indicates much higher IL-2-dependent activation of S6 in CD8+ versus CD4+ effector T cells. This finding correlates with much more extensive IL-2-dependent T cell expansion by activated CD8+ T cells. Moreover, activation of S6 in CD8+ T cells is resolved into two populations, i.e., high versus low activation of S6. This finding suggests that IL-2R signaling may mark CD8+ T cell subsets with distinct properties and might reflect their propensity to develop into effector versus memory cells.
IL-2 signaling in Treg cells differs from T effectors in that Treg cells essentially fail to activate the PI3K-Akt pathway through high expression of PTEN (phosphatase and tensin homolog) protein (Bensinger et al., 2004; Walsh et al., 2006). Indeed, even though Treg cells express the high-affinity IL-2R, increased PTEN amounts at least partially account for the failure of Treg cells to directly undergo IL-2-dependent proliferation in vitro. Accordingly, blocking PTEN in Treg cells restored direct IL-2 responsiveness (Walsh et al., 2006). Moreover, antagonizing PI3K, Akt, or mTOR or alternatively promoting these pathways (either with drugs or genetically) favor or oppose Treg cell production, respectively (Crellin et al., 2007; Delgoffe et al., 2009; Liu et al., 2009; Patton et al., 2006; Sauer et al., 2008). With respect to IL-2R signaling, in practical terms, Treg cells behave as if only the Stat5 pathway is activated and are resistant to rapamycin, which acts on mTOR, a downstream target of the PI3K pathway. Thus, the use of rapamycin to inhibit the proliferation of contaminating conventional effector cells facilitates the growth of nTreg and iTreg cells in vitro (Battaglia et al., 2006; Kang et al., 2008; Long and Buckner, 2008).
Expression of IL-2 and IL-2R by Effector and Regulatory T cells
Expression of the high-affinity IL-2R is critical for endowing T cells to respond to low concentrations of IL-2 that is transiently available in vivo. IL-2Rα expression is absent on naive and memory T cells but is induced after antigen activation. IL-2Rβ is constitutively expressed by NK, NKT, and memory CD8+ T cells but is also induced on naive T cells after antigen activation. γc is much less stringently regulated and is constitutively expressed by all lymphoid cells. Once the high-affinity IL-2R is induced by antigen, IL-2R signaling upregulates the expression of IL-2Rα in part through Stat5-dependent regulation of Il2ra transcription (Kim et al., 2001). This process represents a mechanism to maintain expression of the high-affinity IL-2R and sustain IL-2 signaling while there remains a source of IL-2. Most Treg cells are characterized by apparent constitutive expression of the high-affinity IL-2R. Nevertheless, autoantigens likely provide TCR-dependent activation signals for its expression, while IL-2 actively maintains Treg IL-2Rα expression (Yu et al., 2009). This latter process is likely the result of direct action of Stat5 and Foxp3, the latter in complex with the transcription factor Runx1, on Il2ra transcription (Ono et al., 2007; Zheng et al., 2007b). Treg cells, therefore, utilize distinct and overlapping mechanisms with effector cells to maintain IL-2Rα expression.
Although very high expression of IL-2Rα is readily achieved upon activation of conventional T cells in vitro, when T cells from mouse or man are directly evaluated ex vivo, the IL-2Rαlo phenotype predominately associates with effector T cells (Kalia et al., 2010; Miyara et al., 2009). During an acute antigen-driven immune response in vivo, high expression of CD25 is only very transiently observed, consistent with a short duration where they receive optimal TCR and IL-2R signals to maintain expression of the high-affinity IL-2R. In contrast, the IL-2Rαhi phenotype marks Treg cells. Thus, purification of CD4+ CD25hi T cells represents a strong enrichment for Foxp3+ Treg cells. The relatively high levels of the high affinity IL-2R by Treg cells is consistent with receiving chronic autoantigen and IL-2 dependent signals. Physiologically, Treg cells preferentially utilize IL-2 after antigenic challenge (O’Gorman et al., 2009).
The capacity to secrete IL-2 by effector cells represents a hall-mark feature which distinguishes them from Treg cells. Direct repression of Il2 by Foxp3 interacting with Runx1 and NFAT prevents Treg cells from producing IL-2 (Ono et al., 2007; Wu et al., 2006). Upon activation, conventional CD4+ and CD8+ T cells, the latter to a lesser extent, produce IL-2. During an immune response, IL-2 is consumed in an autocrine or paracrine manner by cells in close proximity bearing the high-affinity IL-2R. The induction of IL-2 production is under strict transcriptional and posttranscriptional regulation that depends on activation of NF-κB and NFAT, among other transcriptional activators, and AU-rich repeats in the 3′ untranslated region of IL-2 mRNA marking it for rapid degradation (Jain et al., 1995). Additionally, IL-2 transcription is repressed in activated T cells by T-bet and Blimp-1 (Gong and Malek, 2007; Hwang et al., 2005; Martins et al., 2008). Naive T cells lack expression of Blimp-1, but it is upregulated by antigen- and IL-2-dependent signaling (Kallies et al., 2006; Martins et al., 2006). Blimp-1 expression is particularly high in chronically activated T cells, which are characterized by their inability to produce IL-2 (Shin et al., 2009). Thus, Blimp-1-mediated IL-2 repression represents a negative feedback loop to limit IL-2R signaling and may represent a failsafe mechanism to limit IL-2-dependent responses in situations where antigen, the primary inductive signal for IL-2 and IL-2R, is not eliminated, potentially avoiding catastrophic expansion of the antigen-reactive T cells.
IL-2 in the Development of Primary Immunity
In response to infectious agents, T cells undergo clonal expansion and differentiate into effector T cells. Once the pathogen is cleared, a contraction period follows where the majority of the T cells undergo apoptosis. A small proportion survives and persists as memory T cells that protect the host long-term against reinfection with the same pathogen (Harty and Badovinac, 2008; Williams and Bevan, 2007). This expansion phase readily and substantially occurs without IL-2 in vivo. This point has been convincingly established using Il2−/− and Il2ra−/− mice in settings that avoid autoimmune complications related to the nonredundant role of IL-2 for Treg cell development (Malek, 2008). Furthermore, IL-2-independent primary responses resolve infectious challenges indicative of robust T cell expansion and effector development (Yu et al., 2003). What accounts for substantial IL-2-independent expansion has not yet been established and still occurs after blocking all γc-dependent cytokines (Decaluwe et al., 2010; Jin et al., 2006; Lantz et al., 2000). IL-2-independent clonal expansion and effective primary immunity may be the result of TCR signaling on its own or in conjunction with inflammatory signals. Nevertheless, when substantial IL-2 is present, this level of clonal expansion is augmented (D’Souza and Lefrançois, 2003; Yu et al., 2003). Some evidence also indicates that IL-2 driven responses are more prominent within tissues at the site of infection (D’Souza et al., 2002).
Two subpopulations of CD8+ T cells, short-lived effector cells (SLECs) and memory precursor effector cells (MPECs), are present in the expansion phase in response to viral and bacterial infections (Kaech and Wherry, 2007). SLECs and MPECs are distinguished from each other by their phenotype, level of effector activity, and potential to give rise to memory cells (Figure 1). High IL-2R signaling is essential for an optimal number of SLECs, including their differentiation into terminal effector (TE) cells, that maximally express effector molecules such as IFN-γ and granzyme B, but produce low amounts of IL-2 (Kalia et al., 2010; Mitchell et al., 2010; Obar et al., 2010; Pipkin et al., 2010). This step depends upon the transcriptional repressor Blimp-1, which promotes the SLEC and TE fate and directly represses IL-2 transcription (Kallies et al., 2009; Rutishauser et al., 2009). Thus, even though vigorous protective immunity develops without IL-2, optimal primary responses are shaped by IL-2R signaling.
Figure 1. A Model of IL-2 and Its Contribution to CD8+ T Cell Immune Responses.

After activation, CD8+ T cells undergo clonal expansion yielding a large population of effector cells that readily develop without IL-2. Most SLECs and TE require high amounts of IL-2 to differentiate into TE cells. MPECs are IL-2 independent and give rise to CM and EM precursors. A low amount of IL-2 during the expansion phase is required for proper development of long-lived EM, but not CM, cells. Contraction proceeds largely in the absence of IL-2R signaling. The surviving EM and CM cells form the pool CD8+ memory cells. Memory recall responses depend on IL-2 in two ways for optimal development of EM cells during priming and repopulation of the secondary recalled effector pool that expresses maximal functional activity. The colored heat map (white, IL-2 independent; orange, IL-2 levels, low to high) for the IL-2 requirement applies to an IL-2R signaling gradient observed through various stages of CD8+ T cell differentiation.
