

Abstract
Because of unsatisfactory treatment options for colon cancer, there is a need to develop novel preventive approaches for this malignancy. One such strategy is through chemoprevention by the use of non-toxic dietary substances and botanical products. Delphinidin, an anthocyanidin in pigmented fruits and vegetables, possesses strong antioxidant and anti-inflammatory properties. In the present study, we investigated the antiproliferative and proapoptotic properties of delphinidin in human colon cancer HCT116 cells. We found that treatment of cells with delphinidin (30–240 μM; 48 h) resulted in (i) decrease in cell viability (ii) induction of apoptosis, (iii) cleavage of PARP, (iv) activation of caspases-3, -8, and -9, (v) increase in Bax with a concomitant decrease in Bcl-2 protein, and (vi) G2/M phase cell cycle arrest. NF-κB provides a mechanistic link between inflammation and cancer, and is a major factor controlling the ability of both pre-neoplastic and malignant cells to resist apoptosis-based tumor surveillance mechanisms. We therefore, determined the effect of delphinidin on NF-κB signaling pathway. The immunoblot, ELISA and EMSA analysis demonstrated that the treatment of HCT116 cells with delphinidin resulted in the inhibition of (i) IKKα, (ii) phosphorylation and degradation of IκBα, (iii) phosphorylation of NF-κB/p65 at Ser536, (iv) nuclear translocation of NF-κB/p65, (v) NF-κB/p65 DNA binding activity, and (vi) transcriptional activation of NF-κB. Our results suggest that delphinidin treatment of HCT116 cells suppressed NF-κB pathway, resulting in G2/M phase arrest and apoptosis. We suggest that delphinidin could have potential in inhibiting colon cancer growth.
Keywords: apoptosis, cell cycle arrest, delphinidin, NF-κB
INTRODUCTION
Colon cancer is the third most common cancer in men and women and accounts for 10% of all cancers related deaths in the United States. Current treatments for this malignancy include chemotherapy, radiotherapy and surgery all of which are associated with a high risk of complications and are not always successful [1]. Because of unsatisfactory treatment options for colon cancer, there is a need to develop novel preventive treatment approaches for this malignancy. For a variety of reasons, naturally occurring botanicals and dietary substances are gaining increasing attention as cancer chemopreventive agents. There is considerable emphasis in identifying novel botanicals that selectively induces apoptosis and growth arrest of cancer cells without affecting normal cells with the hope of slowing cancer progression [2].
The transcription factor nuclear factor-kappa B (NF-κB) controls the expression of genes involved in many critical physiological responses, including immune and acute phase inflammatory responses, cell adhesion, differentiation, oxidative stress and apoptosis. NF-κB is composed of two subunits, p65 and p50, and is normally sequestered in the cytosol by an inhibitory protein, IκBα. Exposure of cells to a variety of extracellular stimuli leads to the rapid phosphorylation, ubiquitination, and ultimately proteolytic degradation of IκBα, resulting in the release of NF-κB from its inhibitory protein to translocate to the nucleus where it regulates transcription of various genes [3]. Increased NF-κB activity has been demonstrated in colon cancer, which is believed to enhance cancer cell survival by inhibiting apoptosis. Persistent activation of NF-κB in colon cancer can lead to the production of pro-inflammatory molecules, resulting in colitis and ultimately, colon cancer [3–8]. The inflammatory disease of the colon cancer, Crohn’s disease and ulcerative colitis, are associated with the constitutive activation of NF-κB. Recent studies have demonstrated that NF-κB activation is closely correlated with resistance to the apoptosis induced by various chemotherapeutic agents in colorectal cancer cells [7–10]. Therefore, inhibition of NF-κB signaling pathway provides attractive targets for new chemopreventive and chemotherapeutic approaches.
Fruits, vegetables, and common beverages, as well as several herbs and plants with diversified pharmacological properties, have been shown to be rich sources of phytochemicals with the potential to prevent human cancers [11–14]. Delphinidin (Figure 1A) is the major anthocyanidin present in pigmented fruits such as pomegranate, berries and dark grapes and vegetables such as egg plant, tomato, carrot, purple sweet potato, red cabbage and red onion and possesses strong antioxidant and anti-inflammatory properties [15]. Recently, we have shown that delphinidin protects HaCaT keratinocytes and mouse skin against UVB-mediated oxidative stress and apoptosis [15]. It has also been demonstrated that anthocyanins like delphinidin and cyanidin induced cell cycle arrest and apoptosis in different human cell lines [16].
Figure 1.
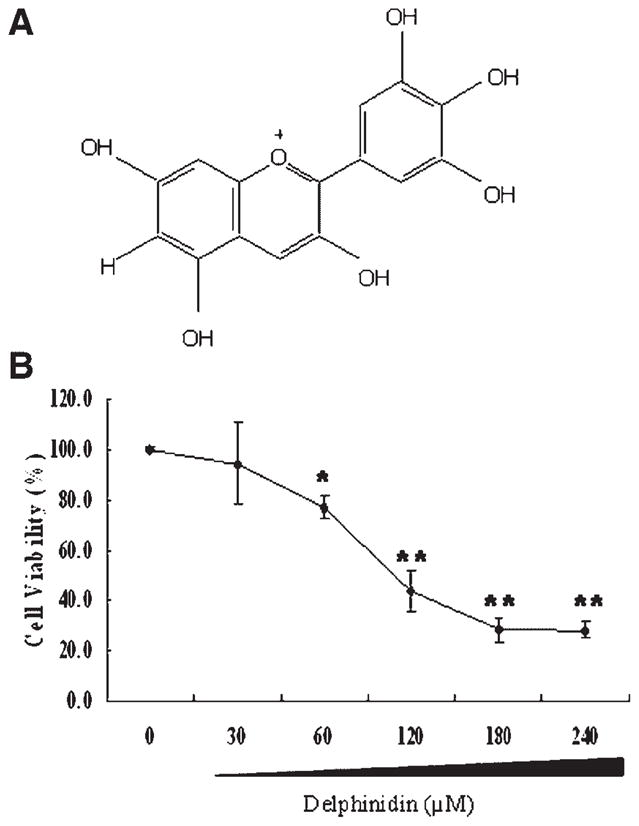
(A) Chemical structure of delphinidin. (B) Effect of delphinidin on the viability of HCT116 cells. Cells were treated with specified concentrations of delphinidin for 48 h, and cell viability was determined by MTT assay as described in Materials and Methods Section. The values are represented as the percentage cell inhibition where vehicle treated cells were regarded as 100%. The data represents the mean ± SD of three independent experiment each conducted in triplicate.
