

Abstract
Although studies have shown alterations in cerebral metabolism after traumatic brain injury (TBI), clinical data in the developing brain is limited. We hypothesized that post-traumatic metabolic changes occur early (<24 h) and persist for up to 1 week. Immature rats underwent TBI to the left parietal cortex. Brains were removed at 4 h, 24 h, and 7 days after injury, and separated into ipsilateral (injured) and contralateral (control) hemispheres. Proton nuclear magnetic resonance (NMR) spectra were obtained, and spectra were analyzed for N-acetyl-aspartate (NAA), lactate (Lac), creatine (Cr), choline, and alanine, with metabolite ratios determined (NAA/Cr, Lac/Cr). There were no metabolic differences at any time in sham controls between cerebral hemispheres. At 4 and 24 h, there was an increase in Lac/Cr, reflecting increased glycolysis and/or decreased oxidative metabolism. At 24 h and 7 days, there was a decrease in NAA/Cr, indicating loss of neuronal integrity. The NAA/Lac ratio was decreased (∼15–20%) at all times (4 h, 24 h, 7 days) in the injured hemisphere of TBI rats. In conclusion, metabolic derangements begin early (<24 h) after TBI in the immature rat and are sustained for up to 7 days. Evaluation of early metabolic alterations after TBI could identify novel targets for neuroprotection in the developing brain.
Key words: N-Acetyl aspartate, development, lactate, mitochondria, NMR spectroscopy
Introduction
Traumatic brain injury (TBI) is a leading concern in children and young adults in the United States, with more than 3,000 deaths and 29,000 hospitalizations annually (Centers for Disease Control and Prevention). Present intensive care management does not offer any neuroprotective therapies that have correlated with improved long-term outcome. Even after recovery, many children face significant neurologic sequelae, which potentially persist into adulthood (Hawley, 2003; Keenan et al., 2007). In an effort to minimize long-term morbidity, it is essential to understand the basic mechanisms occurring in the brain following injury. Alterations in cerebral metabolism after TBI have been described in both clinical and experimental studies in all ages (Ashwal et al., 2000; Bartnik et al., 2005; Kochanek et al., 2000; Thomas et al., 2000; Yoshino et al., 1991). These associated changes in metabolism may contribute to the long-term learning, memory, and psychologic consequences seen after TBI in the pediatric population.
Proton (1H) nuclear magnetic resonance (NMR) spectroscopy provides a non-invasive method of evaluating alterations in brain metabolism and provides insight into the structural and metabolic integrity of brain tissue (Ashwal et al., 2006b; Barkovich et al., 1999; Pascual et al., 2007). The majority of data from in vivo 1H-NMR spectroscopy of brain after TBI is from the adult population, where the extent of metabolic change is related to the severity of injury and correlates with neurologic outcome (Brooks et al., 2001; Garnett et al., 2000). At this time, the pediatric studies are limited. A series of studies by Ashwal and colleagues have used NMR spectroscopy to evaluate TBI in the pediatric population. They reported an association between abnormal metabolite ratios and the presence of lactate with a poor neurologic outcome in infants and children following closed head injury (Ashwal et al., 2000, 2004; Holshouser et al., 1997). However, given the instability of critically ill pediatric patients, the mean time of a patient's scan was 6–9 days after injury. This time point may be beyond the optimal time for neuroprotective measures. The limitations in obtaining data noted in human studies can be overcome through the use of an animal model. Use of an animal model allows for evaluation of metabolism early (<24 h) after injury, prior to irreversible cell death. In adult rats, ex vivo NMR studies have demonstrated early (<4 h) increases in lactate in the injured hemisphere after controlled cortical impact (CCI) injury (Bartnik et al., 2005; Schuhmann et al., 2003). To our knowledge, no study to date has used the advanced technology of 1H-NMR spectroscopy to describe the time course of cerebral metabolic alterations following TBI in the developing brain. This study tested the hypothesis that in a clinically relevant rat model of pediatric TBI, alterations in brain metabolism occur early after injury and are sustained for at least 1 week.
Methods
Animals
This study was approved by the University of Maryland Animal Care and Use Committee. All care and handling of the rats were in compliance with the National Institutes of Health guidelines. In order to model pediatric TBI, immature (postnatal days 16–17), male Sprague-Dawley rats (Charles River Laboratories) weighing 32–40 g were used in all studies and were housed with littermates both before surgery and after recovery from anesthesia.
Traumatic Brain Injury Model
Brain trauma was induced using an established rat model of TBI with modifications for immature rats previously described by Robertson et al. (2007). Surgical anesthesia was induced in a Plexiglas chamber using isoflurane inhalation (4% for induction). Once anesthetized, animals were secured in a stereotactic device, receiving maintenance anesthesia (2% isoflurane) via nose cone with 30% oxygen for the remainder of the surgery. A rectal probe and heating blanket were used to maintain a rectal temperature of 37.0 ± 0.5°C. A midline incision was made over the skull, and a high-speed dental drill was used for creating a left parietal craniotomy. A brain temperature probe was placed in the contralateral temporalis muscle with cranial temperature maintained at 37.0 ± 0.5°C using a heating lamp. Rats were allowed a 30-min period of stable brain and rectal temperatures prior to TBI.
CCI injury was generated with a 6-mm flat-tipped impactor at 5 m/sec velocity, 50 msec duration, and depth of 1.5 mm. Following impact, the craniotomy was resealed using an acrylic mixture, and the surgical site was closed with interrupted sutures. After surgery was completed, anesthesia was discontinued, and the rat was awakened and returned to the dam with littermates. A separate group of sham animals underwent the same procedure without sustaining cortical injury.
Brain Preparation
At specified time points after injury (4 h, 24 h, 7 days), the animals were euthanized by decapitation. Brains were rapidly removed, placed on a cooled brain matrice, and the most rostral ∼3 mm of brain tissue (uninjured) was excised and discarded. The remaining peritrauma segment was quickly separated into ipsilateral (ipsi, trauma) and contralateral (contra, control) hemispheres. These peritrauma segments were rapidly frozen in liquid nitrogen (Viant et al., 2005) and stored at −80°C until analysis.
Brain tissue was weighed and homogenized with ice cold 7% perchloric acid (PCA, Tenbroeck, 7-mL tissue grinder) and extracted for NMR spectroscopy as described by Richards et al. (2007). Homogenates were transferred to 15-mL Corex round bottomed tubes, and centrifuged at 5,000g for 5 min (Beckman J2-MC, Rotor 25.5, 4°C). The supernatants were then removed and saved. The PCA pellets were extracted with 2 mL of ice-cold deionized water, resuspended by vortexing, and centrifuged as above. The combined supernatants were neutralized to a pH of 6.5–7.0, centrifuged at 5,000g for 10 min to remove any particulate matter, shell frozen and lyophilized overnight. Lyophilized samples were stored at −70°C until spectra were obtained. The pellet from the PCA extraction was saved for protein assay.
