

Abstract
Traumatic brain injury (TBI) triggers a massive glutamate efflux, hyperactivation of N-methyl-d-aspartate receptors (NMDARs) and neuronal cell death. Previously it was demonstrated that, 15 min following experimentally induced closed head injury (CHI), the density of activated NMDARs increases in the hippocampus, and decreases in the cortex at the impact site. Here we show that CHI-induced alterations in activated NMDARs correlate with changes in the expression levels of the major NMDARs subunits. In the hippocampus, the expression of NR1, NR2A, and NR2B subunits as well as the GluR1 subunit of the AMPA receptor (AMPAR) were increased, while in the cortex at the impact site, we found a decrease in the expression of these subunits. We demonstrate that CHI-induced increase in the expression of NMDAR subunits and GluR1 in the hippocampus, but not in the cortex, is associated with an increase in NR2B tyrosine phosphorylation. Furthermore, inhibition of NR2B-phosphorylation by the tyrosine kinase inhibitor PP2 restores the expression of this subunit to its normal levels. Finally, a single injection of PP2, prior to the induction of CHI, resulted in a significant improvement in long-term recovery of motor functions observed in CHI mice. These results provide a new mechanism by which acute trauma contributes to the development of secondary damage and functional deficits in the brain, and suggests a possible role for Src tyrosine kinase inhibitors as preoperative therapy for planned neurosurgical procedures.
Key words: closed head injury, MAPK, NMDA receptor, NR2B, tyrosine phosphorylation
Introduction
Traumatic brain injury (TBI) is a devastating disease, and a leading cause of mortality and morbidity among young people in the Western world (Waxweiler et al., 1995). TBI survivors suffer from emotional, cognitive, and motor disturbances, leading to decreased quality of life. Among the processes triggered by TBI is the derangement of glutamatergic neurotransmission, including impaired synaptic plasticity and excitotoxicity (Albensi et al., 2000; Sanders et al., 2000; Yi and Hazell, 2006). An acute increase in extracellular glutamate levels has been detected in both experimental brain trauma models and in human patients (Bullock et al., 1995; Hovda et al., 1995; Nilsson et al., 1990), which can lead to overstimulation of glutamate receptors (Alessandri et al., 1996; Baker et al., 1993; Brown et al., 1998; Lynch and Dawson, 1994; Palmer et al., 1993; Yi and Hazell, 2006). The excess glutamate results in an increased influx of calcium into the cell, which is mainly mediated through a specific post-synaptic glutamate receptor ion channel, the N-methyl-d-aspartate receptor (NMDAR). Therefore, this receptor was thought to play a key role in the mechanism of secondary excitotoxic damage after TBI (Faden et al., 1989; Hayes et al., 1988; Miller et al., 1990), although this theory was not universally accepted (Obrenovitch and Urenjak, 1997; Obrenovitch et al., 2000).
NMDARs are heteromeric assemblies of a core NR1 subunit with different modulatory NR2 (A-D) and less common NR3 (A&B) subunits (Sucher et al., 1996; Sun et al., 1998). Among the intracellular processes that regulate NMDAR channel function is tyrosine phosphorylation of its NR2 subunits by members of the Src family of protein tyrosine kinases (Src-PTKs), which enhances channel function (Salter and Kalia, 2004). Physiological and pharmacological studies indicate that tyrosine phosphorylation of the NMDAR by Src-PTKs is crucial for several forms of learning and for the induction of long-term potentiation (LTP) in both hippocampus (Grant et al., 1992; Lu et al., 1998) and cortex (Rosenblum et al., 1996; Rosenblum et al., 1997). Broad-spectrum Src-PTKs inhibitors prevent tetanus-induced LTP but do not alter pre-existing potentiation (O'Dell et al., 1991), implying that Src-PTKs are involved in induction rather than maintenance of LTP. Studies on Src-PTKs signaling in models of cerebral ischemia have revealed that transient ischemia induces an increase in tyrosine phosphorylation of NR2A and NR2B (Cheung et al., 2000, 2003; Takagi et al., 1997, 1999). The tyrosine residue on position 1472 (Y1472) in the NR2B intracellular C-terminal tail is hyperphosphorylated in post-ischemic rats, and phosphorylation of Y1472 is reduced by inhibition of Src-PTKs (Cheung et al., 2003). Furthermore, Src-PTKs inhibitors suppress NMDAR-mediated excitotoxicity in vitro (Hashimoto et al., 2003), and provide neuroprotection against surgically induced brain injury (Jadhav et al., 2007a,b), or MK-801-induced cortical injury (Dickerson and Sharp, 2006) in vivo. These results implicate a Src-mediated pathway and tyrosine phosphorylation of NMDARs in the pathophysiological mechanism of neuronal death caused by different types of brain injury.
It was previously shown that the density of activated NMDARs following experimentally induced closed head injury (CHI) in mice, changed as a function of time post-injury and brain region. Using a radiolabeled noncompetitive NMDAR open channel blocker, MK801, a dramatic increase in NMDAR in the hippocampus was observed, 15 min post-injury, while in the cortical area at the impact site a reduction of MK801-labled NMDAR was found (Biegon et al., 2004). However, at later time points post-injury (hours to days), activated NMDARs and NR1, NR2A, and NR2B subunit expression declined gradually in both hippocampus and cortex (Biegon et al., 2004). These data raise the possibility that the early region-specific differential changes observed following CHI may result from alteration in the expression of NMDARs, their function, or both through a phosphorylation-dependent mechanism. Therefore, the aim of this study was to investigate the involvement of Src-PTKs mediated phosphorylation of NMDARs in the early biochemical changes in a mouse model of CHI.
Here we show that 15 min post-CHI, the expression of NR1, NR2A, and NR2B, as well as the GluR1 subunit of the AMPA receptor (AMPAR) increased in the hippocampus and decreased in the cortex. These changes were associated with an increase in NR2B tyrosine phosphorylation and activation of ERK signaling in the hippocampus but not in the cortex. Moreover, inhibition of NR2B-phosphorylation restored the expression of these subunits in the hippocampus but not in the cortex. Finally, to correlate these changes with functional outcome, we show that a single in vivo injection of Src-PTKs specific inhibitor improves the long-term recovery of motor functions observed in CHI mice. Taken together, these results suggest that tyrosine phosphorylation of NR2B may contribute to the early alterations in NMDAR function or in signaling pathways in the post-traumatic brain and may be related to pathogenic events leading to neuronal damage.
Methods
Reagents
Goat anti-NR2B polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-NR2A polyclonal antibody (Upstate Biotechnology, Waltham, MA), mouse anti-NR1 monoclonal antibody (Zymed Laboratories, San Francisco, CA), rabbit anti-(pY1472) NR2B polyclonal antibodies, anti-pY polyclonal antibodies, rabbit anti-GluR1 polyclonal antibodies (Chemicon, Temecula, CA), mouse anti-phospho-p44/42 MAPK(pERK) monoclonal antibody and rabbit anti-p44/42 MAPK (ERK) polyclonal antibody (Cell Signaling, Danvers, MA) were used for immunoblotting experiments. Monoclonal anti-transferrin receptor (TfR) antibodies were a generous gift from Dr. Aroeti B. (Hebrew university of Jerusalem, Israel). PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo[3,4-d]pyrimidine) was purchased from Cal-biochem (La Jolla, CA).
Animals and Trauma Model
The study was performed according to the guidelines of the Institutional Animal Care Committee of the Hebrew University (Jerusalem, Israel). Male Sabra mice of the Hebrew University strain, 8–10 weeks old and weighing 35–45 g, were used in this study. Animals were bred in a specific pathogen-free (SPF) environment; kept in cages, with four to six mice per cage, under standard conditions of temperature and light; and fed with food and water ad libitum. Twelve to 18 animals per group were used for the biochemical and behavioral experiments. Experimental CHI was induced by using a modified weight-drop device developed previously (Chen et al., 1996). Briefly, under isoflurane anesthesia, a midline longitudinal incision was made, skin was retracted, and the skull was exposed. The left anterior frontal area was identified, and a tipped Teflon cone (2-mm diameter) was placed 1 mm lateral to the midline, in the midcoronal plane. The head was fixed and a weight, calibrated according to the age and weight of the animal, was dropped on the cone from a fixed height, resulting in a focal injury to the left hemisphere. After trauma, the animals received supporting oxygenation with 95% O2 for no longer than 2 min and were returned to their cages. Sham controls received anesthesia and skin incision only.
