

This work uses natural genetic diversity to study species-wide connectivity between metabolites, enzymes, and biomass. The resulting network analysis, based on 129 Arabidopsis accessions, shows that biomass can be predicted by two independent integrative metabolic biomarkers: preferential investment in photosynthetic machinery and optimization of carbon use.
Abstract
Natural genetic diversity provides a powerful resource to investigate how networks respond to multiple simultaneous changes. In this work, we profile maximum catalytic activities of 37 enzymes from central metabolism and generate a matrix to investigate species-wide connectivity between metabolites, enzymes, and biomass. Most enzyme activities change in a highly coordinated manner, especially those in the Calvin-Benson cycle. Metabolites show coordinated changes in defined sectors of metabolism. Little connectivity was observed between maximum enzyme activities and metabolites, even after applying multivariate analysis methods. Measurements of posttranscriptional regulation will be required to relate these two functional levels. Individual enzyme activities correlate only weakly with biomass. However, when they are used to estimate protein abundances, and the latter are summed and expressed as a fraction of total protein, a significant positive correlation to biomass is observed. The correlation is additive to that obtained between starch and biomass. Thus, biomass is predicted by two independent integrative metabolic biomarkers: preferential investment in photosynthetic machinery and optimization of carbon use.
INTRODUCTION
The rate of plant growth depends on the rate of photosynthetic carbon (C) assimilation and on developmental programs that influence how efficiently C is converted into biomass. These processes are sometimes termed source and sink strength, respectively. The molecular and genetic determinants of these parameters are not well understood. Reverse genetics has been widely used to study the relationship between the expression individual enzymes and the rate of photosynthesis (Stitt et al., 2010b). A 2-fold reduction of ribulose-1,5-bis-phosphate carboxylase/oxygenase (Rubisco; Quick et al., 1991; Mate et al., 1993; Stitt and Schulze, 1994), sedoheptulosebisphosphatase (Harrison et al., 1998), and aldolase (Ald) (Haake et al., 1998, 1999) and a somewhat larger decrease of transketolase (TK) (Henkes et al., 2001) lead to a perceptible inhibition of photosynthesis in various species. Phosphoglycerate kinase (PGK), NADP-dependent glyceraldehyde-3-phosphate dehydrogenase (NADP-GAPDH), triose phosphate isomerase (TPI), and phosphoribulokinase can be reduced 5- to 10-fold without a perceptible impact on pathway flux (Paul et al., 1995; Price et al., 1995; Stitt, 1995; Paul and Pellny, 2003; Stitt et al., 2010a). The contributions of individual enzymes to the control of flux depend on the conditions at the moment and on the prehistory of the plant (Stitt and Schulze, 1994; Stitt and Sonnewald, 1995). A similar picture has emerged for sucrose and starch synthesis (Stitt, 1995; Stitt and Sonnewald, 1995; Paul and Foyer, 2001). Photosynthetic metabolism operates as an integrated network, in which metabolite levels and fluxes are determined by interactions between many enzymes. Analogous conclusions have been reached from experimental analyses of metabolism in bacteria, yeast, and mammals.
The relationship between photosynthetic C gain and growth is also complex (Gifford and Evans, 1981; Sparks et al., 2001). For example, inhibition of photosynthesis due to decreased expression of Rubisco leads to a strong inhibition of growth in nitrogen-replete conditions but not in nitrogen-limited conditions (Stitt and Schulze, 1994). More generally, free air CO2 elevation studies reveal that higher rates of photosynthesis rarely lead to a commensurate increase in biomass (Long et al., 2006; Rogers and Ainsworth, 2006; Leakey et al., 2009).
Natural genetic diversity provides a powerful and complementary resource to analyze complex metabolic networks (Hansen et al., 2008; Stitt et al., 2010a). Accessions or cultivars of a given species have allelic diversity for hundreds or thousands of genes, allowing a systems-level study of how metabolic networks respond to many independent perturbations. Natural genetic diversity is often investigated using inbred lines (ILs), which uncover inherent genetic diversity by resorting two genomes. ILs allow coarse mapping of quantitative trait loci (QTL), although these still represent relatively large genomic regions and cloning of the underlying genes (e.g., Fridman et al., 2000) is laborious. A complementary approach is to survey large panels of genotypes from wild populations or crop breeding programs to identify correlations between traits on a species-wide basis. Association mapping can be performed with candidate genes (Baxter et al., 2008; Sulpice et al., 2009) or genome-wide scans (Buckler and Thornsberry, 2002; Aranzana et al., 2005) to identify loci that may influence the traits, although this requires rigorous correction for population structure (Nordborg et al., 2005; Zhao et al., 2007).
There have been numerous studies of natural variation in the levels of central metabolites, such as nitrate, amino acid, starch, and sugars (Loudet et al., 2002, 2003; Calenge et al., 2006), and secondary metabolites (Kliebenstein et al., 2002; Sønderby et al., 2007; Wentzell et al., 2007). The number of metabolites investigated can be increased using metabolite profiling (Fiehn et al., 2000; Sumner et al., 2003; von Roepenack-Lahaye et al., 2004; Kopka, 2006). This has allowed hundreds of metabolite QTL to be detected in tomato (Solanum lycopersicum; Schauer et al., 2006) and Arabidopsis thaliana (Kliebenstein et al., 2002, 2006; Keurentjes et al., 2006; Lisec et al., 2008; Rowe et al., 2008) IL populations.
Using ILs populations, it has been previously shown (Schauer et al., 2006; Meyer et al., 2007) that rosette biomass in Arabidopsis and fruit yield in tomato correlate negatively with metabolites. While the correlation to rosette biomass was very weak for individual metabolites, a highly significant correlation was obtained when multivariate analysis was used on the entire metabolite profile (Meyer et al., 2007). This indicates that part of the genetic variation for biomass affects the balance between resource availability, and the developmental programs that determine how rapidly metabolites are used for growth. (Sulpice et al., 2009) used multivariate analysis to show that the starch content at the end of the day is an integrator of the metabolic response in Arabidopsis. Starch correlated strongly and negatively with rosette biomass in a population of 92 genotypically diverse accessions. Starch accumulates during the light period and is remobilized to support metabolism and growth at night. These results imply that there is genetic variation in C allocation and that this contributes to the differences in biomass between Arabidopsis accessions.
