

Abstract
The use of transgenic markers in pluripotent stem cells allows the facile isolation of transient cell populations that appear at certain phases of embryonic development. Here we describe a procedure for deriving cardiac progenitors from mouse pluripotent stem cells carrying a GFP reporter under the control of an Nkx2.5 enhancer sequence. The cells are propagated under standard conditions and differentiated using the hanging-droplet method with medium optimized for commitment to the cardiac lineage. Cardiac progenitors are isolated from the differentiation culture using fluorescence-activated cell sorting (FACS) and can be cultured further for functional characterization and experimentation. The protocols described here can be applied to both embryonic and induced pluripotent stem cells and can easily be adapted to transgenic lines carrying other cardiac cell lineage reporters.
Keywords: mouse embryonic stem cells, induced pluripotent stem cells, cardiac progenitor cells, in vitro differentiation
Introduction
In recent years, there has been growing interest among basic and clinical scientists in harnessing the tremendous developmental potential of pluripotent stem cells for understanding fundamental biological processes and disease pathogenesis. These cells, derived either from pre-implantation stage embryos of human or animal origin (embryonic stem (ES) cells), from neonatal or adult testis (mature adult germline stem cells (maGSC)), or by molecular reprogramming of differentiated somatic cells (induced pluripotent stem cell (iPS) cells), are believed to share similar transcriptional profiles and molecular phenotypes in culture. The ability of these cells to differentiate in vitro into all lineages of the three germ layers has allowed the isolation and phenotypic characterization of various populations of progenitor and transient-amplifying cells prior to their terminal differentiation. It is believed that by identifying and isolating lineage-specific stem/progenitor cells from pluripotent stem cells, one may then be able to translate the developmental potential of these cells into therapeutic applications for a wide range of neurological, cardiovascular, and hematological diseases. The objective of this unit is to describe, in sufficient detail, our methods for identifying and isolating cardiac progenitor cells from in vitro differentiated mouse ES and iPS cells. It is anticipated that all readers with standard training in cell and molecular biology will be able to accomplish this by following the steps described here.
Before we describe our basic protocol for isolating cardiac progenitor cells from pluripotent stem cells, it is worth mentioning the rationale and advantages for using in vitro differentiated ES or iPS cells rather than isolating them directly from developing embryos, First, working with early stage embryos can be technically difficult and costly, and the number of cells obtained may be insufficient for further experimentation. Pluripotent stem cells, on the other hand, can be expanded in large numbers to satisfy the cell number required for most studies. Second, the rapid doubling time of pluripotent stem cells allows for a short turnaround time between experiments. Third, pluripotent stem cells can be genetically modified to carry lineage specific reporters and/or mutations, which would allow for the in vitro isolation of specific cell-types of interest that recapitulate disease phenotypes without incurring the time and cost associated with generating genetically-modified animals. These advantages, coupled with the potential uses of pluripotent stem cells in cell-based therapies, account for the growing popularity of these cells in regenerative biology and medicine.
In this unit we describe the isolation and characterization of mouse ES and iPS cell-derived Nkx2.5+ cardiac progenitor cells. While a number of cardiac stem/progenitor cell populations from mouse and human ES cells have been described (Moretti et al., 2006, Kattman et al., 2006, Yang et al., 2008, Bu et al., 2009, Domian et al., 2009), the biological relationships between these cell populations remain to be clarified. Moreover, the protocols used by each laboratory to purify cardiac progenitor cells vary significantly. For example, the use of Flk1 to isolate multipotent cardiovascular progenitor cells relies on the addition of a cocktail of growth factors during differentiation (Kattman et al., 2006). When mouse ES cells are differentiated in the presence of serum and the absence of growth factor cocktail, very few cardiomyogenic Flk1+ cells can be isolated (Wu, S.M. unpublished data). Our approach here takes advantage of the availability of a committed cardiac progenitor cell marker, Nkx2.5, which has been well described to mark the first identifiable heart-forming cells in the developing embryo (Wu et al., 2006). Furthermore, we have shown that mouse ES cell-derived Nkx2.5+ cells can give rise specifically to cardiomyocytes and smooth muscle cells both in vivo and in vitro (Wu et al., 2006). Although the protocol described here is based on studies using a transgenic Nkx2.5-eGFP fluorescence reporter to identify cardiac progenitor cells, the methods for culturing, differentiating, and progenitor cell isolation can be applied to other ES and iPS cells carrying appropriate lineage or surface markers.
The first protocol describes the general culturing and in vitro differentiation of mouse ES cells by the hanging droplet method. Subsequent protocols describe the isolation, continued culture, and characterization of the FACS-purified eGFP+ cardiac progenitor cells from Nkx2.5-eGFP ES cells.
NOTE: This unit assumes that the user has some experience with the growth and maintenance of undifferentiated mouse embryonic stem (ES) cells; for example protocol, see Tremml et al., 2008.
NOTE: The following procedures are performed in a Class II biological hazard flow hood, using sterile equipment and solutions and proper aseptic technique.
NOTE: All incubations are performed in a humidified 37°C, 5% CO2 incubator unless otherwise noted.
NOTE: All centrifugations are done in an Eppendorf Model 5810R benchtop centrifuge.
Basic Protocol 1: Culturing and in vitro differentiation of pluripotent stem cells
This protocol is adapted from the well-described hanging-droplet method of ES differentiation (Samuelson et al., 2006), with slight modifications designed to enhance cardiac lineage differentiation (Wobus et al., 1991, Takahashi et al., 2003). It can be adapted to generate large quantities of cardiac progenitor cells by scaling up accordingly. For high-throughput screening assays, a monolayer differentiation protocol using lineage-labeled ES or IPS cells in a 96-well plate format can be used (see Alternative Protocol 1). Additionally, a hybrid protocol that incorporates both methods may be used for other applications. In such a protocol, cells differentiated in hanging-droplet cultures are dissociated at the desired time point and re-plated into 6-, 12-, 24-, or 96-well plates for further treatments and characterization (see Basic Protocol 2).
