

Abstract

Enantiomerically enriched potassium β-trifluoroboratoamides were synthesized as air stable solids in greater than 95:5 dr using pseudoephedrine as the chiral auxiliary. With these chiral nucleophiles, Suzuki-Miyaura cross-coupling reactions were carried out with various aryl- and hetaryl chlorides in good to excellent yields. Moreover the diastereoselectivities were preserved throughout the Suzuki–Miyaura cross-coupling reactions.
α−Chiral β-arylated carbonyl compounds represent an important class of organic molecules. The most common approaches to these materials are benzylation of enolates1 and conjugate additions of arylmetallics to α,β-unsaturated carbonyl precursors (Scheme 1).2 Catalytic asymmetric hydrogenation of α−substituted α,β-unsaturated carbonyl substrates is also a viable entry.3 With the availability of many chiral auxiliaries, the benzylation approach is a highly attractive one. However, drawbacks include a relative dearth of commercially available benzylic halide precursors and a lack of chemoselectivity toward other embedded functional groups during the alkylation process. The 1,4-addition approach ultimately requires an enantioselective protonation of the enolate, a process that is highly substrate dependent and somewhat variable in its efficacy.4 Catalytic asymmetric hydrogenation methods have similar challenges to those of the enantioselective protonation, and in addition require access to stereodefined trisubstituted alkene substrates.
Scheme 1.

Strategies for the Synthesis of α-Chiral β-Arylated Carbonyls
An alternative approach would derive from the cross-coupling of β-metallo carbonyl substrates with aryl- and hetaryl halides. Recently, we demonstrated the Suzuki–Miyaura cross-coupling reactions of β-trifluoroborato ketones, esters, and amides, which led to the construction of β-aryl/hetaryl carbonyls.5 The β-trifluoroborato carbonyl reagents are attractive cross-coupling partners because of their functional group tolerance and minimal toxicity.6 As a family, potassium organotrifluoroborates have been shown to possess increased stability compared to their boronic acid/boronate counterparts in Suzuki–Miyaura reactions, including an indefinite stability to ambient moisture and a low tendency to protodeboronate during cross-coupling.7
Related cross-coupling reagents have been utilized in similar contexts. Thus, Negishi cross-coupling has proven particularly effective for the construction of arylated alanyl derivatives utilizing enantiomerically enriched α-chiral organozinc coupling partners.8 However, zinc homoenolates are generally unstable to moisture and air, and consequently they must be prepared in situ and maintained under an inert atmosphere. Furthermore, these organozinc reagents exhibit reactivity with a variety of electrophiles, and thus may not be tolerant of other functional groups in desired cross-coupling transformations.9
We envisioned that an extension of our previous work on β-trifluoroboratoamides to their enantiomerically enriched counterparts, prepared by employing chiral auxiliaries, would be a valuable tool in asymmetric synthesis. We report herein a protocol for the synthesis of enantioenriched α-chiral β-trifluoroborato amide substrates and their cross coupling with a variety of aryl- and hetaryl chlorides (Scheme 2). To the best of our knowledge, this chiral reagent class has not previously been prepared or cross-coupled.
Scheme 2.

Synthesis and Suzuki-Miyaura Cross-coupling of Chiral β-Trifluoroboratoamides
One of the concerns in being able to conduct the desired transformation is retention of the stereochemical integrity of the α stereocenter under the aqueous basic conditions at elevated temperatures required for the organotrifluoroborate cross-coupling. The use of enantiomerically enriched materials in Suzuki–Miyaura cross-coupling reactions where the stereocenter resides α to a carbonyl is limited to only a few examples.10 In only one of these examples did the stereogenic center reside in the organoboron component.
To prepare the enantiomerically enriched β-trifluoroborato amides 6a–d, (1S, 1S)-(+)-pseudoephedrine was used as the chiral auxiliary11 in a modification of a Matteson protocol.12 Thus the chirality was successfully installed via alkylation of the enolate using iodomethylpinacolboronate. The alkylated products 5a–d were obtained in relatively high yields. The diastereomeric ratios of 5a–d were determined to be >95:5 by 1H NMR of the pinacolboronate intermediate. Subsequent addition of KHF2 provided enantiomerically enriched potassium trifluoroborato amides 6a–d in moderate to excellent yields. The prepared β-trifluoroborato amides 6a–d were moisture and air stable solids and thus stored on the bench indefinitely without detectable epimerization or decomposition (Scheme 3).