Metabolic interconversions are catalyzed by enzymes. The question arises whether the genetic variation in metabolite levels can be explained by changes in the activities of one or several enzymes. If this were the case, it would provide an attractive strategy to link the changes in metabolite levels to genetic variation at loci that encode enzymes or regulate the expression of enzymes. Enzyme activities exhibit considerable natural genetic variation. Analyses of a maize (Zea mays) IL population detected QTL for sucrose phosphate synthase (SPS) and invertase activity, which colocated with structural genes for the enzymes (Causse et al., 1995; Prioul et al., 1999; Thevenot et al., 2005). A study of six enzymes from primary metabolism and four enzymes from secondary metabolism in an Arabidopsis recombinant inbred lines (RIL) population detected several enzyme activity QTL that colocalized with structural genes (Mitchell-Olds and Pedersen, 1998). This study also found strong correlations between the activities of five glycolytic enzymes (PGI, PGM, G6PDH, FBPase, and glucose-6-phosphatase), and coarse mapping detected a trans-QTL for three of the enzymes, which did not colocate with their structural genes and might represent a joint regulator of these enzymes. A recent study of 15 enzymes from central metabolism in a Landsberg erecta (Ler) × Cvi RIL population detected a total of 15 QTL for nine of the enzymes. Some colocated with structural genes and expression QTL, while others were regulated by trans-acting loci (Keurentjes et al., 2008). However, few studies have investigated enzyme activities and metabolites from central metabolism in parallel, and those that did only investigated a small number of traits (Keurentjes et al., 2008). This is partly for technical reasons. It is still a challenge to obtain quantitative information about large numbers of proteins (Baerenfaller et al., 2008) or enzyme activities.
The relationship between enzyme activities and metabolite levels could be rather nonintuitive. As already outlined, empirical studies show that a change in the level of a single enzyme can result in complex changes of metabolites and fluxes. Theoretical considerations point to the same conclusion. In a simple linear pathway, metabolite levels will change inversely relative to flux at the step(s) where regulatory mechanisms operate to alter enzyme activities (Rolleston, 1972; Newsholme and Start, 1973). This expectation has been explicitly included in algorithms to detect sites at which genetic regulation may affect metabolism (Rowe et al., 2008). However, the relationship between metabolites and flux becomes more complicated if the pathway is regulated at multiple steps (Fell and Thomas, 1995; Hofmeyer and Cornish-Bowden, 2000) or by feedback or feedforward loops (Hofmeyr and Cornish-Bowden, 2000; Curien et al., 2003). It becomes even more complex in networks that contain branch points, cycles, and redundant reactions (Kacser and Burns, 1973; Fell and Thomas, 1995; Hofmeyr and Cornish-Bowden, 2000; Curien et al., 2003; Junker et al., 2007). Furthermore, if there is a coordinated increase of all the enzymes in a pathway, flux will increase but the levels of metabolic intermediates will remain essentially unaltered (Kacser and Acerenza, 1993). The extent of the connectivity between enzyme activities and metabolites remains an open question. It may depend on the complexity of the pathway and the extent to which genetic variation generates coordinated or noncoordinated changes of enzyme activities.
We have robotized activity assays for >20 enzymes from central metabolism in Arabidopsis leaves (Gibon et al., 2004b, 2006) and subsequently extended the platform to cover 35 enzymes from different pathways in central metabolism. The assays are optimized to measure the maximum catalytic activity (Newsholme and Start, 1973), which can be seen as a proxy for the amount of an enzyme (Piques et al., 2009; Stitt et al., 2010a). This allows us to study natural variation in enzyme activities in central metabolism in a more systematic manner than was previously possible. In the following study, we profile the maximum catalytic activities of 35 enzymes in the rosettes of 129 Arabidopsis accessions, including all 92 accessions used by Sulpice et al. (2009). Our aims are (1) to document the extent of genetic variation for activities of a large number of enzymes in central metabolism at a species-wide scale and (2) to ask whether natural genetic diversity results in coordinated or uncoordinated changes of the activities of the various enzymes. We then combine the information about enzyme activities with a data set for metabolite levels and biomass in the same material (Sulpice et al., 2009) and examine (3) how much connectivity exists between enzyme activities and metabolite levels, (4) whether enzyme activities provide predictive information about biomass, and (5) whether this prediction is redundant with or additive to the prediction provided by analyses of metabolite levels. We show that there is considerable genetic variation in enzyme activities and that the majority of the changes in enzyme activity are highly correlated with each other, whereas there is little connectivity between enzyme activities and metabolite levels. Furthermore, enzyme activities can be used to derive an integrative parameter that is significantly correlated with biomass, independent of the prediction provided by starch and which, together with starch, explains almost a third of the species-wide variation in biomass in Arabidopsis.
RESULTS
Parameters Investigated
A set of 129 Arabidopsis accessions, including all 92 accessions analyzed in Sulpice et al. (2009), was selected from a larger set of 406 accessions to maximize genotypic diversity (Sulpice et al., 2007). The accessions and their passport data are listed in Supplemental Data Set 1A online. They were grown in short-day conditions at 20°C to focus on the response during vegetative growth with limiting C and excess N and harvested at the end of the light period when internal resources like starch and amino acids have accumulated to their diurnal maxima. A list of measured metabolites and enzymes, with abbreviations and classifications into pathways, is provided in Supplemental Data Sets 1B and 1C online.
Enzyme maximum activities were measured in stopped assays, using sensitive cycling assays to measure the products (Gibon et al., 2004b). This allows assays to be performed at very high extract dilutions, which minimizes interference by other components of the extract. For all assays, it was checked that the reaction was linear with time and the amount of extract added and that substrate levels supported maximum activity of the enzyme. Maximum activities provide information about the enzymatic capacity for a given reaction and will overestimate the activity in vivo, which also depends on the levels of substrates, inhibitors, activators, and, in some cases, posttranslational modification. The maximum activities of this set of enzymes correlate well with summed protein levels for the gene family, determined by mass spectroscopy (Piques et al., 2009). Most enzyme activities are stable during the diurnal cycle, even though there are often marked diurnal changes in the levels of the encoding transcripts (Gibon et al., 2004b, 2006). The results obtained for enzyme activities from one harvest point in the diurnal cycle should therefore be representative of the activity across the complete cycle. For two enzymes, an additional assay was performed to provide information about posttranslational regulation (RubisCOin and NADPMDHin, see below).