Analogous to the expansion phase of the primary response, contraction of antigen-specific T cells proceeds normally in the absence of IL-2R signaling. This corresponds to the very transient nature of CD25 expression, which is undetectable at the peak of the expansion phase, rendering the cells unresponsive to IL-2 (Kalia et al., 2010). However, increased IL-2Rβ expression is retained on most CD8+ T cells at the peak of the response, rendering them responsive to IL-15, a cytokine that also utilizes IL-2Rβ and γc for signal transduction. MPECs receive survival signals through both the IL-7R and IL-15R, whereas SLECs lack IL-7R expression and become dependent only on IL-15. However, IL-15 does not sustain SLEC maintenance or homeostatic turnover (Joshi et al., 2007), probably because of unavailability of the cytokine in their microenvironment. Consistent with this interpretation, agonist IL-2-anti-IL-2 or IL-15-anti-IL15 complexes improve the survival and limits contraction of SLECs (Blattman et al., 2003; Rubinstein et al., 2008; Yajima et al., 2006). Thus, early memory cells derived from MPECs preferentially survive the contraction phase, leading to memory formation, while SLECs and the remaining MPECs ultimately undergo apoptosis because of low Bcl-2 expression and increased amounts of Bim (Prlic and Bevan, 2008; Sanjabi et al., 2009).
IL-2 in Supporting T Cell Memory
After contraction, T effector memory (Tem) (IL-7Rαhi, CCR7lo, CD62Llo, KLRG-1lo, IL-2lo) and T central memory (Tcm) (IL-7Rαhi, CCR7hi, CD62Lhi, KLRG-1neg/lo, IL-2hi) cells are found (Kaech and Wherry, 2007). In vitro exposure of activated CD4+ or CD8+ T cells to IL-2 promoted their development into memory cells after transfer into mice, the latter forming Tcm cells, suggesting a role for IL-2R signaling in memory development (Carrio et al., 2004; Dooms et al., 2007). Direct support for a required role of IL-2 for CD8+ T memory came from mixed bone marrow chimeras yielding a mixture of wild-type (WT) and Il2ra−/− T lymphocytes. After challenged with lymphocytic choriomeningitis virus (LCMV) or Listeria monocytogenes (LM), similar primary responses and memory development were noted for the WT and IL2ra−/− T cells. However, the Il2ra−/− memory CD8+ T cells failed to mount a response upon antigenic rechallenge (Williams et al., 2006). This striking defect was not because of failed expansion of the rechallenged cells, but because of their enhanced cell death and decreased development into IFN-γ-producing cells. Remarkably, providing IL-2 during the primary response yielded normal memory recall responses. This result has led to the concept that IL-2R signaling during the primary response programs subsequent memory recall responses.
What constitutes such IL-2-dependent programming is poorly defined at the cellular level and unexplored at the molecular level. Memory cells are readily detected in the absence of IL-2R signaling, which tends to rule out a role for IL-2 in programming memory survival, for example, by simply upregulation of molecules such as IL-7R or Bcl-2. One possibility is that, during the primary response, IL-2 alters the chromatin structure of certain effector and/or survival genes that are necessary for the recall response, but so far there are no data for this notion. At the cellular level in responses to LCMV by CD8+ T cells, the absence of IL-2R signaling resulted in impaired development of Tem cells, leading to poor recall expansion and secondary effectors with an altered cytokine profile and a phenotype that resembles central memory cells (Mitchell et al., 2010). However, in another related study, only a minimal effect was seen on recall expansion to LM and vaccinia virus, but development of secondary SLECs was impaired (Obar et al., 2010). Thus, along with shaping the Tem pool, IL-2 may contribute to the development of SLECs and TE cells during secondary rechallenge in an analogous manner it promotes these populations in the primary response.
Another issue to consider is Il2ra−/− T cells in the mixed chimera model are competent to respond to IL-15 through transpresentation to IL-2Rβ and γc. IL-15 might redundantly function with IL-2 in memory programming or survival. Such redundancy might vary based on the nature, dose, or route of the infectious agent. The potential for such redundancy also depends on the extent that IL-2 and IL-15 provide quantitative and qualitative equivalent signals for memory programming. Consistent with this hypothesis, in the response to LM, IL-15 not only provides survival signals and preserves homeostatic proliferation of CD8 memory T cells but also appears to play an important role in shaping components of the memory pool, including Tem cells (Sandau et al., 2010; Sanjabi et al., 2009). This result suggests that IL-15 may under some cases redundantly function with IL-2 in memory programming. However, recent work directly addressing the overlap of IL-2 and IL-15 in the response to LCMV indicates that IL-2 promotes effector and effector memory formation, whereas the dominant role for IL-15 is to promote the survival of effector and effector memory T cells (Mitchell et al., 2010). Thus, the potential for some overlap between IL-2 and IL-15 does not does not imply that both cytokines are synonymous during an immune response.
IL-2R Signaling and Effector versus Memory Cell-Fate Decision
As shown in Figure 1, it is not simply the presence or absence of IL-2 that influences T cell immune responses, but the amount of IL-2R signaling. CD8+ T cells cultured in high versus low IL-2 resulted in proliferation that exhibited characteristics of effector and memory cells (Manjunath et al., 2001; Pipkin et al., 2010). Of note, IL-15 functioned in a manner very similar to low IL-2 in supporting memory-like cells, consistent with its weaker signal transduction through IL-2Rβ and γc (Carrio et al., 2004; Cornish et al., 2006). In an analogous manner during an acute viral infection, virus-specific CD8+ T cells that received less IL-2 because of low expression of CD25 have increased persistence and better recall responses than CD25hi cells (Kalia et al., 2010). These data are consistent with a model whereby more extensive IL-2 signaling promotes most SLECs and TE cells whereas low IL-2 or IL-15 supports MPECs and acquisition of memory cell traits.
IL-2-dependent regulation of the transcriptional repressor Blimp-1 represents one mediator controlling the effector versus memory cell fate during the primary response. High Blimp-1 expression is supported by extensive antigen stimulation and strong IL-2R signaling (Gong and Malek, 2007; Kallies et al., 2006; Martins et al., 2006). These conditions also lead to high amounts of T-bet and low amounts of the IL-7R, Eomes, and Bcl-6, which readily support an effector differentiation program leading to increased expression of granzyme B and perforin and the inability to produce IL-2, the latter because of direct repression by Blimp-1 (Intlekofer et al., 2005; Joshi et al., 2007; Martins et al., 2008; Pipkin et al., 2010). Correspondingly, in CD8+ T cells that lack Blimp-1, the development of SLECs is impaired and the resulting effector cells produce increased IL-2 (Kallies et al., 2009; Rutishauser et al., 2009). In contrast, low IL-2R signaling is associated with increased expression of Bcl-6, IL-7Rα, and Eomes and low expression of Blimp-1 and T-bet, conditions that support the development of memory CD8 T cells. Blimp-1 represses Bcl-6, and reciprocally, Bcl-6 represses Blimp1 expression (Martins and Calame, 2008). Thus, high versus low IL-2R signaling leads to TE versus memory cell fates in part by controlling the amounts of these two key transcriptional regulators. IL-2-dependent immunity, therefore, results in optimal development of SLECs, TE, and memory cells. An interesting issue, therefore, concerns the environmental cues and niches that apparently simultaneously support these distinct cell-fate outcomes that require distinct amounts of IL-2R signaling.
The upstream mediators of IL-2R signaling that activate or repress these transcription factors are not known. Rapamycin-sensitive mTOR, downstream of the PI3K-AKT pathway, represents a candidate because it modulates the expression of both T-bet and Eomes (Delgoffe et al., 2009; Rao et al., 2010). Agents that inhibit mTOR block T effector cell production while increasing memory CD8+ T cells (Araki et al., 2009; Pearce et al., 2009), suggesting that memory development may be independent of IL-2-dependent activation of the PI3K-AKT pathway. Notably, this pathway is impaired in T cells activated with IL-15 (Cornish et al., 2006), whose signaling readily supports CD8+ memory T cell development. In addition, mTOR is also necessary for CD4+ effector Th1, Th2, and Th17 cells while repressing Treg cell formation (Delgoffe et al., 2009; Haxhinasto et al., 2008). Thus, much remains to be learned concerning the transcriptional programs controlling effector and memory cell fates, including how they relate to IL-2 and IL-15 signaling thresholds and their receptor proximal pathways.