Preparation of Brain Homogenates and Subcellular Fractionation
At 15 min after CHI, the animals were decapitated. The brains were removed and the cortex below the weight impact site on the skull (typically 3–4 mm in diameter; Fig. 1), and the ipsilateral hippocampus were dissected immediately and frozen in liquid nitrogen. The same brain regions were taken from sham animals. Samples (pools of three animals) were homogenized in homogenization buffer (HB) containing 320 mM sucrose, 10 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1 mM EGTA, protease inhibitor cocktail (Sigma), and the phosphatase inhibitors 1 mM NaVO4 and 5 mM NaF. Sub-cellular fractionation was performed as previously described (Yaka et al., 2003a). Briefly, homogenates were centrifuged at 1000g for 5 min to remove nuclei and large debris (P1). The supernatant (S1) was centrifuged at 10,000g for 30 min to obtain a crude synaptosomal fraction (P2) and the cytosolic and light membrane fraction (S2). The pellet was re-suspended in solubilization buffer (SB) containing 1% sodium dodecyl sulfate (SDS), 10 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1 mM EGTA, protease inhibitor cocktail (Sigma), and the phosphatase inhibitors 1 mM NaVO4 and 5 mM NaF.
FIG. 1.

Morphology of closed head injury (CHI) mouse brain. (Left) Whole brain of mouse removed immediately after performing the weight-drop procedure applied to the left hemisphere, demonstrating the impact area. (Right) Coronal slice (2–3 mm) was prepared from the same injured brain, at a plane indicated by the arrow. Note the hemorrhage developed around the impact site (arrow) at the cortical surface. Cortical or hippocampal tissues from the impact level (3–4 mm in diameter) were dissected and used for biochemical analysis. The scale is in mm.
Western Blot Analysis
Tissue samples were boiled with SDS–loading buffer for 10 min at 95°C prior to loading onto gels. Equal protein amounts (50 μg protein/lane) were used in each lane of the gel, and proteins were separated using 10% SDS—polyacrylamide gel electrophoresis (PAGE) and transferred to a nitrocellulose membrane. Membranes were incubated in blocking buffer (5% milk in 0.5% phosphate-buffered saline [PBS]-Tween) for 1 h at room temperature. The membranes were incubated overnight at 4°C with appropriate primary antibodies and then incubated (2 h at room temperature) with appropriate horseradish peroxidase (HRP)–conjugated secondary antibodies.
PP2 Treatment Protocol
PP2 (0.03 mg/kg in dimethyl sulfoxide [DMSO] diluted 1:1000 in sterile PBS) or vehicle (DMSO at the same dilution) were injected intraperitioneally (i.p.) right before induction of CHI, to allow its penetration through the blood–brain barrier (BBB), which was breached as a result of this procedure. One group (CHI and sham mice) was sacrificed 15 min post-injury, and another group was kept for 24 days to assess the effect of PP2 on motor function using the Neurological Severity Score (NSS) and biochemical analysis thereafter.
Neurobehavioral Evaluation
Mice were evaluated by a blinded examiner, using a set of 10 tasks, collectively known as NSS, which tests reflexes, alertness, coordination, and motor abilities (Yatsiv et al., 2002). One point is awarded for failure to perform a particular task; thus, a normal mouse scores 0. NSS was evaluated at 1 h (NSS1h) following CHI to define severity of injury, and at 1, 2, 3, 7, 17, and 24 days following CHI. The extent of recovery was calculated as the difference between NSS1h and that at any other time, as shown: dNSS = NSS (1 h) − NSS (t).
Statistical Analysis
All data are expressed as mean ± standard deviation (SD). Digitized images of the bands corresponding to glutamate receptor subunits, phosphorylated NR2B and ERK, were quantitatively analyzed by densitometry, with NIH Image version 1.61 providing peak areas. Values were expressed as percent of control. Statistical analysis was performed using Mann-Whitney test for significant differences. For behavioral experiments, significance was determined using the non-parametric Mann-Whitney test for dNSS values. All p values of <0.05 were considered significant for all comparisons.
Results
CHI Differentially Affects the Expression of NMDAR Subunits
Our first aim was to determine whether the changes observed earlier by quantitative autoradiography of the open channel blocker MK801 reflect alteration in the expression of NMDAR subunits following acute CHI. CHI was induced as described previously (Yaka et al., 2007), and the level of NMDAR subunits in the hippocampus at 15 min post-CHI was measured by Western blot analysis. As shown in Figure 2A, we found a significant increase in NR1, NR2A, and NR2B subunits in the total homogenates in the hippocampus. However, the expression of these subunits significantly decreased in the cortex within the impact site (Fig. 2B). Next, we determined whether the observed changes in NMDAR subunits are reflected in the synaptic membrane, the primary location of NMDAR. At 15 min post-CHI, we performed subcellular fractionation and compared the levels of NMDAR subunits in the crude synaptosomal fraction (P2) and cytosolic and light membranes fraction (S2) from both hippocampus and cortex. As shown in Figure 3, the expression of NR1, NR2A, and NR2B was significantly increased in the hippocampus in both synaptosomal and cytosolic fractions (Fig. 3A) and decreased in the cortex in the same fractions (Fig. 3B), similar to the results obtained in the total homogenates (Fig. 2A,B). In order to examine whether CHI-induced alterations in NMDAR expression is specific, we tested two other residents of glutamatergic synapses, the GluR1 subunit of the AMPAR, and the transferrin receptor (TfR). As shown in Figure 3C,D, the immunoreactivity of GluR1 was increased significantly in the hippocampus and decreased in the cortex in the membranal and cytosolic fractions, similar to NMDAR subunits. However, no significant change in transferrin receptor was evident in both regions and in all fractions that were tested. Taken together, these results suggest that CHI alters the expression of the glutamate ion channels NMDAR and AMPAR but not other membranal receptors such as transferrin receptor, in a region-specific manner and these changes are reflected in the crude synaptosomal membranes.
FIG. 2.

Closed head injury (CHI) induces an increase in the hippocampus and a decrease in cortical N-methyl-d-aspartate receptor (NMDAR) subunits. (A) At 15 min following CHI, the hippocampus ipsilateral to the injured side was removed, homogenized, and total protein was isolated. Samples from sham and CHI mice (50 μg/lane) were resolved on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Membranes were probed with anti-NR1, anti-NR2A, and anti-NR2B antibodies. Histogram depicts the level of proteins and presented as mean ± SD percent of (sham) control (n = 6 pools of 3 animals in each pool). **p < 0.01; *p < 0.05; significantly different from sham (Mann-Whitney test). (B) At 15 min following CHI, the cortex ipsilateral to the injured side was removed, homogenized and total protein were isolated and analyzed as in A. Histogram depicts the level of proteins presented as mean ± SD percent of (sham) control (n = 4 pools of 3 animals in each pool). **p < 0.01; *p < 0.05; significantly different from sham (Mann-Whitney test).
FIG. 3.

Closed head injury (CHI)–induced alterations of N-methyl-d-aspartate receptor (NMDAR) and AMPA receptor (AMPAR) subunits are localized to the synaptic membranes. (A) At 15 min following CHI, the hippocampus ipsilateral to the injured side was removed, homogenized, and synaptosomal (P2) and light membranes (S2) fractions were isolated. Samples from sham and CHI mice (50 μg/lane) were resolved on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Membranes were probed with anti-NR1, anti-NR2A, and anti-NR2B antibodies. Histogram depicts the level of proteins presented as mean ± SD percent of (sham) control (n = 6 pools of 3 animals in each pool). **p < 0.01; *p < 0.05; significantly different from sham (Mann-Whitney test). (B) At 15 min following CHI, the cortex ipsilateral to the injured side was removed, homogenized and total protein were isolated and analyzed as in A. Histogram depicts the level of proteins presented as mean ± SD percent of (sham) control (n = 4 pools of 3 animals in each pool). **p < 0.01; *p < 0.05; significantly different from sham (Mann-Whitney test). (C) Animals were treated as in A, and membranes containing hippocampal proteins were probed with anti-GluR1 and anti-transferrin receptor (TfR) antibodies. Histogram depicts the level of proteins presented as mean ± SD percent of (sham) control (n = 4 pools of 3 animals in each pool). **p < 0.01; significantly different from sham (Mann-Whitney test). (D) Cortical samples were analysed as in C. Histogram depicts the level of proteins, presented as mean ± SD percent of (sham) control (n = 4 pools of 3 animals in each pool). **p < 0.01; significantly different from sham (Mann-Whitney test).
