

Abstract
In the nearly 50 years since the description of Williams syndrome by Williams et al. in 1961, the focus of scientific inquiry has shifted from identification, definition, and description of the syndrome in small series to genotype-phenotype correlation, pathophysiologic investigation in both humans and in animal models, and therapeutic outcomes in large cohorts. Study of this rare syndrome has provided insight into the structure and function of the extracellular matrix, has contributed to understanding of genomic structure and rearrangement, and is beginning to elucidate genetic underpinnings of learning, language, and behavior. The results of current research not only recommend interventions that can be implemented now, but also identify areas requiring additional investigation, and suggest future therapeutic approaches.
Keywords: Williams syndrome, Williams-Beuren syndrome, supravalvar aortic stenosis, elastin
INTRODUCTION
Williams syndrome (WS) (OMIM 194050) is a multisystem disorder caused by the deletion of 26 contiguous genes, including elastin (ELN) (OMIM 130160) on chromosome 7q11.23. WS is a genomic disorder with an incidence of 1/7500 [Strømme et al.,2002] that occurs due to nonallelic homologous recombination (NAHR) in a region of chromosome 7 containing blocks of low copy repeats with high sequence homology that predispose to rearrangements during meiosis [Dutly and Schinzel,1996; Urban et al., 1996]. The Williams syndrome chromosome region (WSCR) is 1.55Mb-1.8Mb, the size depending on which blocks of low copy repeats are involved in NAHR. The WS phenotype is characterized by dysmorphic facial features, intellectual disability, elastin arteriopathy, short stature, connective tissue abnormalities, infantile hypercalcemia, and a unique personality and cognitive profile. This review will summarize the historical background, genetic etiology, phenotype and medical complications of WS in order to introduce the papers in this issue of the Seminars in Medical Genetics.
Historical Background
The first cases of Williams syndrome were included in early reports detailing the clinical characteristics of children who had infantile hypercalcemia, short stature, and variable congenital malformations [Fanconi et al., 1952]. After an epidemic of infantile hypercalcemia in Britain resolved with adjustment of vitamin D supplementation in food, Stapleton et al. [1957] noted that there was a group of infants with persistent symptoms including failure to thrive, developmental delay, and systolic murmurs. The next decade saw cardiology reports describing a condition with dysmorphic facial features, supravalvar aortic stenosis (SVAS) (OMIM 185500) and mental retardation [Williams et al., 1961; Beuren et al., 1962]. Isolated SVAS, a rare cause of left ventricular outflow tract obstruction, had been described by Chevers [1842], and more recently Eisenberg et al. [1964] reported autosomal dominant inheritance. In 1963, geneticists recognized that SVAS could segregate in families, but also could occur sporadically as part of a syndrome that included mental retardation [Merritt et. al., 1963]. Astute clinicians observed that the facial features of idiopathic hypercalcemia of infancy (IHC) and syndromic SVAS were similar [Black and Carter, 1963; Hooft et al., 1963]. The connection was proven when Garcia et al. [1964] described SVAS in a child who had documented IHC. The early recognition of hypercalcemia in WS led to a hypothesis that hypersensitivity to vitamin D caused the syndrome. Friedman and Roberts [1966] found that rabbits exposed prenatally to high doses of vitamin D developed aortic lesions and abnormal shortened craniofacies. (Interestingly, because high doses of vitamin D inhibit tropoelastin deposition, the observed phenotype was likely due to a secondary elastin deficiency.) These first reports of WS also made note of the characteristic behavioral phenotype, describing loquacity, anxiety, and friendliness [Beuren et al.,1964; von Arnim and Engel, 1964].