Scheme 3.

Preparation of α-Chiral β-Trifluoroboratoamides
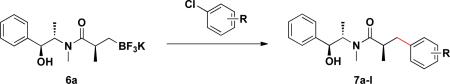
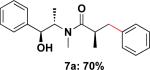
With the enantiomerically enriched β-trifluoroborato amides in hand, we first investigated their application in Suzuki–Miyaura cross-coupling reactions employing 6a with chlorobenzene to determine optimal conditions. After screening several ligands, bases, solvents, and reaction times, the combination of 5 mol % of Pd(OAc)2 and 10 mol % of RuPhos in the presence of three equivalents of K2CO3 in toluene/H2O (4:1, 0.25 M) for 22 h emerged as the best conditions, leading to 7a in 70% isolated yield (Table 1, entry 1).
Table 1.
Pd-Mediated Cross-Coupling of 6a with Various Aryl Chlorides
 | |
|---|---|
| entry | producta yield |
| 1 |
|
| 2 |
|
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
|
| 7 |
|
| 8 |
|
| 9 |
|
| 10 |
|
| 11 |
|
| 12 |
|
5% Pd(OAc)2, 10% RuPhos, 3 equiv K2CO3, toluene/H2O, 85 °C, 22 h
b 5% Pd(OAc)2, 10% SPhos, 3 equiv K2CO3, toluene/H2O, 85 °C, 22 h
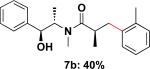
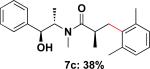
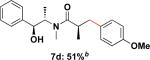
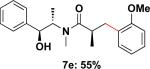
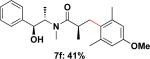
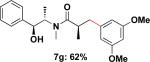
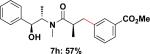
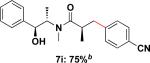
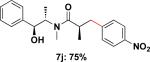
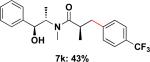
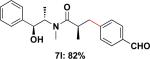
Employing this set of reaction conditions, a variety of aryl chlorides were studied as the coupling partner. Electron-neutral, -donating, and -withdrawing groups on the aryl ring were all tolerated during the cross-coupling reactions. The ortho-, meta-, and para-substituted electrophiles could all be used as suitable coupling partners (Table 1). In certain cases, SPhos was found to be a superior ligand to RuPhos (entries 4 and 9). With increased steric hindrance at the ortho-position, the yields dropped dramatically, and generally more hindered electrophiles provided lower yields than less hindered ones (entries 1–3). Electron-rich electrophiles, sometimes known to be poor coupling partners in Suzuki reactions,13 proceeded well under the same conditions (entries 4–7). Electrophiles containing electron-poor functional groups gave relatively better yields (entries 8–12). Moreover, various functional groups, such as esters, nitriles, nitro groups, and aldehydes survived the reaction conditions (entries 8–10 and 12). Of note, these functional groups, especially the aldehyde in 7l, would not be compatible with enolate alkylation routes to the same target structure.
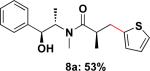
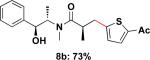
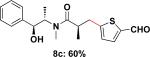
To expand the substrate scope, we also investigated the use of hetaryl chlorides as the coupling partners. Under the same reaction conditions, nitrogen, sulfur or oxygen-containing hetaryl electrophiles could be cross-coupled with chiral β-trifluoroborato amide 6a to give the desired products in good to moderate yields (Table 2).
Table 2.
Pd-Mediated Cross-Coupling of 6a with Various Hetaryl Chlorides
 | |
|---|---|
| entry | producta yield |
| 1 |
|
| 2 |
|
| 3 |
|
| 4 |
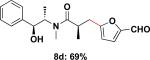
|
| 5 |
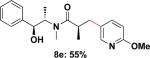
|
| 6 |
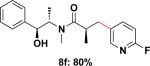
|
5% Pd(OAc)2, 10% RuPhos, 3 equiv K2CO3, toluene/H2O, 85 °C, 22 h
In addition to these studies, Suzuki–Miyaura cross-coupling reactions were conducted with β-trifluoroborato amides 6b–d possessing different substituents α to the carbonyl. Again in this series, sterics critically affect the overall yield of the process. As the steric bulk of the substituent at the α-carbon increased, the reactions required longer times to go to completion and the yields were dramatically reduced (Table 3).