Materials
-
-
Nkx2.5-eGFP transgenic ES or IPS cells in standard culture (DMEM-ES medium (see recipe) plus feeder layer of growth-arrested mouse embryonic fibroblasts (MEFs)); passaged fewer than 30 times since original derivation
-
-
0.25% trypsin/EDTA GIBCO 25200-056
-
-
0.1% gelatin solution (see recipe)
-
-
Differentiation medium (see recipe)
-
-
10cm cell culture plate
-
-
150 × 15mm slippable monoplate petri dishes Fisher 08-757-14
-
-
10–100µl multichannel pipet
MEF depletion from cell culture (2 days)
When ES or IPS cells reach 50–70% confluency in one well of a 6-well plate, aspirate medium and add 0.5ml of 0.25% trypsin/EDTA to each well of cells. Incubate cells for 5 minutes, or until cells are in single-cell suspension as visualized under a microscope.
Add 2ml of IMDM-ES medium to each well to inactivate the trypsin.
Centrifuge cells at 1000 rpm for 3 minutes. Aspirate supernatant and re-suspend cell pellet in IMDM-ES medium (~1ml).
Add 10ml of IMDM-ES medium to a 10cm cell culture plate pre-coated for 20min with 0.1% gelatin. Add re-suspended ES cells to the plate and pipet gently to redistribute cells evenly throughout the plate. Incubate for 2 days.
EB differentiation (Days 0–16)
At the end of 2 days, dissociate cells from the 10cm plate using 2ml of 0.25% trypsin/EDTA. Incubate for 5 minutes or until cells have lifted from the plate and inactivate the trypsin with 8ml of differentiation medium.
Centrifuge cells at 1000 rpm for 3 minutes. Aspirate supernatant and re-suspend cell pellet in differentiation medium (~1ml).
Count live cells. Dilute cell suspension in differentiation medium to a final concentration of 200,000 cells/mL.
Using a multichannel pipet set at 11uL (to compensate for ~1uL loss with each pipet action), pipet ~250 droplets onto the bottom of a 15cm petri dish (without gelatin coating) (Fig. 1a). The number of cells from a 10cm IMDM-ES culture (~1.5×107) is sufficient for ~25 of these 15cm plates.
Replace the lid of each plate, and turn the plate upside-down. The droplets will hang from the bottom of the plate (Fig. 1b). This configuration allows the cells to aggregate and form embryoid bodies (EBs). The day that the droplets are made is counted as Day 0, with the subsequent differentiation days (e.g. Day 1) counted from this time point.
Incubate the cells as hanging droplets for 2 days. On Day 2, turn the plates right side up and add 10ml of differentiation medium to each 15cm petri dish. Swirl to ensure even coverage of the plate. The EBs should be barely visible as small white specks floating in suspension. A well-formed EB should be spherically-shaped and have a smooth, bright border.