We hypothesized that delphinidin may be a useful chemotherapeutic agent against colon cancer. To test this hypothesis, we determined the efficacy of delphinidin as a growth inhibitory agent against human colon cancer HCT116 cells and delineated the mechanisms of this effect.
MATERIALS AND METHODS
Materials
Delphinidin (>98% pure) was purchased from Extrasynthase (Lyon, France). Anti-PARP116, anti-caspase-8,-9, anti-p53 and anti-p27Kip1 antibodies were procured from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-cyclin B1, anti-IKKα, anti-Bcl-2, anti-cdc2, anti-phospho-NF-κB/p65 and anti-p21WAF1/Cip1 antibodies were procured from Cell Signaling Technology (Beverly, MA). Anti-Bax and anti- NF-κB/p65 was procured from Upstate Biotechnology (Lake Placid, NY). Anti-PARP p85 antibody was procured from Promega (Madison, WI). Anti-mouse or anti-rabbit secondary antibody horse-radish peroxidase conjugate was obtained from Amersham Life Science, Inc. (Arlington Heights, IL). Annexin-V-FLOUS staining kit was purchased from Roche Diagnostic Corporation (Indianapolis, IN). APO-DIRECT apoptosis kit was obtained from Phoenix Flow systems (San Diego, CA). Trans-AM NF-κB/p65 enzyme-linked immunosorbent assay (ELISA) kit was purchased from Active Motif North America (Carlsbad, CA). Light-shift™ chemiluminescent electrophoretic mobility shift assay (EMSA) kit was obtained from Pierce (Rockford, IL). The BCA™ protein assay kit was purchased from Pierce. Novex precast Tris-Glycine gels were obtained from Invitrogen (Carlsbad, CA). All other chemicals, unless otherwise stated, were obtained from Sigma (St. Louis, MO).
Cell Culture
The human colon cancer HCT116 cells were obtained from American Type Culture Collection (Rockville, MD). HCT116 cells were cultured in DMEM (ATCC, Manassas, VA) containing 10% fetal bovine serum, 100 U/mL of penicillin, and 100 μg/mL of streptomycin (Gibco/BRL, San Francisco, CA). All cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Treatment of Cells
Delphinidin (dissolved in DMSO) was used for the treatment of cells. The final concentration of DMSO used was 0.1% (v/v) for each treatment. The cells (50–60% confluent) were treated with delphinidin (30–240 μM) for 48 h in DMEM medium after which the media was removed and cells were washed with phosphate-buffered saline (PBS) and then harvested.
MTT Assay for Cellular Viability
Cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma Co.) assay. The cells were plated at 1 × 104 cells per well 200 μL of complete culture medium and treated with designed concentrations of delphinidin (30–240 μM) in 96 well microtiter plates for 48 h at 37°C in a humidified chamber. Each concentration of delphinidin was repeated in 10 wells. After incubation for specified times at 37°C in a humidified incubator, MTT reagent (50 μL, 2 mg/mL in PBS) was added to each well and incubated for 4 h. The microtiter plate containing the cells was centrifuged at 1800 rpm for 5 min at 4°C. The MTT solution was removed from the wells by aspiration and the formazan crystals were dissolved in DMSO (150 μL). Absorbance was recorded on a microplate reader at 570 nm wavelength. The effect of delphinidin on growth inhibition was assessed as the percentage of inhibition in cell growth where vehicle-treated cells were taken as 100% viable. IC50 values were determined from three independent experiments.
Apoptosis Assessment by Annexin-V/Propidium Iodide Staining
The annexin-V-Fluos staining kit was used for the detection of apoptotic and necrotic cells according to vendor’s protocol. This kit uses a dual-staining protocol in which the apoptotic cells are stained with annexin-V (green fluorescence), and the necrotic cells are stained with propidium iodide (PI) (red fluorescence). Cells were grown to about 40% confluency and treated with delphinidin (30–240 μM) for 48 h. The fluorescence was measured by a Zeiss 410 confocal microscope (Thornwood, NY). Confocal images of green annexin-FITC fluorescence were collected using 488 nm excitation light from an argon/krypton laser, a 560 nm dichroic mirror, and a 514–540 nm bandpass barrier filter. Images of red PI fluorescence were collected using a 568 nm excitation light from the argon/krypton laser, a 560 nm dichroic mirror, and a 590 nm-long pass filter.
Quantification of Apoptosis
For quantification of apoptosis, the cells were treated with delphinidin (30–240 μM) for 48 h. The cells were trypsinized, washed with PBS, and processed for labeling with fluorescenin-tagged deoxyuridine triphosphate nucleotide and PI by use of an APO-DIRECT apoptosis kit as per manufacturer’s protocol. The labeled cells were then analyzed by flow cytometry.
DNA Cell Cycle Analysis by Flow Cytometry
The effect of delphinidin treatment on distribution of cells in different phases of the cell cycle was analyzed by flow cytometry. Briefly, after treatment of cells with delphinidin (30–240 μM) for 48 h, the cells were trypsinized and thereafter washed twice with cold PBS, and centrifuged. The pellet was resuspended in 50 μL cold PBS and 450 μL cold ethanol for 1 h at 4°C. The cells were centrifuged at 110g for 5 min, pellet washed twice with cold PBS, suspended in 500 μL PBS, and incubated with 5 μL RNAase (20 μg/mL final concentration) at 37°C for 30 min. The cells were then chilled over ice for 10 min and stained with PI (50 μg/mL final concentration) for 1 h for analysis by flow cytometry. Flow cytometry was performed with a FACScan (Becton Dickinson, Heidelberg, Germany). A minimum of 10 000 cells per sample were collected, and the DNA histograms were further analyzed by using Modi-FitLT software (Verily Software House, Topsham, ME) for cell cycle analysis.