IL-2 and nTreg Cell Development
The early observation that CD25 is a relatively good, albeit not exclusive, marker for Treg cells was not a coincidence because these cells express and depend on signals from the high-affinity IL-2R for their development, homeostasis, and function. The first evidence that IL-2R functioned during nTreg cell development was that thymic-targeted expression of IL-2Rβ in IL-2Rβ-deficient mice restored thymic and peripheral Treg numbers and prevented their severe autoimmunity (Malek et al., 2002). After Foxp3 was defined as a specific marker of Treg cells, an important role for IL-2R signaling in the thymus was questioned because Foxp3+ T cells were detected in IL-2- and IL-2R-deficient mice (D’Cruz and Klein, 2005; Fontenot et al., 2005b). However, defective Treg cell production was also noted in Stat5-deficient mice (Snow et al., 2003; Yao et al., 2007), and this effect was attributed to failed IL-2R signaling in the thymus and periphery (Burchill et al., 2007), emphasizing the important role of this signaling pathway in Treg cells. Moreover, in normal mice, thymic Treg numbers are reduced after treatment with IL-2 mAb (Bayer et al., 2005). Further study revealed that IL-2R-deficient mice contained immature Treg cells that express a lower amount of Foxp3 and lack expression of CD25 (Bayer et al., 2007). This finding is important because low expression of Foxp3 is not able to induce the Treg cell program (Wan and Flavell, 2007). Additionally, forced expression of Foxp3 in IL-2Rβ-deficient mice restored Treg cell production and prevented autoimmunity (Soper et al., 2007), functionally linking IL-2R signaling with control of Foxp3 expression. Failed action of IL-10, TGF-β, and CTLA4 have also been linked to the lymphoproliferative disorder and severe autoimmunity associated with absent IL-2R signaling (Brandenburg et al., 2008; Carrier et al., 2007; Hwang et al., 2004; Tsuji-Takayama et al., 2008). These all represent activities of Treg cell-mediated suppression and may represent functional defects ascribed to impaired immature Treg cells.
As shown in Figure 2, developing thymocytes that received instructive TCR and CD28 signals developed into Treg cells after signaling through the IL-2R (Burchill et al., 2008; Lio and Hsieh, 2008; Tai et al., 2005). CD28 may also contribute to Treg cell development by maximizing IL-2 production. Thus, there is now ample evidence for an IL-2-dependent step during nTreg development that in part is due to direct regulation of Foxp3 and CD25 expression through Stat5 binding to respective enhancer elements of these genes (Yao et al., 2007; Zorn et al., 2006) to influence growth and survival signaling by the maturing Treg cells. The extent IL-2 controls other properties of Treg cells remains to be determined.
Figure 2. Model of IL-2-Dependent Control of Thymic Treg Development.

IL-7 acts at either the DN or DP stage to help open Foxp3. TGFβ provides a survival signal to rescue negatively selected cells from apoptosis, permitting cells to divert to the Treg cell lineage. IL-2 acts through activation of Stat5 as a terminal developmental signal to promote maturation of immature Foxp3lo precursor cells.
Although some Foxp3-bearing cells are detected in the thymus from mice deficient in IL-2 or TGF-βR, Treg cells are absent in mice double-deficient for IL-2 and TGFβRI (Liu et al., 2008), demonstrating that these two cytokines cooperate in Treg cell development. TGF-β promotes Treg cell development by providing survival signal by constraining negative selection through increasing Bcl-2 and decreasing Bim expression (Ouyang et al., 2010). Low amount of Foxp3 associated with immature Treg cells in IL-2-IL-2R-deficient mice is also attributed in part to signaling from another γc-dependent cytokine because Foxp3+ T cells are absent within the reduced T cell pool of γc-deficient mice (Fontenot et al., 2005b). Mice doubly deficient in IL-2- and IL-7-dependent signaling recapitulate the Treg cell phenotype of γc-deficient animals (Bayer et al., 2008; Vang et al., 2008), definitively establishing IL-7 as the other γc cytokine to function in nTreg cell development. IL-7 also activates Stat5. The development of immature Treg cells because of only the IL-7R signaling might be attributed to less efficient Stat5-driven Treg cell instructive signals at the same developmental step regulated by IL-2. However, domain swapping experiments of IL-2Rβ and IL-7Rα in vivo do not favor this notion because a chimeric receptor consisting of the extracytoplasmic domain of IL-7R and the cytoplasmic domain of IL-2Rβ did not support Treg cell maturation in IL-2Rβ-deficient mice (Yu and Malek, 2006). Treg cell maturation was supported, however, by a chimeric receptor consisting of extracytoplasmic domain of IL-2Rβ and the cytoplasmic domain of IL-7Rα, demonstrating that IL-7R signaling is competent for Treg cell development when it is ligated by IL-2. Thus, too little IL-7 is available in the niche where Treg cells mature even when signaling is normalized to that delivered by IL-2.
These data support a role for niche-dependent IL-2R signaling during thymic development where Stat5 signaling must occur at a late stage, i.e., after positive selection, of thymic development because Foxp3+ cells are not seen until after this development stage (Figure 2). The thymus contains very few IL-2 producing cells, which are most numerous at the cortical-medullary junction, suggesting that this thymic location may be where IL-2 instructive signals are delivered. In contrast, the thymus also produces limiting amounts of IL-7 (Munitic et al., 2004), which is most prominent within the cortex and cortical-medullary junction where essential IL-7R signaling occurs at the CD4CD8 “double-negative” stage and later for commitment to the CD8 lineage (Alves et al., 2009; Park et al., 2010). If IL-7R signaling is not synonymous with the IL-2 niche, the paradox is how does solely signaling through the IL-7R lead to immature Foxp3lo thymocytes. At this juncture, our favored explanation is that IL-7-dependent Stat5 activation at the “double-negative” or at the DP stage prior to the Treg commitment stage begins to open the Foxp3 locus, but Foxp3 expression is delayed until sufficient TCR signals promote the Foxp3lo phenotype. Even though IL-2R signaling on its own, without IL-7R, supports Treg cell maturation, such Foxp3lo cells are expected to be exquisitely sensitive to limiting IL-2 within the thymus to ensure Treg cell development and self-tolerance. Such high sensitivity of Treg cells to low IL-2R signaling is readily illustrated by the capacity of lL-2Rβ signaling mutants to fully support Treg thymic maturation despite only low transient IL-2-dependent Stat5 activation (Yu et al., 2009).
IL-2 in nTreg Cell Homeostasis and Function
IL-2 is also the major cytokine contributing to the homeostasis of peripheral Treg cells. This homeostatic activity is first noted in neonatal mice where IL-2 rapidly amplifies the numbers of Treg cells in the lymph nodes (Bayer et al., 2005). Conventional T cell development occurs during fetal development, while Treg cell development is delayed until the early neonatal period in mice (Fontenot et al., 2005a). Furthermore, this first wave of conventional T cell development occurs more quickly than in adults, with a contracted period of negative selection, resulting in a higher probability of thymic export of autoreactive T cells. By day 3 of life, the neonatal lymph nodes contain essentially only conventional T cells, but by day 7, the proportion of Treg cells is at the normal 5%–10% of CD4+ T lymphocytes. This rapid increase in Treg cells probably represents a necessary catch-up phase to suppress the autoreactive T cells already present in the periphery of neonatal mice. IL-2 from autoreactive T cells represents the likely source of IL-2 to fuel this Treg expansion (Setoguchi et al., 2005) and helps to select the most favorable Treg TCR specificities through adaptive homeostasis.
The homeostatic proliferative rate of peripheral Treg cells in the adult lymphoid compartment is set at a much higher rate than conventional naive or memory T cells (Bayer et al., 2007). Treg cells, therefore, are constantly sensing their environment for autoantigens and IL-2 to maintain their numbers to balance an equally high death rate. The involvement of IL-2R signaling in Treg cell homeostasis is illustrated by lower Treg cell numbers after anti-IL-2R blockade (Bayer et al., 2005; Setoguchi et al., 2005). Importantly, this reduction in Treg cell numbers leads to increased instances of autoimmune disease in autoimmune-prone strains (Setoguchi et al., 2005). Spontaneous autoimmunity in NOD mice is genetically dependent upon several autoimmune Idd loci, and one of these (Idd3) maps to the IL-2 gene. One aspect of the Idd3 region is somewhat lower production of IL-2, which contributes to autoimmune-mediated diabetes though impaired maintenance of Treg cells, particularly in the inflamed pancreas (Yamanouchi et al., 2007). Thus, IL-2 therapy using anti-IL-2-IL-2 complexes directed at the high-affinity IL-2R, which increases Treg cell numbers, protects NOD mice from diabetes and experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis (Tang et al., 2008; Webster et al., 2009). However, there is a fine balance for IL-2 therapy favoring tolerance over autoimmunity because excessive IL-2 signaling accelerated type 1 diabetes in NOD mice or favored auto-aggressive memory T cells over Treg cells to a neoautoantigen (Tang et al., 2008; Waithman et al., 2008).