Prior to spectroscopy, samples were resuspended in 0.8 mL of D20, containing 0.02% sodium 3-trimethylsilyl proprionate-2,2,3,3-d4 (TMSP) as internal concentration standard. Samples were neutralized to pH 6.5–7.0 prior to placement in NMR tube (Wilmad-LabGlass, Buena, NJ).
Protein Assay of PCA Extracted Pellet
The PCA pellet was digested overnight with 1N NaOH, and the protein content was determined by the method of Lowry et al. (1951).
1H-NMR Spectroscopy
1H-NMR spectra were obtained using a Varian Inova 500-MHz spectrometer with a broad-band detection probe. Spectra were acquired using a pulse angle of 30 degrees, an acquisition time of 2.8 sec, relaxation delay of 1 sec, and a total of 256 repetitions. A line broadening of 1 Hz was used. Optimum shims were obtained before each spectra was run.
Chemical shifts were identified relative to the TMSP internal standard at 0.0 ppm and by comparison with previously reported values (Harris et al., 1997). The known amount of TMSP added to samples was used to calculate the concentration of compounds identified in the 1H spectra, including lactate (Lac), N-acetyl-aspartate (NAA), total creatine (Cr), choline (Cho), and alanine (Ala), as described by Harris et al. (1997). The peak height of each metabolite relative to the peak height of creatine was determined (Gasparovic et al., 2001; Richards et al., 2007; Hazany et al., 2007) and data used for statistical analysis. Other metabolite ratios (e.g., NAA/Lac, Lac/Ala) were also calculated. The ipsilateral (injured) side was compared with the contralateral (control) side from the same animal. Comparisons were also made between different times in the metabolite ratios of samples from the ipsilateral hemisphere of injured rats. In cases of doublets, such as lactate and alanine, the two peak heights were quantified, and the mean height calculated. The mean height was used for further statistical analysis of ratios.
Absolute Concentration of Metabolites
Quantitative analysis of the metabolites was obtained using the methodology previously described by Harris et al. (1997), by integrating the peak areas from the one dimensional spectra relative to TMSP using the following calculation to yield the final concentration of metabolite:
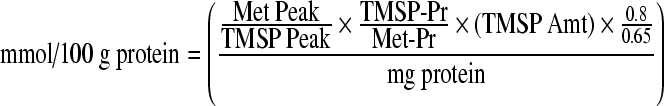 |
“Met Peak” denotes the metabolite of interest's peak height on the spectra, while “TMSP Peak” is the height of TMSP (standard). “TMSP-Pr” and “Met-Pr” are the number of protons per resonance of TSP and metabolite of interest, respectively. “TMSP Amt” is the micromoles of TMSP added to the NMR sample. The ratio 0.8/0.65 is a volume correction factor, as the lyophilized sample is suspended in a volume of 0.8 mL, and then 0.65 mL of this solution is placed in NMR tubes for spectroscopic analysis. “Mg protein” is the total protein content in each NMR sample.
Statistical Analysis
All values were reported as the mean ± SEM for the ipsilateral (injured) and contralateral (uninjured) peritrauma regions at 4 h, 24 h, and 7 days post-injury (or sham). A paired t-test was used to compare the ipsilateral to the contralateral hemispheres in each animal. Comparison across groups or times (e.g., comparison of metabolic ratios at three time points in injured hemispheres) were performed with one-way analysis of variance (ANOVA) testing with post-hoc Holm-Sidak comparisons, or with Kruskal-Wallis one-way ANOVA on ranks with post-hoc Dunn's comparisons. A probability level of <0.05 was considered to be significant.
Results
High-quality 1H-NMR spectra were obtained with good signal-to-noise ratio from perchloric acids extracts of brain from TBI and sham-operated rats. Representative spectra from both the ipsilateral (injured) and contralateral (control) sides of brain are shown in Figure 1. In sham-operated controls, there were no significant differences at any time point between the ipsilateral and contralateral sides of brains in any of the ratios evaluated.
FIG. 1.

Representative 1H spectra from ipsilateral hemisphere (A), contralateral hemisphere (B), or ipsilateral sham control (C) at 24 h after traumatic brain injury (TBI) or sham surgery. The spectra demonstrate good signal-to-noise ratio, with readily measurable peaks for key metabolites. NAA, N-acetyl-aspartate; Lac, lactate; Cr, creatine; Cho, choline; ala, alanine; TMSP, trimethylsilyl proprionate, internal standard.
N-Acetyl-Aspartate and Lactate Relationship
Evaluation of both NAA and lactate were made using creatine as a reference, as total creatine (creatine + phosphocreatine) is relatively constant in the brain after injury (Yeo et al., 2006) and during rat brain development (Burri et al., 1990). At 4 h after TBI (Fig. 2A), a significant increase in the Lac/Cr ratio was found on the injured side (Lac/Cr ipsi = 1.25 ± 0.11, contra = 1.03 ± 0.08, p < 0.01). At 24 h after TBI (Fig. 2B), the injured side showed a persistence of the increase in Lac/Cr (ipsi = 1.31 ± 0.11, contra = 1.05 ± 0.08, p < 0.01), accompanied by a significant decrease in the NAA/Cr ratio on the injured side (NAA/Cr ipsi = 0.75 ± 0.01, contra = 0.81 ± 0.02, p < 0.01). Seven days after injury (Fig. 2C), the decrease in NAA/Cr in the ipsilateral side persisted (ipsi = 0.68 ± 0.01, contra = 0.74 ± 0.02, p < 0.01). In contrast to the prolonged decrease in NAA/Cr, the elevation in Lac/Cr seen in the ipsilateral side of the brain at 4 and 24 h was no longer present. There were no differences in the NAA/Cr or Lac/Cr ratios between the contralateral hemisphere of injured rats and both hemispheres of sham controls at any time points.
FIG. 2.

Ipsilateral and contralateral hemispheric ratios of N-acetyl-aspartate to creatine (NAA/Cr) and lactate to creatine (Lac/Cr) at 4 h (A), 24 h (B), and 7 days (C) after traumatic brain injury (TBI). The Lac/Cr ratio was increased ipsilateral to injury at 4 h (A; *p < 0.05 vs. contralateral). At 24 h, the NAA/Cr ratio had decreased and Lac/Cr remained increased ipsilateral to injury (B; *p < 0.05 vs. contralateral). By 7 days, the ipsilateral NAA/Cr remained decreased (C; *p < 0.05 vs. contralateral).