Figure 1 shows the locations of the enzymes in metabolism. Many enzymes can be unambiguously assigned to one pathway. When an enzyme operates in two or more pathways or subcellular compartments, it is assigned to the pathway that is associated with the highest activity of the enzyme (see legend to Figure 1).
Figure 1.

Assignment of Enzymes to Pathways and Average Maximum Enzyme Activities across All the Accessions.
Rubisco and NADP-GAPDH are specific for the Calvin-Benson cycle, AGP for starch synthesis, cFBP and SPS for sucrose synthesis, PFK, PFP, NADH-GAPDH, PK, and PEPC for glycolysis, G6PDH for the oxidative pentose phosphate (OPP), and INV, SuSy, FK, and GK for sucrose breakdown and the use of hexose sugars. TPI, Ald, and PGK are involved in the Calvin-Benson cycle and glycolysis and TK in the Calvin-Benson cycle and the OPP pathway. TPI, Ald, PGK, and TK are assigned to the Calvin-Benson cycle because the major isoforms of these enzymes are located in the chloroplast stroma (Heldt, 1997) and because fluxes are 10- to 100-fold higher in photosynthesis than in glycolysis (Geiger and Servaites, 1994; Gibon et al., 2004a). UGP is involved in the synthesis and breakdown of sucrose, and PGI and PGM are involved in the synthesis and breakdown of sucrose and starch. The assays distinguished plastidic PGI (pPGI) and cytosolic (cPGI) (Gibon et al., 2004b). UGP was assigned to sucrose synthesis. Several TCA cycle enzymes have additional functions. CS and NAD- and NADP-ICDH are required to generate 2-oxoglutarate, which is the C-acceptor during nitrate and ammonium assimilation. NADH-MDH is involved in the synthesis of malate and, together with NADP-MDH, in metabolite cycles that allow redox equivalents to be exchanged between the NADP system in the plastid and the NAD systems in the cytosol and the mitochondrion (Scheibe, 2004). The maximum catalytic activities of each enzyme, averaged across all accessions, are shown by color coding (see figure for scale). All abbreviations are defined in Supplemental Data Set 1C online. The subscript “in” for Rubisco and NADP-MDH indicates “initial activity.”
Genetic Variation of the Traits Analyzed
Average, minimum, and maximum values and the coefficient of variation (CV) were calculated for each parameter and accession (see Supplemental Figure 1A and Supplemental Data Set 1D online). The CV was small for structural components (chlorophyll and protein) and major central metabolites (total amino acids, sucrose, and starch), larger for rosette fresh weight (FW), reducing sugars, and the major organic acids (malate and fumarate) (20 to 30%), and highest for low molecular weight metabolites determined by gas chromatography–mass spectrometry (GC-MS) (average 45%). Some enzyme activities showed low (TPI, PGM, and Rubisco), many showed intermediate, and some showed higher (GK, FK, G6PDH, INV, NAD-ICDH, and NADP-ICDH) CV values, although not as high as the CV for some of the small metabolites. These results resemble those in (Cross et al., 2006) but cover many more accessions and metabolic parameters.
Due to the large number of plants assayed and the parameters measured, the whole set of accessions was partitioned into eight subsets (called experiments). Each subset of accessions was grown in the same conditions in growth cabinets. Each accession was analyzed in two to four different experiments, and analyses of sugars and FW in the reference accession Columbia-0 (Col-0) were included in all experiments. Two-way analysis of variance (ANOVA) was performed to estimate how much of the variation is determined by the genotype (G) or experimental (E) fluctuations. The F-statistic for the genotype effect and the variances explained by the genotype are given in Supplemental Data Set 1D online. For almost all traits, the genotype effect was highly significant. Due to missing data, a proper model could not be fitted for three traits (salicylate, guanidine, and nicotinate; see Supplemental Data Set 1D online).
Averaged across all traits, 38% of the variation was genetically determined. Genetic variation for enzyme activities ranged from 20% (Rubisco) up to 57% (acid invertase), with an average of 34%. This resembles the range found in earlier studies with smaller sets of enzymes (Mitchell-Olds and Pedersen, 1998; Cross et al., 2006; Keurentjes et al., 2008). Metabolites determined by GC-MS showed higher genetic heritability (average 48%). This is comparable to or higher than in other studies (Rowe et al., 2008; Schauer et al., 2008). Relatively low values were observed for protein (21%), chlorophyll b (20%), chlorophyll a (28%), and starch (21%) (see also Cross et al., 2006). Genetic variance was highly significant for most traits; exceptions are some metabolites and several enzyme activities (Rubisco, RubisCOin, Ald, PGK, CS, and NADP-MDHin).
Absolute Enzyme Activities
We first asked if enzymes in a given pathway have similar maximum activities and if these differ from those of enzymes in other pathways. The heat map in Figure 1 displays the average activity for each enzyme across all the accessions. Calvin-Benson cycle enzymes have high activities (between 6727 and 15,6000 nmol/min gFW). Especially high activities were found for TPI and PGK, while the other activities were in the range 6727 to 10,852 nmol/min gFW. Intermediate activities are found for enzymes involved in starch (700 to 1190 nmol/min gFW), sucrose (111 to 4094 nmol/min gFW), and amino acid (1148 to 6236 nmol/min gFW) synthesis. Enzymes involved in the sucrose degradation have low activities (112 to 1223 nmol/min gFW), as do most dedicated enzymes in glycolysis (PFK, PFP, PK, and PEPC) and the tricarboxylic acid (TCA) cycle (CS, aconitase, NAD-ICDH, and NADP-ICDH). The activities of NADH-GAPDH, NAD-MDH, and fumarase are comparable to those of the Calvin-Benson cycle enzymes, indicating they might have other functions, in addition to catalyzing fluxes in glycolysis and the TCA cycle.