Over the next 3–5 days, if the ES cells have successfully differentiated, a significant portion (>50%) of the EBs will adhere to the bottom of the plate while continuing to enlarge. The first GFP+ cells can be detected as early as Day 5; these represent the earliest cardiac progenitors. Beating clusters containing GFP+ cells (Fig. 1c) will become visible under fluorescence microscope starting at Day 7. With proper re-feeding of the culture plates (see step 8), these clusters can continue to beat for the next 8 to 10 days. At desired time points during the course of differentiation, EBs may be isolated for flow cytometric analysis, fluorescence-activated cell sorting (FACS), immunocytochemistry, gene expression analysis, and other assays (see Basic Protocol 2).
Differentiating cells secrete cytokines, growth factors, and other as yet poorly defined chemical signals into the culture medium that are essential for proper differentiation. Therefore, the culture media should not be removed before the cells have become committed to their respective lineages (usually by Day 7). However, for continued EB growth and development beyond this time point, the medium will need to be replenished every 2 to 3 days. At Day 7, remove as much of the existing medium as possible through aspiration, taking care to not aspirate any unattached EBs. Add back 10ml of fresh differentiation medium. For optimal results, repeat every 2 to 3 days.
Figure 1.
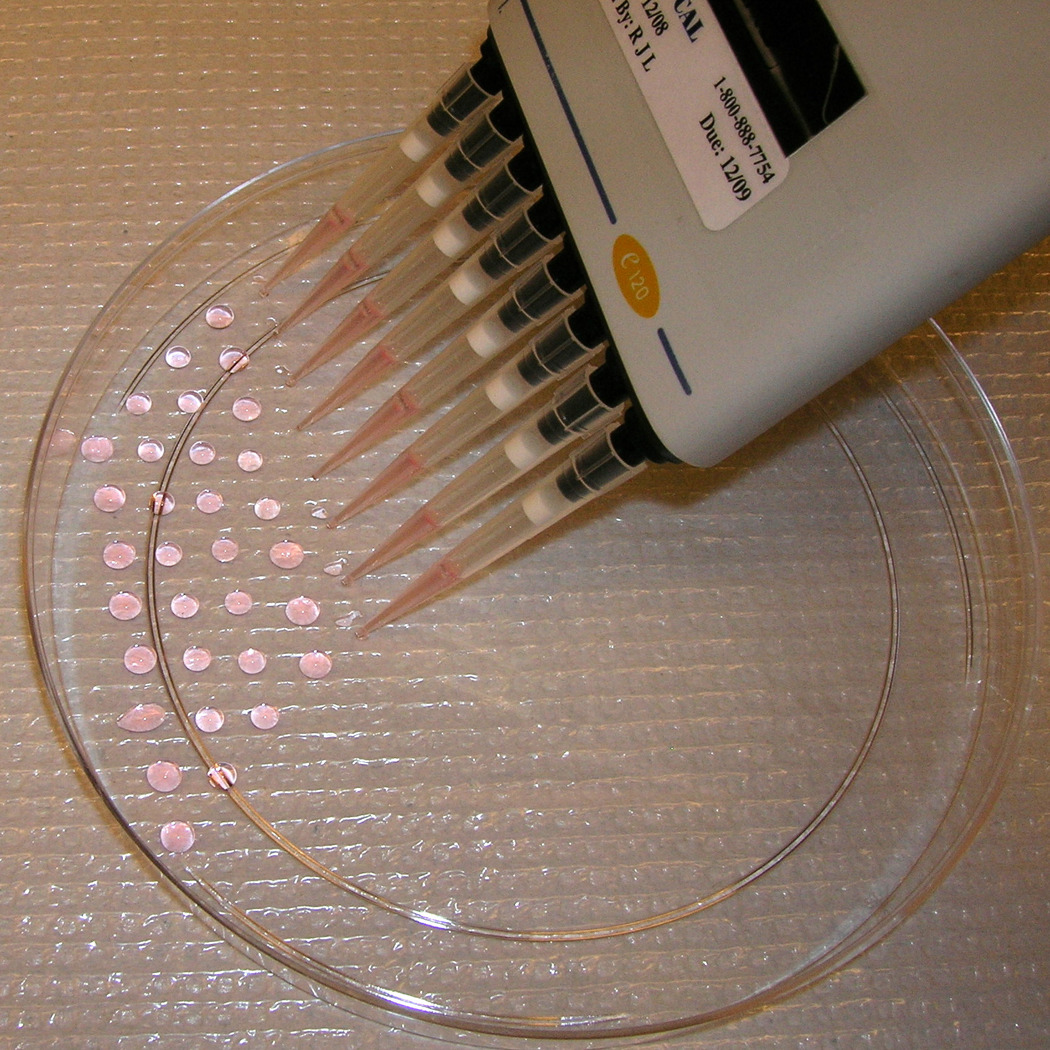
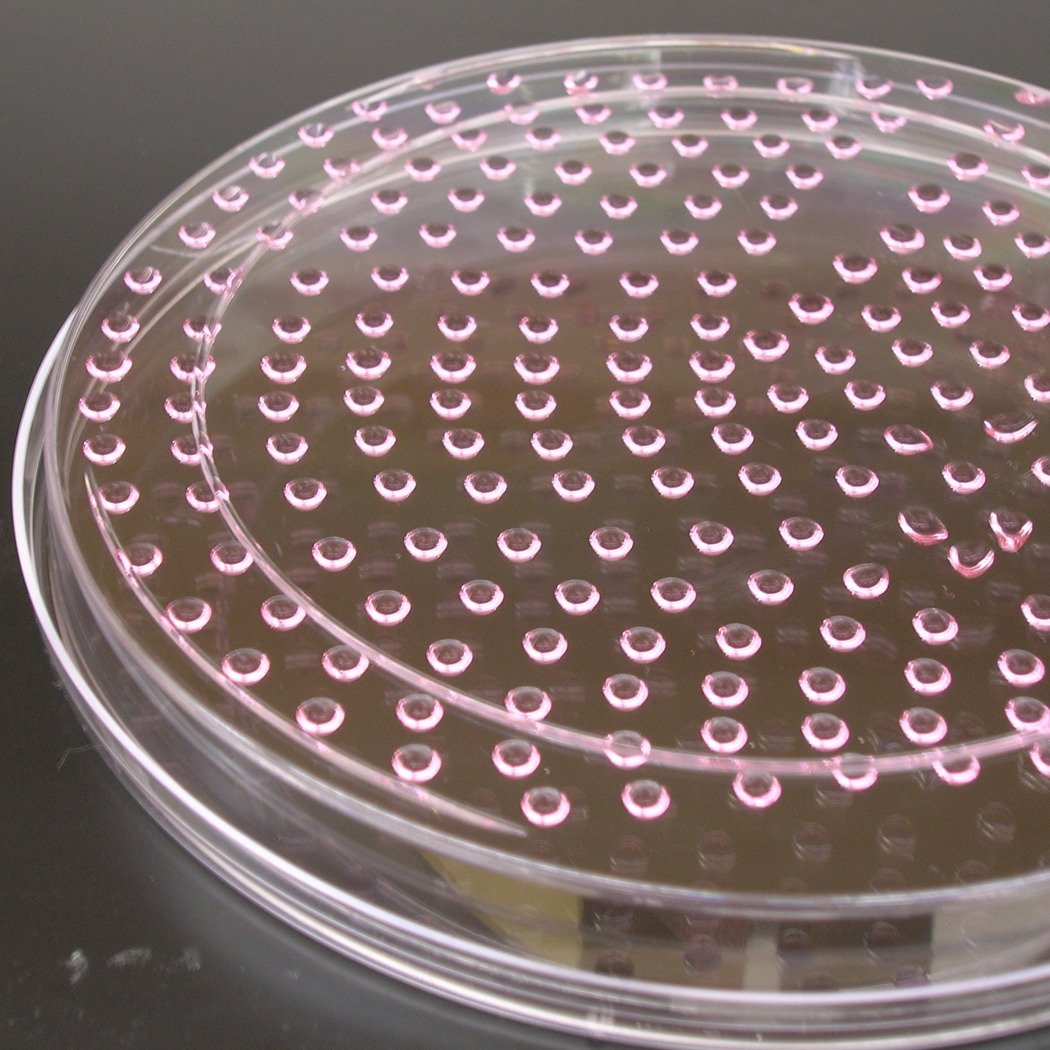
Embryoid body formation from hanging droplet. (a) Pipetting 11µL droplets of cells onto the bottom of a 15cm petri dish. (b) The dish is inverted to generate hanging droplets. The droplet density shown here (~250 droplets/plate) yields the maximum number of EBs per plate. (c) A representative area of beating eGFP+ cells within an EB.
Basic Protocol 2: Preparation of differentiation culture for flow cytometry analysis or FACS
This protocol describes the dissociation of EBs into a single cell suspension to be used for flow cytometry analysis or FACS. Dissociation is also recommended for isolating particular EBs or areas in the differentiation culture for long-term culture (see Basic Protocol 4).
Materials
-
-
Collagenase/DNase solution (see recipe)
-
-
HBS: Hepes-buffered saline (see recipe)
-
-
Flow cytometry buffer (see recipe)
-
-
Propidium iodide solution (1mg/mL) Invitrogen P3566
-
-
40µm cell strainers
Mechanical dissociation of EBs
Using a 10ml serological pipet, pick up the medium in an EB culture plate and eject it back onto the plate to dislodge EBs that have adhered to the bottom. Be careful not to generate excess foam during this process. The EBs should detach after a few rounds of pipetting (Fig. 2a). Of note, during the later stages of differentiation (e.g. beyond Day 10), EBs become increasingly adherent to the plate and will require more vigorous pipetting to detach.
Using the 10mL serological pipet, transfer detached EBs into a 15ml or 50ml conical tube and let stand for 10 minutes, until the EBs have settled to the bottom of the tube (Fig. 2b). Since the EBs are denser than medium, it is usually not necessary to centrifuge the tubes. Carefully aspirate the medium above the EBs.