To facilitate comparison between our study and others in developmental acute brain injury, we also determined the ratio of NAA to lactate at each of the time points. The NAA/Lac ratio was significantly decreased (11–19%) in the injured cortex compared to the contralateral cortex (p < 0.05 vs. contralateral) at all times points (4 h, 24 h, and 7 days; Fig. 3). At 4 h after TBI, the NAA/Lac ratio was ∼17% lower on the ipsilateral side (0.62 ± 0.04) compared to contralateral side of brain (0.75 ± 0.04, p < 0.001). At 24 h after TBI, the NAA/Lac ratio remained lower (∼19%) on the ipsilateral side (0.60 ± 0.01) compared to contralateral side of brain (0.74 ± 0.01, p < 0.001). At 7 days after TBI, the reduction in NAA/Lac persisted in the ipsilateral side (0.76 ± 0.03 vs. 0.86 ± 0.03, p < 0.01).
FIG. 3.

The N-acetyl-aspartate to lactate (NAA/Lac) ratio was decreased ipsilateral to injury at all time points studied in injured rats (4 h, 24 h, and 7 days; all *p < 0.05 ipsilateral vs. contralateral).
The complete time course of each metabolite ratio from TBI and sham rats is displayed in Figure 4. Differences between ipsilateral and contralateral hemisphere ratios in TBI rats as described above are displayed in this figure. Additionally, there were differences in metabolite ratios across times in TBI rats in the ipsilateral hemisphere. Both the NAA/Cr and Lac/Cr ratios were lower at 1 week than at 4 or 24 h in TBI ipsilateral samples (Fig. 4). The NAA/Lac ratio was higher at 1 week than at 24 h in TBI ipsilateral samples (Fig. 4).
FIG. 4.

Full time-course plots of the trends in metabolic ratios of N-acetyl-aspartate to creatine (NAA/Cr; A), lactate to creatine (Lac/Cr; B), and N-acetyl-aspartate to lactate (NAA/Lac; C) in traumatic brain injury (TBI; solid lines) and sham (dashed lines) rats, with ipsilateral samples represented by closed marks and contralateral with open marks (*p < 0.05 TBI ipsilateral vs. contralateral). Both the Naa/Cr and Lac/Cr ratios were lower at 1 week than at 4 or 24 h in TBI ipsilateral samples (πp < 0.05 1 week vs. 4 and 24 h). The NAA/Lac ratio was higher at 1 week than at 24 h in TBI ipsilateral samples (δp < 0.05 1 week vs. 24 h).
Absolute Concentrations of NAA, Lactate, and Creatine
The absolute concentrations of NAA, lactate, and creatine were calculated and expressed as mmol/100 g brain protein as shown in Table 1. The absolute concentration of NAA was significantly decreased in the injured (ipsilateral) hemisphere compared to the control (contralateral) hemisphere of brain at all three time points after TBI (4 h, 24 h, 7 days; p < 0.05 vs. contralateral). Interestingly, at 24 h after TBI, there was a significant decrease in the absolute concentration of creatine noted in the ipsilateral side. Although the ratio of lactate to other metabolites was altered by TBI as noted above, the absolute amount of lactate in brain was not different in the ipsilateral and contralateral sides of brain. There were no differences in absolute metabolite concentrations between ipsilateral and contralateral sides of brain in sham rats.
Table 1.
Total Concentration (mmol/100 g Protein) of N-Acetyl-Aspartate (NAA), Lactate (Lac), and Creatine (Cr)
| |
NAA |
Lac |
Cr |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Trauma |
Sham |
Trauma |
Sham |
Trauma |
Sham |
||||||
| Ipsi | Contra | Ipsi | Contra | Ipsi | Contra | Ipsi | Contra | Ipsi | Contra | Ipsi | Contra | |
| 4 h | 4.65 ± 0.45* | 5.52 ± 0.50 | 4.52 ± 0.41 | 5.09 ± 0.58 | 3.69 ± 0.37 | 3.62 ± 0.34 | 3.70 ± 0.29 | 3.47 ± 0.33 | 3.07 ± 0.37 | 3.55 ± 0.48 | 4.22 ± 1.02 | 4.39 ± 1.29 |
| 2 h | 5.19 ± 0.27* | 6.41 ± 0.48 | 5.49 ± 0.47 | 5.91 ± 0.27 | 4.33 ± 0.19 | 4.37 ± 0.28 | 3.69 ± 0.10 | 3.62 ± 0.08 | 3.48 ± 0.20* | 3.97 ± 0.25 | 3.46 ± 0.28 | 3.69 ± 0.24 |
| 7 days | 5.75 ± 0.21* | 6.35 ± 0.17 | 6.83 ± 0.08 | 6.8 ± 0.11 | 3.81 ± 0.13 | 3.78 ± 0.16 | 3.89 ± 0.13 | 3.69 ± 0.12 | 4.21 ± 0.17 | 4.22 ± 0.14 | 4.66 ± 0.31 | 4.37 ± 0.14 |
Ipsi, ipsilatera; contra, contralateral.
p < 0.05 ipsilateral versus contralateral (bold).
Choline and Alanine
To gather additional information about post-traumatic cerebral metabolism, we also measured two other important metabolites, choline and alanine. There was a significant increase in Cho/Cr ratio in the ipsilateral side of brain 7 days after TBI (Fig. 5). However, the Cho/Cr ratio in both TBI hemispheres was lower than the ratio from both sham samples at 7 days (Fig. 5), suggesting loss of lipid from both sides of the injured brain.
FIG. 5.

Time-course plot of the choline to creatine (Cho/Cr) ratio in traumatic brain injury (TBI; solid lines) and sham (dashed lines) rats, with ipsilateral samples represented by closed marks and contralateral with open marks. There was a significant increase in the Cho/Cr ratio at 7 days in TBI rats ipsilateral to injury (*p < 0.05 vs. TBI contralateral).
Consistent with the increases in Lac/Cr, we also found an increase in Ala/Cr ratio in the ipsilateral side of brain 4 h after TBI (ipsi = 0.11 ± 0.02 vs. contra = 0.08 ± 0.01, p < 0.05), which was not seen in sham rats. In order to evaluate the reason for ipsilateral increases in Lac/Cr, we also examined lactate to alanine (Lac/Ala) ratios, since this ratio can change in parallel to lactate to pyruvate ratios. There was no difference in the Lac/Ala ratio between ipsilateral and contralateral hemispheres in injured rats at any time point (4 h, 24 h, 7 days; data not shown).
Discussion
Magnetic resonance spectroscopy provides a noninvasive method to evaluate alterations in cerebral metabolism after injury. Hence, it could be a valuable tool in determining the optimal time for neuroprotective measures to be utilized in humans. To our knowledge, this is the first study to use 1H NMR spectroscopy to elucidate the time course of clinically relevant markers of structural and metabolic integrity of the brain after TBI in the immature rat. We determined that there was a significant increase in the ratio of Lac/Cr at 4 h and 24 h after TBI in the injured hemisphere relative to the contralateral side, which was not seen in the sham controls. Likewise, there was a significant decrease in the ratio of NAA/Cr in the injured hemisphere at both 24 h and 7 days after injury. The decreases in NAA/Cr were also correlated with a decrease in the absolute concentration of NAA present in the tissue examined. Finally, there were TBI-induced alterations in the Ala/Cr and Cho/Cr ratios at 4 h and 7 days, respectively.