Expression profiling of Treg cells with and without impaired IL-2R signaling showed enrichment of genes involved in cellular growth and death, consistent with a role for IL-2 in homeostasis (Fontenot et al., 2005b; Yu et al., 2009). Similar to Treg cell development, mice that harbor mutant IL-2Rs with impaired, but not absent, Stat5 activation contain a normal number and proportion of peripheral Treg cells (Yu et al., 2009). Such mice are also protected from lethal autoimmunity but exhibit symptoms of organ-specific autoimmunity as they age. These findings indicate that relatively normal Treg cell homeostasis is maintained with suboptimal IL-2R signaling, but susceptibility to autoimmune disease increases. Normal Treg cell numbers in a setting of impaired IL-2R signaling are attributed to impaired homeostatic proliferation that is compensated for by enhanced survival (Bayer et al., 2007). However, in a competitive setting in the periphery, Treg cells with lower IL-2R signaling poorly compete with WT Treg cells. This finding further supports the critical role of IL-2 in shaping Treg cell homeostasis and suggests that other yet unidentified factors cooperate with low Stat5 activation to maintain an outwardly normal peripheral Treg cell compartment.
A main function of Foxp3 is to reinforce and maintain Treg cell suppressive function (Gavin et al., 2007). IL-2 through induction of Stat5, which directly contributes to Foxp3 transcription, is intimately linked to Treg cell fitness not only at the level of homeostasis but also through broadly maintaining the suppressive program. Interfering with IL-2 signaling, therefore, represents an obvious risk for autoimmunity. For example, an increase in protein phosphatase N2 (PTPN2), a negative regulator of IL-2R signaling, lowers IL-2R signaling, leading to lower levels of Foxp3 in Treg cells from T1D patients (Long et al., 2010). Furthermore, polymorphisms linked to IL-2, IL-2Rα, and IL-2Rβ are associated with type 1 diabetes, celiac diseases, multiple sclerosis, Grave’s disease, and rheumatoid arthritis (Gregersen and Olsson, 2009; Todd, 2010).
The IL-2-IL-2R genetic risk for autoimmunity is not the result of absent IL-2R signaling but may represent increased sensitivity to IL-2 by autoreactive cells, promoting effector potency and/or memory programming or impaired responsiveness to IL-2 by Treg cells, lowering their suppressive potential. Normal Foxp3 expression is found in mice with mutant IL-2Rs, which readily explains overt proper Foxp3-dependent Treg cell fitness (Yu et al., 2009). Indeed, gene-expression profiling of Treg cells with impaired IL-2R signaling revealed not only normal amounts of Foxp3, but normal expression of TGF-β and CTLA4, key mediators of Treg cell suppression. An important conclusion from this result is that Treg cells are generally resistant to substantial interference with IL-2R signaling. Thus, how impaired IL-2 signaling represents a risk for autoimmune disease is more complex than simply interfering with IL-2-dependent regulation of Foxp3. Interestingly, this same gene profiling also revealed important IL-2-dependent gene expression changes in Treg cells, and many genes were identified that required more stringent IL-2R signaling (Yu et al., 2009). Globally, 20% of these differentially expressed genes overlapped with the Treg cell signature (Hill et al., 2007), further linking IL-2R signaling with important aspects of Treg cells independent of Foxp3 expression. Moreover, IL-10 and granzyme B, two other mediators of Treg cell suppression, were lower. These tendencies may contribute to development of organ specific autoimmunity in this mouse model and potentially reflects factors that contribute to development of human autoimmune disease.
IL-2-Dependent Adaptive Homeostasis
The inability of Treg cells to produce IL-2 renders them dependent on paracrine IL-2 for their development, homeostasis, and function. The main IL-2-producing cells are activated conventional T cells, although some evidence also suggests that dendritic cells (DCs) produce IL-2 (Granucci et al., 2003), but the physiological role of this IL-2 has not been definitively established. It seems unlikely that T cells activated by foreign antigen are the main source of IL-2 for nTreg cells because the availability of IL-2 for Treg cells becomes unpredictable and may require IL-2 to act at a distance from Treg cells where the concentration of cytokine becomes limiting. Rather, autoreactive T cells themselves represent a potential consistent and predictable source of IL-2 (Setoguchi et al., 2005). In such a model (Figure 3), autoreactive T cells recognize autoantigens presented by a DC and begin to produce IL-2. Treg cells specific for the same or distinct self-specificity are recruited to the same DC and readily bind IL-2 and induce IL-2R signaling to promote the necessary IL-2-dependent functions. Treg cells, in turn, suppress autoreactive T cells, inhibiting further activation and IL-2 production, thereby maintaining immune tolerance. Such a mechanism links the autoreactive T cell and suppressive Treg cell to bring them in close proximity to receive IL-2 and mediates suppression through direct effects on the DC and/or the autoreactive T cell. This mechanism selects Treg cells with the best TCR specificities to readily survive and, as such, adapts Treg cells toward suppression of relevant autoreactive T cells. If DCs also produce lL-2, either on their own or with IL-2 from autoreactive T cells, Treg and autoreactive T cells remain closely linked within the same cellular microenvironment to promote IL-2-dependent Treg cell functions and immune tolerance.
Figure 3. Model of Adaptive Homeostasis of Peripheral Treg Cells.
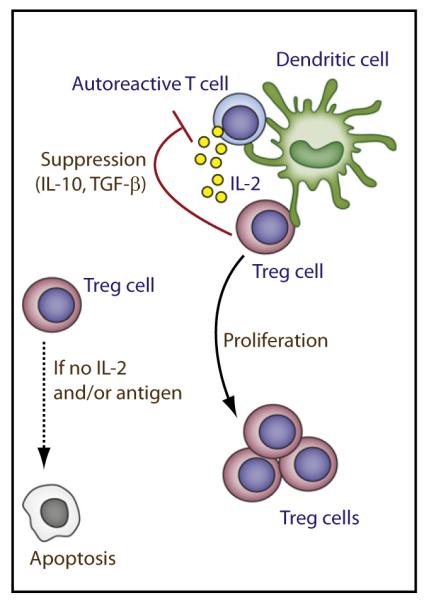
Focusing Treg and autoreactive T cells (Tauto) though recognition of autoantigens on DCs place them in close proximity for the Treg cells to receive an IL-2-dependent signal. This interaction promotes homeostatic proliferation and survival of Treg cells and enhances aspects of their functional program. The Treg and autoreactive cells may recognize the same self antigen or distinct self antigens on the DC. Although the TCR antigenic specificity of Tauto and Treg may be identical, their fine specificity, i.e., V region sequences, may vary. Treg cells that fail to receive TCR and/or IL-2 signaling for an extended period of time undergo apoptosis. Thus, Treg cells adapt and are selectively maintained to self antigens that, in part, depend on the autoreactive T cells that they suppress.
Two lines of evidence support the above model, which we coin as adaptive Treg cell homeostasis. First, this model mirrors the basis by which Treg cells suppress T cell responses in vitro, i.e., blockade of IL-2 secretion by activated T cells (Thornton et al., 2004). Such inhibition depends on initial IL-2 from the activated T cells that act on the Treg cells, which, in turn, suppress IL-2 transcription by the activated cells through a yet poorly defined molecular mechanism. In this setting, feedback blockade of IL-2 transcription results in low amounts of IL-2. The main IL-2-dependent activities of Treg cells, therefore, are tethered to weak and transit amount of IL-2R signaling. Indeed, the observation that Treg cell development, homeostasis, and function are largely normal in vivo even after extensive mutations of key IL-2Rβ cytoplasmic tyrosine residues (Yu et al., 2009), which markedly interfere with receptor proximal signaling, is consistent with this view. Second, IL-2 is essential for WT Treg cell expansion and their maintenance after transfer into neonatal IL-2Rβ-deficient mice (Malek et al., 2002). In this setting, the TCR specificities of the donor Treg cells undergo extensive reshaping and narrowing. Moreover, this adaptation of the TCR repertoire varies greatly between each recipient while still controlling autoimmunity (Adeegbe et al., 2010). This result links the requirement for IL-2 with a variable host component, which is easily accounted for by distinct specificities expressed by autoreactive T cells. Indeed, the TCR repertoire of individual untreated IL-2- or IL-2Rβ-deficient mice exhibit distinct spectratyping patterns, consistent with outgrowth of unique clones of autoreactive T cells (Adeegbe et al., 2010; Zheng et al., 2008a).
The high expression of the high-affinity IL-2R by Treg cells favors their utilization of IL-2 over the adjacent autoreactive T cells. This preferential consumption of IL-2 has been suggested to represent another key mechanism by which Treg cells mediate suppression and immune tolerance (Pandiyan et al., 2007). However, as discussed above, antigen-driven T cell activation, expansion, and effector differentiation readily occurs in vivo without IL-2-IL-2R. Therefore, depriving IL-2 from autoreactive T cells is not likely to be sufficient on its own to effectively suppress their response. Relevant to this point, the severe lethal autoimmunity associated with IL-2Rβ-deficient mice, which contain auto-aggressive T cells that cannot respond to IL-2 and IL-15, are “cured” from this disease by WT Treg cells (Malek et al., 2002). Tolerance in this setting readily occurs yet consumption of IL-2 by the Treg cells is irrelevant. Thus, tolerance is critically dependent upon suppressive mechanisms independent of passive utilization of IL-2. Nevertheless, preferential Treg-mediated IL-2 consumption may favor immune tolerance through interfering with terminal effector development or memory generation.