Figure 2.


Harvesting EBs for isolation and analysis of cardiac progenitor cells. (a) After dissociation from the culture plate, EBs (indicated by arrows) appear as large particles in suspension. The plate shown is at Day 7 of differentiation. (b) The contents of the plate shown in (a) are transferred to a 15ml conical tube, and the EBs are allowed to settle to the bottom. Larger tubes (e.g. 50mL conical tube) may be used when pooling multiple plates.
Collagenase/DNase digestion of EBs (used for Day 6 EBs and later)
For each 15cm plate of EBs, add 0.5ml collagenase/DNase solution to the dislodged cells and pipet gently up and down 30 times with a 1000µL pipet to facilitate the breaking of EBs into a single-cell suspension.
Incubate tubes in a 37°C water bath for 1 hour. Repeat triturition every 15min during incubation.
Preparation of cells for flow cytometry applications
To remove collagenase/DNase solution after incubation, dilute with HBS at a minimum of 5 times the volume of the previously added collagenase/DNase solution.
Centrifuge the cells for 3 minutes at 1000rpm. Aspirate supernatant.
Re-suspend cell pellet in flow cytometry buffer, or the appropriate medium or buffer for other applications. For most flow cytometry experiments, a ratio of 0.5ml flow cytometry buffer to a maximum of 5 million cells is appropriate. Add 1µL of propidium iodide (PI) solution for every 0.5ml buffer. Some visible clumps may remain after digestion and re-suspension, especially during the later stages of differentiation. To remove these, filter the cell suspension through a 40µm cell strainer.