Preparation of Whole Cell Lysate
After treatment of cells with delphinidin, the medium was aspirated and the cells were washed twice in PBS (10 mM, pH 7.4). The cells were incubated in 0.2 mL ice-cold lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 1 mM ethyleneglycol-bis(aminoethylether)-tetraacetic acid [EGTA], 1 mM ethylenediaminetetraacetic acid [EDTA], 20 mM NaF, 100 mM Na3VO4, 0.5% Nonidet P-40 [NP-40], 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride [PMSF] [pH 7.4]) with freshly added protease inhibitor cocktail (Protease Inhibitor Cocktail Set III; Calbiochem, La Jolla, CA) for 20 min. The cells were then centrifuged at 14 000g for 25 min at 4°C, and the supernatant (whole cell lysate) was stored at −80°C. The protein concentration was determined by the BCA protein assay kit.
Preparation of Cytosolic and Nuclear Lysates
After treatment of cells with delphinidin, the medium was aspirated and the cells were washed twice in PBS (10 mM, pH 7.4). The cells were incubated in 0.2 mL ice-cold lysis buffer (10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid [HEPES], 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol [DTT], 1 mM PMSF [pH 7.9]) with freshly added protease inhibitor cocktail (Protease Inhibitor Cocktail Set III; Calbiochem) for 15 min, after which 12.5 μL of 10% NP-40 was added and the contents were mixed on a vortex and then centrifuged for 1 min (14 000g) at 4°C. The supernatant was saved as cytosolic lysate and stored at −80°C. The nuclear pellet was resuspended in 50 μL of ice-cold nuclear extraction buffer (20 mM HEPES, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF [pH 7.9]) with freshly added protease inhibitor cocktail for 30 min with intermittent mixing. The cells were centrifuged for 5 min (14 000g) at 4°C, and the supernatant (nuclear extract) was stored at −80°C. The protein concentration was determined by the BCA protein assay kit.
Immunoblot Analysis
For immunoblot analysis, 40 μg of the protein was resolved over 12% polyacrylamide gels and transferred onto a nitrocellulose membrane. The blot containing the transferred protein was blocked in blocking buffer (5% nonfat dry milk, 1% Tween-20 in 20 mM Tris-buffered saline, pH 7.6) for 2 h at room temperature, incubated with appropriate monoclonal primary antibody in blocking buffer for 2 h at room temperature followed by incubation with anti-mouse or anti-rabbit secondary antibody horse-radish peroxidase for 2 h and then washed three times and detected by chemiluminescence and autoradiography using Hyperfilm obtained from Amersham Biosciences.
Enzyme-Linked Immunosorbent Assay (ELISA)
The commercially available Trans-AM kit used for the assay of NF-κB/p65 uses an oligonucleotide containing NF-κB consensuses site (5′-GGGACTTT-CC-3′) that binds to the nuclear extract and can detect NF-κB, which can recognize an epitope on p65 activated and bound to its target DNA. In the absence of the competitive binding with the wild-type or mutated consensus oligonucleotide, 30 μL of binding buffer was added to each well in duplicate. Alternatively, 30 μL of binding buffer containing 20 pmol (2 μL) of appropriate oligonucleotide, in duplicate, was added to the corresponding wells. Ten micrograms of protein from the nuclear lysate of each sample diluted in 20 μL lysis buffer was loaded per well. For positive control, 20 μL of lysis buffer containing 1 μL of control cell extract per well, and for blank, 20 μL of lysis buffer per well was used. The plate was sealed with the adhesive film and incubated for 1 h at room temperature with mild agitation, after which the well was washed three times with 200 mL of 1× wash buffer and 100 mL of diluted primary antibody (1:1000 dilution in 1× antibody-binding buffer) was added to each well and incubated at room temperature for 1 h without agitation. The wells were washed again three times with 1× wash buffer and 100 μL of diluted horse-radish peroxidase-conjugate antibody (1:1000 dilution in 1× antibody-binding buffer) was added to each well and incubated for 1 h. The wells were again washed four times with 1× wash buffer followed by the addition of 100 μL of developing solution. The content was incubated for 5 min at room temperature. This was followed by an addition of 100 μL of stop solution to each well, and the absorbance was read within 5 min at 450 nm.
Electrophoretic Mobility Shift Assay (EMSA)
EMSA for NF-κB was performed using a Light-shift™ chemiluminecent EMSA kit (Pierce) by following the manufacturer’s protocol. To start with, DNA was biotin labeled using the Biotin 3′ endlabeling kit (Pierce). Briefly, in a 50 μL reaction buffer, 5 pmol of double-stranded NF-κB oligonucleotide 5′-AGT TGA GGG GAC TTT CCC AGG C-3′; 3′-TCA ACT CCC CTG AAA GGG TCC G-5′ was incubated in a microfuge tube with 10 μL of 5× teminal deoxynucleotidyl transferase (TdT) buffer, 5 μL of 5 M biotin-N4-CTP, 10 U of diluted TdT, and 24 μL of ultra purewater, and incubated at 37°C for 30 min. For supershift assay, the reaction mixture was incubated for an additional 20 min with or without antibody for NF-κB/p65 (1 μg, Upstate, Inc.). The reaction was stopped with 2.5 μL of 0.2 M EDTA. To extract labeled DNA, 50 μL of chloroform/isoamyl alcohol (24:1) was added to each tube and centrifuged briefly at 13 000g. The top aqueous phase containing the labeled DNA was removed and saved for binding reactions. Each binding reaction contained 1× binding buffer (100 mM Tris, 500 mM KCl, 10 mM dithiothreitol, pH 7.5), 2.5% glycerol, 5 mM MgCl2, 50 ng/μL poly (dl-dC), 0.05% NP-40, 5 μg of nuclear extract, and 20–40 fm of biotin-endlabeled target DNA. The content was incubated at room temperature for 20 min. To this reaction mixture, 5 μL of 5× loading buffer was added, subjected to gel electrophoresis on a native polyacrylamide gel, and transferred to a nylon membrane. When the transfer was complete, DNA was crosslinked to the membrane at 120 mJ/cm2 using a UV crosslinker equipped with 254 nm bulbs. The biotin endlabeled DNA was detected using streptavidin-horseradish peroxidase conjugate and a chemiluminescent substrate. The membrane was exposed to X-ray film (XAR-5 Amersham Life Science, Inc.) and developed using a Kodak film processor.