Table 3.
Cross-Coupling Reaction of Various Potassium β-Trifluoroborato Amides with Chlorobenzene
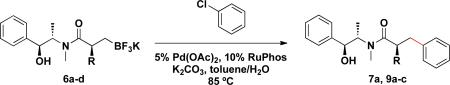
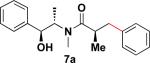
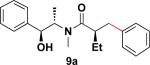
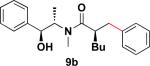
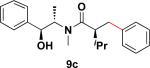
22 h
48 h
The degree of stereoretention in the cross-coupled products was determined using the simple one-step protocol developed by Myers employing triflic anhydridepyridine to form oxazolium triflate derivatives 11 by cyclization of pseudoephedrine amides 10. The diastereoselectivities of the final products 11 were determined by 1H NMR, with the peaks associated with the protons at the α stereocenter easily detectable (eq 1).14 For all substituents at the α-carbon examined, the diastereomeric ratios were greater than 95:5.15 Unfortunately, the more highly functionalized products displayed in Tables 1 and 2 were incompatible with the triflic acid, and their diastereoselectivities could not be determined in this manner. The stereochemistry of the major diastereomer of 7a was determined by comparison to the known compound.11b The stereochemistry of the remainder of the products was assigned by analogy. Through these studies, we concluded that the diastereoselectivities were preserved throughout the cross-coupling reactions.14
 |
(1) |
In conclusion, the potassium salts of enantiomerically enriched α-chiral β-trifluoroboratoamides were successfully synthesized using pseudoephedrine as a chiral auxiliary. With these air- and moisture stable reagents, Suzuki–Miyaura cross-coupling reactions were studied with various aryl- and hetaryl chlorides, providing an umpolung approach to these valuable materials that is complementary to previously reported protocols. Many of the compounds prepared would be challenging to synthesize by other methods such as direct alkylations with benzylic halides because of the reactivity of the functional groups incorporated (e.g., aldehydes and ketones). Additionally, for alkylation protocols the relative difficulty in accessing the requisite benzylic halides in both aryl and heteroaryl systems (as compared to the vast array of commerically available aryl- and hetaryl chlorides) also recommends the current procedure. The transformation developed herein provides materials of reliably high diastereoselectivity, a feature that is not always achieved in conjugate addition/enantioselective protonation or asymmetric hydrogenation approaches. The current method thus represents a valuable addition to existing approaches to α-chiral β-arylated carbonyl target structures.
Supplementary Material
Acknowledgment
We thank the NIGMS (R01 GM035249) for their support of this research. We also acknowledge Dr. Rakesh Kohli (University of Pennsylvania) for obtaining HRMS data.
Footnotes
Supporting Information Available Experimental procedures and spectral data of all compounds synthesized. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Evans DA. In: Asymmetric Synthesis. Morrison JD, editor. Vol. 3. Academic Press; New York: 1984. pp. 1–100. [Google Scholar]; b Lutomski KA, Meyers AI. In: Asymmetric Synthesis. Morrison JD, editor. Vol. 3. Academic Press; New York: 1984. pp. 213–274. [Google Scholar]
- 2.Posner GH. Organic Reactions. Vol. 19. Wiley; New York: 1972. pp. 1–113. [Google Scholar]
- 3.a Sugimura T, Uchida T, Watanabe J, Kubota T, Okamoto Y, Misaki T, Okuyama T. J. Cat. 2009;262:57. [Google Scholar]; b Lu W–J, Chen Y–W, Hou X–L. Angew. Chem. Int. Ed. 2008;47:10133. doi: 10.1002/anie.200803872. [DOI] [PubMed] [Google Scholar]; c Lu S–M, Bolm C. Angew. Chem. Int. Ed. 2008;47:8920. doi: 10.1002/anie.200803709. [DOI] [PubMed] [Google Scholar]; d Li S, Zhu S–F, Zhang C–M, Song S, Zhou Q-L. J. Am. Chem. Soc. 2008;130:8584. doi: 10.1021/ja802399v. [DOI] [PubMed] [Google Scholar]