Increases in cerebral lactate after TBI typically begin early (minutes to hours) after injury in adult animal models of both fluid percussion (Cohen et al., 1991; Inao et al., 1988; McIntosh et al., 1987) and CCI (Brooks et al., 2001; Schuhmann et al., 2003). Increased lactate can result from several potential mechanisms, such as post-injury increases in glycolysis (Kawamata et al., 1995; Yoshino et al., 1991) and reduced oxidative metabolism, with oxidative injury to key metabolic enzymes (Bogaert et al., 2000; Kochanek et al., 2006; Martin et al., 2005; Opii et al., 2007; Richards et al., 2006; Robertson et al., 2007). Another source of lactate after TBI is from infiltration of inflammatory cells, such as macrophages (Lopez-Villegas et al., 1995; Schuhmann et al., 2003; Petroff et al., 1992). In addition, astrocytes can produce lactate, serving as a post-traumatic energy substrate for nearby neurons (Pellerin et al., 1998; Schuhmann et al., 2003; Soustiel et al., 2005; McKenna et al., 1994).
Regardless of the sources of lactate, increases in this metabolite have been associated with poor neurologic outcome in human TBI studies using 1H spectroscopy (Marino et al., 2007; Brooks et al., 2001; Ashwal et al., 1997; Holshouser et al., 1997; Ross et al., 1998). Pediatric studies showed that the detection of lactate in the uninjured tissue was strongly correlated with poor neurologic outcome up to several years after injury (Ashwal et al., 2000; Brenner et al., 2003). Other forms of pediatric acute brain injury, such as perinatal asphyxia (Barkovich et al., 1999; Malik et al., 2002; Shu et al., 1997) and inflicted head injury (Makoroff et al., 2005) have linked increased brain lactate with poor neurologic outcome. Using our immature rat model, Lac/Cr ratio is elevated at both 4 and 24 h in the ipsilateral hemisphere, resolving by 7 days. Future studies will evaluate times between 24 h and 7 days. Since extraction time was comparable amongst all samples, any ex vivo production of lactate during the brain extraction (Serres et al., 2004) should not have influenced our results.
Similar to the increases in Lac/Cr, we also found increases in the Ala/Cr ratio in the ipsilateral side of brain 4 h after TBI. Alanine is commonly produced by transamination of pyruvate, and is often elevated with increased glycolysis and/or TCA cycle metabolism impairment. Furthermore, deamination of alanine in the astrocyte produces pyruvate, which is rapidly converted to lactate (Zwingmann et al., 2001). Therefore, it is not surprising that increases in the Lac/Cr ratio were observed at the same time (4 h) as early increases in the Ala/Cr ratio. Since pyruvate is not detectable by 1H-NMR spectroscopy, we could not assess the lactate to pyruvate ratio. We used alanine, which is formed by the transamination of pyruvate, since changes in pyruvate can be reflected as parallel changes in alanine. There was no difference in Lac/Ala ratios between hemispheres after TBI, supporting the hypothesis that decreased oxidative metabolism, rather than hyperglycolysis, is a main contributor to increases in Lac/Cr ratios.
NAA is a key metabolite in the nervous system (Clark, 1998). Studies have demonstrated that NAA is synthesized intramitochondrially, in an energy-dependent fashion (Meijer and van Dam, 1974). Found in all areas of the brain, it is the second most abundant amino acid in the central nervous system after glutamate (Clark, 1998; Tallan et al., 1956). It has been used clinically as a measure of neuronal or mitochondrial integrity, and decreased concentration of NAA is found in many neurodegenerative diseases (Davies et al., 1995; Federico et al., 1997; Godbolt et al., 2006). The precise role of NAA continues to be investigated. It is a postulated source of acetate for lipid and myelin synthesis of oligodendrocytes (Satrustegui et al., 2007). As the immature rat brain develops, rapid increases in NAA are detected during the time of peak myelination (D'Adamo and Yatsu, 1966; Bates et al., 1989; Burri et al., 1990). Using purified rat brain mitochondria (Patel and Clark, 1979), the synthesis and transport of NAA out of mitochondria increased with brain development in concert with myelination. NAA may also act as an osmolyte involved in fluid balance in the brain, thus providing protection to neurons (Taylor et al., 1995). Alterations in NAA observed by 1H-NMR are well described in clinical studies in adults after TBI (Holshouser et al., 2006; Marino et al., 2007; Choe et al., 1995; Garnett et al., 2000; Ross et al., 1998), and reductions were predictive of impairment in cognitive function (Brooks et al., 2000; Friedman et al., 1999).
Pediatric TBI studies have described reductions in the NAA/Cr ratio, but few analyses were performed <72 h after TBI, with a mean time from injury to spectra acquisition of 4–5 days in infants and 6–9 days in children (Ashwal et al., 2000; Brenner et al., 2003). Early reductions (at < 14 days) in NAA/Cr were correlated with long-term impairments in cognitive function (Ashwal et al., 2000; Brenner et al., 2003). This correlation persisted even when spectra were obtained up to 3 years after injury (Hunter et al., 2005). As the developing brain may have unique responses to TBI (Robertson et al., 2004, 2006), it is important to use a developmental rat model to study the age-dependent metabolic response. Data from the present study suggest that alterations in NAA/Cr occur very early (<24 h) after TBI and persist for at least 7 days, which may represent neuronal loss or loss of mitochondrial metabolic integrity in developing brain.
Consistent with decreased NAA/Cr ratios, we found reduced absolute concentrations of NAA in the ipsilateral hemisphere of brain at all three times (4 h, 24 h, 7 days) after TBI. We also saw decreases in the absolute concentration of creatine in the ipsilateral hemisphere at 24 h after TBI. Total creatine (creatine plus phosphocreatine) is generally considered to be stable in brain, making it ideal for comparative spectroscopy ratios. However, decreases in the total creatine have been reported after TBI in tissue near the contusion (Schuhmann et al., 2003). Therefore, it is possible that creatine synthesis was decreased, or degradation was increased in the first 24 h after injury in our study. We did not detect hemispheric differences in lactate concentration in the brains of TBI rats, despite significant increases in the ipsilateral Lac/Cr ratio at 4 and 24 h. Although the reasons for this apparent discrepancy between the concentration data and ratios are not clear, reduction in creatine at 24 h may have contributed to increases in Lac/Cr ratio. It is also important to recognize the complex nature of lactate balance in brain which includes synthesis and possible transport between and within astrocytes and neurons (McKenna et al., 2006). The intracellular content of lactate is influenced by changes in the glycolytic rate, activity of the lactate and pyruvate dehydrogenase enzymes, TCA cycle activity and the rate of lactate efflux from the brain (Dienel and Cruz, 2003; McKenna et al., 2006; O'Brien et al., 2007). In addition, aspects of lactate balance can change during brain maturation, since the expression of lactate dehydrogenase (LDH) isozymes is developmentally regulated (McKenna et al., 2006). It is important to note that most clinical trials correlating spectroscopic measurements with outcome have used metabolite ratios (e.g., NAA/Cr, Cho/Cr), giving our metabolic ratio data greater clinical relevance. Pediatric TBI studies have not quantified brain lactate concentration.