Correlation Matrices
A correlation matrix was generated by performing Spearman rank correlation analysis for all pairs of measured traits across the whole population. This analysis used average values calculated from all raw determinations for a given trait/accession pair, without considering the experiment effect. Spearman’s rank correlation coefficients (Rs) and accompanying false discovery rate (FDR)–corrected P values (pBH; Benjamini-Hochberg) are provided in Supplemental Data Sets 2A and 2B online. The FDR corrected P values are summarized as a heat map in Figure 2A and Supplemental Figure 1B online for the matrix obtained when traits were expressed on a FW and a total protein basis, respectively. The traits are organized into seven classes: biomass, large structural components, carbohydrates, organic acids, amino acids, secondary metabolites, and enzyme activities. Derived traits (reducing sugars, total sugars, and total amino acids) and the initial activities of Rubisco and NADP-MDH were excluded from the matrices.
Figure 2.

Connectivities between Metabolic Traits Expressed on a Fresh Weight Basis.
(A) Correlation matrices based on Spearman correlation coefficients between structural components, metabolites, and enzyme activities in the accessions. Spearman rank correlation analysis was performed on the mean values for all pairs of measured traits across the whole population. Relationships that are significant at pBH < 0.001, pBH < 0.01, and pBH < 0.05 are indicated by dark, medium, and light shading with positive and negative correlations being distinguished by blue and orange, respectively. Data used for every trait analyzed are based on at least two independent experiments. The original data are given in Supplemental Data Set 1E online. For the display, the traits were organized into seven classes: 1, biomass; 2, structural components; 3, carbohydrates; 4, organic acids; 5, amino acids; 6, miscellaneous metabolites; 7, enzyme activities. The data are expressed on a FW basis. A similar picture emerged when they were expressed on a protein basis (see Supplemental Figure 1B online)
(B) Cartographic representation of the primary metabolism network of Arabidopsis. All trait-trait correlations that were significant at pBH < 0.01 were visualized using an algorithm that identifies functional modules within complex networks (Guimera and Amaral, 2005; Guimera et al., 2005). Blue and red lines signify positive and negative correlations.
The matrices display five general features. First, there are many more positive than negative correlations. On a FW basis, there were 361 positive and eight negative correlations (from a total of 5356 trait pairs) at a significance threshold of pBH < 0.001 (Table 1). When traits were expressed on a protein basis, the corresponding numbers were 161 positive and 0 negative correlations. Second, most of the positive correlations are between traits from the same major trait class (85% at a threshold of pBH < 0.001). Third, all of the negative correlations are between traits from different classes. Fourth, the same pattern emerges, irrespective of whether traits are expressed on a FW (Figure 2A) or a protein (see Supplemental Figure 1B online) basis. In particular, the high frequency of positive correlations between enzyme activities is retained when the activities are expressed on a protein basis. This excludes a potentially trivial explanation for the coordinated changes of enzyme activities, which would be that they are due to a change in the overall protein content. Fifth, as previously seen (Meyer et al., 2007; Sulpice et al., 2009), many metabolites correlate negatively with biomass, especially when they are expressed on a FW basis. Some enzymes show a positive correlation with biomass, a feature that becomes especially marked when the activities are expressed on a protein basis.
Table 1.
Number of Correlations between Different Types of Metabolic Parameters
| Number of Correlationsa |
||||
| Parameter Pair | Positive | Negative | Total No. of Pairs | % with Significant Correlation |
| All | 161 | 1 | 5356 | 3.0 |
| Enzyme-enzyme | 66 | 0 | 666 | 9.9 |
| Metabolite-metabolite | 70 | 0 | 1891 | 3.7 |
| Amino acid–amino acid | 40 | 0 | 276 | 14.5 |
| Enzyme-metabolite | 3 | 0 | 2294 | 0.1 |
Derived metabolic traits (reducing sugars, total sugars, and total amino acids) were excluded, as were initial activities of Rubisco and NADP-MDH.
pBH < 0.001.
Global Network
A complementary overview of the enzyme-metabolite-biomass network was obtained by extracting all significant trait-trait correlations (pBH < 0.01) and visualizing them using an algorithm that identifies functional modules in complex networks (Guimera and Amaral, 2005; Guimera et al., 2005). The resulting network (Figure 2B) contained two large, strongly connected modules and three smaller, more weakly connected modules. It captures many of the features identified by stepwise analysis of the correlation matrix.
Most of the enzymes are in the largest and most interconnected module. The Calvin-Benson cycle enzymes and many of the enzymes from biosynthetic pathways are tightly interconnected, while others are linked by only a few edges (NR, G6PDH, and enzymes for sucrose degradation). This module contains chlorophyll, but no low molecular weight metabolites. The second large module contains only metabolites. The 26 metabolites in this module include 15 amino acids, 2-oxoglutarate, and some secondary metabolites like sinapate and putrescine, which are synthesized from amino acids. One of the small loosely interconnected modules contained a set of metabolites (several amino acids, organic acids, carbohydrates, ascorbate, and dehydroascorbate) and three enzymes (GLDH, NAD-ICDH, and CS). A second loosely connected module contains five metabolites (including malate and fumarate) and two enzymes (SPS and NADP-MDHin). A third loosely connected module contains salicylate, starch and sucrose, glucose and fructose, total amino acids, three individual amino acids (Ala, Gly, and homoserine), total protein, and rosette FW. Some metabolites in this module (total amino acids, starch, and sucrose) are connected to chlorophyll and individual enzymes in the large enzyme module.
Coordinated Changes of Enzyme Activities
The following sections explore the correlation matrix in more detail, starting with enzyme activities. Key information is summarized in Table 2. RubisCOin and NADP-MDHin (the subscript “in” indicates “initial activity”) were excluded from this analysis because they provide information about enzyme regulation, rather than the amount of enzyme (see below). Ald was excluded because measurements were available for only 58 of the 129 accessions.
Table 2.