- 4.a Fehr C. Angew. Chem. Int. Ed. 1996;35:2567. [Google Scholar]; b Navarre L, Martinez R, Genet J–P, Darses S. J. Am. Chem. Soc. 2008;130:6159. doi: 10.1021/ja710691p. [DOI] [PubMed] [Google Scholar]; c Navarre L, Darses S, Genet J–P. Angew. Chem. Int. Ed. 2004;43:719. doi: 10.1002/anie.200352518. [DOI] [PubMed] [Google Scholar]; d Cardillo G, Gentilucci L, Tolomelli A, Tomasini C. Tetrahedron. 1999;55:6231. [Google Scholar]; e Moss RJ, Wadsworth KJ, Chapman CJ, Frost CG. Chem. Commun. 2004:1984. doi: 10.1039/b406905f. [DOI] [PubMed] [Google Scholar]; f Hargrave JD, Herbert J, Bish G, Frost CG. Org. Biomol. Chem. 2006;4:3235. doi: 10.1039/b606977k. [DOI] [PubMed] [Google Scholar]; g Frost CG, Penrose SD, Lambshead K, Raithby PR, Warren JE, Gleave R. Org. Lett. 2007;9:2119. doi: 10.1021/ol070603g. [DOI] [PubMed] [Google Scholar]; h Sibi MP, Tatamidani H, Patil K. Org. Lett. 2005;7:2571. doi: 10.1021/ol050630b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a Molander GA, Petrillo DE. Org. Lett. 2008;10:1795. doi: 10.1021/ol800357c. [DOI] [PubMed] [Google Scholar]; b Molander GA, Jean-Gérard L. J. Org. Chem. 2009;74:1297. doi: 10.1021/jo802453m. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Molander GA, Jean-Gérard L. J. Org. Chem. 2009;74:5446. doi: 10.1021/jo900968h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a Molander GA, Canturk B. Angew. Chem. Int. Ed. 2009;48:9240. doi: 10.1002/anie.200904306. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457. [Google Scholar]
- 7.a Darses S, Genêt J-P. Chem. Rev. 2008;108:288. doi: 10.1021/cr0509758. [DOI] [PubMed] [Google Scholar]; b Molander GA, Ellis N. Acc. Chem. Res. 2007;40:275. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]; c Stefani HA, Cella R, Vieira AS. Tetrahedron. 2007;63:3623. [Google Scholar]; d Molander GA, Figueroa R. Aldrichimica Acta. 2005;38:49. [Google Scholar]; e Darses S, Genêt J-P. Eur. J. Org. Chem. 2003:4313. [Google Scholar]; f Molander GA, Sandrock DL. Curr. Opin. Drug Disc. Dev. 2009;12:811. [PMC free article] [PubMed] [Google Scholar]; h Molander GA, Jean-Gérard L. In: Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine, 2/e. Hall DG, editor. Wiley-VCH; Weinheim: 2010. [Google Scholar]
- 8.Rilatt I, Caggiano L, Jackson RFW. Synlett. 2005:2701. [Google Scholar]
- 9.Nakamura E, Aoki S, Sekiya K, Oshino H, Kuwajima I. J. Am. Chem. Soc. 1987;109:8056. [Google Scholar]
- 10.a Roush WR, Koyama K, Curtin ML, Moriarty KJ. J. Am. Chem. Soc. 1996;118:7502. [Google Scholar]; b Lee CB, Chou T-C, Zhang X-G, Wang Z-G, Kuduk SD, Chappell MD, Stachel SJ, Danishefsky SJ. J. Org. Chem. 2000;65:6525. doi: 10.1021/jo000617z. [DOI] [PubMed] [Google Scholar]; c Prieto M, Mayor S, Rodríguez K, Lloyd-Williams P, Giralt E. J. Org. Chem. 2007;72:1047. doi: 10.1021/jo0621266. [DOI] [PubMed] [Google Scholar]
- 11.a Myers AG, Yang BH, Chen H, Gleason JL. J. Am. Chem. Soc. 1994;116:9361. [Google Scholar]; b Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL. J. Am. Chem. Soc. 1997;119:6496. [Google Scholar]
- 12.Matteson DS, Man H-W. J. Org. Chem. 1994;59:5734. (b) Evans oxazolidinones and the Oppolzer sultam were also studied in Suzuki–Miyaura cross-coupling reactions. However, although the organotrifluoroborates could be prepared, both auxiliaries were cleaved during cross-couling events.
- 13.Littke AF, Fu GC. Angew. Chem., Int. Ed. Engl. 2002;41:4176. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 14.Myers AG, Chain WJ. Org. Lett. 2007;9:355. doi: 10.1021/ol0628762. [DOI] [PubMed] [Google Scholar]
- 15.See Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.