Trauma also induced changes in Cho/Cr ratios in this pediatric TBI model. Although there was a relative increase in the ipsilateral Cho/Cr ratio at 7 days, Cho/Cr ratios were lower in both TBI hemispheres compared to sham. Choline is primarily a membrane marker, and increases in choline on spectroscopy are thought to represent gliosis, membrane breakdown and/or inflammation (Ashwal et al., 2006a; Brooks et al., 2001). Acutely, elevations in choline also have value in detecting diffuse axonal injury (Holshouser et al., 2005). Longer-term (days) increases in choline can be secondary to gliosis (Garnett et al., 2001), while long-term decreases can represent demyelination following injury (Liu et al., 2006). In pediatric and adult TBI studies, increases in Cho/Cr ratios were associated with poor neurologic outcome (Ashwal et al., 2000; Brenner et al., 2003; Hunter et al., 2005; Shutter et al., 2004; Brooks et al., 2000; Garnett et al., 2000). We found relative ipsilateral increases at 7 days, which could be consistent with gliosis in the injured hemisphere. However, this could be underestimated by the lower ratios of Cho/Cr in both hemispheres in injured rats, possibly representing diffuse, bilateral demyelination after TBI (Liu et al., 2006). Future studies could more directly evaluate the course and severity of white matter damage in this immature rat model.
In conclusion, the most important observation of the present study is that there are significant alterations in metabolite ratios very early after TBI in developing brain and that some alterations persist for at least 7 days. We found early (4 and 24 h) increases in Lac/Cr and delayed (24 h, 7 days) decreases in NAA/Cr, with significant decreases in the NAA/Lac ratios at all times studied after CCI in immature rats. The present study and others (Pascual et al., 2007) demonstrate that the use of NMR spectroscopy in clinically relevant animal models of pediatric TBI can allow description of the time course and severity of metabolic alterations after injury. Importantly, altered metabolic ratios at 4 and 24 h occur prior to significant cell death, reflecting potentially reversible metabolic dysfunction of intact cells. Future studies could use this methodology to develop and test early neuroprotective strategies targeting cerebral energy failure in the developing brain after TBI.
Acknowledgments
This work was supported by the National Institutes of Health (grant K08 NS42805, HD 16596) and the University of Maryland Department of Pediatrics (SRIS grants).
Author Disclosure Statement
No competing financial interests exist.
References
- Ashwal S. Holshouser B.A. Tomasi L.G. Shu S. Perkin R.M. Nystrom G.A. Hinshaw D.B., Jr. 1H-Magnetic resonance spectroscopy–determined cerebral lactate and poor neurological outcomes in children with central nervous system disease. Ann. Neurol. 1997;41:470–481. doi: 10.1002/ana.410410410. [DOI] [PubMed] [Google Scholar]
- Ashwal S. Holshouser B.A. Shu S.K. Simmons P.L. Perkin R.M. Tomasi L.G. Knierim D.S. Sheridan C. Craig K. Andrews G.H. Hinshaw D.B. Predictive value of proton magnetic resonance spectroscopy in pediatric closed head injury. Pediatr. Neurol. 2000;23:114–125. doi: 10.1016/s0887-8994(00)00176-4. [DOI] [PubMed] [Google Scholar]
- Ashwal S. Holshouser B. Tong K. Serna T. Osterdock R. Gross M. Kido D. Proton MR spectroscopy detected glutamate/glutamine is increased in children with traumatic brain injury. J. Neurotrauma. 2004;21:1539–1552. doi: 10.1089/neu.2004.21.1539. [DOI] [PubMed] [Google Scholar]
- Ashwal S. Babikian T. Gardner-Nichols J. Freier M.C. Tong K.A. Holshouser B.A. Susceptibility-weighted imaging and proton magnetic resonance spectroscopy in assessment of outcome after pediatric traumatic brain injury. Arch. Phys. Med. Rehabil. 2006a;87:S50–S58. doi: 10.1016/j.apmr.2006.07.275. [DOI] [PubMed] [Google Scholar]
- Ashwal S. Holshouser B.A. Tong K.A. Use of advanced neuroimaging techniques in the evaluation of pediatric traumatic brain injury. Dev. Neurosci. 2006b;28:309–326. doi: 10.1159/000094157. [DOI] [PubMed] [Google Scholar]
- Barkovich A.J. Baranski K. Vigneron D. Partridge J.C. Hallam D.K. Hajnal B.L. Ferriero D.M. Proton MR spectroscopy for the evaluation of brain injury in asphyxiated, term neonates. AJNR Am. J. Neuroradiol. 1999;20:1399–1405. [PMC free article] [PubMed] [Google Scholar]
- Bartnik B.L. Sutton R.L. Fukushima M. Harris N.G. Hovda D.A. Lee S.M. Upregulation of pentose phosphate pathway and preservation of tricarboxylic acid cycle flux after experimental brain injury. J. Neurotrauma. 2005;22:1052–1065. doi: 10.1089/neu.2005.22.1052. [DOI] [PubMed] [Google Scholar]
- Bates T.E. Williams S.R. Gadian D.G. Bell J.D. Small R.K. Iles R.A. 1H NMR study of cerebral development in the rat. NMR Biomed. 1989;2:225–229. doi: 10.1002/nbm.1940020509. [DOI] [PubMed] [Google Scholar]
- Bogaert Y.E. Sheu K.F. Hof P.R. Brown A.M. Blass J.P. Rosenthal R.E. Fiskum G. Neuronal subclass-selective loss of pyruvate dehydrogenase immunoreactivity following canine cardiac arrest and resuscitation. Exp. Neurol. 2000;161:115–126. doi: 10.1006/exnr.1999.7250. [DOI] [PubMed] [Google Scholar]
- Brenner T. Freier M.C. Holshouser B.A. Burley T. Ashwal S. Predicting neuropsychologic outcome after traumatic brain injury in children. Pediatr. Neurol. 2003;28:104–114. doi: 10.1016/s0887-8994(02)00491-5. [DOI] [PubMed] [Google Scholar]