Connectivities of Enzyme Activities
| No. of Correlations to Other Enzymes |
||||||||
| Enzymes Individual Enzymes | Rs, Enzyme Activity versus Total Protein | pBH < 0.01 | pBH < 0.05 | Specific Activitya | Average Maximum Activity | Average Amount as % of Total Protein | Rs (% Protein in Enzyme versus % Protein in All-Rubisco) | Rs (% Protein in Enzyme versus % Protein in All Enzymes) |
| Rubisco | 0.32 | 13 | 18 | 1,600 | 8,268 | 28.38 | 0.491 | 0.964 |
| PGK | 0.24 | 10 | 16 | 983,684 | 77,735 | 0.45 | 0.548 | 0.405 |
| NADP-GAPDH | 0.16 | 15 | 19 | 80,000 | 10,852 | 0.78 | 0.703 | 0.453 |
| TPI | 0.40 | 17 | 23 | 6,858,936 | 155,855 | 0.13 | 0.663 | 0.500 |
| TK | 0.16 | 7 | 16 | 22,000 | 8,402 | 2.08 | 0.698 | 0.436 |
| Glycerate kinase | 0.27 | 13 | 19 | 54,600 | 2,607 | 0.26 | 0.474 | 0.339 |
| NADP-MDH | 0.20 | 16 | 19 | 32,527 | 1,159 | 0.19 | 0.498 | 0.336 |
| cFBP* | 0.21 | 0 | 2 | 44,194 | 111 | 0.00 | 0.184 | 0.220 |
| cPGI | 0.25 | 6 | 17 | 495,000 | 1,836 | 0.00 | 0.442 | 0.392 |
| PGM | 0.41 | 8 | 16 | 82,500 | 4,042 | 0.26 | 0.415 | 0.489 |
| UGP* | 0.20 | 3 | 12 | 435,058 | 4,095 | 0.06 | 0.386 | 0.376 |
| SPS* | −0.06 | 4 | 9 | 44,750 | 510 | 0.06 | 0.366 | 0.317 |
| AGP | 0.24 | 12 | 24 | 39,175 | 700 | 0.13 | 0.454 | 0.373 |
| pPGI | 0.33 | 9 | 18 | 495,000 | 1,192 | 0.00 | 0.423 | 0.325 |
| PFK | 0.11 | 3 | 10 | 219,380 | 176 | 0.00 | 0.312 | 0.133 |
| PFP | 0.09 | 1 | 4 | 23,150 | 161 | 0.06 | 0.363 | 0.212 |
| NAD-GAPDH | 0.17 | 9 | 17 | 105,000 | 16,756 | 0.91 | 0.672 | 0.498 |
| PK | 0.25 | 12 | 18 | 88,388 | 1,006 | 0.06 | 0.440 | 0.190 |
| PEPC | −0.03 | 10 | 17 | 30,653 | 744 | 0.13 | 0.549 | 0.401 |
| NAD-MDH | 0.28 | 18 | 24 | 800,000 | 53,492 | 0.39 | 0.639 | 0.486 |
| Fumarase | 0.00 | 2 | 3 | 293,012 | 4,423 | 0.06 | 0.226 | 0.114 |
| NADP-ICDH | 0.17 | 6 | 12 | 24,254 | 1,466 | 0.32 | 0.141 | 0.133 |
| CS | 0.01 | 2 | 3 | n.a. | 190 | n.a. | n.a. | n.a. |
| NAD-ICDH* | 0.07 | 3 | 6 | 11,000 | 55 | 0.00 | 0.225 | 0.025 |
| NR* | 0.13 | 1 | 1 | 55,000 | 409 | 0.06 | 0.160 | 0.222 |
| GS | 0.20 | 9 | 16 | 22,627 | 1,148 | 0.26 | 0.476 | 0.292 |
| GOGAT | 0.20 | 0 | 0 | 40,800 | 1,803 | 0.26 | 0.099 | 0.195 |
| AlaAT | 0.24 | 11 | 18 | 45,573 | 5,942 | 0.71 | 0.601 | 0.441 |
| AspAT | 0.37 | 11 | 16 | 420,000 | 6,236 | 0.06 | 0.461 | 0.246 |
| Shikimate DH | 0.23 | 12 | 17 | 86,000 | 600 | 0.06 | 0.480 | 0.430 |
| GLDH | −0.01 | 4 | 14 | 62,680 | 576 | 0.06 | 0.483 | 0.210 |
| G6PDH* | 0.04 | 0 | 2 | 62,075 | 268 | 0.00 | 0.243 | 0.130 |
| Susy* | 0.05 | 0 | 3 | 180,000 | 122 | 0.00 | 0.194 | 0.190 |
| INV | −0.04 | 0 | 1 | 115,566 | 1,223 | 0.06 | 0.227 | 0.260 |
| FK | 0.09 | 4 | 9 | 10,500 | 117 | 0.06 | 0.270 | 0.247 |
| GK | 0.13 | 3 | 7 | 43,240 | 112 | 0.00 | 0.262 | 0.326 |
| Grouped in pathways | 0.00 | |||||||
| All enzymes Rubisco | 6.8 | 12.2 | 7.99 | 1.000 | 0.672 | |||
| All enzymes | 7.0 | 12.3 | 36.36 | 0.672 | 1.000 | |||
| Calvin-Benson cycle | 13.7 | 18.7 | 31.82 | 0.606 | 0.991 | |||
| Calvin-Benson cycle minus Rubisco | 13.2 | 18.2 | 3.44 | 0.870 | 0.553 | |||
| Sucrose synthesis | 6.0 | 13.0 | 0.39 | 0.583 | 0.586 | |||
| Starch synthesis | 12.5 | 23.0 | 0.13 | 0.493 | 0.405 | |||
| Respiration | 9 | 13.6 | 1.95 | 0.805 | 0.580 | |||
| Amino acid metabolism | 11.3 | 16.3 | 1.43 | 0.645 | 0.487 | |||
| Sucrose breakdown | 6.3 | 9.5 | 0.13 | 0.364 | 0.367 | |||
The Rs values for correlations between enzyme activity (nmol/min·gFW) and protein content (mg/gFW) and the number of correlations between enzyme pairs are from Supplemental Data Set 2A online. The measured maximum enzyme activities (nmol/min·g FW; from Supplemental Data Set 1E online) were divided by literature values for specific activities (a; nmol/min.mg protein; see Piques et al., 2009 for sources) to estimate the amount of protein invested in each enzyme (mg protein/gFW). The average values for a given enzyme across all accessions are shown. The individual values for each enzyme were summed in each accession and used to calculate the total amount of protein invested in enzymes in that accession. The Rs values are given for the regression plot between the amount of protein in a given enzyme and the summed estimate of protein in all enzymes and for the corresponding plot but omitting Rubisco from the sum of protein. Rs values are coded according to significance (italic and bold for pBH < 0.05 and 0.01, respectively). *enzyme determined in 62 to 92 accessions over 129. n.a., not available.
In nmol/min·mg−1 purified enzyme.