In the 1970s, larger series defined the WS phenotype [Beuren 1972; Jones and Smith, 1975]. However, by 1984, there were few reports of adults with WS. A mother’s questions, “What happens when they grow up? Do they grow up?” inspired me to initiate a study of the natural history of WS during my fellowship in medical genetics and dysmorphology at the University of Utah [Morris et al., 1988, Morris et al., 1990]. Subsequently, a colleague, Dr. Cynthia Moore at Indiana University, had observed some members of a family with autosomal dominant SVAS who had learning disabilities and some facial features reminiscent of WS. We wondered if WS could represent an “iceberg dominant” with WS being the most severe manifestation SVAS. A visit to Indiana University revealed a 25-year archive of families evaluated by their genetics program, including several multigenerational families with SVAS. Dr. Gregory Ensing, pediatric cardiologist at Indiana University, performed echocardiograms to classify family members in several SVAS kindreds as affected or unaffected. Samples were collected for linkage analysis performed by Amanda Ewart in the molecular genetics laboratory of Dr. Mark Keating at the University of Utah. Linkage to the elastin gene was established [Ewart et al.,1993a]. Subsequently, one of the SVAS families was found to have a 6;7 translocation that disrupted the elastin gene [Curran et al., 1993; Morris et al., 1993], demonstrating that elastin mutation caused SVAS. Ewart et al. [1993b] then discovered that WS was associated with ELN deletion, a finding that was rapidly confirmed [Lowery et al., 1995, Mari et al., 1995], and led to the first laboratory test for the disorder. The family that had prompted the genetic study was later found to have a ~500 kb deletion in the WSCR that included the elastin gene [Morris et al., 2003]. Currently, diagnostic testing for the deletion may be accomplished by fluorescent in situ hybridization (FISH), multiplex ligation-dependent probe amplification (MPLA), or chromosome microarray.
GENETICS AND GENOTYPE-PHENOTYPE CORRELATION
In most families, WS is a sporadic occurrence, but familial cases including male-to-male transmission have been reported (Fig 1) [Morris et al, 1993; Mulik et al., 2004; Sadler et al., 1993]. Farwig et al. describe the first study of genetic counseling in adults with Williams syndrome in this issue. The genomic structure of the WS region predisposes to rearrangements secondary to meiotic NAHR, and thus can lead to deletion, duplication, or inversion of the WS region [Schubert, 2009]. Individuals who have duplication of the WS region have a phenotype that includes facial asymmetry, long nasal columnella, speech difficulty, and separation anxiety [Somerville et al., 2005; Berg et al., 2007; van der Aa et al., 2009]. The fact that both deletion and duplication of the WSCR is associated with anxiety suggests that there is a dosage sensitive gene in the region that contributes to anxiety disorders [Osborne and Mervis, 2007]. An individual who has the inversion polymorphism has a normal phenotype, but has an increased chance to have offspring with WS or duplication of the WS region [Osborne et al.,2001; Bayes et al., 2003]. In this issue, Hobart et al. report the population incidence of the inversion polymorphism and supply recurrence risks for genetic counseling.
Figure 1.

Mother and daughter with Williams syndrome: Left, mother age 30 years (note graying hair), daughter age 23 months; Center, daughter age 17 years; Right, mother age 46 years.
Individuals with typical WS who have the 1.8Mb deletion that includes NCF1 are less likely to have hypertension [Del Campo, et al, 2006]. Those with much longer deletions typically have a more severe phenotype; if the deletion includes MAG12, severe cognitive disability and infantile spasms are more likely [Marshall et al., 2008]. In contrast, individuals with shorter deletions in the WSCR have a subset of WS signs and symptoms, depending on which genes are deleted. Study of individuals with short deletions has provided some insight regarding the possible roles for various genes in the WS phenotype, but the small numbers of such individuals has made interpretation of the findings challenging [Sharp, 2009]. Dr. Osborne discusses the challenges of genotype-phenotype correlation in WS, details the characteristics of knock-out mouse models, and discusses how further study of these animals will contribute to our understanding of the genes in the region and their phenotypic contribution. The role of the elastin gene in WS is proven: its deletion is responsible for the connective tissue phenotype of WS, which includes hoarse voice, some of the facial features (periorbital fullness and full cheeks in infancy), soft skin, lax ligaments, elastin arteriopathy (most commonly supravalvar aortic stenosis), hernias, bowel and bladder diverticuli, and joint abnormalities. The most significant cause of morbidity and mortality in WS is the cardiovascular disease. SVAS is present in ~70% and requires surgical correction in ~30%, usually before age 5 years [Collins et al., 2010]. Arterial narrowing in the pulmonary circulation usually improves over time, though infants who have biventricular outflow tract obstruction are at particularly high risk for negative outcome [Burch et al., 2008]. Although ELN haploinsufficiency is present in all individuals with WS, the connective tissue signs and symptoms can be quite variable. Some phenotypic differences are due to age, gender [Sadler et al., 2001], or treatment of the individual, but genetic factors that may contribute to the variability have not been identified. An association of alpha 1 antitrypsin deficiency carrier status with scoliosis and joint dislocation is reported in this issue.