Luciferase Assay
To determine the effect of delphinidin on NF-κB transcriptional activity, HCT116 cells were seed in 100 mm dishes at a density of 1 × 105 cells/mL. For transient transfections, HCT116 cells were harvested, washed once in cold phosphate-buffered saline, and resuspended in the specified electroporation buffer to a final concentration of 1 × 106 cells/mL. One microgram of luciferase reporter plasmid and 50 ng of Renilla reporter plasmid were mixed with 0.1 mL of Amexa Nucleofector solution (Amaxa, Cologne, Germany). HCT 116 cells were nucleofected using the Nucleofection Kit V. After electrophoration, cells were immediately transferred to complete medium, and cultured in 24 well plates at 37°C for 24 h. After 24 h post-transfection, the cultures were treated with various concentrations of compounds tested. After 48 h incubation, 24 well plates was washed twice cold phosphate-buffered saline. And then cells were collected. Lysates were made after washing with PBS and then incubating with 100 μL of Passive Lysis Buffer (Promega) for 15 min on a shaker at room temperature. Luciferase activity was measured by using the Dual-Luciferase Reporter Assays System (Promega) according to the manufacturer’s protocol. The firefly luciferase activity was normalized to the renilla luciferase activity for variability in transfection efficiency.
Statistical Analysis
Each experiment was performed at least three times. Results are expressed as the mean value ± standard deviation (SD). Statistical analysis was performed using Student’s t-test and statistical significance is expressed as *P <0.05, **P <0.01.
RESULTS
Delphinidin Inhibits HCT116 Cell Growth
The effect of delphinidin on cell viability was determined employing the MTT assay. As shown in Figure 1B, delphinidin (30–240 μM; 48 h) treatment to HCT116 cells resulted in significant growth inhibition. Delphinidin treatment resulted in 6%, 23%, 57%, 72%, and 72% decrease in cell viability at 30, 60, 120, 180, and 240 μM, respectively (Figure 1B). The IC50 value of delphinidin for HCT116 cells was found to be 110 μM.
Delphinidin Induces Apoptosis of HCT116 Cells
We performed Annexin-V/Propidium iodide staining in order to determine whether the decrease in observed cell viability is due to induction of apoptosis. Annexin-V specially binds to phosphatidylserine and has been employed for determination of apoptotic cells. When HCT116 cells were stained with Annexin-V/Propidium iodide and examined under a fluorescence microscope, apoptotic cells were found to be increased in delphinidin treated cells in a dose-dependent manner (Figure 2A). The extent of apoptosis was further quantified by flow cytometric analysis. As shown by the data in Figure 2B, delphinidin treatment resulted in 24%, 35%, and 38% increase in apoptosis at 120, 180, and 240 μM, respectively.
Figure 2.

Effect of delphinidin on apoptosis of HCT116 cells. (A) Fluorescence microscopy. The cells were treated with vehicle alone or specified concentration of delphinidin for 48 h. Annexin V-fluos staining kit was used for the detection of apoptosis and necrotic cells. Apoptosis and necrosis were detected by the kit according to the vendor’s protocol using a Zeiss Axiovert 100 microscope as described in Materials and Methods Section. The samples were excited at 330–380 nm, and the image was observed and photographed under a combination of a 400 nm dichroic mirror and the 420 nm high pass filter. Data from a typical experiment repeated three times with similar results are shown; Magnification 200×. (B). TUNEL Assay. The cells treated with delphinidin (30–240 μM; 48 h) by using apoptosis kit obtained from Phoenix Flow Systems (San Diego, CA) as per vendor’s protocol followed by flow cytometry. Cells showing deoxyuridine triphosphate fluorescence above that of control population, as indicated by the line in each histogram, are considered as apoptotic cells and their percentage population is shown in each box. The details are described in Materials and Methods Section.
Delphinidin Induces Activation of Caspases and Cleavage of PARP Protein in HCT116 Cells
To obtain further insight into the mechanism of delphinidin-induced apoptosis, as a first step, we examined whether delphinidin treatment could induce poly(ADP-ribose) polymerase (PARP) cleavage. PARP is a 116 kDa protein that is cleaved into 85 kDa fragment during apoptotic cell death. Thus, the cleavage of PARP is regarded as hallmark for the induction of apoptotic response. As shown in Figure 3A, the native 116 kDa PARP protein was found to be cleaved into its characteristic 85 kDa fragment upon treatment with delphinidin in a dose dependent manner (Figure 3A). Next, we determined the effect of delphinidin on activation of initiator and activator caspases. Caspases are aspartate-specific cysteine protease that play a key role in mediating apoptotic response. Caspases are responsible for many of the biochemical and morphological changes including nuclear membrane breakdown, chromatin condensation, fragmentation and the formation of apoptotic bodies that occur during apoptosis. Caspases are an evolutionarily conserved family of cysteine proteases that are responsible for diverse cellular functions including inflammation and apoptosis [17]. Employing immunoblot analysis we found that 48 h of delphinidin treatment resulted in a decrease in the protein expression of procaspase-3 (effector caspase), procaspase-8, and 9 (initiator caspases), indicating that the proform of caspase is cleaved into active form (Figure 3A).
Figure 3.

Effect of delphinidin on the protein expression of (A) PARP and procaspase-3, 8 and 9 (B) Bax and Bcl-2 in HCT116 cells. The cells treated with delphinidin (30–240 μM; 48 h) were harvested and nuclear lysate was prepared and protein was subjected to SDS–PAGE as detailed in Materials and Methods Section. Equal loading of protein was confirmed by stripping the immunoblot and reprobing it for β-actin. The immunoblot shown here are representative of three independent experiments with similar results. (C) The data were presented in the bar graphs as percentage of Bax/Bcl-2 ratio.