As already noted, many enzymes change in a coordinated manner. This is quantified in Table 2. When activities were expressed on a FW basis, each enzyme had an average of 7.0 and 12.3 (from a maximum of 35) significant correlations with other enzymes at pBH < 0.01 and pBH < 0.05, respectively. A similar picture emerged when activities were expressed on a protein basis; each enzyme had an average of 10 and 16 significant correlations with other enzymes at pBH < 0.01 and pBH < 0.05, respectively. As already noted, this excludes the trivial explanation that the coordinated response is due to changes in total protein.
Comparison of Enzyme Activities and Total Protein
We next explored the relationship between the variation in the activities of individual enzymes and the variation in total leaf protein (leftmost column, Table 2). All parameters are expressed on a FW basis. A Spearman coefficient of regression (Rs) of 0.32 was obtained between Rubisco and protein. The very low Kcat (3 s−1) of Rubisco is compensated for by having a high concentration of Rubisco protein (typically 30 to 40% of total leaf protein; Farquhar et al., 2001; Zhu et al., 2007). It can therefore be expected that changes in Rubisco will result in changes in the total leaf protein content. Chlorophyll binding proteins and other proteins in the thylakoid membranes represent another major component of total leaf protein (Adam et al., 2000). Chlorophyll provides indirect information about the changes of these proteins. Chlorophyll correlated strongly with total protein (Rs = 0.53; see Supplemental Data Set 2A online). Of the other four Calvin-Benson cycle enzymes investigated, TPI had an Rs value of 0.4, PGK of 0.2, and TK and NADP-GAPDH of 0.16 with total protein. Most enzymes involved in sucrose and starch synthesis and nitrogen assimilation also had Rs values of 0.2 to 0.4 with total protein. The changes of total leaf protein between accessions are therefore partly attributable to coordinated changes of Rubisco, other Calvin-Benson cycle enzymes, and enzymes that are required to synthesize the major end products of photosynthesis. Two exceptions were NR (Rs = 0.13) and SPS (Rs = −0.06). Interestingly, these are key regulatory sites in sucrose synthesis and nitrate assimilation (Stitt et al., 1987; Winter and Huber, 2000).
A different picture emerged for glycolysis, the TCA cycle, and sucrose degradation. While Rs values with protein of 0.2 to 0.4 were obtained for NAD-GAPDH and PK, many of the enzymes in these pathways had relatively low Rs values, and some had very low Rs values (PEPC, GLDH, INV, FK, G6PDH, Fumarase, PFP, and Susy), showing that they vary independently of the total leaf protein.
Correlations between Enzymes
We next investigated which enzymes show the highest connectivity to other enzymes. The second column from the left in Table 2 summarizes the number of significant (pBH < 0.01) positive correlations for each individual enzyme. As already noted, at this significance level, on average each enzyme correlates with 7.0 of the other 35 enzymes. The most highly connected enzyme was NAD-MDH, followed by TPI and NADP-MDH. Most enzymes from the Calvin-Benson cycle, starch and sucrose synthesis, nitrogen metabolism, and PEPC, PK, and NADP-ICDH showed above-average connectivity. Poorly connected enzymes included GOGAT, G6PDH, Susy, and INV (no significant correlations), NR and PFP (only one significant correlation), and fumarase (two significant correlations). A qualitatively similar picture emerged at the less stringent filter of P < 0.05 (Table 2, third column from left). Connections between weakly linked enzymes may nevertheless be functionally important. For example, during nitrate assimilation, PEPC is required to synthesize malate as a counteranion (Heldt, 1997). Of the two enzymes that correlate with fumarase, one (NAD-MDH) catalyzes an adjacent reaction in the TCA cycle.
We next investigated whether enzymes are more highly connected to enzymes in the same pathway than to enzymes in other pathways. The initial classification defined 10 pathways (Figure 1; see Supplemental Data Set 1C online), but some were represented by only one to two enzymes. We decreased this to five sets by retaining the pathway sets for the Calvin-Benson cycle, sucrose biosynthesis, and sucrose breakdown, extending nitrogen assimilation and amino acid metabolism to include glycerate kinase, and combining G6PDH, glycolysis, and the TCA cycle as respiration (see Supplemental Data Set 1C online).
We calculated the mean values of Rs for pairwise correlations between enzymes within each pathway and between enzymes in one pathway and each of the other pathways (Figure 3). Calvin-Benson cycle enzymes showed the most highly coordinated changes (i.e., accessions that had high activity of one Calvin-Benson cycle enzyme tended to have high activities of the other Calvin-Benson cycle enzymes). Other pathways showed a less coordinated within-pathway response. Indeed, enzymes assigned to sucrose synthesis, amino acid metabolism, and respiration correlated as well (sucrose synthesis and respiration) or better (amino acid metabolism) with Calvin-Benson cycle enzymes than with enzymes from their own pathway (Figure 3; for details, see Supplemental Data Set 2A online). This may reflect the fact that fluxes through these pathways are dependent on C-fixation in the Calvin-Benson cycle (see below for discussion). The matrix in Supplemental Data Set 2A online reveals that the enzymes for starch synthesis (AGP and PGM) also correlate with Calvin-Benson cycle enzymes. Enzymes involved in sucrose breakdown show a disparate response (Figure 3), both with each other and with enzymes from other pathways. INV and SuSy change independently of each other and of almost all other enzymes analyzed in our study. GK and FK correlate with each other (pBH = 3.48E-05) but change independently of INV and SuSy (see Supplemental Data Set 2A online).
Figure 3.

Correlation Matrix Showing Similarities and Differences in the Response at the Level of Metabolic Pathways.
Information about the enzymes and their pathway assignments is given in Supplemental Data Set 1C online. Some pathways (starch synthesis) were excluded from the analysis because too few enzymes were determined. Enzymes from TCA cycle, glycolysis, and OPP were grouped as “respiration.” Spearman correlation coefficients (Rs) were calculated for each enzyme-enzyme pair. Average values of these coefficients for enzyme-enzyme pairs belonging to the same pathway or to different pathways were calculated. The values range from 0.32 (dark blue) to 0.08 (light blue).