- Brooks W.M. Stidley C.A. Petropoulos H. Jung R.E. Weers D.C. Friedman S.D. Barlow M.A. Sibbitt W.L., Jr. Yeo R.A. Metabolic and cognitive response to human traumatic brain injury: a quantitative proton magnetic resonance study. J. Neurotrauma. 2000;17:629–640. doi: 10.1089/089771500415382. [DOI] [PubMed] [Google Scholar]
- Brooks W.M. Friedman S.D. Gasparovic C. Magnetic resonance spectroscopy in traumatic brain injury. J. Head Trauma Rehabil. 2001;16:149–164. doi: 10.1097/00001199-200104000-00005. [DOI] [PubMed] [Google Scholar]
- Burri R. Bigler P. Straehl P. Posse S. Colombo J.-P. Herschkowitz N. Brain development: 1H magnetic resonance spectroscopy of rat brain extracts compared with chromatographic methods. Neurochem. Res. 1990;15:1009–1016. doi: 10.1007/BF00965747. [DOI] [PubMed] [Google Scholar]
- Choe B.Y. Suh T.S. Choi K.H. Shinn K.S. Park C.K. Kang J.K. Neuronal dysfunction in patients with closed head injury evaluated by in vivo 1H magnetic resonance spectroscopy. Invest. Radiol. 1995;30:502–506. doi: 10.1097/00004424-199508000-00008. [DOI] [PubMed] [Google Scholar]
- Clark J.B. N-Acetyl aspartate: a marker for neuronal loss or mitochondrial dysfunction. Dev. Neurosci. 1998;20:271–276. doi: 10.1159/000017321. [DOI] [PubMed] [Google Scholar]
- Cohen Y. Sanada T. Pitts L.H. Chang L.H. Nishimura M.C. Weinstein P.R. Litt L. James T.L. Surface coil spectroscopic imaging: time and spatial evolution of lactate production following fluid percussion brain injury. Magn. Reson. Med. 1991;17:225–236. doi: 10.1002/mrm.1910170125. [DOI] [PubMed] [Google Scholar]
- D'Adamo A.F., Jr. Yatsu F.M. Acetate metabolism in the nervous system. N-Acetyl-L-aspartic acid and the biosynthesis of brain lipids. J. Neurochem. 1966;13:961–965. doi: 10.1111/j.1471-4159.1966.tb10292.x. [DOI] [PubMed] [Google Scholar]
- Davies S.E. Newcombe J. Williams S.R. McDonald W.I. Clark J.B. High-resolution proton NMR spectroscopy of multiple sclerosis lesions. J. Neurochem. 1995;64:742–748. doi: 10.1046/j.1471-4159.1995.64020742.x. [DOI] [PubMed] [Google Scholar]
- Dienel G.A. Cruz N.F. Neighborly interactions of metabolically-activated astrocytes in vivo. Neurochem. Int. 2003;43:339–354. doi: 10.1016/s0197-0186(03)00021-4. [DOI] [PubMed] [Google Scholar]
- Federico F. Simone I.L. Lucivero V. De Mari M. Giannini P. Iliceto G. Mezzapesa D.M. Lamberti P. Proton magnetic resonance spectroscopy in Parkinson's disease and progressive supranuclear palsy. J. Neurol. Neurosurg. Psychiatry. 1997;62:239–242. doi: 10.1136/jnnp.62.3.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman S.D. Brooks W.M. Jung R.E. Chiulli S.J. Sloan J.H. Montoya B.T. Hart B.L. Yeo R.A. Quantitative proton MRS predicts outcome after traumatic brain injury. Neurology. 1999;52:1384–1391. doi: 10.1212/wnl.52.7.1384. [DOI] [PubMed] [Google Scholar]
- Garnett M.R. Blamire A.M. Corkill R.G. Cadoux-Hudson T.A. Rajagopalan B. Styles P. Early proton magnetic resonance spectroscopy in normal-appearing brain correlates with outcome in patients following traumatic brain injury. Brain. 2000;123:2046–2054. doi: 10.1093/brain/123.10.2046. [DOI] [PubMed] [Google Scholar]
- Garnett M.R. Corkill R.G. Blamire A.M. Rajagopalan B. Manners D.N. Young J.D. Styles P. Cadoux-Hudson T.A. Altered cellular metabolism following traumatic brain injury: a magnetic resonance spectroscopy study. J. Neurotrauma. 2001;18:231–240. doi: 10.1089/08977150151070838. [DOI] [PubMed] [Google Scholar]
- Gasparovic C. Arfai N. Smid N. Feeney D.M. Decrease and recovery of N-acetylaspartate/creatine in rat brain remote from focal injury. J. Neurotrauma. 2001;18:241–246. doi: 10.1089/08977150151070856. [DOI] [PubMed] [Google Scholar]
- Godbolt A.K. Waldman A.D. MacManus D.G. Schott J.M. Frost C. Cipolotti L. Fox N.C. Rossor M.N. MRS shows abnormalities before symptoms in familial Alzheimer disease. Neurology. 2006;66:718–722. doi: 10.1212/01.wnl.0000201237.05869.df. [DOI] [PubMed] [Google Scholar]
- Harris N.G. Plant H.D. Inglis B.A. Briggs R.W. Jones H.C. Neurochemical changes in the cerebral cortex of treated and untreated hydrocephalic rat pups quantified with in vitro 1H-NMR spectroscopy. J. Neurochem. 1997;68:305–312. doi: 10.1046/j.1471-4159.1997.68010305.x. [DOI] [PubMed] [Google Scholar]
- Hawley C.A. Reported problems and their resolution following mild, moderate and severe traumatic brain injury amongst children and adolescents in the UK. Brain Inj. 2003;17:105–129. doi: 10.1080/0269905021000010131. [DOI] [PubMed] [Google Scholar]
- Hazany S. Hesselink J.R. Healy J.F. Imbesi S.G. Utilization of glutamate/creatine ratios for proton spectroscopic diagnosis of meningiomas. Neuroradiology. 2007;49:121–127. doi: 10.1007/s00234-006-0167-z. [DOI] [PubMed] [Google Scholar]
- Holshouser B.A. Ashwal S. Luh G.Y. Shu S. Kahlon S. Auld K.L. Tomasi L.G. Perkin R.M. Hinshaw D.B., Jr. Proton MR spectroscopy after acute central nervous system injury: outcome prediction in neonates, infants, and children. Radiology. 1997;202:487–496. doi: 10.1148/radiology.202.2.9015079. [DOI] [PubMed] [Google Scholar]
- Holshouser B.A. Tong K.A. Ashwal S. Proton MR spectroscopic imaging depicts diffuse axonal injury in children with traumatic brain injury. AJNR Am. J. Neuroradiol. 2005;26:1276–1285. [PMC free article] [PubMed] [Google Scholar]
- Holshouser B.A. Tong K.A. Ashwal S. Oyoyo U. Ghamsary M. Saunders D. Shutter L. Prospective longitudinal proton magnetic resonance spectroscopic imaging in adult traumatic brain injury. J. Magn. Reson. Imaging. 2006;24:33–40. doi: 10.1002/jmri.20607. [DOI] [PubMed] [Google Scholar]
