

Abstract
Williams syndrome (WS) is a multisystem disorder caused by deletion of about 1.55 Mb of DNA (including 26 genes) on chromosome 7q11.23, a region predisposed to recombination due to its genomic structure. Deletion of the Williams syndrome chromosome region (WSCR) occurs sporadically. To better define chance for familial recurrence and to investigate the prevalence of genomic rearrangements of the region, 257 children with WS and their parents were studied. We determined deletion size in probands by metaphase FISH, parent-of-origin of the deleted chromosome by molecular genetic methods, and inversion status of the WSCR in both parents by interphase FISH. The frequency of WSCR inversion in the transmitting parent group was 24.9%. In contrast, the rate of inversion in the non-transmitting parent group (a reasonable estimate of the rate in the general population) was 5.8%. There were no significant gender differences with respect to parent-of-origin for the deleted chromosome or the incidence of the inversion polymorphism. There was no difference in the rate of spontaneous abortion for mothers heterozygous for the WSCR inversion relative to mothers without the inversion. We calculate that for a parent heterozygous for a WSCR inversion, the chance to have a child with WS is about 1 in 1750, in contrast to the 1 in 9500 chance for a parent without an inversion.
Keywords: Williams syndrome, inversion, 7q11.23, genetic counseling
INTRODUCTION
Williams syndrome (WS, OMIM #194050) is a contiguous gene syndrome caused by a microdeletion in chromosome band 7q11.23. WS occurs in approximately 1 in 7,500 births [Strømme et al., 2002] and causes developmental disabilities affecting neurologic function and cardiovascular structure. The submicroscopic deletion in typical or ‘classic’ WS is either 1.55Mb in size comprising 26 genes (95%) or 1.84Mb containing 28 genes (5%) [Osborne et al., 2001; Bayes et al., 2003; Schubert, 2009]. Individuals with deletions larger than 1.84Mb typically have a more severe phenotype [e.g., Mizugishi et al., 1998; Stock et al., 2003; Marshall et al., 2008], while individuals with smaller deletions have been described with variable subsets of WS signs and symptoms [e.g., Morris et al., 2003; Morris, 2006]. The genomic structure of the region predisposes toward chromosomal rearrangements including inversions, deletions, and duplications (elegantly reviewed by Schubert [2009]). The deleted region (Williams Syndrome Chromosome Region, WSCR) is bounded by a complex set of blocks of low-copy repeat (LCR) sequences. Each LCR contains both genes and pseudogenes. The genomic rearrangements created by mispairing of LCRs flanking the WSCR are shown in Figure 1. The most common deletion of the WSCR region is generated by meiotic mispairing of the directly oriented centromeric and medial B block LCRs followed by non-allelic homologous recombination (NAHR) between the B block LCRs. NAHR between centromeric and medial A block LCRs causes the 1.84Mb deletion. Haplotype analysis using polymorphic markers has demonstrated that interchromosomal NAHR is the cause of ⅔ of the deleted chromosomes, while intrachromosomal NAHR, either between sister chromatids or within a chromatid results in ⅓ of the deletions [Dutly and Schinzel, 1996; Baumer et al., 1998; Urban et al., 1996; Thomas et al., 2006]. An inversion of the WSCR occurs within a chromatid during meiosis or mitosis when a centromeric LCR mispairs with a telomeric LCR that is inverted relative to the centromeric block, resulting in NAHR (Figure 2) [Osborne et al., 2001; Bayes et al., 2003; Turner et al., 2008]. The inversion in turn increases the probability of a subsequent meiotic genomic rearrangement, that is deletion of the WSCR. During meiosis, an inversion loop forms to maintain sequence pairing between the inverted and non-inverted chromosome. A crossover event within the loop can result in abnormal products, including dicentric and acentric fragments. With the presence of repetitive elements within the loop, NAHR events can lead to deletion and duplication [Bayes et al., 2003]. An additional predisposing mechanism to deletion—copy number variations of the flanking LCRs in the transmitting parent—has been described for 4.44% of cases [Cusco et al., 2008].
Figure 1.

Map of the WSCR, showing LCRs and sites of NAHR. 1A: Schematic of the William Syndrome Critical Region (WSCR) in context of the local genomic region, showing the LCR blocks and the boxed WSCR and flanking non-boxed genes. Below is indicated the 1.55 Mb deletion found in 95% of individuals with WS and the residual genes flanking the WS deletion region. 1B shows the 1.84 Mb deletion found in 5% of individuals with WS.
Figure 2.


WSCR Inversions. 2A: Schematic of chromatin loops with LCRs indicated. X represents potential recombination sites that create inversions. Below, schematics of 3 chromatids with inversions of the WBCR caused by recombinations at the three potential sites. 2B: A normal chromosome 7 and one with an inverted WSCR in context of the local genomic region, showing the LCR blocks and the boxed WSCR and flanking non-boxed genes, are shown. The red dashed line shows the genomic content that will be retained in the deleted chromosome created by recombination at X. Below is a schematic of the resultant deleted chromosome with the retained genes and LCR block orientation.