Some coordinated responses span pathways. Supplemental Figure 2 online shows two examples. One is a correlation between NADP-MDH, NAD-MDH, NADP-ICDH, TPI, and NAD-GAPDH. These five enzymes are in the large enzyme module of Supplemental Figure 1B online. They are involved in metabolite shuttles that allow redox transfer between different intracellular compartments (see Discussion). A second example is CS, NAD-ICDH, and GLDH. These three enzymes grouped in one of the small modules in Supplemental Figure 1B online. They are involved in organic acid metabolism: CS and NAD-ICDH in the TCA cycle and GLDH in amino acid catabolism, where it generates 2-oxoglutarate.
The average Rs values for the various pathways are <0.5 (Figure 3). This could still allow marked divergence between accessions with respect to the relative levels of enzymes in a pathway. To explore how large this variation is, we calculated the activity ratio for each enzyme-enzyme pair for each pathway subset (Calvin-Benson cycle, sucrose synthesis, nitrogen metabolism, sucrose degradation, and respiration), in each accession, combined the values, and depicted them as a frequency plot (Figure 4; see Supplemental Figure 3 online). For Calvin-Benson cycle enzymes, 96% of the ratios are between 0.7 and 1.4 (Figure 4A). Thus, there are very few cases where the activities of two Calvin-Benson cycle enzymes change >2-fold relative to each other in this large set of accessions. The statistical significance of this result was checked by showing that there is a far larger range of values in a shuffled data set (see Supplemental Figure 3 online). An analogous analysis of the other pathways showed a larger spread of values for pairwise ratios between the constituent enzymes. Nevertheless, shuffling revealed significant coordination in all pathways, except sucrose breakdown where ~25% of the normalized pairwise ratios change >2-fold and some by up to 10-fold (Figure 4B).
Figure 4.

Frequency Histogram of the Enzyme-Enzyme Ratios for Enzymes from Different Pathways across the Entire Set of Accessions.
The enzymes were assigned to five subsets: Calvin-Benson cycle enzymes, sucrose synthesis, nitrogen metabolism, enzymes of sucrose degradation, and respiration. For each enzyme, the activity in a given accession was normalized to the mean activity of that enzyme in all of the accessions. This allowed activity ratios in the same range to be obtained for each enzyme pair; a value of 1 thus represents the average value across all the accessions. The activity ratio was then calculated for each enzyme-enzyme pair from each set in each accession. The values were then summed and depicted as a frequency plot. Significance of the plots obtained was addressed by shuffling 1000 times the normalized activities and comparing the frequency distribution of the standard deviations of the ratios against the standard deviation of the ratios obtained with the real data (see Supplemental Figure 3 online).
(A) Calvin-Benson cycle (Rubisco, PGK, NADP-GAPDH, TPI, and TK).
(B) Sucrose degradation (INV, SuSy, GK, and FK).
(C) Rubisco compared with all other enzymes (white bars) and selected Calvin-Benson cycle enzymes (gray bars, PGK, NADP-GAPDH, TPI, and TK).
(D) NR activity compared with the activity of all other enzymes.
Figures 4C and 4D show the frequency distribution for pairwise activity ratios for two selected enzymes. Rubisco shows only small deviations compared other enzymes. For comparison, the frequency distribution is shown for ratios between Rubisco and four Calvin-Benson cycle enzymes (PGK, NADP-GAPDH, TPI, and TK). A similar frequency distribution is obtained for these four enzymes and for the entire set of enzymes. This emphasizes that Rubisco correlates not only with Calvin-Benson cycle enzymes but also with many other enzymes in central metabolism. NR shows a larger spread of ratios, with almost 20% of the values lying <0.7 or >1.4. The spread of ratios was nevertheless smaller than in shuffled data sets (see Supplemental Figure 3 online).
Correlations between Metabolites
Metabolites are less connected to each other than are enzymes. From a maximum possible number of 61, metabolites have on average 4.4 significant (pBH < 0.01) correlations to other metabolites (Table 1). The lower connectivity is not due to noise. Genetic variance actually explains a larger proportion of the total variation in metabolite levels than in enzyme activities (see Supplemental Data Set 1D online).
Metabolites correlate most frequently with metabolites from the same sector of metabolism. This is most marked for amino acids, with on average 5.5 correlations per amino acid (pBH < 0.01), from a maximum possible number of 23 (Table 1). There are strong correlations between central amino acids like Gln, Glu, Asp, and Asn (see Supplemental Data Set 2A online) and some minor amino acids (Arg and Met). There is a strong correlation between the three aliphatic amino acids (Leu, Ileu, and Val), between β-Ala and 4-aminobutyrate, and between the three aromatic amino acids. Gln, Glu, Asp, and several minor amino acids correlate with 2-oxoglutarate (Table 3), which is the C-acceptor in the GOGAT pathway (see Discussion), and with putrescine and sinapate. These metabolites are in the same module in Figure 2B. Interestingly, Gly, which is formed during photorespiration, is poorly connected with other amino acids. Other examples of metabolite-metabolite correlations include sucrose and starch (Rs = +0.42; PBH = 4.88E-05), glucose and fructose (Rs = +0.59; P = 1.15E-10), raffinose and its precursors (galactinol and myoinositol), ascorbate and dehydroascorbate (pBH = 1.1E-04), and between the latter and threonic acid, which is formed during breakdown of ascorbate (Debolt et al., 2007).
Table 3.