PHENOTYPE AND MEDICAL COMPLICATIONS
The facial gestalt of WS is unique: young children typically have a broad forehead, bitemporal narrowing, depressed nasal root, periorbital fullness, stellate/lacy iris pattern, strabismus, bulbous nasal tip, malar flattening, long philtrum, thick vermilion of the lips, wide mouth, full cheeks, dental malocclusion with small widely-spaced teeth, small jaw, and prominent earlobes (Fig 2). Older children and adults usually have a more gaunt facial appearance with a prominent supraorbital ridges, narrow nasal root of normal anterior prominence, full nasal tip, malar flattening, wide mouth, thick vermilion of the lips, small jaw, dental malocclusion, and a long neck accentuated by sloping shoulders (Fig 3 and 4).
Figure 2.

Young children with Williams syndrome (Left to Right): Asian female, age 19 months; Caucasian male, age 2 years; Hispanic female, age 3 years; African-American female, age 5 years.
Figure 3.

Older children and adult with WS (left to right): Hispanic male, age 9; Caucasian male, age 13; African American male, age 7; Caucasian male, age 30.
Figure 4.
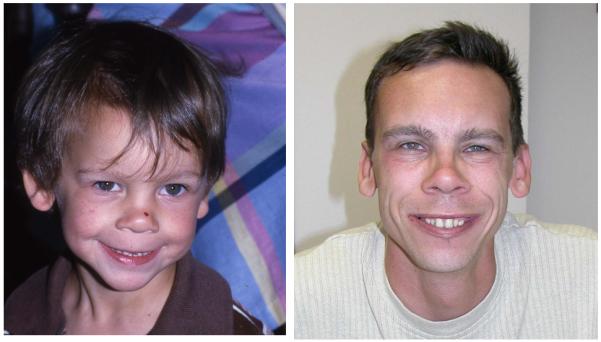
Changing facial phenotype over time in a male with WS: left, age 3.5; right, age 21.
Medical problems in infancy include strabismus, dacrostenosis, hypotonia, developmental delay, feeding difficulties, failure to thrive, hypercalcemia, chronic otitis media, elastin arteriopathy, inguinal hernias, gastroesophageal reflux, lax joints, and constipation. Compared to the family background, individuals with WS have short stature. Older children experience gradual tightening of hamstrings and heel cords, leading to a stiff awkward gait, and have hyperreflexia of the lower extremities [Gagliardi et al., 2007]. Sleep disturbance is a common complaint [Goldman et al., 2009]. The sensory modulation difficulties experienced by children with WS are described in the paper by John and Mervis in this issue. Chronic constipation is common at all ages and should be vigorously treated; there is an increased risk for diverticulosis in WS, and diverticulitis can occur at a young age [Partsch et al., 2005]. Genitourinary problems are often symptomatic and include increased urinary frequency, enuresis, and urinary tract structural anomalies [Sammour et al., 2005]. Despite recognition of WS for almost 50 years, there are certain clinical problems that have been elucidated only recently. Pober et al. describe the high incidence of abnormal glucose metabolism in adults with WS in this issue. Marler et al. report that sensorineural hearing loss, often unrecognized, occurs in the majority of individuals with WS. For a recent review of WS medical complications, the reader is referred to Pober [2010] and Morris et al., [2006], and for a comprehensive discussion of medical management in WS to Morris, [2010].
The WS cognitive profile is characterized by intellectual disability (usually mild) with a relative strength in language and verbal short term memory and an extreme weakness in visuospatial construction [Mervis et al., 1999]. The WS personality is typified by empathy, overfriendliness, attention problems, and anxiety [Mervis et al., 2000]. The cognitive and behavior phenotype of WS is one of the key recognizable elements of the syndrome. In this issue, Mervis and John discuss the cognitive and behavioral phenotype with an emphasis on treatment recommendations. John et al. also describe a new study investigating sensory modulation in Williams syndrome while Woodruff-Borden and colleagues explore the longitudinal course of anxiety in children with WS.
Some of the recommendations that come out of the research reported in this issue have the potential to improve the health and well being of individuals with WS.
Anxiety is a common feature of WS, but few symptomatic children are treated for it. Research is needed to evaluate prevention and intervention programs, and clinicians should refer children for treatment.
Sensory modulation impairment in WS results in problem behaviors and poor adaptive function. Desensitization, sensory integration and occupational therapies should be considered in individual treatment plans.