The deletion that causes WS typically occurs de novo, and almost all of the rare familial cases have been instances of parent- child transmission [Morris et al., 1993; Sadler et al., 1993] or identical twins [Castorina et al., 1997]. However, Scherer et al. [2005] described two families with unaffected parents in which siblings with WS had the same parent of origin for the deletion. In one of these families, the parent of origin had an inversion of the WSCR. A high frequency of WSCR-inversion heterozygosity in parents transmitting the chromosome 7 that is deleted in their offspring with WS has been demonstrated in two studies. Osborne et al. [2001] found 4 inversion heterozygotes among 12 transmitting parents (33%), and Bayes et al., [2003] found evidence of WSCR inversion in the transmitting parent for 21 of 74 individuals with WS (28%).
The question of whether there are significant differences in maternal vs. paternal transmission of the WS deletion has been addressed in seven studies. Thomas et al. [2006] combined their own data with that of the other six studies [Dutly and Schinzel, 1996; Pérez Jurado et al., 1996; Robinson et al., 1996; Urban et al., 1996; Baumer et al., 1998; Bayes et al., 2003], yielding a total of 278 cases. The mother was the parent of origin in 148 cases (53.2%) and the father in 130 (46.8%). This difference in proportion of maternal vs. paternal transmission of the WS deletion was not statistically significant.
In the present study, we provide the first large-sample examination of the transmission of the WS deletion, identifying the parent of origin and the inversion status of both parents of 257 children with WS. Our first goal was to examine the relation between the presence of an inversion of the WSCR in a parent and the likelihood of having a child with WS. A second goal was to determine the rate of inversion of the WSCR in the general population. A third goal was to confirm that mothers and fathers do not differ significantly in their odds of transmitting a chromosome with a WSCR deletion to their offspring. A fourth goal was to determine if there was an increased rate of spontaneous abortion among women who had an inversion of the WS region. The final goal, based on the other four, was to strengthen the basis for genetic counseling of parents of children with WS regarding the recurrence risk of WS.
METHODS
Participants
Participants were 257 couples and their biological child with classic WS (1.55Mb or 1.84 Mb deletion). Couples whose child with WS had a shorter or longer deletion were excluded. Informed consent and specimens were obtained following IRB-approved protocols.
Molecular Cytogenetics
Cytogenetic preparations and DNA extractions were made by standard methods from either fresh whole blood or previously established lymphoblastoid lines. Lymphoblastoid lines were established from most fresh blood specimens. DNA for FISH probes was purified from BACs, PACs, and cosmids according to standard protocols and labeled by incorporation of SpectrumRed and SpectrumGreen labeled nucleotide by nick translation (Vysis, Abbott Molecular, Des Plaines, IL). Standard methods for metaphase and interphase FISH experiments were followed. The results were observed and documented on a Zeiss Axioscop epifluorescence microscope with a Metasystems digital imaging system and Isis imaging software (Altlussheim, Germany).
Probands were evaluated for deletion size using FISH probes across the WSCR to interphase and metaphase chromosomes. WSCR probes included: BCL7b (B-Cell lymphoma 7B), STX1a (syntaxin), ELN (elastin: two probes, for the 3’ and the 5’ ends), LIMK1 (LIM kinase 1), CYLN2 (CLIP2; cytoplasmic linker 2), and GTF2IRD1 (general transcription factor II I repeat). Probes for the WSCR flanking regions included the BAC clones CTB-139P11 (telomeric) and RP11-815K3 (centromeric).
Three color interphase FISH was used to demonstrate the orientation of the parental WSCR, using the methods of Osborne et al. [2001]. For each experiment, the probe outside the WBCR (anchor probe) always had a yellow signal (mixture of red-labeled and green-labeled probes) and one of the probes within the region was labeled green, the other red. The color order observed, yellow-green-red or yellow-red-green, indicated the orientation of the WSCR (Figure 3a). Nuclei were considered informative for the inversion studies if the three probe signals from one or both chromosomes formed distinctly linear patterns (Figure 3b, c). For each parent, a minimum of 30 informative nuclei was scored. For most participants, a single experiment was sufficient to determine inversion status. Inversions were confirmed using anchor probes from the other end of the WSCR to read the orientation in the opposite direction.
Figure 3.