Basic Protocol 3: Flow cytometry analysis and FACS-based purification of cardiac progenitor cells
The Nkx2.5-eGFP marker allows for the quantization and purification of cardiac progenitor cells that develop during EB differentiation. Assessment of the efficiency of cardiac differentiation can be performed using flow cytometry to determine the percentage of cells that have committed to the cardiac lineage (based on the expression of eGFP). After cells have been prepared according to Basic Protocol 2, standard flow cytometry procedures are used to quantitate the percentage of eGFP+ cells with each batch of differentiation. A typical series of gates used for determining the percentage of eGFP+ cells present in differentiated EBs is shown in Fig. 3. We routinely observe 1–5% of eGFP+ cells present in the total PI-negative live-cell population. Fluorescence-activated cell sorting (FACS) can be used to recover the eGFP+ cells for further culture and/or experimentation (see Basic Protocol 4) if desired.
Figure 3.
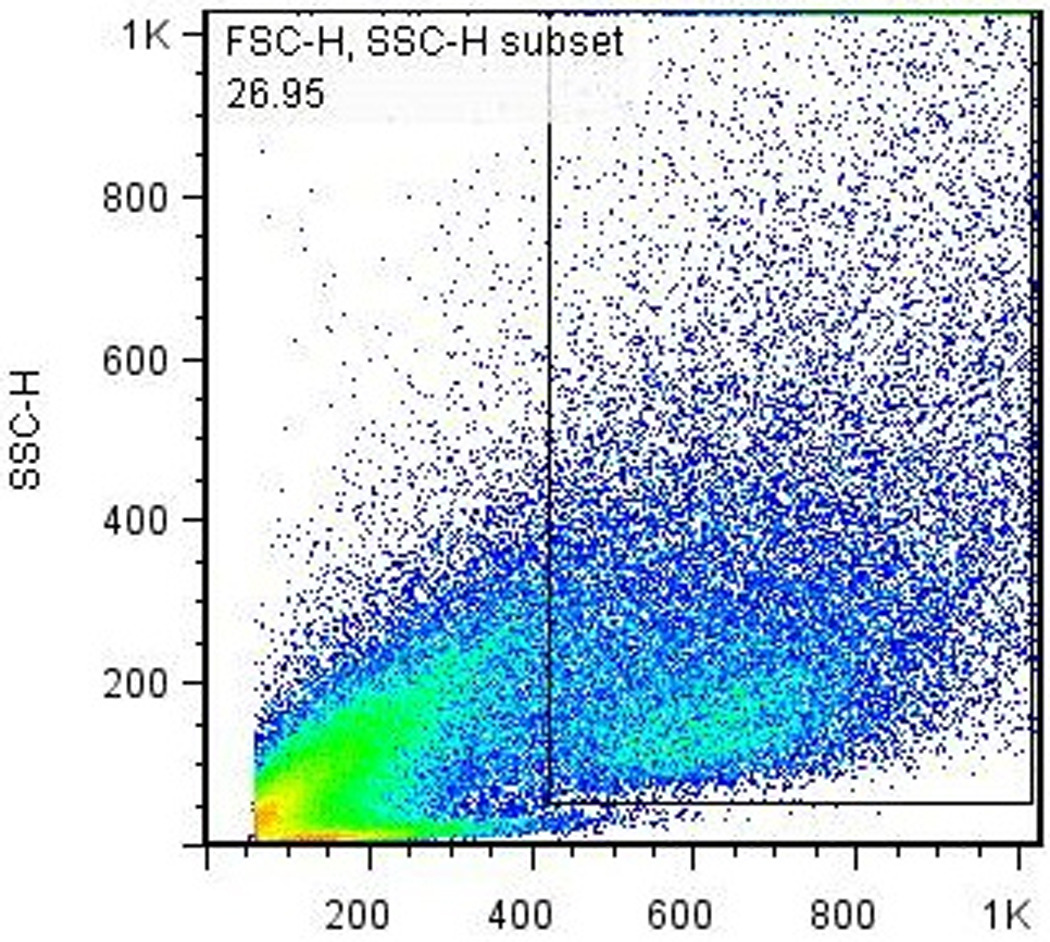
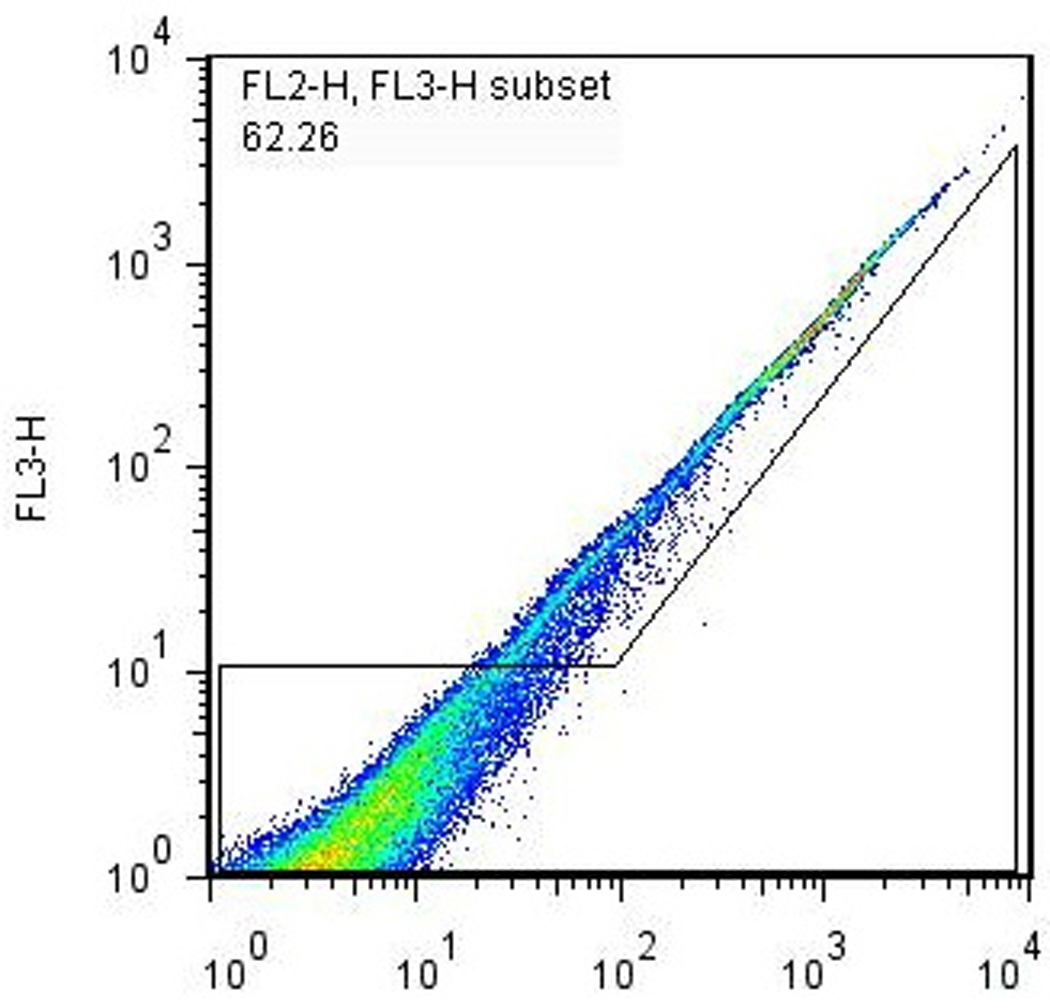
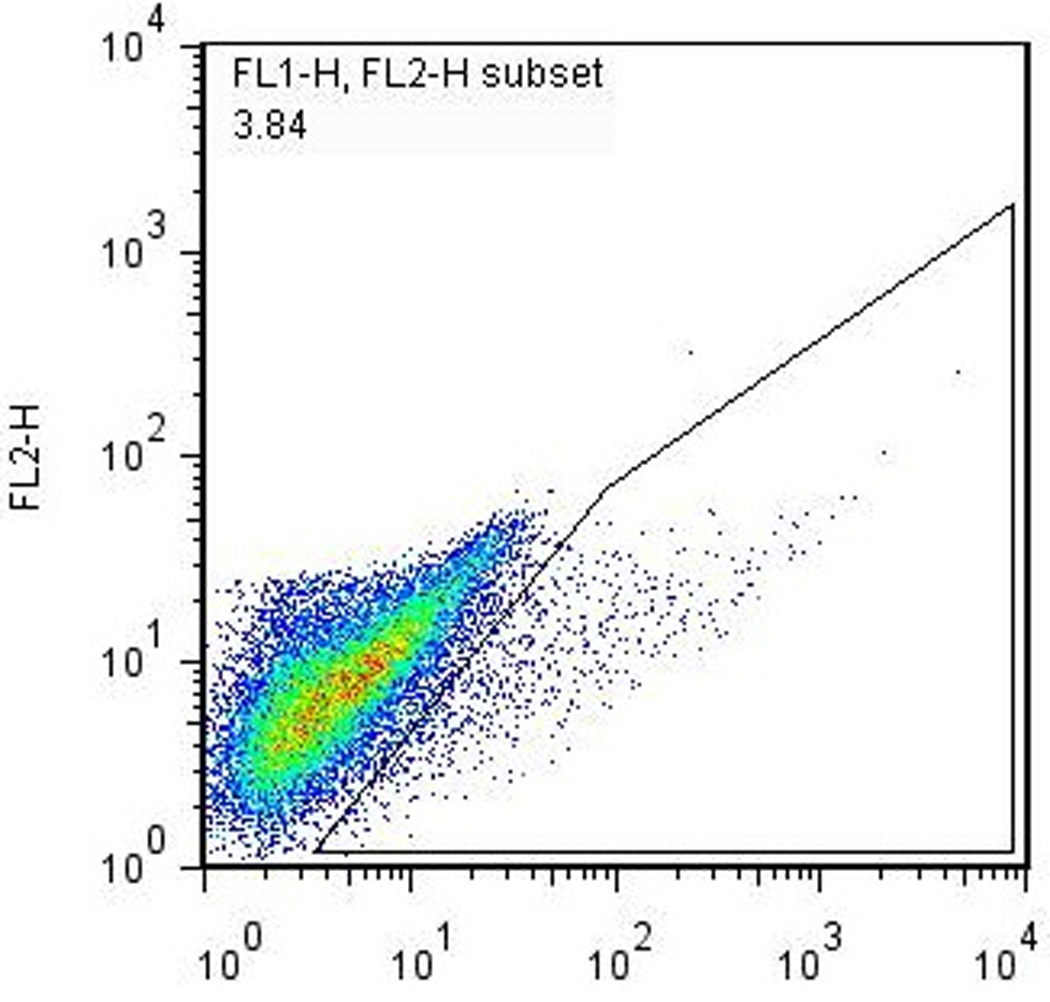
Flow cytometry analysis of differentiated EBs. A single-cell suspension of EB-derived differentiated Nkx2.5-eGFP cells were prepared according to Basic Protocol 2 and analyzed using a FACSCalibur® flow cytometer. (a) Forward- vs. side-scatter plot, gated to exclude noncellular debris. (b) Gating for live (PI-) cells. (c) Gating for GFP+ cells. The percentage of GFP+ cells in this particular analysis, 3.84%, is typical for a normal differentiation using healthy cells.