CHI Increases NR2B Phosphorylation in the Hippocampus
We next hypothesized that CHI-induced changes in the expression of the NMDAR subunits are associated with alteration of its phosphorylation state. To test this hypothesis, we assessed CHI-induced alteration in the phosphorylation state of Y1472 on the NR2B subunit, the major tyrosine phosphorylation site for Src-PTKs identified by Nakazawa et al. (2001). Therefore, we isolated crude synaptosomal (P2) and cytosolic (S2) fractions from the cortex and the hippocampus, and tested NR2B tyrosine phosphorylation using specific antibodies that recognize the phosphorylated tyrosine residue on position 1472 on NR2B. As shown in Figure 4, a significant increase in NR2B tyrosine phosphorylation was observed 15 min post-CHI in both synaptosomal and cytosolic fractions from the hippocampus (Fig. 4A). However, although we found a decrease in NR2B expression in the cortex, we observed no change in NR2B phosphorylation on pY1472 as a result of CHI (Fig. 4B), suggesting that the CHI-induced increase in NMDAR expression in the hippocampus but not in the cortex is associated with an increase in NR2B phosphorylation. Based on these results, we hypothesized that CHI-induced increase in the expression of the NMDAR subunits in the hippocampus is mediated via tyrosine phosphorylation of NR2B. Therefore, inhibition of phosphorylation by the potent Src-PTKs inhibitor, PP2, should restore the expression of NMDAR subunits to their basal levels. To test this hypothesis, PP2 or vehicle were injected intraperitoneally (i.p.) right before injury, and CHI and sham mice were sacrificed 15 min following injury. As shown in Figure 5A, PP2 completely abolished the CHI-induced increase in NR1, NR2A and NR2B in both P2 and S2 fractions, and restored their expression levels to those of sham control animals. To confirm that PP2 inhibits NR2B phosphorylation on tyrosine 1472 we also probed the membranes with anti-pY1472 antibodies and found that the level of phosphorylated NR2B in PP2-treated animals was similar to that found in sham controls (Fig. 5). We next examined whether PP2 will inhibit the increase in the GluR1 subunit of AMPAR induced by CHI. Similar to the NMDAR, a single injection of PP2 completely abolished the increase in GluR1 in the hippocampus (Fig. 5). Interestingly, we found a significant increase in ERK activity (phospho-ERK1/2) in both synaptosomal and cytosolic fractions from the hippocampus, that was completely abolished by PP2 (Fig. 5), raising the possibility that CHI-induced increase in the expression of NMDARs through increased phosphorylation of NR2B is activating the ERK signaling pathway which in turn increases AMPAR.
FIG. 4.
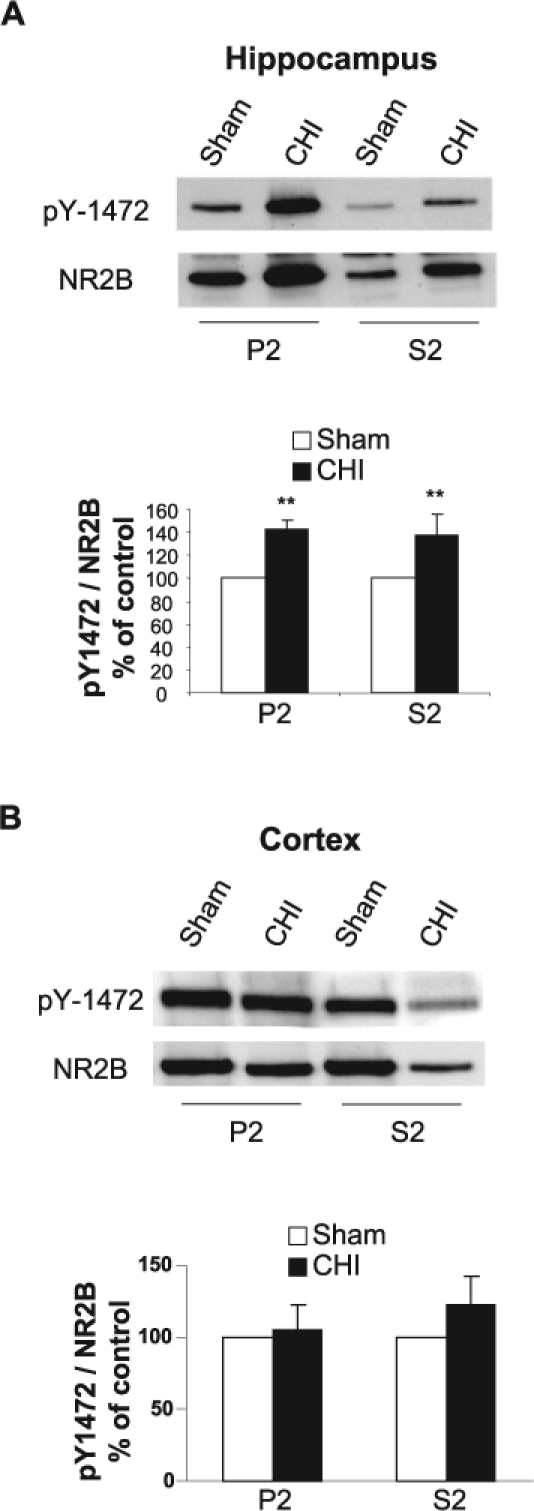
Closed head injury (CHI) induces an increase in tyrosine phosphorylation of the NR2B subunit in the hippocampus but not in the cortex. (A) At 15 min following CHI, the hippocampus ipsilateral to the injured side was removed, homogenized, and synaptosomal (P2) and light membranes (S2) fractions were isolated. Samples from sham and CHI mice (50 μg/lane) were resolved on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Membranes were probed with anti-pY1472 and anti-NR2B antibodies, and the level of phosphorylation was normalized to NR2B. Representative blots are shown from sham (n = 4 pools of 3 animals in each pool) and CHI mice (n = 4 pools of 3 animals in each pool). Histogram depicts the level of NR2B pY-1472, presented as mean ± SD percent of (sham) control (n = 4 pools of 3 animals in each pool). **p < 0.01; significantly different from sham (Mann-Whitney test). (B) At 15 min following CHI, the cortex ipsilateral to the injured side was removed, homogenized, and total protein was isolated and analyzed as in A. Histogram depicts the level of NR2B pY-1472, presented as mean ± SD percent of (sham) control (n = 4 pools of 3 animals in each pool). No statistically significant difference in pY-1472 was detected between sham and CHI mice (Mann-Whitney test).
FIG. 5.