IL-2, iTreg Cells, and Lineage Stability
There is now compelling evidence that peripheral conventional T cells develop or convert to Foxp3+ iTreg cells, which is more prominent in mucosal tissues such as the gut and lungs (Izcue et al., 2009). Environmental cues, primarily mediated by cytokines, sensed by activated T cells importantly dictate whether a cell adopts an effector or regulatory cell fate. In vitro experiments place IL-2 in conjunction with TGF-β as an important mediator of iTreg cell production (Davidson et al., 2007; Zheng et al., 2007a). IL-2 also functions through Stat5 activation to promote Foxp3 expression in iTreg cells, but distinct conserved noncoding regions of Foxp3 are remodeled in iTreg versus nTreg cells (Zheng et al., 2010), raising the possibility of a distinct contribution by IL-2R signaling in these lineage choices. The vitamin A metabolite retinoic acid and TGFβ also readily promote iTreg cells in a Stat5-independent manner, supporting the notion that there is an IL-2-independent pathway for iTreg cells (Elias et al., 2008).
Substantial data support the notion that Th17 and Treg cell-fate choices are interrelated (Littman and Rudensky, 2010), and IL-2R signaling may contribute to this decision. First, IL-2 promotes iTreg cell development, but reciprocally, IL-2-induced Stat5 antagonism enhances Th17 cell development (Laurence et al., 2007). Although IL-2 inhibits Th17 cell development, this does not occur in the presence of IL-1 (Kryczek et al., 2007). Once generated, however, iTreg cells are resistant to Th17 cell conversion by IL-6 (Zheng et al., 2008b). Thus, the cell-fate choice between Th17 and iTreg cells is particularly sensitive to amounts of IL-2 and inflammatory cytokines. Second, Foxp3+ CD25− Treg cells have more plasticity than Foxp3+ CD25+ to dedifferentiate into Th17 effector cells (Komatsu et al., 2009; Zhou et al., 2009). CD25− Treg cells cannot respond to IL-2, suggesting that failed IL-2R signaling contributes to their plasticity. Lastly, the greater plasticity of Dicer-deficient Treg cells may be in part because of impaired miR-155 modulation of SOCS1, which decreases Treg sensitivity to IL-2R signaling (Liston et al., 2008; Zhou et al., 2008). Thus, how aberrant IL-2R signaling leads to autoimmunity requires direct evaluation of its role in promoting auto-aggressive T effector cells and plasticity in Treg cells.
The role of IL-2 in vivo for iTreg cells is less clear. Data are limited and only suggestive concerning a required role for IL-2 in iTreg cell production in vivo. One study showed that providing IL-2 to IL-2-deficient peripheral T cells resulted in production of CD4+CD25+ T cells with suppressive properties (Furtado et al., 2002). However, this finding might simply represent post-thymic maturation of nTreg cells by instructive IL-2R-dependent signals. Another study showed IL-2-dependent conversion of Rag1−/− TCR transgenic T cells after transferred to recipients that ubiquitously expressed the relevant antigen as a transgene (Knoechel et al., 2005). However, this extensive antigen stimulation may not parallel physiological settings. Both studies did not assess Foxp3 directly or examine gut-associated lymphoid tissues, hall-marks of iTreg cells.
Concluding Remarks
The in vivo requirements for IL-2R signaling by effector and memory CD8+ T cells and nTreg cells are known with some precision, but less is understood concerning effector CD4+ T cells, iTreg cells, and T effector lineage plasticity. Furthermore, the molecular basis by which IL-2 shapes immune responses and tolerance is only beginning to be defined. Signal transduction through the IL-2R was intensively explored over 10 years ago, which defined the main receptor proximal pathways and biological outcomes in vitro. Comparatively less is known concerning how the high-affinity IL-2R is ordered within the plasma membrane, how a stable signaling complex is assembled, and the essential IL-2-dependent targets in vivo. It is not just the presence or absence of IL-2R signaling, but graded signaling thresholds that control IL-2-dependent responses. A potential fruitful area of future investigation may be to define the basis by which discrete quantitative and qualitative aspects of IL-2-dependent signaling differentially program Treg and T effector cells. Such studies should put on firmer ground, the basis by which unique and overlapping mechanisms are shaped by IL-2 in Treg, terminal differentiated effector, and memory cells and lead to rational design of immunotherapy to block or enhance immune response to self or foreign antigen.
ACKNOWLEDGMENTS
We thank O. Umland for critically reading this manuscript. Our work is supported by National Institutes of Health grants (R01CA45957; RO1AI055815; P01CA109094; 3R56AI40114).
REFERENCES
- Adeegbe D, Matsutani T, Yang J, Altman NH, Malek TR. CD4(+) CD25(+) Foxp3(+) T regulatory cells with limited TCR diversity in control of autoimmunity. J. Immunol. 2010;184:56–66. doi: 10.4049/jimmunol.0902379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves NL, Richard-Le Goff O, Huntington ND, Sousa AP, Ribeiro VS, Bordack A, Vives FL, Peduto L, Chidgey A, Cumano A, et al. Characterization of the thymic IL-7 niche in vivo. Proc. Natl. Acad. Sci. USA. 2009;106:1512–1517. doi: 10.1073/pnas.0809559106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann MF, Wolint P, Walton S, Schwarz K, Oxenius A. Differential role of IL-2R signaling for CD8+ T cell responses in acute and chronic viral infections. Eur. J. Immunol. 2007;37:1502–1512. doi: 10.1002/eji.200637023. [DOI] [PubMed] [Google Scholar]
- Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J. Immunol. 2006;177:8338–8347. doi: 10.4049/jimmunol.177.12.8338. [DOI] [PubMed] [Google Scholar]
- Bayer AL, Yu A, Adeegbe D, Malek TR. Essential role for interleukin-2 for CD4(+)CD25(+) T regulatory cell development during the neonatal period. J. Exp. Med. 2005;201:769–777. doi: 10.1084/jem.20041179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer AL, Yu A, Malek TR. Function of the IL-2R for thymic and peripheral CD4+CD25+ Foxp3+ T regulatory cells. J. Immunol. 2007;178:4062–4071. doi: 10.4049/jimmunol.178.7.4062. [DOI] [PubMed] [Google Scholar]
- Bayer AL, Lee JY, de la Barrera A, Surh CD, Malek TR. A function for IL-7R for CD4+CD25+Foxp3+ T regulatory cells. J. Immunol. 2008;181:225–234. doi: 10.4049/jimmunol.181.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensinger SJ, Walsh PT, Zhang J, Carroll M, Parsons R, Rathmell JC, Thompson CB, Burchill MA, Farrar MA, Turka LA. Distinct IL-2 receptor signaling pattern in CD4+CD25+ regulatory T cells. J. Immunol. 2004;172:5287–5296. doi: 10.4049/jimmunol.172.9.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattman JN, Grayson JM, Wherry EJ, Kaech SM, Smith KA, Ahmed R. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat. Med. 2003;9:540–547. doi: 10.1038/nm866. [DOI] [PubMed] [Google Scholar]
- Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311:1924–1927. doi: 10.1126/science.1122927. [DOI] [PubMed] [Google Scholar]
- Brandenburg S, Takahashi T, de la Rosa M, Janke M, Karsten G, Muzzulini T, Orinska Z, Bulfone-Paus S, Scheffold A. IL-2 induces in vivo suppression by CD4(+)CD25(+)Foxp3(+) regulatory T cells. Eur. J. Immunol. 2008;38:1643–1653. doi: 10.1002/eji.200737791. [DOI] [PubMed] [Google Scholar]
- Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J. Immunol. 2007;178:280–290. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- Burchill MA, Yang J, Vang KB, Moon JJ, Chu HH, Lio CW, Vegoe AL, Hsieh CS, Jenkins MK, Farrar MA. Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity. 2008;28:112–121. doi: 10.1016/j.immuni.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrier Y, Yuan J, Kuchroo VK, Weiner HL. Th3 cells in peripheral tolerance. II. TGF-β-transgenic Th3 cells rescue IL-2-deficient mice from autoimmunity. J. Immunol. 2007;178:172–178. doi: 10.4049/jimmunol.178.1.172. [DOI] [PubMed] [Google Scholar]
- Carrio R, Bathe OF, Malek TR. Initial antigen encounter programs CD8+ T cells competent to develop into memory cells that are activated in an antigen-free, IL-7- and IL-15-rich environment. J. Immunol. 2004;172:7315–7323. doi: 10.4049/jimmunol.172.12.7315. [DOI] [PubMed] [Google Scholar]