High frequency sensorineural hearing loss, often undetected, is present in most individuals with WS and may worsen with age. Yearly audiologic evaluation, preferential seating, and hearing protection should be routine, and use of a personal listening device in the classroom should be considered. Even though desensitization therapy for specific phobias is helpful in WS, it should not be used to treat phobia for loud noises, as hearing damage may result.
Children with WS should be taught to read using phonics methods, as other forms of reading instruction are not successful.
Abnormalities of glucose metabolism are present in most adults with WS. Oral glucose tolerance tests should be used to monitor for diabetes mellitus, and a regular program of exercise should be prescribed.
Recurrence risk information is now available for individuals who have an inversion of the WS region. In addition, a method for providing genetic counseling to adults with WS is described.
As a group, people with WS have lower adaptive behavior skills than would be expected for their IQ. Despite a social disinhibition, they have difficulty with appropriate social skills. Applied behavior analysis based interventions and social skills training have been successful in this population. It is critically important that children with WS do chores, and be required to master daily living skills if they are to achieve a sense of mastery and reach their full potential.
FUTURE DIRECTIONS
Genotype-phenotype correlation continues to challenge geneticists who are searching for the genes in the WSCR that contribute to anxiety, language, and behavior. Progress will be made through continued study of mouse models, evaluation of individuals with unusual deletions and duplications, functional neuroimaging experiments, and continued elucidation of neurodevelopmental pathways. Advances in understanding the complex developmental role of the extracellular matrix will likely result in new therapies for the connective tissue abnormalities. Since WS was first described, medical treatment has been refined for the condition, but mental health treatment has not been adequately addressed. Therapeutic interventions for cognition, language, and behavior will have to be designed and tested for efficacy.
ACKNOWLEDGMENTS
Preparation of this manuscript was supported by grant R01 NS 35102 from the National Institute of Neurological Disorders and Stroke. I am grateful for the enthusiastic research participation of individuals with Williams syndrome and their families; they advance our understanding of the condition in order to help others. I thank the Williams Syndrome Association for its continuing encouragement, for facilitating research efforts at regional and national conferences, and for fostering collaborations at international WS professional conferences. I am grateful to The Lili Claire Foundation for hosting research clinics. I appreciate the collaboration of numerous colleagues who attend the David W. Smith Malformations and Morphogenesis workshops. Finally, I am grateful to my mentor, Dr. John C. Carey, for continuing advice and friendship.
Biography
Colleen A. Morris, M. D. is Chief of the Genetics Division in the Department of Pediatrics at the University of Nevada School of Medicine. She has studied the natural history and genotype-phenotype correlations of Williams syndrome for 25 years and also has research interests in syndrome delineation and identification and treatment of children with Fetal Alcohol Spectrum Disorders.
REFERENCES