Delphinidin Modulates the Expression of Bcl2 Family Proteins in HCT116 Cells
We next determined the effect of delphinidin treatment on the protein expression of the Bcl-2 family members. Both Bcl-2 (antiapoptotic) and Bax (proapoptotic) proteins are members of large Bcl-2 family. Since cells maintain a fine balance between the concentrations of these two family members, shift in the levels of one of these proteins, that is, decreased expression of Bcl-2 or increased expression of Bax, can be critical for leading them to undergo apoptosis [18]. The immunoblot analysis data showed that the delphinidin treatment of cells increased the expression of Bax with concomintant decrease in the expression of Bcl-2 in a dose-dependent manner (Figure 3B). Thus, delphinidin treatment was found to result in alteration in Bax/Bcl-2 ratio in favor of apoptosis (Figure 3C). These results suggest that delphinidin-induced apoptosis may be mediated via modulation of Bcl-2 family proteins.
Delphinidin Causes G2/M Phase Cell Cycle Arrest in HCT 116 Cells
To investigate whether delphinidin treatment has an effect on the cell cycle regulation in HCT116 cells, we determined its effect on G2/M phase cell cycle distribution by flow cytometry after staining with PI. As shown in Figure 4, concomitant with growth inhibitory effects, delphinidin treatment induced a strong G2/M phase arrest. The distribution of cells in G2/M phase was 14%, 14%, 23%, 34%, and 37% at 30, 60, 120, 180 and 240 μM concentrations of delphinidin, respectively.
Figure 4.

Effect of delphinidin on distribution of cells in different phases of cell cycle in HCT116 cells. The cells treated with delphinidin for 48 h and were collected and stained with PI by using an apoptosis APO-DIRECT kit (Phoenix Flow systems) as per vendor’s protocol followed by flow cytometry. Following FACS analysis, cellular DNA histograms were further analyzed by ModfitLT V3.0. The data are representative example for duplicate tests. The details are described in Materials and Methods Section.
Delphinidin Inhibits Cyclin B and cdc2 and Induces p21WAF1/Cip1 and p53 Protein Expression in HCT 116 Cells
To understand the mechanism underlying G2/M arrest in delphinidin-treated HCT116 cells, we next determined the effect of delphinidin on cell cycle-regulatory molecules, including, cyclin B1 and cell division cycle 2 kinase (cdc2). Immunoblotting revealed that delphinidin treatment resulted in a significant decrease in the protein levels of cyclin B1 and cdc2 in a dose-dependent manner (Figure 5A). Tumor suppressor gene p53 is a key element in the induction of cell cycle arrest and apoptosis following DNA damage or cellular stress in human cells. The p53 dependent cell-cycle arrest requires transactivation of p21WAF1/Cip1 or other cell cycle-related factors. The induction of p21WAF1/Cip1 causes subsequent arrest in the G1/G0 or G2/M phase of the cell cycle by binding of the cyclin-cdk complex [19,20]. We therefore, next determined the effect of delphinidin treatment of cells on the cytoplasmic levels of p21WAF1/Cip1 and p53. As shown by immunoblot analysis in Figure 5B, delphinidin upregulated the expression of tumor suppressor protein p53 and its downstream target molecule p21WAF1/Cip1. These data show the possibility for the involvement of p21WAF1/Cip1 and p53 in delphinidin-induced G2/M phase arrest.
Figure 5.
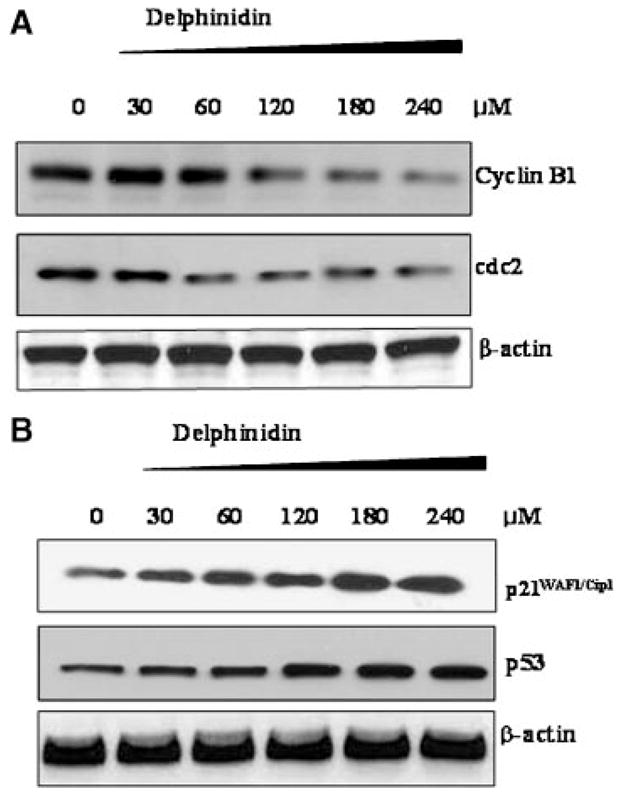
Effect of delphinidin on the protein expression on (A) cyclin B1 and cdc2 and (B) p21 and p53 (B) in HCT116 cells. The cells treated with delphinidin (30–240 μM; 48 h) were harvested and nuclear lysate was prepared and protein was subjected to SDS–PAGE as detailed in Materials and Methods Section. Equal loading of protein was confirmed by stripping the immunoblot and reprobing it for β-actin. The immunoblot shown here are representative of three independent experiments with similar results.