Basic Protocol 4: Culture and functional characterization of cardiac progenitors
This protocol describes the continued culturing and characterization of the Nkx2.5-eGFP+ cardiac progenitor cells isolated by FACS from EB differentiation. There are a wide variety of functional assays that may be used to characterize these progenitor cells and their progeny. We will restrict our description to ones that can be performed using conventional molecular biology tools instead of more specialized techniques such as electrophysiological assessment or contractility assays of cardiomyocytes.
Materials
-
-
Differentiation medium (DM)
-
-
Chamber slides
-
-
Cell culture plates
-
-
0.1% gelatin
-
-
Trizol [Invitrogen 15596026] or other cell lysis solution for RNA isolation
-
-
Antibodies for cardiac troponin T (cTnT) (e.g. 13-11, Neomarkers, Fremont, CA), sarcomeric myosin heavy chain (sarcMHC) (e.g. MF20, Developmental Studies Hybridoma Bank, Iowa City, IA), sarcomeric actinin (sarcActinin) (e.g. clone EA-53, Sigma), smooth muscle actin-α (SMAα) (e.g. clone IA4, DakoCytomation), smooth muscle myosin heavy chain (SM-MHC) (e.g. polyclonal, Biomedical Technologies Inc., Stoughton, MA)
Culturing of Nkx2.5+ cardiac progenitors after isolation by FACS
During FACS isolation of Nkx2.5-eGFP+ cardiac progenitor cells, the eGFP+ cells should be collected directly into differentiation medium (DM). After the collection is finished and provided that the volume of saline carried over from FACS is less than 10% of total media volume, the cells can be plated directly into gelatin-coated chamber slides for immunocytochemistry, or into 6-, 12-, 24-, or 96-well plates as required for further experimentation. Alternatively, if gene expression analysis is desired, the cells can be either sorted directly into Trizol or pelleted via centrifugation for RNA preparations.
Depending on the experimental condition required and the density of cells in each well, the collected Nkx2.5+ cardiac progenitor cells should be re-fed every 2–3 days with fresh differentiation medium.
If isolated at Day 5 or 6 of EB differentiation, the eGFP+ cardiac progenitor cells will continue to differentiate spontaneously into cardiomyocytes (~60%) or smooth muscle cells (~40%). On rare occasions, there may be CD31+ cells by gene expression or immunocytochemistry but it is unclear if these represent true endocardial cells. The identity of differentiated progenies from purified cardiac progenitor cells can be elucidated by antibody staining for early (sarc. MHC, SMAα) and late (cTnT, SM-MHC) markers for cardiomyocytes and smooth muscle cells, respectively. From differentiation cultures at Day 10 and beyond, staining of cardiac progenitor cell-derived cardiomyocytes with sarcomeric actinin will reveal the presence of striation pattern characteristic for mature cardiomyocytes (Fig. 4).
Figure 4.
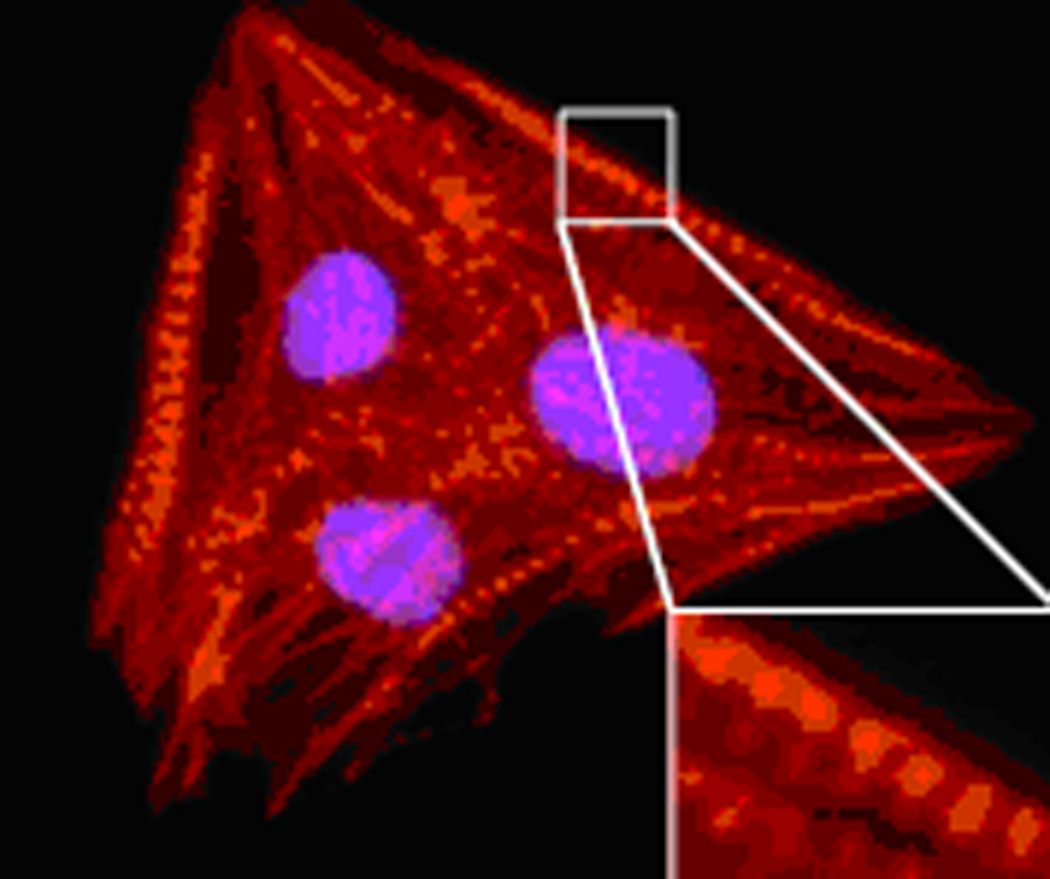
Staining for the sarcomeric structures of ES-derived cardiomyocytes using anti-sarcomeric actinin mouse monoclonal antibody.