- Bayes M, Magano LF, Rivera N, Flores R, Perez Jurado LA. Mutational mechanisms of Williams-Beuren syndrome deletions. Am J Hum Genet. 2003;73:131–151. doi: 10.1086/376565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg JS, Brunetti-Pierri N, Peters SU, Kang SH, Fong CT, Salamone J, Freedenberg D, Hannig VL, Prock LA, Miller DT, Raffalli P, Harris DJ, Erickson RP, Cunniff C, Clark GD, Blazo MA, Peiffer DA, Gunderson KL, Sahoo T, Patel A, Lupski JR, Beaudet al., Cheung SW. Speech delay and autism spectrum behaviors are frequently associated with duplication of the 7q11.23 Williams-Beuren syndrome region. Genet Med. 2007;9:427–441. doi: 10.1097/gim.0b013e3180986192. [DOI] [PubMed] [Google Scholar]
- Beuren AJ. Supravalvular aortic stenosis: A complex syndrome with and without mental retardation. Birth Defects. 1972;8:45–56. [Google Scholar]
- Beuren AJ, Apitz J, Harmjanz D. Supravalvular aortic stenosis in association with mental retardation and a certain facial appearance. Circulation. 1962;27:1235–1240. doi: 10.1161/01.cir.26.6.1235. [DOI] [PubMed] [Google Scholar]
- Beuren AJ, Schulze C, Eberle P, Harmjanz D, Apitz J. The syndrome of supravalvular aortic stenosis, peripheral pulmonic stenosis, mental retardation, and similar facial appearance. Am J Cardiol. 1964;13:471–482. doi: 10.1016/0002-9149(64)90154-7. [DOI] [PubMed] [Google Scholar]
- Black JA, Carter REB. Association between aortic stenosis and facies of severe infantile hyercalcemia. Lancet. 1963;91:745–748. doi: 10.1016/s0140-6736(63)90553-1. [DOI] [PubMed] [Google Scholar]
- Burch TM, McGowan FX, Jr, Kussman BD, Powell AJ, DiNardo JA. Congenital supravalvular aortic stenosi and sudden death associated with anesthesia: what’s the mystery? Anesth Analg. 2008;107:1848–1854. doi: 10.1213/ane.0b013e3181875a4d. [DOI] [PubMed] [Google Scholar]
- Chevers N. Observations on the diseases of the orifice and valves of the aorta. Guys Hospital Reports. 1842;7:387–442. [Google Scholar]
- Curran ME, Atkinson DL, Ewart AK, Morris CA, Leppert MF, Keating MT. the elastin gene is disrupted by a translocation associated wit supravalvular aortic stenosis. Cell. 1993;73:159–168. doi: 10.1016/0092-8674(93)90168-p. [DOI] [PubMed] [Google Scholar]
- Del Campo M, Antonell A, Magano LF, Muñoz FG, Flores R, Bayés M, Pérez Jurado LA. Hemizygosity at the NCF1 gene in patients with Williams-Beuren syndrome decreases their risk of hypertension. Am J Hum Genet. 2006;78:533–542. doi: 10.1086/501073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutly F, Schinzel A. Unequal interchromosomal rearrangements may result in elastin gene deletions causing the Williams-Beuren syndrome. Hum Mol Genet. 1996;5:1893–1898. doi: 10.1093/hmg/5.12.1893. [DOI] [PubMed] [Google Scholar]
- Eisenberg R, Young D, Jacobson B, Boito A. Familial supravalvular aortic stenosis. Am J Dis Child. 1964;108:341–3347. doi: 10.1001/archpedi.1964.02090010343002. [DOI] [PubMed] [Google Scholar]
- Ewart AK, Morris CA, Ensing GJ, Loker J, Moore C, Leppert M, Keating M. A human vascular disorder, suprvalular aortic stenosis, maps to chromosome 7. Proc Natl Acad Sci. 1993a;90:3226–3230. doi: 10.1073/pnas.90.8.3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, Stock AD, Leppert M, Keating MT. Hemizygosity at the elastin locus in a developmental disorder, Williams Syndrome. Nat Genet. 1993b;5:11–16. doi: 10.1038/ng0993-11. [DOI] [PubMed] [Google Scholar]
- Fanconi G, Giradet P, Schlesinger B, Butler N, Blade JS. Chronische Hypercalcaemie kombiniert mit Osteosklerose, Hyperazotaemie, Minderwuchs, und kongenitalen Missbildungen. Helv Paediatr Acta. 1952;7:314–334. [PubMed] [Google Scholar]
- Friedman W, Roberts W. Vitamin D and the supravalvular aortic stenosis syndrome. Circulation. 1966;34:77–86. doi: 10.1161/01.cir.34.1.77. [DOI] [PubMed] [Google Scholar]
- Gagliardi C, Martelli S, Burt MD, Borgatti R. Evolution of neurologic features in Williams syndrome. Pediatr Neurol. 2007;36:301–306. doi: 10.1016/j.pediatrneurol.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Garcia RE, Friedman WF, Kaback MM, Rowe RD. Idiopathic hypercalcemia and supravalvular aortic stenosis. N Engl J Med. 1964;271:117–120. doi: 10.1056/NEJM196407162710302. [DOI] [PubMed] [Google Scholar]