Delphinidin Inhibits Constitutive Activation of NF-κB in HCT116 Cells
NF-κB is known to regulate transcription of genes responsible for growth and survival as well as inhibition of apoptosis. Thus, we hypothesized that delphinidin might be inhibiting the NF-κB activation in HCT116 cells. As shown in Figure 6A, treatment of HCT116 cells with delphinidin (30–240 μM; 48 h) resulted in inhibition of phosphorylation and degradation of IκBα. Expression of I kappa B kinase α (IKKα) has been shown to be necessary for phosphorylation and degradation of IκBα. Immunoblot analysis showed that treatment of cells with delphinidin inhibited activation of IKKα in a dose-dependent manner (Figure 6A). We next assessed the effect of delphinidin on activation and nuclear translocation of NF-κB employing immunoblot analysis, ELISA and EMSA. Phosphorylation of NF-κB has been recently suggested as an additional step in the cascade of events leading to NF-κB activation [21]. As evident from the immunoblot analysis, we found that treatment of cells with delphinidin resulted in a dose-dependent inhibition of phosphorylation of NF-κB/p65 at Ser536 (Figure 6A). Immunoblot analysis data also revealed that treatment of cells with delphinidin inhibited nuclear translocation of NF-κB/p65 in a dose-dependent manner (Figure 6A). To further confirm our results, we also performed ELISA and found that treatment of cells with delphinidin inhibited nuclear translocation of NF-κB/p65 in a dose-dependent manner (Figure 6B). Furthermore, we performed EMSA (supershift) to investigate the effect of delphinidin treatment on NF-κB DNA-binding activity. As shown in Figure 6C, delphinidin treatment significantly inhibited NF-κB DNA-binding activity in a dose-dependent manner (Figure 6C). Next, we investigated the transcriptional activity of NF-κB by luciferase reporter assay. As shown in Figure 6D, the transcriptional activity of NF-κB in delphinidin-treated cells decreased as compared to untreated cells. These data indicate that delphinidin treatment resulted in a significant inhibition of constitutive NF-κB activation.
Figure 6.

Effect of delphinidin on IKKα, phosphorylation and degradation of IκBα and activation of NF-κB in HCT 116 cells. The cells treated with delphinidin (30–240 μM; 48 h) were harvested and nuclear lysate was prepared and protein was subjected to SDS-PAGE as detailed in Materials and Methods Section. (A) Immunoblot analysis of IKKα, IκBα, and NF-κB/p65. Equal loading of protein was confirmed by stripping the immunoblot and reprobing it for β-actin. The immunoblot shown here are representative of three independent experiments with similar results. (B) ELISA and (C) EMSA for NF-κB binding complex was demonstrated by anti-p65 antibody. (D) NF-κB transcriptional activity. At 48 h post-transfection with NF-κB luciferase plasmid, HCT116 cells were treated with the indicated concentration of delphinidin. Luciferase assay was performed as described in Materials and Methods Section.
DISCUSSION
Colon cancer is the second leading cause of cancer death in the USA and Europe [1,22]. The search for new chemopreventive and/or chemotherapeutic agents that are more effective without toxic effects has generated great interest in identifying phytochemicals for their potential use for humans. Several natural compounds, particularly plant products and dietary constituents, have been found to exhibit chemopreventive activities both in vitro and in vivo [2,23].
Anthocyanidins, a glycons of anthocyanins, are abundant in fruits and vegetables, and make a major contribution to the total polyphenol dietary intake [24]. They have been associated with a broad spectrum of beneficial health effects ranging from the treatment of diabetic retinopathy [25], atherosclerosis [26] and various microvascular diseases to potential anti-inflammatory [27], antitumoral [28], antimutagenic and anticarcinogenic [29], and chemopreventive properties [30]. Delphinidin (Figure 1A) is the major anthocyanidin present in pigmented fruits and vegetables and possesses strong antioxidant and anti-inflammatory properties [15].
Apoptosis represents a universal and exquisitely efficient suicide pathway, and is as an ideal way for elimination of damaged cells [31]. Recently, the apoptosis signaling systems have been shown to provide promising targets for the development of novel anticancer agents. Several plant-derived bio-active agents are known to be chemopreventive agents by inducing apoptosis in a number of experimental models of carcinogenesis [32]. It is now widely appreciated that agents capable of inducing apoptosis in cancer cells can potently lead to the development of mechanism-based prevention and treatment approaches for cancer. Therefore, a complete understanding of the mechanism of delphinidin action is important for its development for cancer prevention and/or therapy.
This study was designed to show the chemotherapeutic effects of delphinidin against colon cancer. Treatment of HCT116 cells with delphinidin resulted in decrease in cell viability (Figure 1B). Our results suggested that treatment of cells with delphinidin induced apoptosis (Figure 2). Delphinidin treatment also led to activation of initiator caspase-8 and caspase 9 and downstream effector caspase-3 (Figure 3A). The activation of effector caspase-3, in response to delphinidin treatment also resulted in cleavage of PARP in HCT116 cells (Figure 3A). The ratio between Bcl-2 and Bax has been suggested as a primary event in determining the susceptibility to apoptosis through maintaining the integrity of the mitochondria and inhibiting the activation of caspase cascade [18]. Delphinidin treatment resulted in a significant increase in proapoptotic protein Bax and decrease in antiapoptotic protein Bcl-2 resulting in a shift in Bax/Bcl-2 ratio in favor of apoptosis (Figure 3B and C).
Induction of apoptosis and/or inhibition of cell proliferation are highly correlated with the activation of variety of intracellular signaling pathways leading to arrest of cell cycle in the G1, S, or G2/M phase [31]. Cell cycle regulation and its modulation by various plant-derived agents are gaining widespread attention in recent years. A large number of phytochemicals have been shown to inhibit cell cycle progression of various cancer cells. G2/M transition provides an effective checkpoint in cell cycle progression that is regulated by the sequential activation and deactivation of cdc family proteins and cyclin complexes. The cdc2 interacts with cyclin B1, and activation of the cyclin B/cdc2 complex is required for transition from G2 to M phase of the cell cycle [33]. In this study, we observed that treatment of HCT116 cells with delphinidin resulted in arrest of cells in G2/M phase (Figure 4) and is associated with a decrease in the protein levels of cyclin B1 and cdc2 (Figure 5A). These results suggested that reduced expression of cyclin B1 and cdc2 may be involved in delphinidin-induced G2/M phase arrest, leading to cell growth inhibition and possible apoptotic death.