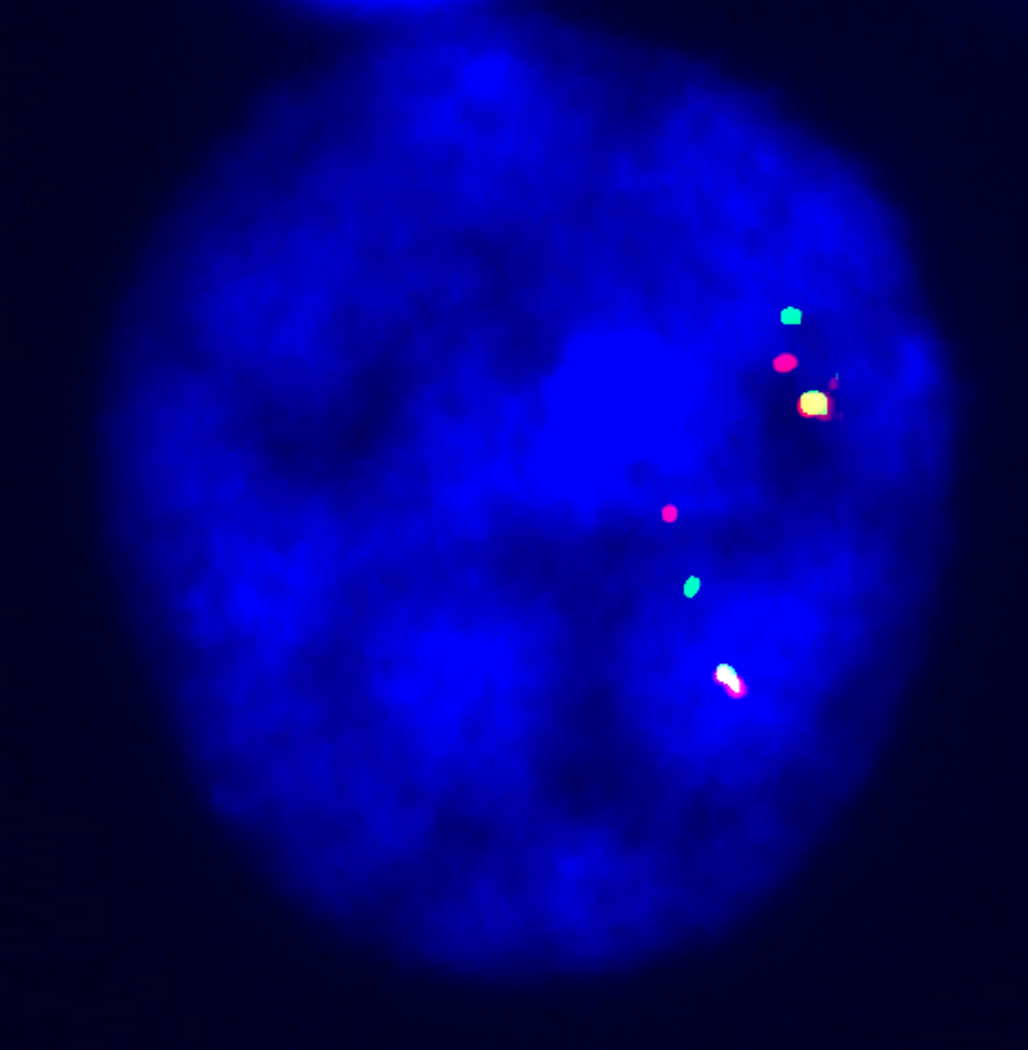
Interphase FISH for the inversion studies. 3A: Color scheme for 3 color interphase FISH used to indicate the orientation of the WS region.
3B: Nucleus showing non-inverted orientation (Y-G-R/Y-G-R). 3C: Nucleus demonstrating an inverted pattern (Y-G-R/Y-R-G) for the WS region.
The parental origin of the deleted chromosome in the child with WS was determined by genotyping polymorphic markers that were known to be located within the WSCR to determine parental haplotypes. Absence of a parent’s marker haplotype was evidence of the origin of the deleted chromosome. The markers included microsatellite repeats and single nucleotide polymorphisms (SNPs). Microsatellite markers, including D7S615, D7S809, D7S1870, and D7S1820, were genotyped on a CEQ8000 fluorescence sequencer (Beckham-Coulter, Fullerton, CA). Primer sequences were those listed in the NCBI UNiSTS database (http://www.ncbi.nlm.nih.gov/). The forward primer for each marker was labeled with one of three WellRed dyes (Proligo Inc, Boulder, CO), in a pattern that allowed multiple markers to be analyzed in a single lane on the sequencer. The genotypes were determined after analysis with CEQ8000 genotyping software. SNPs from the region were analyzed using TaqMan probes on an ABI 7900HT sequence detection system. The parent of origin was called if at least 2 SNP markers showed a particular parent failed to pass alleles to the child with WS or if one microsatellite marker showed a clear distinction.
Maternal Reproductive History
Reproductive histories were obtained from 161 of the 257 mothers. Each of these mothers indicated the number of children to whom she had given birth and the number of spontaneous abortions she had had.