- Cho JH, Kim HO, Surh CD, Sprent J. T cell receptor-dependent regulation of lipid rafts controls naive CD8+ T cell homeostasis. Immunity. 2010;32:214–226. doi: 10.1016/j.immuni.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish GH, Sinclair LV, Cantrell DA. Differential regulation of T-cell growth by IL-2 and IL-15. Blood. 2006;108:600–608. doi: 10.1182/blood-2005-12-4827. [DOI] [PubMed] [Google Scholar]
- Crellin NK, Garcia RV, Levings MK. Altered activation of AKT is required for the suppressive function of human CD4+CD25+ T regulatory cells. Blood. 2007;109:2014–2022. doi: 10.1182/blood-2006-07-035279. [DOI] [PubMed] [Google Scholar]
- D’Cruz LM, Klein L. Development and function of agonist-induced CD25+Foxp3+ regulatory T cells in the absence of interleukin 2 signaling. Nat. Immunol. 2005;6:1152–1159. doi: 10.1038/ni1264. [DOI] [PubMed] [Google Scholar]
- D’Souza WN, Lefrançois L. IL-2 is not required for the initiation of CD8 T cell cycling but sustains expansion. J. Immunol. 2003;171:5727–5735. doi: 10.4049/jimmunol.171.11.5727. [DOI] [PubMed] [Google Scholar]
- D’Souza WN, Schluns KS, Masopust D, Lefrançois L. Essential role for IL-2 in the regulation of antiviral extralymphoid CD8 T cell responses. J. Immunol. 2002;168:5566–5572. doi: 10.4049/jimmunol.168.11.5566. [DOI] [PubMed] [Google Scholar]
- Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting Edge: IL-2 is essential for TGF-β-mediated induction of Foxp3+ T regulatory cells. J. Immunol. 2007;178:4022–4026. doi: 10.4049/jimmunol.178.7.4022. [DOI] [PubMed] [Google Scholar]
- Decaluwe H, Taillardet M, Corcuff E, Munitic I, Law HK, Rocha B, Rivière Y, Di Santo JP. γ(c) deficiency precludes CD8+ T cell memory despite formation of potent T cell effectors. Proc. Natl. Acad. Sci. USA. 2010;107:9311–9316. doi: 10.1073/pnas.0913729107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooms H, Wolslegel K, Lin P, Abbas AK. Interleukin-2 enhances CD4+ T cell memory by promoting the generation of IL-7R α-expressing cells. J. Exp. Med. 2007;204:547–557. doi: 10.1084/jem.20062381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias KM, Laurence A, Davidson TS, Stephens G, Kanno Y, Shevach EM, O’Shea JJ. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood. 2008;111:1013–1020. doi: 10.1182/blood-2007-06-096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Dooley JL, Farr AG, Rudensky AY. Developmental regulation of Foxp3 expression during ontogeny. J. Exp. Med. 2005a;202:901–906. doi: 10.1084/jem.20050784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat. Immunol. 2005b;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- Furtado GC, de Lafaille M.A. Curotto, Kutchukhidze N, Lafaille JJ. Interleukin 2 signaling is required for CD4(+) regulatory T cell function. J. Exp. Med. 2002;196:851–857. doi: 10.1084/jem.20020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffen SL. Signaling domains of the interleukin 2 receptor. Cytokine. 2001;14:63–77. doi: 10.1006/cyto.2001.0862. [DOI] [PubMed] [Google Scholar]
- Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771–775. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- Gong D, Malek TR. Cytokine-dependent Blimp-1 expression in activated T cells inhibits IL-2 production. J. Immunol. 2007;178:242–252. doi: 10.4049/jimmunol.178.1.242. [DOI] [PubMed] [Google Scholar]
- Granucci F, Feau S, Angeli V, Trottein F, Ricciardi-Castagnoli P. Early IL-2 production by mouse dendritic cells is the result of microbial-induced priming. J. Immunol. 2003;170:5075–5081. doi: 10.4049/jimmunol.170.10.5075. [DOI] [PubMed] [Google Scholar]
- Gregersen PK, Olsson LM. Recent advances in the genetics of autoimmune disease. Annu. Rev. Immunol. 2009;27:363–391. doi: 10.1146/annurev.immunol.021908.132653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty JT, Badovinac VP. Shaping and reshaping CD8+ T-cell memory. Nat. Rev. Immunol. 2008;8:107–119. doi: 10.1038/nri2251. [DOI] [PubMed] [Google Scholar]
- Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J. Exp. Med. 2008;205:565–574. doi: 10.1084/jem.20071477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hémar A, Subtil A, Lieb M, Morelon E, Hellio R, Dautry-Varsat A. Endocytosis of interleukin 2 receptors in human T lymphocytes: distinct intracellular localization and fate of the receptor α, β, and γ chains. J. Cell Biol. 1995;129:55–64. doi: 10.1083/jcb.129.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, Mathis D, Benoist C. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. 2007;27:786–800. doi: 10.1016/j.immuni.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Huse M, Lillemeier BF, Kuhns MS, Chen DS, Davis MM. T cells use two directionally distinct pathways for cytokine secretion. Nat. Immunol. 2006;7:247–255. doi: 10.1038/ni1304. [DOI] [PubMed] [Google Scholar]
- Hwang KW, Sweatt WB, Mashayekhi M, Palucki DA, Sattar H, Chuang E, Alegre ML. Transgenic expression of CTLA-4 controls lymphoproliferation in IL-2-deficient mice. J. Immunol. 2004;173:5415–5424. doi: 10.4049/jimmunol.173.9.5415. [DOI] [PubMed] [Google Scholar]
- Hwang ES, Hong JH, Glimcher LH. IL-2 production in developing Th1 cells is regulated by heterodimerization of RelA and T-bet and requires T-bet serine residue 508. J. Exp. Med. 2005;202:1289–1300. doi: 10.1084/jem.20051044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat. Immunol. 2005;6:1236–1244. doi: 10.1038/ni1268. [DOI] [PubMed] [Google Scholar]
- Izcue A, Coombes JL, Powrie F. Regulatory lymphocytes and intestinal inflammation. Annu. Rev. Immunol. 2009;27:313–338. doi: 10.1146/annurev.immunol.021908.132657. [DOI] [PubMed] [Google Scholar]
- Jain J, Loh C, Rao A. Transcriptional regulation of the IL-2 gene. Curr. Opin. Immunol. 1995;7:333–342. doi: 10.1016/0952-7915(95)80107-3. [DOI] [PubMed] [Google Scholar]
- Jin H, Gong D, Adeegbe D, Bayer AL, Rolle C, Yu A, Malek TR. Quantitative assessment concerning the contribution of IL-2Rbeta for superantigen-mediated T cell responses in vivo. Int. Immunol. 2006;18:565–572. doi: 10.1093/intimm/dxh398. [DOI] [PubMed] [Google Scholar]
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, Wherry EJ. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity. 2007;27:393–405. doi: 10.1016/j.immuni.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity. 2010;32:91–103. doi: 10.1016/j.immuni.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Kallies A, Hawkins ED, Belz GT, Metcalf D, Hommel M, Corcoran LM, Hodgkin PD, Nutt SL. Transcriptional repressor Blimp-1 is essential for T cell homeostasis and self-tolerance. Nat. Immunol. 2006;7:466–474. doi: 10.1038/ni1321. [DOI] [PubMed] [Google Scholar]
- Kallies A, Xin A, Belz GT, Nutt SL. Blimp-1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity. 2009;31:283–295. doi: 10.1016/j.immuni.2009.06.021. [DOI] [PubMed] [Google Scholar]
- Kang J, Huddleston SJ, Fraser JM, Khoruts A. De novo induction of antigen-specific CD4+CD25+Foxp3+ regulatory T cells in vivo following systemic antigen administration accompanied by blockade of mTOR. J. Leuk. Biol. 2008;83:1230–1239. doi: 10.1189/jlb.1207851. [DOI] [PubMed] [Google Scholar]
- Kim HP, Kelly J, Leonard WJ. The basis for IL-2-induced IL-2 receptor α chain gene regulation: importance of two widely separated IL-2 response elements. Immunity. 2001;15:159–172. doi: 10.1016/s1074-7613(01)00167-4. [DOI] [PubMed] [Google Scholar]
- Knoechel B, Lohr J, Kahn E, Bluestone JA, Abbas AK. Sequential development of interleukin 2-dependent effector and regulatory T cells in response to endogenous systemic antigen. J. Exp. Med. 2005;202:1375–1386. doi: 10.1084/jem.20050855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc. Natl. Acad. Sci. USA. 2009;106:1903–1908. doi: 10.1073/pnas.0811556106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryczek I, Wei S, Vatan L, Escara-Wilke J, Szeliga W, Keller ET, Zou W. Cutting edge: opposite effects of IL-1 and IL-2 on the regulation of IL-17+ T cell pool IL-1 subverts IL-2-mediated suppression. J. Immunol. 2007;179:1423–1426. doi: 10.4049/jimmunol.179.3.1423. [DOI] [PubMed] [Google Scholar]
- Lantz O, Grandjean I, Matzinger P, Di Santo JP. γ chain required for naïve CD4+ T cell survival but not for antigen proliferation. Nat. Immunol. 2000;1:54–58. doi: 10.1038/76917. [DOI] [PubMed] [Google Scholar]
- Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Le Bras S, Massaad M, Koduru S, Kumar L, Oyoshi MK, Hartwig J, Geha RS. WIP is critical for T cell responsiveness to IL-2. Proc. Natl. Acad. Sci. USA. 2009;106:7519–7524. doi: 10.1073/pnas.0806410106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Létourneau S, van Leeuwen EM, Krieg C, Martin C, Pantaleo G, Sprent J, Surh CD, Boyman O. IL-2/anti-IL-2 antibody complexes show strong biological activity by avoiding interaction with IL-2 receptor α subunit CD25. Proc. Natl. Acad. Sci. USA. 2010;107:2171–2176. doi: 10.1073/pnas.0909384107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lio CW, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity. 2008;28:100–111. doi: 10.1016/j.immuni.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston A, Lu LF, O’Carroll D, Tarakhovsky A, Rudensky AY. Dicer-dependent microRNA pathway safeguards regulatory T cell function. J. Exp. Med. 2008;205:1993–2004. doi: 10.1084/jem.20081062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell. 2010;140:845–858. doi: 10.1016/j.cell.2010.02.021. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat. Immunol. 2008;9:632–640. doi: 10.1038/ni.1607. [DOI] [PubMed] [Google Scholar]
- Liu G, Burns S, Huang G, Boyd K, Proia RL, Flavell RA, Chi H. The receptor S1P1 overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nat. Immunol. 2009;10:769–777. doi: 10.1038/ni.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockyer HM, Tran E, Nelson BH. STAT5 is essential for Akt/p70S6 kinase activity during IL-2-induced lymphocyte proliferation. J. Immunol. 2007;179:5301–5308. doi: 10.4049/jimmunol.179.8.5301. [DOI] [PubMed] [Google Scholar]
- Long SA, Buckner JH. Combination of rapamycin and IL-2 increases de novo induction of human CD4(+)CD25(+)FOXP3(+) T cells. J. Autoimmun. 2008;30:293–302. doi: 10.1016/j.jaut.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long SA, Cerosaletti K, Bollyky PL, Tatum M, Shilling H, Zhang S, Zhang ZY, Pihoker C, Sanda S, Greenbaum C, Buckner JH. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T-cells of type 1 diabetic subjects. Diabetes. 2010;59:407–415. doi: 10.2337/db09-0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek TR. The biology of interleukin-2. Annu. Rev. Immunol. 2008;26:453–479. doi: 10.1146/annurev.immunol.26.021607.090357. [DOI] [PubMed] [Google Scholar]
- Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity. 2002;17:167–178. doi: 10.1016/s1074-7613(02)00367-9. [DOI] [PubMed] [Google Scholar]
- Manjunath N, Shankar P, Wan J, Weninger W, Crowley MA, Hieshima K, Springer TA, Fan X, Shen H, Lieberman J, von Andrian UH. Effector differentiation is not prerequisite for generation of memory cytotoxic T lymphocytes. J. Clin. Invest. 2001;108:871–878. doi: 10.1172/JCI13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins G, Calame K. Regulation and functions of Blimp-1 in T and B lymphocytes. Annu. Rev. Immunol. 2008;26:133–169. doi: 10.1146/annurev.immunol.26.021607.090241. [DOI] [PubMed] [Google Scholar]
- Martins GA, Cimmino L, Shapiro-Shelef M, Szabolcs M, Herron A, Magnusdottir E, Calame K. Transcriptional repressor Blimp-1 regulates T cell homeostasis and function. Nat. Immunol. 2006;7:457–465. doi: 10.1038/ni1320. [DOI] [PubMed] [Google Scholar]
- Martins GA, Cimmino L, Liao J, Magnusdottir E, Calame K. Blimp-1 directly represses Il2 and the Il2 activator Fos, attenuating T cell proliferation and survival. J. Exp. Med. 2008;205:1959–1965. doi: 10.1084/jem.20080526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DM, Ravkov EV, Williams MA. Distinct roles for IL-2 and IL-15 in the differentiation and survival of CD8+ effector and memory T cells. J. Immunol. 2010;184:6719–6730. doi: 10.4049/jimmunol.0904089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, Parizot C, Taflin C, Heike T, Valeyre D, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30:899–911. doi: 10.1016/j.immuni.2009.03.019. [DOI] [PubMed] [Google Scholar]
- Munitic I, Williams JA, Yang Y, Dong B, Lucas PJ, El Kassar N, Gress RE, Ashwell JD. Dynamic regulation of IL-7 receptor expression is required for normal thymopoiesis. Blood. 2004;104:4165–4172. doi: 10.1182/blood-2004-06-2484. [DOI] [PubMed] [Google Scholar]
- Nelson BH, Willerford DM. Biology of the interleukin-2 receptor. Adv. Immunol. 1998;70:1–81. doi: 10.1016/s0065-2776(08)60386-7. [DOI] [PubMed] [Google Scholar]
- O’Gorman WE, Dooms H, Thorne SH, Kuswanto WF, Simonds EF, Krutzik PO, Nolan GP, Abbas AK. The initial phase of an immune response functions to activate regulatory T cells. J. Immunol. 2009;183:332–339. doi: 10.4049/jimmunol.0900691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obar JJ, Molloy MJ, Jellison ER, Stoklasek TA, Zhang W, Usherwood EJ, Lefrançois L. CD4+ T cell regulation of CD25 expression controls development of short-lived effector CD8+ T cells in primary and secondary responses. Proc. Natl. Acad. Sci. USA. 2010;107:193–198. doi: 10.1073/pnas.0909945107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T, Miyachi Y, Tsukada T, Sakaguchi S. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446:685–689. doi: 10.1038/nature05673. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Beckett O, Ma Q, Li MO. Transforming growth factor-β signaling curbs thymic negative selection promoting regulatory T cell development. Immunity. 2010;32:642–653. doi: 10.1016/j.immuni.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat. Immunol. 2007;8:1353–1362. doi: 10.1038/ni1536. [DOI] [PubMed] [Google Scholar]
- Park JH, Adoro S, Guinter T, Erman B, Alag AS, Catalfamo M, Kimura MY, Cui Y, Lucas PJ, Gress RE, et al. Signaling by intrathymic cytokines, not T cell antigen receptors, specifies CD8 lineage choice and promotes the differentiation of cytotoxic-lineage T cells. Nat. Immunol. 2010;11:257–264. doi: 10.1038/ni.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton DT, Garden OA, Pearce WP, Clough LE, Monk CR, Leung E, Rowan WC, Sancho S, Walker LS, Vanhaesebroeck B, Okkenhaug K. Cutting edge: the phosphoinositide 3-kinase p110 δ is critical for the function of CD4+CD25+Foxp3+ regulatory T cells. J. Immunol. 2006;177:6598–6602. doi: 10.4049/jimmunol.177.10.6598. [DOI] [PubMed] [Google Scholar]
- Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan JD, Orekov T, Finkelman FD. Cutting edge: mechanism of enhancement of in vivo cytokine effects by anti-cytokine monoclonal antibodies. J. Immunol. 2008;180:44–48. doi: 10.4049/jimmunol.180.1.44. [DOI] [PubMed] [Google Scholar]
- Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 2010;32:79–90. doi: 10.1016/j.immuni.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prlic M, Bevan MJ. Exploring regulatory mechanisms of CD8+ T cell contraction. Proc. Natl. Acad. Sci. USA. 2008;105:16689–16694. doi: 10.1073/pnas.0808997105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity. 2010;32:67–78. doi: 10.1016/j.immuni.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by γ(c) family cytokines. Nat. Rev. Immunol. 2009;9:480–490. doi: 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein MP, Lind NA, Purton JF, Filippou P, Best JA, McGhee PA, Surh CD, Goldrath AW. IL-7 and IL-15 differentially regulate CD8+ T-cell subsets during contraction of the immune response. Blood. 2008;112:3704–3712. doi: 10.1182/blood-2008-06-160945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, Jacob J, Calame K, Kaech SM. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity. 2009;31:296–308. doi: 10.1016/j.immuni.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatos CA, Doh J, Chakravarti S, Friedman RS, Pandurangi PG, Tooley AJ, Krummel MF. A synaptic basis for paracrine interleukin-2 signaling during homotypic T cell interaction. Immunity. 2008;29:238–248. doi: 10.1016/j.immuni.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75:253–261. doi: 10.1016/0092-8674(93)80067-o. [DOI] [PubMed] [Google Scholar]
- Sadlack B, Löhler J, Schorle H, Klebb G, Haber H, Sickel E, Noelle RJ, Horak I. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur. J. Immunol. 1995;25:3053–3059. doi: 10.1002/eji.1830251111. [DOI] [PubMed] [Google Scholar]