The cyclin dependent kinase (cdk) inhibitor p21WAF1/Cip1 binds and inhibits the cyclin D, E and A-dependent kinases, regulating the G1 to S phase transition of the cell cycle. Studies have demonstrated that cell cycle arrest at G2/M transition by DNA damaging agent is also tightly associated with the induction of p21WAF1/Cip1. p21WAF1/Cip1 has also been reported to influence the outcome of the p53 response to DNA damage and play a protective role in survival signal against apoptosis [34,35]. Tumor suppressor gene p53 is a key element in the induction of cell cycle arrest and apoptosis following DNA damage or cellular stress in human cells [36]. Our results suggested that delphinidin upregulated expression of tumor suppressor protein p53 and its downstream target molecule p21 in HCT116 cells (Figure 5B). The upregulation of p21WAF1/Cip, p53 and the downregulation of cyclin B1 and cdc2 may be one of the molecular mechanisms by which delphinidin inhibited HCT116 cells growth and induced cell-cycle arrest.
Certain dietary chemicals with chemopreventive properties in colorectal cancer cells such as flavonoids, beta-catotene, phenethyl isothiocyanate, sulforaphane and curcumin have been shown to induce apoptosis through a suppression of NF-κB activation [37–40]. In this present study, we investigated the effect of delphinidin on NF-κB pathway. The translocation of NF-κB to the nucleus is preceded by the phosphorylation and the proteolytic degradation of IκBα. We found that delphinidin treatment of HCT116 cells resulted in a dose dependent inhibition of degradation and phosphorylation of IκBα. Studies have shown that IKKα activity is necessary for IκBα protein phosphorylation/degradation. We also found that delphinidin treatment of HCT116 resulted in a dose dependent activation of IKKα (Figure 6A). Treatment of delphinidin to HCT116 cells resulted in inhibition of NF-κB activation, nuclear translocation of NF-κB (Figure 6A and B) and NF-κB DNA-binding activity (Figure 6C) in a dose-dependent manner. As shown Figure 6D, the transcriptional activity of NF-κB in delphinidin-treated HCT116 cells exhibited significant decrease compared to untreated cells. Thus, delphinidin decreased the expression of p65 subunits of NF-κB, reduced IκBαphosphorylation and inhibited nuclear translocation of NF-κB consistent with down-regulation of the NF-κB pathway.
In summary, we demonstrated that delphinidin suppressed growth of HCT116 cells by causing G2/M cell cycle arrest and apoptosis. Our results also suggested that delphinidin-induced apoptosis and cell cycle arrest were associated with inhibition of NF-κB pathway. Taken together, these results suggested that delphinidin may be useful in the chemoprevention and/or treatment of colon cancer.
Acknowledgments
This work was supported by United States Public Health Service Grants R01 CA 78809, R01 CA 101039, R01 CA 120451 and P50 DK065303.
Abbreviations
- NF-κB
nuclear factor-kappa B
- PARP
poly(ADP-ribose) polymerase
- IKKα
I kappa B kinase α
- cdc2
cell division cycle 2 kinase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PI
propidium iodide
- cdk
cyclin dependent kinase
References
- 1.Jemal A, Murray T, Ward E, et al. Cancer statistics. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.Khan N, Afaq F, Mukhtar H. Apoptosis by dietary factors: The suicide solution for delaying cancer growth. Carcinogenesis. 2007;28:233–239. doi: 10.1093/carcin/bgl243. [DOI] [PubMed] [Google Scholar]
- 3.Aggarwal BB, Shishodia S. Suppression of the nuclear factor-kappa B activation pathway by spice-derived phytochemicals: Reasoning for seasoning. Ann NY Acad Sci. 2004;1030:434–441. doi: 10.1196/annals.1329.054. [DOI] [PubMed] [Google Scholar]
- 4.Aggarwal BB. Nuclear factor-kappa B: The enemy within. Cancer Cell. 2004;6:203–208. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Lind DS, Hochwald SN, Malaty J. Nuclear factor kappa B is upregulated in colorectal cancer. Surgery. 2001;130:363–369. doi: 10.1067/msy.2001.116672. [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto Y, Gaynor RB. Therapeutic potential of inhibition of the NF-κB pathway in the treatment of inflammation and cancer. J Clin Invest. 2001;107:135–142. doi: 10.1172/JCI11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scottehus AJG, Baldwin AS., Jr A role for transcription factor NF-κB in intestinal inflammation. Int J Colorectal Dis. 1999;14:18–28. doi: 10.1007/s003840050178. [DOI] [PubMed] [Google Scholar]
- 8.Schmid RM, Adler G. NF-κB/Rel/IκB: Implications in gastrointestinal diseases. Gastroenterology. 2000;118:1208–1228. doi: 10.1016/s0016-5085(00)70374-x. [DOI] [PubMed] [Google Scholar]
- 9.Voboril R, Hochwald SN, Li J, et al. Inhibition of NF-kappa B augments sensitivity to 5-fluorouracil/folinic acid in colon cancer. J Surg Res. 2004;120:178–188. doi: 10.1016/j.jss.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 10.Wang W, McLeod HL, Cassidy J. Disulfiram-mediated inhibition of NF-kappaB activity enhances cytotoxicity of 5-fluorouracil in human colorectal cancer cell lines. Int J Cancer. 2003;104:504–511. doi: 10.1002/ijc.10972. [DOI] [PubMed] [Google Scholar]
- 11.Hong WK, Sporn MB. Recent advances in Chemoprevention of cancer. Science. 1997;278:1073–1077. doi: 10.1126/science.278.5340.1073. [DOI] [PubMed] [Google Scholar]
- 12.Wattenberg LW. An overview of chemoprevention: Current status and future prospects. Proc Soc Exp Biol Med. 1997;216:133–141. doi: 10.3181/00379727-216-44163. [DOI] [PubMed] [Google Scholar]