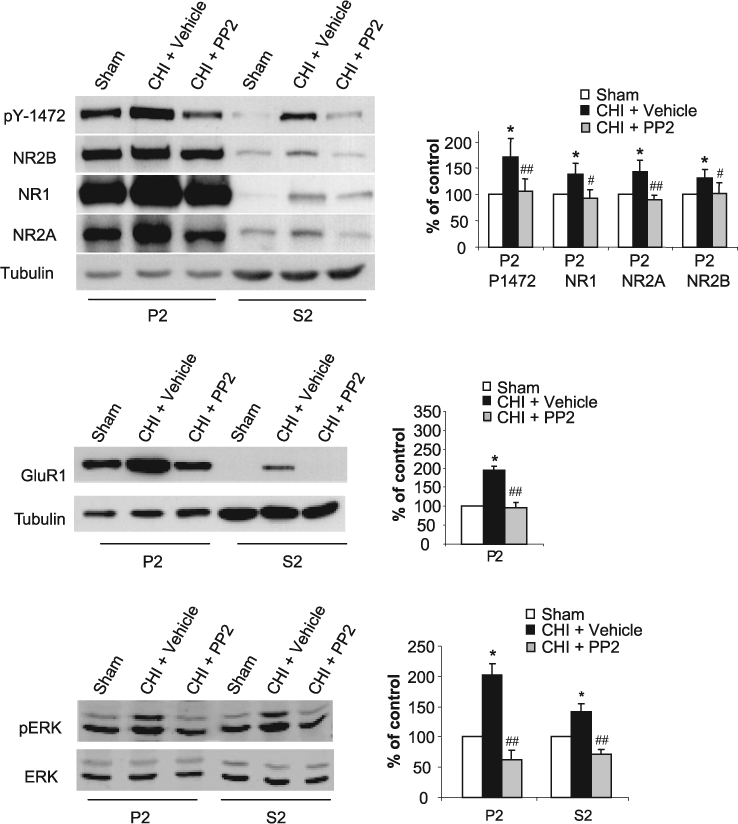
PP2 abolishes closed head injury (CHI)–induced increase in NR2B and ERK phosphorylation, and normalize the amount of N-methyl-d-aspartate receptor (NMDAR) and AMPA receptor (AMPAR) subunits in the hippocampus. Prior to the application of CHI, mice received i.p. injection of vehicle (CHI) or 0.03 mg/kg of PP2, the specific inhibitor of the Src-PTKs (CHI + PP2). After 15 min, the hippocampus ipsilateral to the injured side was removed, homogenized, and synaptosomal (P2) and light membranes (S2) fractions were isolated. Sham animals served as control (Sham). Samples (50 μg/lane) were resolved on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Membranes were probed with anti-pY1472, anti-NR1, anti-NR2A, anti-NR2B (top); anti-GluR1 and anti-TfR (middle); anti-phospho-p44/42 MAPK and anti-p44/42 MAPK antibodies (bottom). Histograms depict the level of NR2B phosphorylation (normalized to total NR2B) and NMDAR subunits in the P2 fraction or GluR1 (normalized to tubulin) in the P2 fraction, and ERK phosphorylation (normalized to total ERK). Data are presented as mean ± SD percent of (sham) control (n = 6 pools of 3 animals in each pool). *p < 0.05, for sham vs. CHI+vehicle; or ##p < 0.01, #p < 0.05 for CHI+PP2 vs. CHI+vehicle (Mann-Whitney test).
Inhibition of Src-PTKs Improve CHI-Induced Motor Dysfunction
If CHI-induced increase in the expression of NMDAR subunits is mediated, at least in part, by NR2B tyrosine phosphorylation in the hippocampus, one may speculate that inhibition of NR2B phosphorylation by PP2 in CHI mice will attenuate the neurological deficits induced by CHI. To test this hypothesis, we evaluated the effects of a single injection of PP2 right before the induction of CHI on neurological function. Animals were randomized into two groups, assigned to receive a single injection of vehicle or PP2, and then CHI was performed. NSS was evaluated at 1 h (NSS1h) following CHI to ensure similar severity of injury, and at 1, 2, 3, 7, 10, 17, and 24 days following CHI. The extent of recovery was calculated as the difference between NSS1h and that at any other time, and defined as dNSS. As shown in Figure 6, dNSS was similar in the two groups up to 7 days post-injury. However, neurological recovery was significantly accelerated in the PP2-treated mice when compared to vehicle-treated mice at 10, 17, and 24 days post-injury. These results suggest that, although PP2 was given at the time of trauma, its beneficial effects are manifested starting from 7–10 days later and are further maintained for at least 2 more weeks.
FIG. 6.
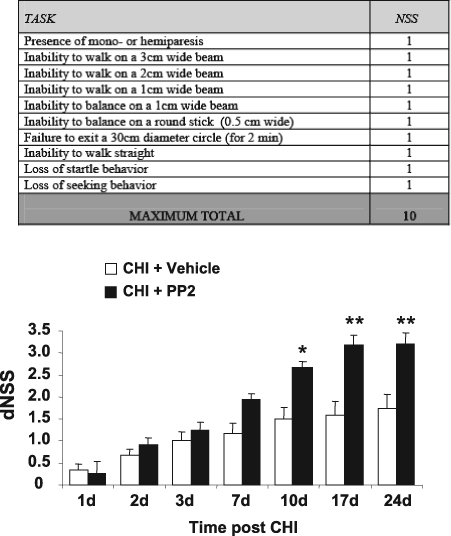
PP2 improves long-term neurological recovery after closed head injury (CHI). PP2 (0.03 mg/kg) or vehicle was administered i.p. prior to the weight-drop procedure (n = 12 in each group). Neurobehavioral status was evaluated by the Neurological Severity Score (NSS), such that an increase in dNSS [dNSS = NSS (1 h) - NSS (t)] represents functional recovery. dNSS was evaluated on 1, 2, 3, 7, 14, and 24 days following injury. Data are presented as mean ± SD. A sustained beneficial effect in PP2-treated compared with vehicle-treated mice was observed, which became significant 10 days after trauma and became more significant up to 24 days. *p < 0.05; **p < 0.01 (Mann-Whitney test).
PP2 Does Not Affect the Levels of Hippocampal and Cortical NMDAR Subunits 24 Days following CHI
In order to examine whether the improvement in the neurological function in PP2-treated CHI mice is due to alterations in NMDAR subunits in the hippocampus, we determined the protein levels of NMDAR subunits taken from the hippocampus and cortex of the same animals following the last NSS test (24 days). As shown in Figure 7, we found no difference in NR1, NR2A, and NR2B from CHI mice compared to sham animals, and PP2-treated animals did not differ from either sham or CHI injected with vehicle. These results suggest that the effect of NR2B phosphorylation on the levels of NMDAR subunits expression is important at early time points following CHI.
FIG. 7.

PP2 does not affect the levels of N-methyl-d-aspartate receptor (NMDAR) subunits 24 days following closed head injury (CHI). At 24 days following CHI, the hippocampus and the cortex ipsilateral to the injury from CHI, CHI+PP2, or sham animals were removed, homogenized, and synaptosomal (P2) and light membranes (S2) fractions were isolated. Hippocampal (left) or cortical (right) samples (50 μg/lane) were resolved on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Membranes were probed with anti-NR1, anti-NR2A, and anti-NR2B antibodies. Histograms depict the level of NMDAR subunits in the P2 and S2 fractions, presented as mean ± SD percent of (sham) control (n = 4 pools of 3 animals in each pool). No significant differences were found between sham and treated mice (Mann-Whitney test).
Discussion
The present study is the first to demonstrate that CHI causes an acute opposite change in the expression of glutamate receptors subunits in the cortex and hippocampus. Shortly (15 min) following CHI, the expression of NMDAR subunits, NR1, NR2A, and NR2B and the GluR1 subunit of AMPAR is increased in the hippocampus and decreased in the cortex at the impact site. The CHI-induced increase in the expression of hippocampal but not in cortical NMDAR subunits, was associated with an increase in NR2B tyrosine phosphorylation and activation of ERK signaling. Furthermore, inhibition of NR2B-phosphorylation by the specific Src-tyrosine kinase inhibitor PP2, restored these subunits to their normal levels and attenuated the increase in ERK phosphorylation following CHI. Finally, a single injection of PP2, prior to the induction of CHI, resulted in a significant improvement in long-term recovery of neurological functions observed in CHI mice.