RESULTS
The frequency of inversions in transmitting and non-transmitting mothers and fathers is reported in Table I. As indicated in the table, 64 of 257 transmitting parents (24.9%) and 15 of 257 non-transmitting parents (5.8%) had an inversion of the WSCR. This percentage provides a reliable estimate of the rate of inversion in the general population. No parent, whether transmitting or non-transmitting, was homozygous for the inversion polymorphism. Of the 257 couples, there were 7 in which both parents had an inversion, 65 in which only one parent had an inversion, and 185 in which neither parent had an inversion.
Table I.
Inversion of the WSCR in 257 Couples who Have a Child with WS
| Parent | Inversion | Total | ||
|---|---|---|---|---|
| Yes | No | |||
| Transmitting | Mother | 29 | 101 | 130 |
| Father | 35 | 92 | 127 | |
| Combined | 64 | 193 | 257 | |
| Non-transmitting | Mother | 8 | 119 | 127 |
| Father | 7 | 123 | 130 | |
| Combined | 15 | 242 | 257 | |
In Table II, the data from Table I are expressed as rates, along with 95% confidence intervals. The confidence intervals were estimated using the modified Wald method [Agresti and Couli, 1998]. A comparison of inversion rates for mothers and fathers indicated no gender differences for either the transmitting parent, χ2(1, 257) = 0.95, p = 0.39, or the non-transmitting parent, χ2(1, 257) = 0.10, p = 0.80. There also were no gender differences in the likelihood of transmitting the WS chromosome, independent of inversion status, χ2(1, 514) = 0.07, p = 0.86. The parent of origin was the mother in 50.6% (130 of 257) of the couples and the father in 49.4% (127 of 257).
Table II.
Rates of inversion of WSCR in parents of children with WS
| Parent | Inversion Rate |
95% Confidence Interval |
||
|---|---|---|---|---|
| Lower | Upper | |||
| Transmitting | Mother | 22.31% | 15.96% | 30.24% |
| Father | 27.56% | 20.51% | 35.93% | |
| Combined | 24.90% | 20.00% | 30.55% | |
| Non-transmitting | Mother | 6.30% | 3.05% | 12.12% |
| Father | 5.38% | 2.44% | 10.89% | |
| Combined | 5.84% | 3.50% | 9.48% | |
These data suggest that a parent with an inversion (whether the mother or the father) has an increased risk of having a child with Williams syndrome. The risk estimates, or odds, of transmitting the WS chromosome are presented in Table III for parents with and without inversions. Data are presented separately for mothers and fathers, as well as combined. The Odds Ratio Test was used to evaluate the statistical significance of the observed increase in risk. A parent with an inversion has a fivefold increased likelihood of having a child with WS (Odds Ratio = 5.35, χ2(1, 514) = 35.9, p < .0001). The risk estimates for mothers and fathers were not statistically different as indicated by the Breslow-Day Test for Homogeneity of Odds Ratios, χ2 (1) = 0.55, p = 0.46.
Table III.
Odds Ratios and Confidence Intervals for Transmission of WS Deletion
| Parent | Odds/Odds Ratio | Value | 95% Confidence Interval |
χ2(1) | p | |
|---|---|---|---|---|---|---|
| Upper | Lower | |||||
| Mother | Odds Ratio | 4.27 | 1.87 | 9.76 | 13.36 | <.0001 |
| Inversion Odds | 3.54 | 1.68 | 7.45 | |||
| No Inversion Odds | 0.83 | 0.75 | 0.92 | |||
| Father | Odds Ratio | 6.69 | 2.84 | 15.72 | 23.11 | <.0001 |
| Inversion Odds | 5.12 | 2.36 | 11.01 | |||
| No Inversion Odds | 0.77 | 0.68 | 0.86 | |||
| Combined | Odds Ratio | 5.35 | 2.96 | 9.68 | 35.91 | <.0001 |
| Inversion Odds | 4.27 | 2.50 | 7.29 | |||
| No Inversion Odds | 0.80 | 0.74 | 0.86 | |||
Strømme et al. [2002] estimated the incidence of WS in the population to be 1 in 7500. Using this figure and the odds computed from our sample, we estimate that a parent with an inversion has an ~ 1/1750 chance of having a child with WS, whereas the estimate for a parent without an inversion is ~ 1 /9500. In families in which one parent has an inversion, siblings of the individual with WS would have a ½ chance of inheriting a chromosome with an inversion and would therefore have an ~ 1/3500 chance of having a child with WS.