Alternative Protocol 1: In vitro differentiation of pluripotent stem cells in a 2-D high-throughput format
For experiments involving chemical or siRNA-based high-throughput screening of compounds or genes that regulate cardiac differentiation of ES or IPS cells, a monolayer differentiation assay in 96-well plate can be employed (Takahashi et al., 2003). If a fluorescence reporter is used to quantitate cardiac differentiation, a number of fluorescence-based high-throughput detection systems such as automated flow-cytometry, automated fluorescent microscopy, or automated fluorescence plate-reader, can used.
Materials
-
-
IMDM-ES medium
-
-
Differentiation medium
-
-
0.25% trypsin/EDTA
-
-
0.1% gelatin solution
-
-
96-well cell culture plates, gelatin-coated
-
-
10–100µL multichannel pipet
-
-
Collagenase/DNase solution
-
-
Polypropylene cluster tubes for flow cytometry Corning Costar 4411
Monolayer differentiation
Prepare ES or IPS cells for differentiation by depleting the MEF layer as described in Basic Protocol 1. Because fewer cells are needed in general for monolayer differentiation compared with EB differentiation, a smaller size cell culture plate (<10cm) may be used for the MEF depletion step. Trypsinize cells at the end of 2 days and re-suspend in differentiation medium. Count live cells.
Using a multichannel pipet, pipet 4000 cells (100 µL of cells from a 40,000 cells/mL suspension in DM) into each well of a gelatin-coated 96-well plate. The day during which this step takes place is counted as Day 0, equivalent to Day 0 of the EB differentiation protocol.
For flow cytometry analysis prior to Day 7, the differentiating cells may be dissociated using 0.25% trypsin/EDTA. After Day 7, it will be necessary to use collagenase/DNase solution. After digestion is complete, inactivate the enzyme by adding medium or HBS as required. Spin down the cells at 1000rpm for 3 minutes, using a centrifuge plate-holder, then aspirate the supernatant. Add 100µL of flow cytometry buffer to each well and pipet gently to re-suspend cells. If a high-throughput flow cytometer is not available, transfer the cell suspensions to a 96-well format rack of cluster tubes and add additional flow cytometry buffer to each tube as needed to meet the minimum sample volume required for the machine.
When using a high-throughput fluorescent microscope or plate-reader, it will be necessary to control for cell number in each well in order to meaningfully compare GFP signals across wells and plates. This can be done using the resazurin assay (Ahmed et al., 1994).
The percentage of eGFP+ cells using this differentiation method should be similar to that obtained through EB differentiation (Basic Protocol 1).
Reagents and Solutions
Unless otherwise noted, all solutions and media are 0.2µm-filter sterilized.
Gelatin 0.1% (w/v)
Dissolve 0.5 g of gelatin (from porcine skin) in 500ml distilled/deionized water and autoclave. Store at room temperature indefinitely.
Gelatinized plates
Dispense 0.1% gelatin solution into tissue culture plates and incubate at room temperature for 15 minutes. At the end of incubation, aspirate excess gelatin solution and let the tissue culture plate surface dry for 5 minutes. Plates may be stored for at least 1–2 months at 4°C until use.
DMEM-ES medium+
| DMEM-High Glucose GIBCO 11965-0692 | 500 mL |
| Non-Essential Amino Acids (10mM) GIBCO 11140-050 | 6.25 mL |
| L-Glutamine0^ (200mM) GIBCO 25030-081 | 6.25 mL |
| Penicillin/Streptomycin (10000U/ml, 10000µg/ml) GIBCO 15070-063 | 12.5 mL |
| Fetal Bovine Serum* GEMINI 100–106 | 94 mL |
| β-mercaptoethanol SIGMA M6250 | 4.4 µL |
| Leukemia Inhibitory Factor (107 units/mL) CHEMICON ESG1107 | 62.5 µL |
IMDM-ES medium+
Differentiation medium+
| IMDM | 500 mL |
| L-Glutamine (200mM) | 6.25 mL |
| Penicillin/Streptomycin (10000U/ml, 10000µg/ml) | 12.5 mL |
| Fetal Bovine Serum* GIBCO 10437-028 | 94 mL |
| Monothioglycerol | 6.5 µL |
| Ascorbic Acid (5mg/mL aqueous solution) SIGMA A4544 | 6.25 mL |
^L-Glutamine is very sensitive to oxidation and must be replenished every 7 days.
*FBS should be lot-tested for minimal induction of differentiation (ES media) or optimal cardiac differentiation results (differentiation media).
+Media are stable for up to 2 weeks at 4°C.
Hepes-buffered saline (HBS)
| 1 M Hepes GIBCO 15630-080 | 2.5 mL |
| 5 M NaCl | 2.68 mL |
| 1 M KCl | 0.5 mL |
| 1 M Na2HPO4 | 70.0 µl |
| ddH2O | 94.25 mL |
Adjust pH to 7.1 as necessary.
Flow cytometry buffer
Fetal bovine serum [GEMINI 100–106] is added to HBS solution to a final FBS concentration of 20%. Store at 4°C for up to 3 months.
Collagenase/DNase solution
Dissolve 50mg each of collagenase A [Roche 11088785103] and collagenase B [Roche 11088823103] in 5ml of flow cytometry buffer. Store at 4°C for up to 4 weeks. Prior to use, supplement an aliquot of the required volume with DNase [Calbiochem 260913] to a final concentration of 10µg/mL.
Commentary
Background
The self-renewal and developmental potential of pluripotent stem cells make them an attractive source for rare cell populations that arise during embryonic development, such as the earliest organ-specific progenitor populations, from which all or a large subset of cell types of a particular organ are derived. It has been demonstrated that such a population exists for the mammalian heart and is marked by positive Nkx2.5, Isl1, and/or Flk-1 expression in mice (Wu et al., 2006, Moretti et al., 2006, Kattman et al., 2006) and KDR-1 or Isl1 expression in differentiated human ES cells (Yang et al., 2008, Bu et al., 2009). These progenitor cells give rise to cardiomyocytes, smooth muscle cells (Wu et al., 2006, Bu et al., 2009), and endothelial cells (Bu et al., 2009), the three primary cell types of the heart. Although cellular markers for isolation of cardiac progenitor cells from in vivo and in vitro contexts have now been identified, the process by which a pluripotent stem cell first becomes committed to a multipotent lineage progenitor remains largely unknown. Further inquiry into these processes holds vast potential for clinical translation, particularly in the areas of congenital heart disease and regenerative therapy for myocardial injury.