- Hunter J.V. Thornton R.J. Wang Z.J. Levin H.S. Roberson G. Brooks W.M. Swank P.R. Late proton MR spectroscopy in children after traumatic brain injury: correlation with cognitive outcomes. AJNR Am. J. Neuroradiol. 2005;26:482–488. [PMC free article] [PubMed] [Google Scholar]
- Inao S. Marmarou A. Clarke G.D. Andersen B.J. Fatouros P.P. Young H.F. Production and clearance of lactate from brain tissue, cerebrospinal fluid, and serum following experimental brain injury. J. Neurosurg. 1988;69:736–744. doi: 10.3171/jns.1988.69.5.0736. [DOI] [PubMed] [Google Scholar]
- Kawamata T. Katayama Y. Hovda D.A. Yoshino A. Becker D.P. Lactate accumulation following concussive brain injury: the role of ionic fluxes induced by excitatory amino acids. Brain Res. 1995;674:196–204. doi: 10.1016/0006-8993(94)01444-m. [DOI] [PubMed] [Google Scholar]
- Keenan H.T. Hooper S.R. Wetherington C.E. Nocera M. Runyan D.K. Neurodevelopmental consequences of early traumatic brain injury in 3-year-old children. Pediatrics. 2007;119:e616–e623. doi: 10.1542/peds.2006-2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochanek P.M. Clark R.S. Ruppel R.A. Adelson P.D. Bell M.J. Whalen M.J. Robertson C.L. Satchell M.A. Seidberg N.A. Marion D.W. Jenkins L.W. Biochemical, cellular, and molecular mechanisms in the evolution of secondary damage after severe traumatic brain injury in infants and children: Lessons learned from the bedside. Pediatr. Crit. Care Med. 2000;1:4–19. doi: 10.1097/00130478-200007000-00003. [DOI] [PubMed] [Google Scholar]
- Kochanek A.R. Kline A.E. Gao W.M. Chadha M. Lai Y. Clark R.S. Dixon C.E. Jenkins L.W. Gelbased hippocampal proteomic analysis 2 weeks following traumatic brain injury to immature rats using controlled cortical impact. Dev. Neurosci. 2006;28:410–419. doi: 10.1159/000094167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.C. Akle V. Zheng W. Kitlen J. O'Steen B. Larner S.F. Dave J.R. Tortella F.C. Hayes R.L. Wang K.K. Extensive degradation of myelin basic protein isoforms by calpain following traumatic brain injury. J. Neurochem. 2006;98:700–712. doi: 10.1111/j.1471-4159.2006.03882.x. [DOI] [PubMed] [Google Scholar]
- Lopez-Villegas D. Lenkinski R.E. Wehrli S.L. Ho W.Z. Douglas S.D. Lactate production by human monocytes/macrophages determined by proton MR spectroscopy. Magn. Reson. Med. 1995;34:32–38. doi: 10.1002/mrm.1910340107. [DOI] [PubMed] [Google Scholar]
- Lowry O.H. Rosebrough N.J. Farr A.L. Randall R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Makoroff K.L. Cecil K.M. Care M. Ball W.S., Jr. Elevated lactate as an early marker of brain injury in inflicted traumatic brain injury. Pediatr. Radiol. 2005;35:668–676. doi: 10.1007/s00247-005-1441-7. [DOI] [PubMed] [Google Scholar]
- Malik G.K. Pandey M. Kumar R. Chawla S. Rathi B. Gupta R.K. MR imaging and in vivo proton spectroscopy of the brain in neonates with hypoxic ischemic encephalopathy. Eur. J. Radiol. 2002;43:6–13. doi: 10.1016/s0720-048x(01)00435-1. [DOI] [PubMed] [Google Scholar]
- Marino S. Zei E. Battaglini M. Vittori C. Buscalferri A. Bramanti P. Federico A. De Stefano N. Acute metabolic brain changes following traumatic brain injury and their relevance to clinical severity and outcome. J. Neurol. Neurosurg. Psychiatry. 2007;78:501–507. doi: 10.1136/jnnp.2006.099796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E. Rosenthal R.E. Fiskum G. Pyruvate dehydrogenase complex: metabolic link to ischemic brain injury and target of oxidative stress. J. Neurosci. Res. 2005;79:240–247. doi: 10.1002/jnr.20293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh T.K. Faden A.I. Bendall M.R. Vink R. Traumatic brain injury in the rat: alterations in brain lactate and pH as characterized by 1H and 31P nuclear magnetic resonance. J. Neurochem. 1987;49:1530–1540. doi: 10.1111/j.1471-4159.1987.tb01024.x. [DOI] [PubMed] [Google Scholar]
- McKenna M.C. Tildon J.T. Stevenson J.H. Hopkins I.B. Energy metabolism in cortical synaptic terminals from weanling and mature rat brain: evidence for multiple compartments of tricarboxylic acid cycle activity. Dev. Neurosci. 1994;16:291–300. doi: 10.1159/000112122. [DOI] [PubMed] [Google Scholar]
- McKenna M.C. Gruetter R. Sonnewald U. Waagepetersen H.S. Schousboe A. Energy metabolism of the brain. In: Siegel G.J., editor; Albers R.W., editor; Brady S.T., editor; Price D.L., editor. Basic Neurochemistry. Elsevier Academic Press; Burlington, MA: 2006. pp. 531–557. [Google Scholar]
- Meijer A.J. van Dam K. The metabolic significance of anion transport in mitochondria. Biochim. Biophys. Acta. 1974;346:213–244. doi: 10.1016/0304-4173(74)90001-9. [DOI] [PubMed] [Google Scholar]
- O'Brien J. Kla K.M. Hopkins I.B. Malecki E.A. McKenna M.C. Kinetic parameters and lactate dehydrogenase isozyme activities support possible lactate utilization by neurons. Neurochem. Res. 2007;32:597–607. doi: 10.1007/s11064-006-9132-9. [DOI] [PubMed] [Google Scholar]
- Opii W.O. Nukala V.N. Sultana R. Pandya J.D. Day K.M. Merchant M.L. Klein J.B. Sullivan P.G. Butterfield D.A. Proteomic identification of oxidized mitochondrial proteins following experimental traumatic brain injury. J. Neurotrauma. 2007;24:772–789. doi: 10.1089/neu.2006.0229. [DOI] [PubMed] [Google Scholar]
- Pascual J.M. Solivera J. Prieto R. Barrios L. Lopez-Larrubia P. Cerdan S. Roda J.M. Time course of early metabolic changes following diffuse traumatic brain injury in rats as detected by 1H NMR spectroscopy. J. Neurotrauma. 2007;24:944–959. doi: 10.1089/neu.2006.0190. [DOI] [PubMed] [Google Scholar]