- 13.Yang CS, Wang ZY. Tea and cancer. J Natl Cancer lnst. 1993;85:1038–1049. doi: 10.1093/jnci/85.13.1038. [DOI] [PubMed] [Google Scholar]
- 14.Agarwal R, Muhktar H. Cancer chemoprevention by polyphenolic compounds present in green tea. Drug News Perspec. 1995;8:216–225. [Google Scholar]
- 15.Afaq F, Syed DN, Malik A, et al. Delphinidin, an anthocyanidin in pigmented fruits and vegetables, protects human HaCaT keratinocytes and mouse skin against UVB-mediated oxidative stress and apoptosis. J Invest Dermatol. 2007;127:222–232. doi: 10.1038/sj.jid.5700510. [DOI] [PubMed] [Google Scholar]
- 16.Lazze MC, Savio M, Pizzala R, et al. Anthocyanins induce cell cycle perturbations and apoptosis in different human cell lines. Carcinogenesis. 2004;25:1427–1433. doi: 10.1093/carcin/bgh138. [DOI] [PubMed] [Google Scholar]
- 17.Rupinder SK, Gurpreet AK, Manjeet S. Cell suicide and caspases. Vascul Pharmacol. 2007;46:383–393. doi: 10.1016/j.vph.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 18.Adams JM, Cory S. The Bcl-2 protein family: Arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 19.Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20:1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- 20.Coqueret O. New roles for p21 and p27 cell cycle inhibitors: A function for each cell compartment? Trends Cell Biol. 2003;13:65–70. doi: 10.1016/s0962-8924(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 21.Jiang X, Takahashi N, Matsui N, Tetsuka T, Okamoto T. The NF-kappa B activation in lymphotoxin beta receptor signaling depends on the phosphorylation of p65 at serine 536. J Biol Chem. 2003;278:919–926. doi: 10.1074/jbc.M208696200. [DOI] [PubMed] [Google Scholar]
- 22.Blijham GH. Chemotherapy of colorectal cancer. Anticancer Drugs. 1991;2:233–245. doi: 10.1097/00001813-199106000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- 24.Prior RL. Fruits and vegetables in the prevention of cellular oxidative damage. Am J Clin Nutr. 2003;78:570S–578S. doi: 10.1093/ajcn/78.3.570S. [DOI] [PubMed] [Google Scholar]
- 25.Boniface R, Robert AM. Effect of anthocyanins on human connective tissue metabolism in the human. Klin Monatsbl Augenheilkd. 1996;209:368–372. doi: 10.1055/s-2008-1035336. [DOI] [PubMed] [Google Scholar]
- 26.Aviram M, Fuhrman B. Wine flavonoids protect against LDL oxidation and atherosclerosis. Ann NY Acad Sci. 2002;957:146–161. doi: 10.1111/j.1749-6632.2002.tb02913.x. [DOI] [PubMed] [Google Scholar]
- 27.Wang H, Nair MG, Chang YC, Booren AM, Gray JI, DeWitt DL. Antioxidant and anti-inflammatory activities of anthocyanins and their aglycon, cyanidin, from tart cherries. J Nat Prod. 1999;62:294–296. doi: 10.1021/np980501m. [DOI] [PubMed] [Google Scholar]
- 28.Koide T, Hashimoto Y, Kamei H, Kojima T, Hasegawa M, Terabe K. Antitumor effect of anthocyanin fractions extracted from red soybeans and red beans in vitro and in vivo. Cancer Biother Radioharm. 1997;12:277–280. doi: 10.1089/cbr.1997.12.277. [DOI] [PubMed] [Google Scholar]
- 29.Galvano F, La Fauci L, Lazzarino G, et al. Cyanidins: Metabolism and biological properties. J Nutr Biochem. 2004;15:2–11. doi: 10.1016/j.jnutbio.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 30.Malik M, Zhao C, Schoene N, Guisti MM, Moyer MP, Magnuson BA. Anthocyanin-rich extract from Aronia melon-carpa E induces a cell cycle block in colon cancer but not normal colonic cells. Nutr Cancer. 2003;46:186–196. doi: 10.1207/S15327914NC4602_12. [DOI] [PubMed] [Google Scholar]
- 31.Singh RP, Dhanalakshmi S, Agarwal R. Phytochemicals as cell cycle modulators: A less toxic approach in halting human cancer. Cell Cycle. 2002;1:156–161. [PubMed] [Google Scholar]
- 32.Taraphdar AK, Roy M, Bhattacharya RK. Natural products as inducers of apoptosis: Implication for cancer therapy and prevention. Curr Sci. 2001;80:1387–1396. [Google Scholar]
- 33.Molinari M. Cell cycle checkpoints and their inactivation in human cancer. Cell Prolif. 2000;33:261–274. doi: 10.1046/j.1365-2184.2000.00191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dulic V, Stein GH, Far DF, Reed SI. Nucelar accumulation of p21Cip1 at the onset of mitosis: A role at the G2/M-phase transition. Mol Cell Biol. 1998;18:546–557. doi: 10.1128/mcb.18.1.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nuculescu AB, III, Chen X, Smeets M, Hengst L, Prives C, Reed SL. Effects of p21(Cip/Waf1) at both the G1/S and the G2/M cell cycle transition; pRb is a critical determinant blocking DNA replication and in preventing endoreduplication. Mol Cell Biol. 1998;18:629–643. doi: 10.1128/mcb.18.1.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harris SL, Lavine AJ. The p53 pathway: Positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- 37.Wenzel U, Kuntz S, Brendel MD, Daniel H. Dietary Flavons is a potent apoptosis inducer in human colon cancer. Cancer Res. 2000;60:3823–3831. [PubMed] [Google Scholar]
- 38.Palozza P, Serini S, Torselo A, et al. Mechanism of activation of caspase cascade during beta-carotene-induced apoptosis in human tumor cells. Nutr Cancer. 2003;47:76–87. doi: 10.1207/s15327914nc4701_10. [DOI] [PubMed] [Google Scholar]
- 39.Palozza P, Serini S, Torselo A, et al. Beta-carotene regulates NF-kappaB DNA-binding activity by a redox mechanism in human leukemia and colon adenocarcinoma cells. J Nutr. 2003;133:381–388. doi: 10.1093/jn/133.2.381. [DOI] [PubMed] [Google Scholar]
- 40.Jeong WS, Kim IW, Hu R, Kong AN. Modulatory properties of various natural chemopreventive agents on the activation of NF-kappa B signaling pathway. Pharm Res. 2004;21:661–670. doi: 10.1023/b:pham.0000022413.43212.cf. [DOI] [PubMed] [Google Scholar]