The high prevalence of the inversion polymorphism in the general population compared to the relatively low incidence of WS has raised the possibility of an increased rate of spontaneous abortion in inversion heterozygotes [Scherer et al., 2005]. To address this possibility, we examined spontaneous abortion rates as a function of the mother’s inversion status in a subsample of 161 couples. In this sample, 24 of the mothers had inversions. For the 24 mothers who had a WSCR inversion, there were 56 pregnancies and the spontaneous abortion rate was 7.14% (95% CI: 2.33% – 17.46%). For the 137 mothers who did not have a WSCR inversion, there were 372 pregnancies and the spontaneous abortion rate was 12.1% (95% CI: 9.14% – 15.83%). The spontaneous abortion rates were not statistically different for the two groups (χ2(1, 161) = 0.74, p = .39). The spontaneous abortion rate for the entire sample was 11.45% (95% CI: 8.75% – 14.83%). Using the 95% confidence interval for comparison, we conclude that the rate estimated for the present sample of 161 mothers did not differ significantly from the spontaneous abortion rate of 10.9% estimated from a population study of 1,221,546 pregnancy outcomes [Andersen et al., 2000].
Consistent with the findings of Tam et al. [2008], none of the parents who had an inversion of the WSCR evidenced any phenotypic abnormality associated with WS. In several families, the inversion of the WSCR was transmitted for multiple generations prior to the birth of a child with WS. For example, in one family, the inversion polymorphism was found in a sibling, parent, grandparent, great-grandparent, and great-aunt of the child with WS.
DISCUSSION
This study provides the first report of a large scale analysis of individuals with WS and their parents aimed at evaluating the relation between the presence of a parental inversion of the WSCR and generation of the WS deletion. We showed that heterozygous presence of this inversion is associated with generation of the WS deletion at a frequency 5.4 times greater than that observed in the absence of the inversion. This finding of increased risk for individuals who have an inversion of the WSCR is consistent with suggestions from earlier studies [Osborne et al., 2001; Bayes et al., 2003]. Our large sample permits a reasonable estimation of the increased chance for offspring with WS for individuals who have the WSCR inversion relative to individuals who do not have this inversion. With respect to sex bias for parent-of-origin of WS, our data set reinforces previous findings that, like most known genomic disorders, there is no statistically significant parent-of-origin gender effect in rate of transmission of the deleted chromosome that causes WS [Stankiewicz and Lupski, 2002; Thomas et al., 2006]. In addition, our data show that there is no gender bias in transmitting parents who have an inversion of the WSCR. Our data also indicate that maternal inversion of the WSCR is not associated with an increased rate of spontaneous abortion.
WSCR deletions and duplications result from the participation of large LCRs in NAHR, with a significant number of occurrences originating from genomic inversion in the transmitting parental chromosome. In addition to playing a major role in determining normal phenotypic variability, genomic elements such as segmental duplications participate in the genesis of many recurrent genomic disorders. An estimated 23% of the copy number variations that cause clinical phenotypes are flanked by segmental duplications of at least 10 kb and at least 95% sequence similarity [Rudd et al., 2009]. Additional pathogenic microdeletions can result from regions of (micro)homology ranging from 2 – 75+ bases [Vissers et al., 2009]. The rate of inversion heterozygosity of the WSCR in the general population (5.8%) is lower than the reported rates for at least two other chromosomal regions associated with genomic disorders. An inversion associated with the Prader-Willi syndrome/Angelman syndrome region at 15q11 is reported to have a population frequency for heterozygosity of about 9% [Gimelli et al., 2003]. Clusters of olfactory receptor genes on chromosome 8p have been reported to participate in generation of cytogenetically-detectable rearrangements as well as submicroscopic inversions. In Europeans, the frequency of heterozygosity for this 8p inversion has been estimated at 26% [Giglio et al., 2001] and it is thought to enhance formation of additional cytogenetic abnormalities. For some genomic disorders, parental inversions are a prerequisite for the occurrence of the duplication or deletion [Sharp, 2009].
The WS deletion usually occurs de novo; in our study ~75% (193/257) of individuals with classic WS have transmitting parents who do not have the inversion. From our large sample, the observed rate of inversion heterozygosity for transmitting parents of 24.9% (64/257) is in general agreement with but slightly lower than the rates reported in previous studies of smaller samples: 33% for a sample of 12 [Osborne et al., 2001] and 28% for a sample of 74 [Bayes et al., 2003]. A person who has a WSCR inversion has a 5.4 fold increased chance of having a child with WS relative to a person who does not have this inversion. Nonetheless, the risk of having a child with WS even for individuals who have a WSCR inversion is still low, ~1/1750. The studies necessary to determine if an individual has an inversion of the WSCR are not commercially available. From a practical standpoint, the lack of commercial availability of these studies may not be important, as the only way that one can know if a particular offspring will have WS is to perform a prenatal diagnostic test for the deletion, and it is not necessary to know parental inversion status for interpretation of results. Of the two reported families with recurrence of WS in a sibship born to unaffected parents, one father had an inversion, but the other recurrence was related to gonadal mosaicism for the WS deletion in the mother [Scherer et al., 2005].