- Goldman SE, Malow BA, Newman KD, Roof E, Dykens EM. Sleep patterns and daytime sleepiness in adolescents and young adults with Williams syndrome. J Intellect Disabil Res. 2009;53:182–188. doi: 10.1111/j.1365-2788.2008.01140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooft C, Vermassen A, Blancquaert A. Observations concerning the evolution of the chronic form of idiopathic Hypercalcemia of children. Helv Paediat Acta. 1963;2:138–147. [PubMed] [Google Scholar]
- Jones KL, Smith DW. The Williams elfin facies syndrome. J Pediatr. 1975;86:718–723. doi: 10.1016/s0022-3476(75)80356-8. [DOI] [PubMed] [Google Scholar]
- Lowery MC, Morris CA, Ewart A, Brothman L, Zhu XL, Leonard CO, Carey JC, Keating MT, Brothman AR. Strong correlations of elastin deletions, detected by FISH, with Williams syndrome: Evaluation of 235 patients. Am J Hum Genet. 1995;57:49–53. [PMC free article] [PubMed] [Google Scholar]
- Mari A, Amati F, Mingarelli R, Giannotti A, Sebastio G, Colloridi V, Novelli G, Dallapiccola B. Analysis of the elastin gene in 60 patients with clinical diagnosis of Williams syndrome. Hum Genet. 1995;96:444–448. doi: 10.1007/BF00191804. [DOI] [PubMed] [Google Scholar]
- Marshall CR, Young EJ, Pani AM, Freckmann M, Lacassie Y, Howald C, Fitzgerald KK, Peippo M, Morris CA, Shane K, Priolo M, Morimoto M, Kondo I, Manguoglu E, Berker-Karauzum S, Edery P, Hobart HH, Mervis CB, Zuffardi O, Reymond A, Kaplan P, Tassabehji M, Gregg RG, Scherer SW, Osborne LR. Infantile spasms is associated with deletion of the MAGI2 gene on chromosome 7q11.23-q21.11. Am J Hum Genet. 2008;83:106–111. doi: 10.1016/j.ajhg.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt DA, Palmar CG, Lurie PR, Petry EL. Supravalvular aortic stenosis: genetic and clinical Studies (abstract) J Lab Clin Med. 1963;62:995. [Google Scholar]
- Mervis CB, Morris CA, Bertrand J, Robinson BF. Williams syndrome: Findings from an integrated program of research. In: Tager-Flusberg H, editor. Neurodevelopmental Disorders. MIT Press; Cambridge, MA: 1999. [Google Scholar]
- Mervis CB, Robinson BF, Bertrand J, Morris CA, Klein-Tasman BP, Armstrong SC. The Williams Syndrome Cognitive Profile. Brain and Cognition. 2000;44:604–628. doi: 10.1006/brcg.2000.1232. [DOI] [PubMed] [Google Scholar]
- Morris CA. Williams syndrome. In: Cassidy SB, Allanson JE, editors. Management of genetic syndromes. 3rd ed. Wiley; Hoboken, NJ: 2010. [Google Scholar]
- Morris CA, Dilts C, Dempsey SA, Leonard CO, Blackburn B. The natural history of Williams syndrome: Physical characteristics. J Pediatr. 1988;113:318–326. doi: 10.1016/s0022-3476(88)80272-5. [DOI] [PubMed] [Google Scholar]
- Morris CA, Lenhoff HM, Wang PP, editors. Williams-Beuren Syndrome, Research, Evaluation, and Treatment. The John Hopkins University Press; 2006. [Google Scholar]
- Morris CA, Leonard CO, Dilts C, Demsey SA. Adults with Williams syndrome. Am J Med Genet Suppl. 1990;6:102–107. doi: 10.1002/ajmg.1320370619. [DOI] [PubMed] [Google Scholar]
- Morris CA, Loker J, Ensing G, Stock AD. Supravalvular aortic stenosis cosegregates with a familial 6:7 translocation which disrupts the elastin gene. Am J Med Genet. 1993;46:737–744. doi: 10.1002/ajmg.1320460634. [DOI] [PubMed] [Google Scholar]
- Morris CA, Mervis CB, Hobart HH, Gregg RG, Bertrand J, Ensing GJ, Sommer A, Moore CA, Hopkin RJ, Spallone PA, Keating MT, Osborne L, Kimberley KW, Stock AD. GTF2I hemizygosity implicated in mental retardation in Williams syndrome: genotype-phenotype analysis of five families with deletions in the Williams syndrome region. Am J Med Genet. 2003;123:45–59. doi: 10.1002/ajmg.a.20496. [DOI] [PubMed] [Google Scholar]
- Morris CA, Thomas IT, Greenberg F. Williams syndrome: Autosomal dominant inheritance. Am J Med Genet. 1993;47:478–481. doi: 10.1002/ajmg.1320470409. [DOI] [PubMed] [Google Scholar]