- Patel T.B. Clark J.B. Synthesis of N-acetyl-L-aspartate by rat brain mitochondria and its involvement in mitochondrial/cytosolic carbon transport. Biochem. J. 1979;184:539–546. doi: 10.1042/bj1840539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L. Pellegri G. Bittar P.G. Charnay Y. Bouras C. Martin J.L. Stella N. Magistretti P.J. Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev. Neurosci. 1998;20:291–299. doi: 10.1159/000017324. [DOI] [PubMed] [Google Scholar]
- Petroff O.A. Graham G.D. Blamire A.M. al Rayess M. Rothman D.L. Fayad P.B. Brass L.M. Shulman R.G. Prichard J.W. Spectroscopic imaging of stroke in humans: histopathology correlates of spectral changes. Neurology. 1992;42:1349–1354. doi: 10.1212/wnl.42.7.1349. [DOI] [PubMed] [Google Scholar]
- Richards E.M. Rosenthal R.E. Kristian T. Fiskum G. Postischemic hyperoxia reduces hippocampal pyruvate dehydrogenase activity. Free Radic. Biol. Med. 2006;40:1960–1970. doi: 10.1016/j.freeradbiomed.2006.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards E.M. Fiskum G. Rosenthal R.E. Hopkins I. McKenna M.C. Hyperoxic reperfusion after global ischemia decreases hippocampal energy metabolism. Stroke. 2007;38:1578–1584. doi: 10.1161/STROKEAHA.106.473967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson C.L. Bucci C.J. Fiskum G. Mitochondrial response to calcium in the developing brain. Brain Res. Dev. Brain Res. 2004;151:141–148. doi: 10.1016/j.devbrainres.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Robertson C.L. Soane L. Siegel Z.T. Fiskum G. The potential role of mitochondria in pediatric traumatic brain injury. Dev. Neurosci. 2006;28:432–446. doi: 10.1159/000094169. [DOI] [PubMed] [Google Scholar]
- Robertson C.L. Saraswati M. Fiskum G. Mitochondrial dysfunction early after traumatic brain injury in immature rats. J. Neurochem. 2007;101:1248–1257. doi: 10.1111/j.1471-4159.2007.04489.x. [DOI] [PubMed] [Google Scholar]
- Ross B.D. Ernst T. Kreis R. Haseler L.J. Bayer S. Danielsen E. Bluml S. Shonk T. Mandigo J.C. Caton W. Clark C. Jensen S.W. Lehman N.L. Arcinue E. Pudenz R. Shelden C.H. 1H MRS in acute traumatic brain injury. J. Magn. Reson. Imaging. 1998;8:829–840. doi: 10.1002/jmri.1880080412. [DOI] [PubMed] [Google Scholar]
- Satrustegui J. Contreras L. Ramos M. Marmol P. del Arco A. Saheki T. Pardo B. Role of aralar, the mitochondrial transporter of aspartate-glutamate, in brain N-acetylaspartate formation and Ca2+ signaling in neuronal mitochondria. J. Neurosci. Res. 2007;85:3359–3366. doi: 10.1002/jnr.21299. [DOI] [PubMed] [Google Scholar]
- Schuhmann M.U. Stiller D. Skardelly M. Bernarding J. Klinge P.M. Samii A. Samii M. Brinker T. Metabolic changes in the vicinity of brain contusions: a proton magnetic resonance spectroscopy and histology study. J. Neurotrauma. 2003;20:725–743. doi: 10.1089/089771503767869962. [DOI] [PubMed] [Google Scholar]
- Serres S. Bezancon E. Franconi J.-M. Merle M. Ex vivo analysis of lactate and glucose metabolism in the rat brain under different states of depressed activity. J. Biol. Chem. 2004;279:47881–47889. doi: 10.1074/jbc.M409429200. [DOI] [PubMed] [Google Scholar]
- Shu S.K. Ashwal S. Holshouser B.A. Nystrom G. Hinshaw D.B., Jr. Prognostic value of 1H-MRS in perinatal CNS insults. Pediatr. Neurol. 1997;17:309–318. doi: 10.1016/s0887-8994(97)00140-9. [DOI] [PubMed] [Google Scholar]
- Shutter L. Tong K.A. Holshouser B.A. Proton MRS in acute traumatic brain injury: role for glutamate/glutamine and choline for outcome prediction. J. Neurotrauma. 2004;21:1693–1705. doi: 10.1089/neu.2004.21.1693. [DOI] [PubMed] [Google Scholar]
- Soustiel J.F. Glenn T.C. Shik V. Boscardin J. Mahamid E. Zaaroor M. Monitoring of cerebral blood flow and metabolism in traumatic brain injury. J. Neurotrauma. 2005;22:955–965. doi: 10.1089/neu.2005.22.955. [DOI] [PubMed] [Google Scholar]
- Tallan H.H. Moore S. Stein W.H. N-Acetyl-L-aspartic acid in brain. J. Biol. Chem. 1956;219:257–264. [PubMed] [Google Scholar]
- Taylor D.L. Davies S.E. Obrenovitch T.P. Doheny M.H. Patsalos P.N. Clark J.B. Symon L. Investigation into the role of N-acetylaspartate in cerebral osmoregulation. J. Neurochem. 1995;65:275–281. doi: 10.1046/j.1471-4159.1995.65010275.x. [DOI] [PubMed] [Google Scholar]
- Thomas S. Prins M.L. Samii M. Hovda D.A. Cerebral metabolic response to traumatic brain injury sustained early in development: a 2-deoxy-D-glucose autoradiographic study. J. Neurotrauma. 2000;17:649–665. doi: 10.1089/089771500415409. [DOI] [PubMed] [Google Scholar]
- Viant M.R. Lyeth B.G. Miller M.G. Berman R.F. An NMR metabolomic investigation of early metabolic disturbances following traumatic brain injury in a mammalian model. NMR Biomed. 2005;18:507–516. doi: 10.1002/nbm.980. [DOI] [PubMed] [Google Scholar]
- Yeo R.A. Phillips J.P. Jung R.E. Brown A.J. Campbell R.C. Brooks W.M. Magnetic resonance spectroscopy detects brain injury and predicts cognitive functioning in children with brain injuries. J. Neurotrauma. 2006;23:1427–1435. doi: 10.1089/neu.2006.23.1427. [DOI] [PubMed] [Google Scholar]
- Yoshino A. Hovda D.A. Kawamata T. Katayama Y. Becker D.P. Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: evidence of a hyper- and subsequent hypometabolic state. Brain Res. 1991;561:106–119. doi: 10.1016/0006-8993(91)90755-k. [DOI] [PubMed] [Google Scholar]
- Zwingmann C. Richter-Landsberg C. Leibfritz D. 13C isotopomer analysis of glucose and alanine metabolism reveals cytosolic pyruvate compartmentation as part of energy metabolism in astrocytes. Glia. 2001;34:200–212. doi: 10.1002/glia.1054. [DOI] [PubMed] [Google Scholar]