The present study is based on a previous observation, which demonstrated that the number of activated NMDARs following CHI dramatically increased in the hippocampus 15 min post-injury, while in cortical areas adjacent to the impact site a reduction of MK801-labled NMDARs was found. However, it was shown that activated NMDAR were decreased in both hippocampus and cortex at time points between 1 h and up to 24 h, but only the cortical decrease were sustained beyond 24 h (Biegon et al., 2004). These findings were supported by Kumar et al. (2002), who demonstrated a decrease in hippocampal NMDAR subunits protein levels which lasted 6–12 h following cortical impact injury. Consistent with Biegon et al. (2004), Osteen et al. (2004) reported a decrease in NMDAR subunits protein levels in the cortex, 1–4 days following lateral fluid percussion with no change in hippocampal NMDAR subunits. Since phosphorylation of NMDAR plays a major role in receptor expression and activity (Chen and Leonard, 1996; Lu et al., 1998; Wang and Salter, 1994; Wang et al., 1996; Yaka et al., 2002, 2003b; Yu et al., 1997), these findings raised the possibility that CHI can affect the number of NMDARs, their function, or both, in a region-specific manner through a phosphorylation-dependent mechanism. The present results thus corroborate our findings on the alteration in the number of activated NMDARs following CHI and suggest that tyrosine phosphorylation of NR2B may contribute to the early alterations in NMDAR function or in signaling pathways in the post-traumatic brain and may be related to pathogenic events leading to neurological deficits.
We observed that CHI resulted in an increase in the amount of the NMDAR subunits NR1, NR2A, and NR2B in the total homogenates in the hippocampus, which may be due to new protein synthesis of these subunits. Since the increase in NMDAR subunits is too rapid to reflect transcriptional activation, we proposed that it is due to activation of the translation machinery locally at the dendritic domain, in a transcription-independent manner, as we have previously shown for NMDAR in ventral tegmental area (VTA) slices following short stimulation with cocaine (Schilstrom et al., 2006). In this regard, Kumar et al. (2002), who reported decreases in NMDAR subunits protein levels a few hours after brain injury, also did not observe changes in gene expression as measured by reverse transcription—polymerase chain reaction (RT-PCR). However, this possibility requires further in depth investigation. Nevertheless, we show that the increase in the expression and synaptic distribution of NMDAR subunits are mediated via tyrosine phosphorylation of the NR2B subunit. These results raise the possibility that phosphorylation can also play a role in receptor trafficking in and out of the synaptic membrane.
NMDAR activity can be modulated by tyrosine kinases and phosphatases (Salter and Kalia, 2004); however, it is not known how this modulation is achieved. Several groups have provided evidence that phosphorylation of the NMDAR may regulate movement of the receptor in and out of the synaptic membrane (Besshoh et al., 2005; Dunah and Standaert, 2001; Grosshans et al., 2002; Lan et al., 2001; Vissel et al., 2001). For example, in striatal synaptosomes, dopamine D1 receptor activation leads to NMDAR movement to the membrane, and this effect seems to be dependent on tyrosine phosphorylation by Fyn kinase (Dunah et al., 2004; Dunah and Standaert, 2001). Consistent with these reports, it was shown that NR2A and NR2B tyrosine phosphorylation is positively correlated with an increase in the number of these receptors in the post-synaptic density (PSD) of adult rats exposed to an ischemic challenge (Besshoh et al., 2005). Indeed, transient global ischemia induces several pathophysiological changes that may lead to neuronal cell death and one of them is a rapid and sustained increase in the tyrosine phosphorylation of the NMDAR subunits NR2A and NR2B in the rat hippocampus, through their enhanced association with Src and Fyn kinases (Takagi et al., 1997, 1999). Additional studies provide support for the hypothesis that specific tyrosine residues are involved in NMDAR trafficking and function (Lavezzari et al., 2003, 2004; Roche et al., 2001; Vissel et al., 2001). It was shown that the C-terminus of the NR2B subunit contains the internalization motif YEKL, which is the binding site for the clathrin adaptor AP-2. The tyrosine (Y1472) within this YEKL motif is phosphorylated by the Src family tyrosine kinases, which in turn inhibits the binding of AP-2 and promotes surface expression of NMDAR (Lavezzari et al., 2003). In agreement with these studies, recent work by Snyder and colleagues suggests that dephosphorylation of NR2B-Y1472 correlates with amyloid-β-induced internalization of the NMDAR in dissociated cortical neurons (Snyder et al., 2005). Tyrosine phosphorylation can influence NMDAR membrane surface expression in the adult rat hippocampus, and in particular Y1472 on the NR2B subunit, may be involved in the localization of the receptor in the synaptic membrane (Goebel et al., 2005). Because NR2B-Y1472 phosphorylation and NR2B surface expression are positively correlated after treatment with tyrosine phosphatase inhibitor (Hallett et al., 2006), these data are consistent with the hypothesis that membrane retention of NR2B may be enhanced by phosphorylation at Y1472 and thus may be due to a decrease in clathrin-mediated internalization of the receptor. These findings clearly show that phosphorylation of key residues, such as Y1472, may act as a “switch” to regulate endocytosis. Therefore, it is possible that a CHI-induced increase in NR2B phosphorylation at Y1472 is one of the initial steps that contribute to hyperactivation of NM-DAR following trauma, leading to neural damage. However, which kinase among the Src-family PTKs is responsible for this modulation remains to be resolved. A likely candidate is Fyn kinase, which was previously shown to regulate NM-DAR function in several animal models, including ischemia (Cheung et al., 2000; Dunah et al., 2004; Takagi et al., 1999; Yaka et al., 2003b).
Calcium influx through the NMDAR activates the Ras/ERK pathway, and ERK in the brain is activated in response to various NMDAR-related stimuli, including LTP (English and Sweatt, 1996), long-term memory (Brambilla et al., 1997), and ischemia (Farnsworth et al., 1995). It was shown that CHI in mice induces an increase in Ras-GTP and phosphorylation of ERK in the contused hemisphere, which was observed already at 10 min after injury and remained elevated even 2 h later. It was also found that CHI-induced increase in phospho-ERK is inhibited by the NMDAR open channel blocker MK-801 and by the Ras inhibitor, FTS (Shohami et al., 2003). In addition, calcium influx through NMDAR in cultured striatal neurons was shown to mediate the phosphorylation of ERK, which was inhibited by PP2 (Crossthwaite et al., 2004). In agreement with these findings, we found a significant increase in ERK activity in both synaptosomal and cytosolic fractions in the hippocampus, which was completely abolished by PP2. NMDAR opening and rise in post-synaptic calcium concentration during repetitive synaptic activity are critical during synaptic plasticity. A number of studies indicate that these inducing stimuli lead to regulated trafficking of post-synaptic AMPA-sensitive glutamate receptors into the synapse during LTP, and out of the synapse during long-term depression (LTD) (Luscher et al., 2000; Malinow et al., 2000; Scannevin and Huganir, 2000; Sheng and Lee, 2001). It was also shown that NMDAR-dependent hippocampal LTP, is mediated via activation of ERK signaling and insertion of GluR1 into the synapse (Zhu et al., 2002). Therefore it is tempting to speculate that the increase in hippocampal GluR1 seen in the present study could be a result of hyperactivation of NMDAR through increases in NR2B phosphorylation and as a result, induces NMDAR-dependent activation of ERK signaling. This in turn causes the insertion of GluR1 to the synapse. Thus, the inhibition of NR2B phosphorylation “turns off” the trigger, thereby preventing downstream signaling induced by CHI. However, temporal dissection of this mechanism is yet to be explored.
In contrast, we observed a decrease in the expression of the NMDAR subunits and GluR1 in the cortex at the impact site, which was not associated with alteration of NR2B phosphorylation state. The rapid decrease in cortical NMDAR subunits after injury, which was also observed by others within 1 h (Giza et al., 2006), would seem to be too quick to be explained by neuronal death. Recently, a new mechanism of autoregulation of the NMDAR in cultured neurons induced by agonist overactivation was suggested. Activation of NMDARs induces a specific and rapid decrease in the levels of full-length NR2A (Gascon et al., 2008) and full-length NR2B subunits (Simpkins et al., 2003), paralleled with calpain-dependent increase in the truncated forms of these subunits lacking the c-terminal domains. Since our antibodies are directed towards the c-terminal of both NR2A and NR2B, which recognize only the full-length proteins, our results in the cortex may reflect a decrease in the full-length proteins, consistent with these studies. However, we cannot exclude the possibility that an increase in the truncated forms of theses receptors occurred in our samples as well. In conclusion, we hypothesized that the cortex is more vulnerable to excitotoxicity than the hippocampus because of its proximity to the impact site, while phosphorylation is the most likely mechanism to explain the CHI-induced increase of NMDAR subunits in the hippocampus which is not directly affected by the impact.