From a genetic counseling perspective, the results of the present study are useful for providing information to families who have had a child with WS. Explaining the genomic nature of the chromosome rearrangement can reassure parents that nothing they did caused the deletion. It also is important to emphasize to families that the inversion polymorphism is not associated with phenotypic abnormality. The chance for recurrence of a child with WS can be discussed and put into perspective, for example by comparing the 1/1750 risk for an inversion carrier to have a child with WS with the higher population risk for having a child with Down syndrome. Because an inversion also predisposes to duplication of the WSCR, families should be counseled regarding that possibility.
Duplication of the WSCR would result only from interchromosomal rearrangements and would therefore be expected to occur with a ratio of 3 deletions (2 deletions by interchromosomal rearrangement and 1 by interchromatid rearrangement) for every 2 duplications. Turner et al. [2008] demonstrated that the ratio of WSCR deletion to duplication in sperm is 2:1. Duplications of the region have been reported [Somerville et al., 2005, Osborne and Mervis, 2007] but the phenotype of the WSCR duplication is still being delineated as additional affected individuals are identified [Kriek et al., 2006; Berg et al., 2007; Van der Aa et al., 2009]. Even though duplications of the WSCR are predicted to occur at a lower rate than deletions in the general population [Turner et al., 2008], it is likely that individuals heterozygous for the WSCR inversion have an equal chance for having a child with a duplication as for having a child with WS. This is because NAHR occurring between an inverted chromosome and its noninverted homologue would give rise to one chromosome with a deletion of the WSCR and one chromosome with the duplication of the WSCR.
For families seeking prenatal diagnosis in subsequent pregnancies, it will be important to use methodology that will detect both deletion and duplication of the WSCR. FISH is the most commonly employed technique for prenatal detection of deletions, but is not ideal for detecting duplications of the WSCR. Either chromosome microarray or multiplex ligation-dependent probe amplification (MPLA) can be used to detect deletion or duplication of the WSCR. For most families, parental inversion status is unknown, so the range of recurrence (1/1750 to 1/9500) will be discussed by genetic counselors. Gonadal mosaicism for the WSCR deletion has been reported once [Scherer et al., 2005], adding to the complexity of the counseling.
Because genomic rearrangements are common, it is likely that prenatal diagnosis for genomic disorders will be offered more broadly in the future. Inversion of the WSCR is only one structural genomic variation that can lead to pathogenic copy number variation in offspring. Detection of common predisposing structural rearrangements across the genome should be a goal of future prenatal screening efforts.
In summary, the present study establishes the population prevalence of the WSCR inversion at 5.8%, compared with the 24.9% prevalence in transmitting parents, and allows calculation of the recurrence risk for WS (1/1750 for inversion heterozygotes, 1/9500 for others). We confirm that mothers and fathers do not differ significantly in their odds of transmitting a chromosome with a WSCR deletion to their offspring and note that women with an inversion of the WSCR do not have an increased rate of spontaneous abortion. This investigation of a large sample advances information important for genetic counseling for WS.
ACKNOWLEDGMENTS
This work was supported by NINDS R01 NS35102. The authors are grateful to the participants and for the cooperation of the Williams Syndrome Association. We thank Lucy Osborne for her assistance, including the gift of FISH probes.
REFERENCES
- Agresti A, Couli B. Approximate is better than ‘exact” for interval estimation of binomial proportions. The American Statistician. 1998;52:119–126. [Google Scholar]
- Andersen A, Wohlfahrt J, Christens P, Olsen J, Melbye M. Maternal age and fetal loss: population based register linkage study. British Medical Journal. 2000;320:1708–1712. doi: 10.1136/bmj.320.7251.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumer A, Dutly F, Balmer D, Reigel M, Tükel T, Krajewska-Walasek M, Schintzel AA. High level of unequal meiotic crossovers at the origin of the 22q11.2 and 7q11.23 deletions. Hum Mol Genet. 1998;7:887–894. doi: 10.1093/hmg/7.5.887. [DOI] [PubMed] [Google Scholar]