- Mulik VV, Temple KI, Howe DT. Two pregnancies in a woman with Williams syndrome. Br J Obstet Gynaecol. 2004;111:511–512. doi: 10.1111/j.1471-0528.2004.00109.x. [DOI] [PubMed] [Google Scholar]
- Osborne LR, Li M, Pober B, Chitayat D, Bodurtha J, Mandel A, Costa T, Grebe T, Cox S, Tsui LC, Scherer SW. A 1.5 million-base pair inversion polymorphism in families with Willimas-Beuren syndrome. Nat Genet. 2001;29:321–325. doi: 10.1038/ng753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne LR, Mervis CB. Rearrangements of the Williams-Beuren syndrome locus: molecular basis and implications for speech and language development. Expert Rev Mol Med. 2007;9:1–16. doi: 10.1017/S146239940700035X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partsch CJ, Siebert R, Caliebe A, Gosch A, Wessel A, Pankau R. Sigmoid diverticulitis in patients with Williams-Beuren syndrome: relatively high prevalence and high complication rate in young adults with the syndrome. Am J Med Genet. 2005;137:52–54. doi: 10.1002/ajmg.a.30865. [DOI] [PubMed] [Google Scholar]
- Pober BR. Williams-Beuren Syndrome. N Engl J Med. 2010;362:239–252. doi: 10.1056/NEJMra0903074. [DOI] [PubMed] [Google Scholar]
- Sadler LS, Pober BR, Grandinetti A, Scheiber D, Fekete G, Sharma AN, Urban Z. Differences by sex in cardiovascular disease in Williams syndrome. J Pediatr. 2001;139:849–852. doi: 10.1067/mpd.2001.118889. [DOI] [PubMed] [Google Scholar]
- Sadler LS, Robinson LK, Verdaasdonk KR, Gingell R. The Williams syndrome: evidence for possible autosomal dominant inheritance. Am J Med Genet. 1993;47:468–470. doi: 10.1002/ajmg.1320470406. [DOI] [PubMed] [Google Scholar]
- Stapleton T, MacDonald WB, Lightwood R. The pathogenesis of idiopathic hypercalcemia in infancy. Amer J Clin Nutr. 1957;5:533–542. doi: 10.1093/ajcn/5.5.533. [DOI] [PubMed] [Google Scholar]
- Strømme P, Bjornstad P, Ramstad K. Prevalence estimation of Williams syndrome. J Child Neurol. 2002;17:269–271. doi: 10.1177/088307380201700406. [DOI] [PubMed] [Google Scholar]
- Schubert C. The genomic basis of the Williams-Beuren syndrome. Cell Mol Life Sci. 2009;66:1178–1197. doi: 10.1007/s00018-008-8401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ. Emerging themes and new challenges in defining the role of structural variation in human disease. Hum Mutat. 2009;30:135–144. doi: 10.1002/humu.20843. [DOI] [PubMed] [Google Scholar]
- Somerville MJ, Mervis CB, Young EJ, Seo E-J, del Campo M, Bamforth S, Peregrine E, Loo W, Lilley M, Pérez-Jurado LA, Morris CA, Scherer SW, Osborne LR. Severe expressive-language delay related to duplication of the Williams-Beuren locus. N Engl J Med. 2005;353:1694–1701. doi: 10.1056/NEJMoa051962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbán Z, Helms C, Fekete G, Csiszár K, Bonnet D, Munnich A, Donis-Keller H, Boyd CD. 7q11.23 deletions in Williams syndrome arise as a consequence of unequal meiotic crossover. Am J Hum Genet. 1996;59:958–962. [PMC free article] [PubMed] [Google Scholar]
- Van der Aa N, Rooms L, Vandeweyer G, van den Ende J, Reyniers E, Fichera M, Romano C, Delle Chiaie B, Mortier G, Menten B, Destrée A, Maystadt I, Männik K, Kurg A, Reimand T, McMullan D, Oley C, Brueton L, Bongers EM, van Bon BW, Pfund R, Jacquemont S, Ferrarini A, Martinet D, Schrander-Stumpel C, Stegmann AP, Frints SG, de Vries BB, Ceulemans B, Kooy RF. Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur J Med Genet. 2009;52:94–100. doi: 10.1016/j.ejmg.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Von Arnim G, Engel P. Mental retardation related to hypercalcemia. Dev Med Child Neurol. 1964;6:366–377. doi: 10.1111/j.1469-8749.1964.tb08138.x. [DOI] [PubMed] [Google Scholar]
- Williams JCP, Barratt-Boyes BG, Lowe JB. Supravalvular aortic stenosis. Circulation. 1961;24:1311–1318. doi: 10.1161/01.cir.24.6.1311. [DOI] [PubMed] [Google Scholar]