Several studies show that competitive or noncompetitive inhibitors of NMDAR given at early time points improved outcome in animal models of TBI and stroke (Chen et al., 1991; Di and Bullock, 1996; Kroppenstedt et al., 1998). Recently, we have shown that early administration (24 h post-injury) of D-cycloserine (DCS), a co-agonist of NMDAR, improves the neurological outcome seen 2 weeks following CHI, which was mediated through subsequent changes (e.g., increase in BDNF expression and LTP) that take a longer time to manifest (Yaka et al., 2007). These results suggest that the timing of intervention is critical for the expression of neurological improvement seen at later time point following injury. Here we show that early inhibition of phosphorylation of a specific subunit (NR2B), rather than full blockade of the whole receptor, is sufficient to improve neurological outcome following CHI, and suggests a possible role for Src tyrosine kinase inhibitors as preoperative therapy for planned neurosurgical procedures.
Acknowledgments
We thank Prof. Esther Shohami for helpful discussions and critical reading of the manuscript. This research was supported by the Israel Science Foundation (grant no. 292/05 R.Y.), Ministry of Health (grant no. 6029-6 R.Y.), and the National Institutes of Health (grant no. R01 NS 050285-01 A2, to A.B.).
Author Disclosure Statement
No conflicting financial interests exist.
References
- Albensi B.C. Sullivan P.G. Thompson M.B. Scheff S.W. Mattson M.P. Cyclosporin ameliorates traumatic brain-injury-induced alterations of hippocampal synaptic plasticity. Exp. Neurol. 2000;162:385–389. doi: 10.1006/exnr.1999.7338. [DOI] [PubMed] [Google Scholar]
- Alessandri B. Landolt H. Langemann H. Gregorin J. Hall J. Gratzl O. Application of glutamate in the cortex of rats: a microdialysis study. Acta Neurochir. Suppl. 1996;67:6–12. doi: 10.1007/978-3-7091-6894-3_2. [DOI] [PubMed] [Google Scholar]
- Baker A.J. Moulton R.J. MacMillan V.H. Shedden P.M. Excitatory amino acids in cerebrospinal fluid following traumatic brain injury in humans. J. Neurosurg. 1993;79:369–372. doi: 10.3171/jns.1993.79.3.0369. [DOI] [PubMed] [Google Scholar]
- Besshoh S. Bawa D. Teves L. Wallace M.C. Gurd J.W. Increased phosphorylation and redistribution of NMDA receptors between synaptic lipid rafts and post-synaptic densities following transient global ischemia in the rat brain. J. Neurochem. 2005;93:186–194. doi: 10.1111/j.1471-4159.2004.03009.x. [DOI] [PubMed] [Google Scholar]
- Biegon A. Fry P.A. Paden C.M. Alexandrovich A. Tsenter J. Shohami E. Dynamic changes in N-methyl-D-aspartate receptors after closed head injury in mice: implications for treatment of neurological and cognitive deficits. Proc. Natl. Acad. Sci. USA. 2004;101:5117–5122. doi: 10.1073/pnas.0305741101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R. Gnesutta N. Minichiello L. White G. Roylance A.J. Herron C.E. Ramsey M. Wolfer D.P. Cestari V. Rossi-Arnaud C. Grant S.G. Chapman P.F. Lipp H.P. Sturani E. Klein R. A role for the Ras signalling pathway in synaptic transmission and long-term memory. Nature. 1997;390:281–286. doi: 10.1038/36849. [DOI] [PubMed] [Google Scholar]
- Brown J.I. Baker A.J. Konasiewicz S.J. Moulton R.J. Clinical significance of CSF glutamate concentrations following severe traumatic brain injury in humans. J. Neuro-trauma. 1998;15:253–263. doi: 10.1089/neu.1998.15.253. [DOI] [PubMed] [Google Scholar]
- Bullock R. Zauner A. Myseros J.S. Marmarou A. Woodward J.J. Young H.F. Evidence for prolonged release of excitatory amino acids in severe human head trauma. Relationship to clinical events. Ann. N. Y. Acad. Sci. 1995;765:290–298. doi: 10.1111/j.1749-6632.1995.tb16586.x. [DOI] [PubMed] [Google Scholar]
- Chen C. Leonard J.P. Protein tyrosine kinase-me-diated potentiation of currents from cloned NMDA receptors. J. Neurochem. 1996;67:194–200. doi: 10.1046/j.1471-4159.1996.67010194.x. [DOI] [PubMed] [Google Scholar]
- Chen M. Bullock R. Graham D.I. Frey P. Lowe D. McCulloch J. Evaluation of a competitive NMDA antagonist (D-CPPene) in feline focal cerebral ischemia. Ann. Neurol. 1991;30:62–70. doi: 10.1002/ana.410300112. [DOI] [PubMed] [Google Scholar]
- Chen Y. Constantini S. Trembovler V. Weinstock M. Shohami E. An experimental model of closed head injury in mice: pathophysiology, histopathology, and cognitive deficits. J. Neurotrauma. 1996;13:557–568. doi: 10.1089/neu.1996.13.557. [DOI] [PubMed] [Google Scholar]
- Cheung H.H. Takagi N. Teves L. Logan R. Wallace M.C. Gurd J.W. Altered association of protein tyrosine kinases with postsynaptic densities after transient cerebral ischemia in the rat brain. J. Cereb. Blood Flow Metab. 2000;20:505–512. doi: 10.1097/00004647-200003000-00009. [DOI] [PubMed] [Google Scholar]
- Cheung H.H. Teves L. Wallace M.C. Gurd J.W. Inhibition of protein kinase C reduces ischemia-induced tyrosine phosphorylation of the N-methyl-d-aspartate receptor. J. Neurochem. 2003;86:1441–1449. doi: 10.1046/j.1471-4159.2003.01951.x. [DOI] [PubMed] [Google Scholar]
- Crossthwaite A.J. Valli H. Williams R.J. Inhibiting Src family tyrosine kinase activity blocks glutamate signalling to ERK1/2 and Akt/PKB but not JNK in cultured striatal neurones. J. Neurochem. 2004;88:1127–1139. doi: 10.1046/j.1471-4159.2004.02257.x. [DOI] [PubMed] [Google Scholar]
- Di X. Bullock R. Effect of the novel high-affinity glycine-site N-methyl-D-aspartate antagonist ACEA-1021 on 125I-MK-801 binding after subdural hematoma in the rat: an in vivo autoradiographic study. J. Neurosurg. 1996;85:655–661. doi: 10.3171/jns.1996.85.4.0655. [DOI] [PubMed] [Google Scholar]
- Dickerson J. Sharp F.R. Atypical antipsychotics and a Src kinase inhibitor (PP1) prevent cortical injury produced by the psychomimetic, noncompetitive NMDA receptor antagonist MK-801. Neuropsychopharmacology. 2006;31:1420–1430. doi: 10.1038/sj.npp.1300878. [DOI] [PubMed] [Google Scholar]
- Dunah A.W. Sirianni A.C. Fienberg A.A. Bastia E. Schwarzschild M.A. Standaert D.G. Dopamine D1-dependent trafficking of striatal N-methyl-D-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol. Pharmacol. 2004;65:121–129. doi: 10.1124/mol.65.1.121. [DOI] [PubMed] [Google Scholar]
- Dunah A.W. Standaert D.G. Dopamine D1 receptor-dependent trafficking of striatal NMDA glutamate receptors to the postsynaptic membrane. J. Neurosci. 2001;21:5546–5558. doi: 10.1523/JNEUROSCI.21-15-05546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English J.D. Sweatt J.D. Activation of p42 mitogen-activated protein kinase in hippocampal long-term potentiation. J. Biol. Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- Faden A.I. Demediuk P. Panter S.S. Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- Farnsworth C.L. Freshney N.W. Rosen L.B. Ghosh A. Greenberg M.E. Feig L.A. Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature. 1995;376:524–527. doi: 10.1038/376524a0. [DOI] [PubMed] [Google Scholar]