- Bayés M, Magano LF, Rivera N, Flores R, Pérez-Jurado LA. Mutational mechanisms of Williams-Beuren syndrome deletions. Am J Hum Genet. 2003;73:131–151. doi: 10.1086/376565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg JS, Brunetti-Pierri N, Peters SU, Kang SH, Fong CT, Salamone J, Freedenberg D, Hannig VL, Prock LA, Miller DT, Raffalli P, Harris DJ, Erickson RP, Cunniff C, Clark GD, Blazo MA, Peiffer DA, Gunderson KL, Sahoo T, Patel A, Lupski JR, Beaud Cheung SW, et al. Speech delay and autism spectrum behaviors are frequently associated with duplication of the 7q11.23 Williams-Beuren syndrome region. Genet Med. 2007;9:427–441. doi: 10.1097/gim.0b013e3180986192. [DOI] [PubMed] [Google Scholar]
- Castorina P, Selicorni A, Bedeschi F, Dalprà L, Larizza L. Genotype-phenotype correlation in two sets of monozygotic twins with Williams syndrome. Am J Med Genet. 1997;69:107–111. doi: 10.1002/(sici)1096-8628(19970303)69:1<107::aid-ajmg21>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Cusco I, Corominas R, Bayes M, Flores R, Rivera-Brugus N, Campuzano V, Pérez-Jurado LA. Copy number variation at the 7q11.23 segmental duplications is a susceptibility factor for the Williams-Beuren syndrome deletion. Genome Res. 2008;18:683–694. doi: 10.1101/gr.073197.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutly F, Schinzel A. Unequal interchromosomal rearrangements may result in elastin gene deletions causing the Williams-Beuren syndrome. Hum Mol Genet. 1996;5:1893–1898. doi: 10.1093/hmg/5.12.1893. [DOI] [PubMed] [Google Scholar]
- Giglio S, Broman KW, Matsumoto N, Calvari V, Gimelli G, Neumann T, Ohashi H, Voullaire L, Larizza D, Giorda R, Weber JL, Ledbetter DH, Zuffardi O. Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am J Hum Genet. 2001;68:874–883. doi: 10.1086/319506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimelli G, Pujana MA, Patricelli MG, Russo S, Giardino D, Larizza L, Cheung J, Armengol Ll, Schintzel A, Estivill X, Zuffardi O. Genomic inversion of human chromosome 15q11–q13 in mothers of Angelman syndrome patients with class II (BP2/3) deletions. Hum Mol Genet. 2003;12:849–858. doi: 10.1093/hmg/ddg101. [DOI] [PubMed] [Google Scholar]
- Kriek M, White SJ, Szuhai K, Knijnenburg, van Ommen G-JB, den Dunnen JT, Breuning MH. Copy number variation in regions flanked (or unflanked) by LCRs among patients with developmental delay and/or congenital malformations; detection of reciprocal and partial Williams-Beuren duplications. Eur J Hum Genet. 2006;14:180–189. doi: 10.1038/sj.ejhg.5201540. [DOI] [PubMed] [Google Scholar]
- Marshall CR, Young EJ, Pani AM, Freckmann ML, Lacassie Y, Howald C, Fitzgerald KK, Peippo M, Morris CA, Shane K, Priolo M, Morimoto M, Kondo I, Manguoglu E, Berker-Karauzum S, Edery P, Hobart HH, Mervis CB, Zuffardi O, Reymond A, Kaplan P, Tassabehji M, Gregg RG, Scherer SW, Osborne LR. Infantile spasms is associated with deletion of the MAGI2 gene on chromosome 7q11.23–q21.11. Am J Hum Genet. 2008;83:106–111. doi: 10.1016/j.ajhg.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizugishi K, Yamanaka K, Kuwajima K, Kondo I. Interstitial deletion of chromosome 7q in a patient with Williams syndrome and infantile spasms. J Hum Genet. 1998;43:178–181. doi: 10.1007/s100380050064. [DOI] [PubMed] [Google Scholar]
- Morris CA. Genotype-phenotype correlation in Williams-Beuren syndrome. In: Morris CA, Lenhoff HM, Wang PP, editors. Williams-Beuren syndrome: Research, evaluation, and treatment. Baltimore, MD: Johns Hopkins University Press; 2006. pp. 58–82. [Google Scholar]
- Morris CA, Mervis CB, Hobart HH, Gregg RG, Bertrand J, Ensing GJ, Sommer A, Moore CA, Hopkin RJ, Spallone PA, Keating MT, Osborne L, Kimberley KW, Stock AD. GTF2I hemizygosity implicated in mental retardation in Williams syndrome: Genotype-phenotype analysis of five families with deletions in the Williams syndrome region. Am J Med Genet. 2003;123A:45–59. doi: 10.1002/ajmg.a.20496. [DOI] [PubMed] [Google Scholar]
- Morris CA, Thomas IT, Greenberg F. Williams syndrome: autosomal dominant inheritance. Am J Med Genet. 1993;47:478–481. doi: 10.1002/ajmg.1320470409. [DOI] [PubMed] [Google Scholar]