Critical parameters
The developmental potential of ES or IPS cells is strongly affected by a number of different factors, the most important of which is their ability to remain uniformly undifferentiated prior to initiating differentiation. During propagation, the cells should always be maintained in fresh media and be passaged before reaching 80% confluency in the culture dish. As the passage number increases (>30), the quality of karyotype deteriorates and the differentiation capacity of ES and IPS cells declines. It is advisable to use lower-passage cells for experimentation. Cells that remain undifferentiated should exhibit small size and round shape with large nuclear-to-cytoplasmic ratio. When cultured on feeder MEF, they should aggregate into larger colonies with raised borders and refractile surfaces. They should be free of any contamination, including mycoplasma. Typical signs of poor quality cells include the appearance of large numbers of cellular fragments floating in the culture medium, the presence of vacuoles on cell surface or within each cell, and colonies with irregular borders and flattened cell morphology. If the culture media turns yellow or yellow-orange in color earlier than usual, a suspicion should be raised for the presence of mycoplasma contamination. It has been our experience that cells exhibiting these defects do not differentiate well and should be discarded.
The efficiency of differentiation into the cardiac lineage is also influenced by the particular lot of fetal bovine serum used to make differentiation medium. Several different lots should be tested, and the one that yields the highest percentage of eGFP+ cells at Days 7–10 of differentiation should be used exclusively for future experiments.
Finally, cell density at the outset of differentiation is also an important factor contributing to the yield of cardiac progenitors. For both EB and monolayer differentiation protocols, the stated number of cells per droplet or well, respectively, have been empirically determined to maximize the number of eGFP+ cells obtained. In adapting the protocols of this unit for ES or IPS lines carrying different transgenic cardiac markers, it may be necessary to adjust the cell seeding density for optimal differentiation.
Anticipated Results
The protocols outlined in this unit can be scaled up to yield large numbers (105 – 106) of Nkx2.5+ cardiac progenitors for a variety of applications directed towards investigating early cardiac development and the therapeutic potential of progenitor populations and their derivatives. Upon continued culture, Nkx2.5+ progenitor cells should spontaneously differentiate into beating cardiomyocytes and smooth muscle cells.
References
- Ahmed SA, Gogal RM, Walsh JE. A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes: an alternative to [3H]thymidine incorporation assay. J Immunol Methods. 1994;170:211–224. doi: 10.1016/0022-1759(94)90396-4. [DOI] [PubMed] [Google Scholar]
- Bu L, Jiang X, Martin-Puig S, Caron L, Zhu S, Shao Y, Roberts DJ, Huang PL, Domian IJ, Chien KR. Human Isl1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature. 2009;460:113–117. doi: 10.1038/nature08191. [DOI] [PubMed] [Google Scholar]
- Domian IJ, Chiravuri M, van der Meer P, Feinberg AW, Shi X, Ying C, Wu SM, Parker KK, Chien KR. Committed ventricular progenitors in the Islet-1 lineage expand and assemble into functional ventricular heart muscle. Science. 2009;326:426–429. doi: 10.1126/science.1177350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kattman SJ, Huber TL, Keller GM. Multipotent flk-1+ cardiovascular progenitor cells give rise to the cardiomyocyte, endothelial, and vascular smooth muscle lineages. Dev.Cell. 2006;11:723–732. doi: 10.1016/j.devcel.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Moretti A, Caron L, Nakano A, Lam JT, Bernshausen A, Chen Y, Qyang Y, Bu L, Sasaki M, Martin-Puig S, Sun Y, Evans SM, Laugwitz KL, Chien KR. Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell. 2006;127:1151–1165. doi: 10.1016/j.cell.2006.10.029. [DOI] [PubMed] [Google Scholar]
- Samuelson LC, Metzger JM. Differentiation of Embryonic Stem (ES) Cells Using the Hanging Drop Method. Cold Spring Harbor Protocols. 2006 doi: 10.1101/pdb.prot4485. doi:10.1101/pdb.prot4485. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Lord B, Schulze PC, Fryer RM, Sarang SS, Gullans SR, Lee RT. Ascorbic acid enhances differentiation of embryonic stem cells into cardiac myocytes. Circulation. 2003;107:1912–1916. doi: 10.1161/01.CIR.0000064899.53876.A3. [DOI] [PubMed] [Google Scholar]
- Tremml G, Singer M, Malavarca R. Culture of Mouse Embryonic Stem Cells. Current Protocols in Stem Cell Biology. 2008 doi: 10.1002/9780470151808.sc01c04s5. doi: 10.1002/9780470151808.sc01c04s5. [DOI] [PubMed] [Google Scholar]
- Wobus AM, Wallukat G, Hescheler J. Pluripotent mouse embryonic stem cells are able to differentiate into cardiomyocytes expressing chronotropic responses to adrenergic and cholinergic agents and Ca2+ channel blockers. Differentiation. 1991;48:173–182. doi: 10.1111/j.1432-0436.1991.tb00255.x. [DOI] [PubMed] [Google Scholar]
- Wu SM, Fujiwara Y, Cibulsky SM, Clapham DE, Lien CL, Schultheiss TM, Orkin SH. Developmental origin of a bipotential myocardial and smooth muscle cell precursor in the mammalian heart. Cell. 2006;127:1137–1150. doi: 10.1016/j.cell.2006.10.028. [DOI] [PubMed] [Google Scholar]
- Yang L, Soonpaa MH, Adler ED, Roepke TK, Kattman SJ, Kennedy M, Henckaerts E, Bonham K, Abbott GW, Linden RM, Field LJ, Keller GM. Human cardiovascular progenitor cells develop from a KDR+ embryonic stem cell-derived population. Nature. 2008;453:524–528. doi: 10.1038/nature06894. [DOI] [PubMed] [Google Scholar]