- Gascón S. Sobrado M. Roda J.M. Rodríguez-Peña A. Díaz-Guerra M. Excitotoxicity and focal cerebral ischemia induce truncation of the NR2A and NR2B subunits of the NMDA receptor and cleavage of the scaffolding protein PSD-95. Mol. Psychiatry. 2008;13:99–114. doi: 10.1038/sj.mp.4002017. [DOI] [PubMed] [Google Scholar]
- Giza C.C. Maria N.S. Hovda D.A. N-Methyl-D-aspartate receptor subunit changes after traumatic injury to the developing brain. J. Neurotrauma. 2006;23:950–961. doi: 10.1089/neu.2006.23.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel S.M. Alvestad R.M. Coultrap S.J. Browning M.D. Tyrosine phosphorylation of the N-methyl-D-aspartate receptor is enhanced in synaptic membrane fractions of the adult rat hippocampus. Brain Res. Mol. Brain Res. 2005;142:65–79. doi: 10.1016/j.molbrainres.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Grant S.G. O'Dell T.J. Karl K.A. Stein P.L. Soriano P. Kandel E.R. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science. 1992;258:1903–1910. doi: 10.1126/science.1361685. [DOI] [PubMed] [Google Scholar]
- Grosshans D.R. Clayton D.A. Coultrap S.J. Browning M.D. LTP leads to rapid surface expression of NMDA but not AMPA receptors in adult rat CA1. Nat. Neurosci. 2002;5:27–33. doi: 10.1038/nn779. [DOI] [PubMed] [Google Scholar]
- Hallett P.J. Spoelgen R. Hyman B.T. Standaert D.G. Dunah A.W. Dopamine D1 activation potentiates striatal NMDA receptors by tyrosine phosphorylation-dependent subunit trafficking. J. Neurosci. 2006;26:4690–4700. doi: 10.1523/JNEUROSCI.0792-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto R. Fujimaki K. Jeong M.R. Christ L. Chuang D.M. Lithium-induced inhibition of Src tyrosine kinase in rat cerebral cortical neurons: a role in neuroprotection against N-methyl-D-aspartate receptor-mediated excitotoxicity. FEBS Lett. 2003;538:145–148. doi: 10.1016/s0014-5793(03)00167-4. [DOI] [PubMed] [Google Scholar]
- Hayes R.L. Jenkins L.W. Lyeth B.G. Balster R.L. Robinson S.E. Clifton G.L. Stubbins J.F. Young H.F. Pre-treatment with phencyclidine, an N-methyl-D-aspartate antagonist, attenuates long-term behavioral deficits in the rat produced by traumatic brain injury. J. Neurotrauma. 1988;5:259–274. doi: 10.1089/neu.1988.5.259. [DOI] [PubMed] [Google Scholar]
- Hovda D.A. Lee S.M. Smith M.L. Von Stuck S. Bergsneider M. Kelly D. Shalmon E. Martin N. Caron M. Mazziotta J., et al. The neurochemical and metabolic cascade following brain injury: moving from animal models to man. J. Neurotrauma. 1995;12:903–906. doi: 10.1089/neu.1995.12.903. [DOI] [PubMed] [Google Scholar]
- Jadhav V. Matchett G. Hsu F.P. Zhang J.H. Inhibition of Src tyrosine kinase and effect on outcomes in a new in vivo model of surgically induced brain injury. J. Neurosurg. 2007a;106:680–686. doi: 10.3171/jns.2007.106.4.680. [DOI] [PubMed] [Google Scholar]
- Jadhav V. Solaroglu I. Obenaus A. Zhang J.H. Neuroprotection against surgically induced brain injury. Surg. Neurol. 2007b;67:15–20. doi: 10.1016/j.surneu.2006.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroppenstedt S.N. Schneider G.H. Thomale U.W. Unterberg A.W. Protective effects of aptiganel HCl (Cerestat) following controlled cortical impact injury in the rat. J. Neurotrauma. 1998;15:191–197. doi: 10.1089/neu.1998.15.191. [DOI] [PubMed] [Google Scholar]
- Kumar A. Zou L. Yuan X. Long Y. Yang K. N-Methyl-D-aspartate receptors: transient loss of NR1/NR2A/NR2B subunits after traumatic brain injury in a rodent model. J. Neurosci. Res. 2002;67:781–786. doi: 10.1002/jnr.10181. [DOI] [PubMed] [Google Scholar]
- Lan J.Y. Skeberdis V.A. Jover T. Zheng X. Bennett M.V. Zukin R.S. Activation of metabotropic glutamate receptor 1 accelerates NMDA receptor trafficking. J. Neurosci. 2001;21:6058–6068. doi: 10.1523/JNEUROSCI.21-16-06058.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavezzari G. McCallum J. Dewey C.M. Roche K.W. Subunit-specific regulation of NMDA receptor endocytosis. J. Neurosci. 2004;24:6383–6391. doi: 10.1523/JNEUROSCI.1890-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavezzari G. McCallum J. Lee R. Roche K.W. Differential binding of the AP-2 adaptor complex and PSD-95 to the C-terminus of the NMDA receptor subunit NR2B regulates surface expression. Neuropharmacology. 2003;45:729–737. doi: 10.1016/s0028-3908(03)00308-3. [DOI] [PubMed] [Google Scholar]
- Lu Y.M. Roder J.C. Davidow J. Salter M.W. Src activation in the induction of long-term potentiation in CA1 hippocampal neurons. Science. 1998;279:1363–1367. doi: 10.1126/science.279.5355.1363. [DOI] [PubMed] [Google Scholar]
- Luscher C. Nicoll R.A. Malenka R.C. Muller D. Synaptic plasticity and dynamic modulation of the postsy-naptic membrane. Nat. Neurosci. 2000;3:545–550. doi: 10.1038/75714. [DOI] [PubMed] [Google Scholar]
- Lynch D.R. Dawson T.M. Secondary mechanisms in neuronal trauma. Curr. Opin. Neurol. 1994;7:510–516. doi: 10.1097/00019052-199412000-00007. [DOI] [PubMed] [Google Scholar]
- Malinow R. Mainen Z.F. Hayashi Y. LTP mechanisms: from silence to four-lane traffic. Curr. Opin. Neurobiol. 2000;10:352–357. doi: 10.1016/s0959-4388(00)00099-4. [DOI] [PubMed] [Google Scholar]
- Miller L.P. Lyeth B.G. Jenkins L.W. Oleniak L. Panchision D. Hamm R.J. Phillips L.L. Dixon C.E. Clifton G.L. Hayes R.L. Excitatory amino acid receptor subtype binding following traumatic brain injury. Brain Res. 1990;526:103–107. doi: 10.1016/0006-8993(90)90254-9. [DOI] [PubMed] [Google Scholar]
- Nakazawa T. Komai S. Tezuka T. Hisatsune C. Umemori H. Semba K. Mishina M. Manabe T. Yamamoto T. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J. Biol. Chem. 2001;276:693–699. doi: 10.1074/jbc.M008085200. [DOI] [PubMed] [Google Scholar]
- Nilsson P. Hillered L. Ponten U. Ungerstedt U. Changes in cortical extracellular levels of energy-related metabolites and amino acids following concussive brain injury in rats. J. Cereb. Blood Flow Metab. 1990;10:631–637. doi: 10.1038/jcbfm.1990.115. [DOI] [PubMed] [Google Scholar]
- Obrenovitch T.P. Urenjak J. Is high extracellular glutamate the key to excitotoxicity in traumatic brain injury? J. Neurotrauma. 1997;14:677–698. doi: 10.1089/neu.1997.14.677. [DOI] [PubMed] [Google Scholar]
- Obrenovitch T.P. Urenjak J. Zilkha E. Jay T.M. Excitotoxicity in neurological disorders–the glutamate paradox. Int. J. Dev. Neurosci. 2000;18:281–287. doi: 10.1016/s0736-5748(99)00096-9. [DOI] [PubMed] [Google Scholar]
- O'Dell T.J. Kandel E.R. Grant S.G. Long-term potentiation in the hippocampus is blocked by tyrosine kinase inhibitors. Nature. 1991;353:558–560. doi: 10.1038/353558a0. [DOI] [PubMed] [Google Scholar]
- Osteen C.L. Giza C.C. Hovda D.A. Injury-induced alterations in N-methyl-D-aspartate receptor subunit composition contribute to prolonged 45calcium accumulation following lateral fluid percussion. Neuroscience. 2004;128:305–322. doi: 10.1016/j.neuroscience.2004.06.034. [DOI] [PubMed] [Google Scholar]