- Sandau MM, Kohlmeier JE, Woodland DL, Jameson SC. IL-15 regulates both quantitative and qualitative features of the memory CD8 T cell pool. J. Immunol. 2010;184:35–44. doi: 10.4049/jimmunol.0803355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjabi S, Mosaheb MM, Flavell RA. Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity. 2009;31:131–144. doi: 10.1016/j.immuni.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, Knight ZA, Cobb BS, Cantrell D, O’Connor E, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc. Natl. Acad. Sci. USA. 2008;105:7797–7802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J. Exp. Med. 2005;201:723–735. doi: 10.1084/jem.20041982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Blackburn SD, Intlekofer AM, Kao C, Angelosanto JM, Reiner SL, Wherry EJ. A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity. 2009;31:309–320. doi: 10.1016/j.immuni.2009.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow JW, Abraham N, Ma MC, Herndier BG, Pastuszak AW, Goldsmith MA. Loss of tolerance and autoimmunity affecting multiple organs in STAT5A/5B-deficient mice. J. Immunol. 2003;171:5042–5050. doi: 10.4049/jimmunol.171.10.5042. [DOI] [PubMed] [Google Scholar]
- Soper DM, Kasprowicz DJ, Ziegler SF. IL-2Rbeta links IL-2R signaling with Foxp3 expression. Eur. J. Immunol. 2007;37:1817–1826. doi: 10.1002/eji.200737101. [DOI] [PubMed] [Google Scholar]
- Stauber DJ, Debler EW, Horton PA, Smith KA, Wilson IA. Crystal structure of the IL-2 signaling complex: paradigm for a heterotrimeric cytokine receptor. Proc. Natl. Acad. Sci. USA. 2006;103:2788–2793. doi: 10.1073/pnas.0511161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Kündig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, Schmits R, Simard JJL, Ohashi PS, Griesser H, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor β. Science. 1995;268:1472–1476. doi: 10.1126/science.7770771. [DOI] [PubMed] [Google Scholar]
- Tai X, Cowan M, Feigenbaum L, Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat. Immunol. 2005;6:152–162. doi: 10.1038/ni1160. [DOI] [PubMed] [Google Scholar]
- Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, Piccirillo CA, Salomon BL, Bluestone JA. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity. 2008;28:687–697. doi: 10.1016/j.immuni.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton AM, Donovan EE, Piccirillo CA, Shevach EM. Cutting edge: IL-2 is critically required for the in vitro activation of CD4 +CD25+ T cell suppressor function. J. Immunol. 2004;172:6519–6523. doi: 10.4049/jimmunol.172.11.6519. [DOI] [PubMed] [Google Scholar]
- Todd JA. Etiology of type 1 diabetes. Immunity. 2010;32:457–467. doi: 10.1016/j.immuni.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Tsuji-Takayama K, Suzuki M, Yamamoto M, Harashima A, Okochi A, Otani T, Inoue T, Sugimoto A, Toraya T, Takeuchi M, et al. The production of IL-10 by human regulatory T cells is enhanced by IL-2 through a STAT5-responsive intronic enhancer in the IL-10 locus. J. Immunol. 2008;181:3897–3905. doi: 10.4049/jimmunol.181.6.3897. [DOI] [PubMed] [Google Scholar]
- Vámosi G, Bodnár A, Vereb G, Jenei A, Goldman CK, Langowski J, Tóth K, Mátyus L, Szöllösi J, Waldmann TA, Damjanovich S. IL-2 and IL-15 receptor α-subunits are coexpressed in a supramolecular receptor cluster in lipid rafts of T cells. Proc. Natl. Acad. Sci. USA. 2004;101:11082–11087. doi: 10.1073/pnas.0403916101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vang KB, Yang J, Mahmud SA, Burchill MA, Vegoe AL, Farrar MA. IL-2, -7, and -15, but not thymic stromal lymphopoeitin, redundantly govern CD4+Foxp3+ regulatory T cell development. J. Immunol. 2008;181:3285–3290. doi: 10.4049/jimmunol.181.5.3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waithman J, Gebhardt T, Davey GM, Heath WR, Carbone FR. Cutting edge: Enhanced IL-2 signaling can convert self-specific T cell response from tolerance to autoimmunity. J. Immunol. 2008;180:5789–5793. doi: 10.4049/jimmunol.180.9.5789. [DOI] [PubMed] [Google Scholar]
- Walsh PT, Buckler JL, Zhang J, Gelman AE, Dalton NM, Taylor DK, Bensinger SJ, Hancock WW, Turka LA. PTEN inhibits IL-2 receptor-mediated expansion of CD4+ CD25+ Tregs. J. Clin. Invest. 2006;116:2521–2531. doi: 10.1172/JCI28057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445:766–770. doi: 10.1038/nature05479. [DOI] [PubMed] [Google Scholar]
- Wang X, Rickert M, Garcia KC. Structure of the quaternary complex of interleukin-2 with its α, β, and gammac receptors. Science. 2005;310:1159–1163. doi: 10.1126/science.1117893. [DOI] [PubMed] [Google Scholar]
- Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, Grey ST, Sprent J. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J. Exp. Med. 2009;206:751–760. doi: 10.1084/jem.20082824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor α chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3:521–530. doi: 10.1016/1074-7613(95)90180-9. [DOI] [PubMed] [Google Scholar]
- Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu. Rev. Immunol. 2007;25:171–192. doi: 10.1146/annurev.immunol.25.022106.141548. [DOI] [PubMed] [Google Scholar]
- Williams MA, Tyznik AJ, Bevan MJ. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature. 2006;441:890–893. doi: 10.1038/nature04790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–387. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- Yajima T, Yoshihara K, Nakazato K, Kumabe S, Koyasu S, Sad S, Shen H, Kuwano H, Yoshikai Y. IL-15 regulates CD8+ T cell contraction during primary infection. J. Immunol. 2006;176:507–515. doi: 10.4049/jimmunol.176.1.507. [DOI] [PubMed] [Google Scholar]
- Yamanouchi J, Rainbow D, Serra P, Howlett S, Hunter K, Garner VE, Gonzalez-Munoz A, Clark J, Veijola R, Cubbon R, et al. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat. Genet. 2007;39:329–337. doi: 10.1038/ng1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Kanno Y, Kerenyi M, Stephens G, Durant L, Watford WT, Laurence A, Robinson GW, Shevach EM, Moriggl R, et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood. 2007;109:4368–4375. doi: 10.1182/blood-2006-11-055756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A, Malek TR. The proteasome regulates receptor-mediated endocytosis of interleukin-2. J. Biol. Chem. 2001;276:381–385. doi: 10.1074/jbc.M007991200. [DOI] [PubMed] [Google Scholar]
- Yu A, Malek TR. Selective availability of IL-2 is a major determinant controlling the production of CD4+CD25+Foxp3+ T regulatory cells. J. Immunol. 2006;177:5115–5121. doi: 10.4049/jimmunol.177.8.5115. [DOI] [PubMed] [Google Scholar]
- Yu A, Zhou J, Marten N, Bergmann CC, Mammolenti M, Levy RB, Malek TR. Efficient induction of primary and secondary T cell-dependent immune responses in vivo in the absence of functional IL-2 and IL-15 receptors. J. Immunol. 2003;170:236–242. doi: 10.4049/jimmunol.170.1.236. [DOI] [PubMed] [Google Scholar]
- Yu A, Zhu L, Altman NH, Malek TR. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity. 2009;30:204–217. doi: 10.1016/j.immuni.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL-2 is essential for TGF-β to convert naive CD4+CD25− cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J. Immunol. 2007a;178:2018–2027. doi: 10.4049/jimmunol.178.4.2018. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007b;445:936–940. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
- Zheng L, Sharma R, Kung JT, Deshmukh US, Jarjour WN, Fu SM, Ju ST. Pervasive and stochastic changes in the TCR repertoire of regulatory T-cell-deficient mice. Int. Immunol. 2008a;20:517–523. doi: 10.1093/intimm/dxn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SG, Wang J, Horwitz DA. Cutting edge: Foxp3+CD4 +CD25+ regulatory T cells induced by IL-2 and TGF-β are resistant to Th17 conversion by IL-6. J. Immunol. 2008b;180:7112–7116. doi: 10.4049/jimmunol.180.11.7112. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463:808–812. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Jeker LT, Fife BT, Zhu S, Anderson MS, McManus MT, Bluestone JA. Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J. Exp. Med. 2008;205:1983–1991. doi: 10.1084/jem.20080707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–655. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, Bellucci R, Raderschall E, Canning C, Soiffer RJ, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006;108:1571–1579. doi: 10.1182/blood-2006-02-004747. [DOI] [PMC free article] [PubMed] [Google Scholar]