- Osborne LR, Li M, Pober B, Chitayat D, Bodurtha J, Mandel A, Costa T, Grebe T, Cox S, Tsui L-C, Scherer SW. A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat Genet. 2001;29:321–325. doi: 10.1038/ng753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne LR, Mervis CB. Rearrangements of the Williams-Beuren syndrome locus: molecular basis and implications for speech and language development. Expert Rev Mol Med. 2007;9(15):1–16. doi: 10.1017/S146239940700035X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez Jurado LA, Peoples R, Kaplan P, Hamel BCJ, Francke U. Molecular definition of the chromosome 7 deletion in Williams syndrome and parent-of-origin effects on growth. Am J Hum Genet. 1996;59:781–792. [PMC free article] [PubMed] [Google Scholar]
- Rudd MK, Keene J, Bunke B, Kaminsky EB, Adam MP, Mulle JG, Ledbetter DH, Martin CL. Segmental duplications mediate novel, clinically relevant chromosome rearrangements. Hum Mol Genet. 2009;18:2957–2962. doi: 10.1093/hmg/ddp233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler LS, Robinson LK, Verdaasdonk KR, Gingell R. The Williams syndrome: evidence for possible autosomal dominant inheritance. Am J Med Genet. 1993;47:468–470. doi: 10.1002/ajmg.1320470406. [DOI] [PubMed] [Google Scholar]
- Scherer SW, Gripp KW, Lucena J, Nicholson L, Bonnefont J-P, Pérez-Jurado LA, Osborne LR. Observation of a parental inversion variant in a rare Williams-Beuren syndrome family with two affected children. Hum Genet. 2005;117:383–388. doi: 10.1007/s00439-005-1325-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert C. The genomic basis of the Williams-Beuren syndrome. Cell Mol Life Sci. 2009;66:1178–1197. doi: 10.1007/s00018-008-8401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ. Emerging themes and new challenges in defining the role of structural variation in human disease. Hum Mutat. 2009;30:135–144. doi: 10.1002/humu.20843. [DOI] [PubMed] [Google Scholar]
- Somerville MJ, Mervis CB, Young EJ, Seo E-J, del Campo M, Bamforth S, Peregrine E, Loo W, Lilley M, Pérez-Jurado LA, Morris CA, Scherer SW, Osborne LR. Severe expressive-language delay related to duplication of the Williams-Beuren locus. N Engl J Med. 2005;353:1694–1701. doi: 10.1056/NEJMoa051962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Genomic architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- Stock AD, Spallone PA, Dennis TR, Netski D, Morris CA, Mervis CB, Hobart HH. Heat shock protein 27 gene: chromosomal and molecular location and relationship to Williams syndrome. Am J Med Genet. 2003;120A:320–325. doi: 10.1002/ajmg.a.20055. [DOI] [PubMed] [Google Scholar]
- Strømme P, Bjørnstad PG, Ramstad K. Prevalence estimation of Williams syndrome. J Child Neurol. 2002;17:269–271. doi: 10.1177/088307380201700406. [DOI] [PubMed] [Google Scholar]
- Tam E, Young EJ, Morris CA, Marshall CR, Loo W, Scherer SW, Mervis CB, Osborne LR. The common inversion of the Williams-Beuren syndrome region at 7q11.23 does not cause clinical symptoms. Am J Med Genet. 2008;146A:1797–1806. doi: 10.1002/ajmg.a.32360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas NS, Durkie M, Potts G, Sandford R, Van Zyl B, Youings S, Dennis NR, Jacobs PA. Parental and chromosomal origins of microdeletion and duplication syndromes involving 7q11.23, 15q11–q13 and 22q11. Eur J Hum Genet. 2006;14:831–837. doi: 10.1038/sj.ejhg.5201617. [DOI] [PubMed] [Google Scholar]
- Turner DJ, Miretti M, Rajan D, Fiegler H, Carter NP, Blayney ML, Beck S, Hurles ME. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbán Z, Helms C, Fekete G, Csiszár K, Bonnet D, Munnich A, Donis-Keller H, Boyd CD. 7q11.23 deletions in Williams syndrome arise as a consequence of unequal meiotic crossover. Am J Hum Genet. 1996;59:958–962. [PMC free article] [PubMed] [Google Scholar]
- Van der Aa N, Rooms L, Vandeweyer G, van den Ende J, Reyniers E, Fichera M, Romano C, Delle Chiaie B, Mortier G, Menten B, Destrée A, Maystadt I, Männik K, Kurg A, Reimand T, McMullan D, Oley C, Brueton L, Bongers EM, van Bon BW, Pfund R, Jacquemont S, Ferrarini A, Martinet D, Schrander-Stumpel C, Stegmann AP, Frints SG, de Vries BB, Ceulemans B, Kooy RF. Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur J Med Genet. 2009;52:94–100. doi: 10.1016/j.ejmg.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Vissers LE, Bhatt SS, Janssen IM, Xia Z, Lalani SR, Pfundt R, Derwinska K, de Vries BB, Gilissen C, Hoischen A, Nesteruk M, Wisniowiecka-Kowalnik B, Smyk M, Brunner HG, Cheung SW, van Kessel AG, Veltman JA, Stankiewicz P. Rare pathogenic microdeletions and tandem duplications are microhomology-mediated and stimulated by local genomic architecture. Hum Mol Genet. 2009;18:3579–3593. doi: 10.1093/hmg/ddp306. [DOI] [PubMed] [Google Scholar]