- Palmer A.M. Marion D.W. Botscheller M.L. Swedlow P.E. Styren S.D. DeKosky S.T. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J. Neurochem. 1993;61:2015–2024. doi: 10.1111/j.1471-4159.1993.tb07437.x. [DOI] [PubMed] [Google Scholar]
- Roche K.W. Standley S. McCallum J. Dune Ly C. Ehlers M.D. Wenthold R.J. Molecular determinants of NMDA receptor internalization. Nat. Neurosci. 2001;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- Rosenblum K. Berman D.E. Hazvi S. Lamprecht R. Dudai Y. NMDA receptor and the tyrosine phosphorylation of its 2B subunit in taste learning in the rat insular cortex. J. Neurosci. 1997;17:5129–5135. doi: 10.1523/JNEUROSCI.17-13-05129.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum K. Dudai Y. Richter-Levin G. Long-term potentiation increases tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit 2B in rat dentate gyrus in vivo. Proc. Natl. Acad. Sci. USA. 1996;93:10457–10460. doi: 10.1073/pnas.93.19.10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter M.W. Kalia L.V. Src kinases: a hub for NMDA receptor regulation. Nat. Rev. Neurosci. 2004;5:317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- Sanders M.J. Sick T.J. Perez-Pinzon M.A. Dietrich W.D. Green E.J. Chronic failure in the maintenance of long-term potentiation following fluid percussion injury in the rat. Brain Res. 2000;861:69–76. doi: 10.1016/s0006-8993(00)01986-7. [DOI] [PubMed] [Google Scholar]
- Scannevin R.H. Huganir R.L. Postsynaptic organization and regulation of excitatory synapses. Nat. Rev. Neurosci. 2000;1:133–141. doi: 10.1038/35039075. [DOI] [PubMed] [Google Scholar]
- Schilstrom B. Yaka R. Argilli E. Suvarna N. Schumann J. Chen B.T. Carman M. Singh V. Mailliard W.S. Ron D. Bonci A. Cocaine enhances NMDA receptor-mediated currents in ventral tegmental area cells via dopamine D5 receptor-dependent redistribution of NMDA receptors. J. Neurosci. 2006;26:8549–8558. doi: 10.1523/JNEUROSCI.5179-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M. Lee S.H. AMPA receptor trafficking and the control of synaptic transmission. Cell. 2001;105:825–828. doi: 10.1016/s0092-8674(01)00406-8. [DOI] [PubMed] [Google Scholar]
- Shohami E. Yatsiv I. Alexandrovich A. Haklai R. Elad-Sfadia G. Grossman R. Biegon A. Kloog Y. The Ras inhibitor S-trans, trans-farnesylthiosalicylic acid exerts long-lasting neuroprotection in a mouse closed head injury model. J. Cereb. Blood Flow Metab. 2003;23:728–738. doi: 10.1097/01.WCB.0000067704.86573.83. [DOI] [PubMed] [Google Scholar]
- Simpkins K.L. Guttmann R.P. Dong Y. Chen Z. Sokol S. Neumar R.W. Lynch D.R. Selective activation induced cleavage of the NR2B subunit by calpain. J. Neurosci. 2003;23:11322–11331. doi: 10.1523/JNEUROSCI.23-36-11322.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder E.M. Nong Y. Almeida C.G. Paul S. Moran T. Choi E.Y. Nairn A.C. Salter M.W. Lombroso P.J. Gouras G.K. Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Sucher N.J. Awobuluyi M. Choi Y.B. Lipton S.A. NMDA receptors: from genes to channels. Trends Pharmacol. Sci. 1996;17:348–355. [PubMed] [Google Scholar]
- Sun L. Margolis F.L. Shipley M.T. Lidow M.S. Identification of a long variant of mRNA encoding the NR3 subunit of the NMDA receptor: its regional distribution and developmental expression in the rat brain. FEBS Lett. 1998;441:392–396. doi: 10.1016/s0014-5793(98)01590-7. [DOI] [PubMed] [Google Scholar]
- Takagi N. Cheung H.H. Bissoon N. Teves L. Wallace M.C. Gurd J.W. The effect of transient global ischemia on the interaction of Src and Fyn with the N-methyl-D-as-partate receptor and postsynaptic densities: possible involvement of Src homology 2 domains. J. Cereb. Blood Flow Metab. 1999;19:880–888. doi: 10.1097/00004647-199908000-00007. [DOI] [PubMed] [Google Scholar]
- Takagi N. Shinno K. Teves L. Bissoon N. Wallace M.C. Gurd J.W. Transient ischemia differentially increases tyrosine phosphorylation of NMDA receptor subunits 2A and 2B. J. Neurochem. 1997;69:1060–1065. doi: 10.1046/j.1471-4159.1997.69031060.x. [DOI] [PubMed] [Google Scholar]
- Vissel B. Krupp J.J. Heinemann S.F. Westbrook G.L. A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat. Neurosci. 2001;4:587–596. doi: 10.1038/88404. [DOI] [PubMed] [Google Scholar]
- Wang Y.T. Salter M.W. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature. 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]
- Wang Y.T. Yu X.M. Salter M.W. Ca2+-independent reduction of N-methyl-D-aspartate channel activity by protein tyrosine phosphatase. Proc. Natl. Acad. Sci. USA. 1996;93:1721–1725. doi: 10.1073/pnas.93.4.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxweiler R.J. Thurman D. Sniezek J. Sosin D. O'Neil J. Monitoring the impact of traumatic brain injury: a review and update. J. Neurotrauma. 1995;12:509–516. doi: 10.1089/neu.1995.12.509. [DOI] [PubMed] [Google Scholar]
- Yaka R. Biegon A. Grigoriadis N. Simeonidou C. Grigoriadis S. Alexandrovich A.G. Matzner H. Schumann J. Trembovler V. Tsenter J. Shohami E. D-Cycloserine improves functional recovery and reinstates long-term potentiation (LTP) in a mouse model of closed head injury. FASEB J. 2007;21:2033–2041. doi: 10.1096/fj.06-7856com. [DOI] [PubMed] [Google Scholar]
- Yaka R. Phamluong K. Ron D. Scaffolding of Fyn kinase to the NMDA receptor determines brain region sensitivity to ethanol. J. Neurosci. 2003a;23:3623–3632. doi: 10.1523/JNEUROSCI.23-09-03623.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaka R. Tang K.C. Camarini R. Janak P.H. Ron D. Fyn kinase and NR2B-containing NMDA receptors regulate acute ethanol sensitivity but not ethanol intake or conditioned reward. Alcohol Clin. Exp. Res. 2003b;27:1736–1742. doi: 10.1097/01.ALC.0000095924.87729.D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaka R. Thornton C. Vagts A.J. Phamluong K. Bonci A. Ron D. NMDA receptor function is regulated by the inhibitory scaffolding protein, RACK1. Proc. Natl. Acad. Sci. USA. 2002;99:5710–5715. doi: 10.1073/pnas.062046299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsiv I. Morganti-Kossmann M.C. Perez D. Dinarello C.A. Novick D. Rubinstein M. Otto V.I. Rancan M. Kossmann T. Redaelli C.A. Trentz O. Shohami E. Stahel P.F. Elevated intracranial IL-18 in humans and mice after traumatic brain injury and evidence of neuroprotective effects of IL-18-binding protein after experimental closed head injury. J. Cereb. Blood Flow Metab. 2002;22:971–978. doi: 10.1097/00004647-200208000-00008. [DOI] [PubMed] [Google Scholar]
- Yi J.H. Hazell A.S. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem. Int. 2006;48:394–403. doi: 10.1016/j.neuint.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Yu X.M. Askalan R. Keil G.J., 2nd Salter M.W. NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science. 1997;275:674–678. doi: 10.1126/science.275.5300.674. [DOI] [PubMed] [Google Scholar]
- Zhu J.J. Qin Y. Zhao M. Van Aelst L. Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]