Correlations of Organic Acids with Amino Acids
| Citrate | 2-OG | Succinate | Fum | Malate | Pyruvate | Glycerate | |
| AA | 0.794 | 0.066 | 0.777 | −0.966 | 0.673 | 0.525 | 0.353 |
| Gly | 0.630 | 0.791 | 0.915 | −0.311 | −0.279 | 0.452 | 0.798 |
| l-Ala | 0.666 | 0.429 | 0.254 | 0.526 | −0.935 | 0.085 | −0.780 |
| Ser | −0.278 | 0.134 | −0.848 | 0.374 | −0.985 | 0.417 | 0.005 |
| Glu | −0.121 | 0.000 | 0.781 | 0.083 | 0.979 | 0.649 | 0.377 |
| Gln | −0.878 | 0.000 | −0.571 | −0.635 | −0.575 | 0.525 | 0.062 |
| Asp | 0.875 | 0.001 | −0.777 | 0.049 | 0.890 | 0.734 | 0.016 |
| Asn | -0.025 | 0.000 | −0.048 | 0.375 | 0.886 | 0.832 | 0.865 |
| Arg | −0.178 | 0.000 | −0.124 | −0.668 | 0.608 | 0.770 | 0.359 |
| Pro | −0.986 | 0.735 | −0.889 | 0.215 | 0.928 | 0.744 | 0.007 |
| Ile | −0.192 | 0.004 | 0.102 | 0.043 | 0.688 | 0.967 | 0.749 |
| Leu | −0.687 | 0.207 | 0.307 | 0.250 | 0.852 | 0.516 | 0.997 |
| Val | −0.241 | 0.005 | 0.436 | 0.056 | 0.918 | 0.147 | 0.511 |
| Lys | −0.236 | 0.023 | −0.082 | −0.574 | −0.472 | 0.858 | −0.923 |
| Phe | 0.336 | 0.054 | 0.356 | 0.989 | −0.053 | 0.740 | 0.194 |
| Thr | −0.928 | 0.347 | 0.773 | −0.842 | −0.062 | 0.176 | 0.014 |
| Trp | −0.256 | 0.068 | −0.753 | 0.728 | −0.525 | 0.302 | 0.359 |
| Tyr | −0.332 | 0.000 | −0.875 | 0.709 | −0.720 | 0.611 | 0.533 |
| Protein | 0.806 | 0.453 | 0.307 | −0.842 | 0.890 | 0.955 | 0.586 |
The results are extracted from Supplemental Data Set 2A online. The table shows the FDR-corrected (Benjamini-Hochberg) P values of the Spearman rank correlation. Significant P values are given in italics, and P values below 0.001 are presented in bold. A negative sign indicates when R had a negative value.
Relationships between Enzyme Activities and Metabolite Levels
There are very few correlations between enzyme activities and metabolites. The matrix contains 62 metabolites and 37 enzymes, allowing 2294 pair-pair correlations between enzymes and metabolites. There are only three and one (pBH < 0.001) significant correlations when the traits are compared on a FW (Table 1) and protein (see Supplemental Data Set 2B online) basis, respectively.
Figure 5 lists the enzymes and metabolites with the largest number of cross-trait class correlations. Among the enzymes, PGM and SPS showed the largest number of correlations with metabolites. Rubisco was correlated with three major C-containing products of photosynthesis (sucrose, starch, and malate). Among the metabolites, myo-inositol, β-Ala, Lys, and mannose had the largest number of significant correlations to enzymes.
Figure 5.

Correlation between Enzyme Activities and Metabolites.
The display shows selected examples of significant Spearman correlation coefficients between enzyme activities and metabolites. The number indicates the R value and the color shading the P value (light, medium, and dark shading represent pBH <0.05, <0.01, and <0.001, respectively). A full list of all correlation coefficients is provided in Supplemental Data Set 2A online. Blue colors are for positive correlations. The yellow shading is used for a negative correlation with pBH < 0.01.
A trivial explanation for the lack of connectivity would be that many of the enzymes and metabolites come from different pathways. To test this possibility, we generated a series of smaller within-pathway matrixes, which contained enzymes and metabolites from the same metabolic sector (starch synthesis, sucrose metabolism, hexose metabolism, nitrogen assimilation, amino transferase reactions, aromatic amino acid biosynthesis, malate, and fumarate metabolism). We also searched for correlations between Calvin-Benson cycle enzymes and the major carbohydrate products of CO2 fixation (starch and sugars). Table 4 summarizes significant Rs values between enzymes and metabolites in these small matrices, using a relaxed P value (pBH < 0.01). The frequency of correlations was no higher than in the entire data matrix.
Table 4.
Frequency of Correlations between Enzymes and Metabolites in the Same Metabolic Sequence
| Total Matrix |
Defined Metabolic Segment |
|||||
| Number of Significant Correlations | Total No. of Metabolite-Enzyme Comparisons | % of Significant Correlations | No. of Correlations between Enzymes and Metabolites | No. of Possible Enzyme-Metabolite Pairs | % of Significant Correlations | |
| Calvin-Benson cycle and carbohydrates | 1 | 520 | 0.2 | 0 | 24 | 0 |
| Starch synthesis | 0 | 161 | 0 | 0 | 2 | 0 |
| Sucrose metabolism | 3 | 347 | 0.9 | 0 | 5 | 0 |
| Hexose metabolism | 0 | 297 | 0 | 0 | 9 | 0 |
| Nitrogen assimilation | 1 | 421 | 0.2 | 0 | 15 | 0 |
| Aminotransferases | 0 | 309 | 0 | 0 | 10 | 0 |
| Aromatic amino acid synthesis | 1 | 272 | 0.4 | 0 | 8 | 0 |
| Malate and fumarate metabolism | 1 | 260 | 0.4 | 0 | 6 | 0 |
Several sets of enzymes and metabolites were identified in which the metabolites are the immediate or near-immediate substrates or products of enzymes: starch synthesis (pPGI, AGP, and starch), sucrose metabolism (cPGI, UGP, SPS, SuSy, INV, and sucrose), hexose metabolism (INV, GK, FK, sucrose, glucose, and fructose), nitrogen assimilation (NR, GS, GOGAT, NAD-IDH, NADP-ICDH and Gln, and 2-oxoglutarate and Glu), central amino transferase reactions (AspAT, AlaAT and Glu, 2-oxoglutarate, pyruvate, Asp, and Ala), aromatic amino acid synthesis (TK, shikimateDH, shikimate, Phe, Trp, and Tyr), and malate and fumarate metabolism (NAD-MDH, NADP-MDH, fumarase, malate, and fumarate). The six Calvin-Benson cycle enzymes were also compared with starch, sucrose, glucose, and fructose. For each set identified, all significant (pBH < 0.01) correlations were between enzyme-metabolite pairs in the trait set. This is compared with the number of correlations that these traits have in the data matrix without the initial activities.
Another explanation for the lack of connectivity between metabolites and single enzymes would be that the level of a metabolite may be determined by several enzymes. Multivariate analysis like partial least squares (PLS) regression can be used to predict one trait from a linear combination of other traits (termed predictors) (Wold, 1975; Barker and Rayens, 2003). PLS identifies the combination of the original predictors (e.g., metabolites and enzymes) that has maximum covariance with the trait of interest. We tested, for each metabolite, whether PLS regression against the entire set of enzyme activities would provide a better fit than regression against single enzymes. We did not find an improvement in any case.
It is also possible that groups of enzymes interact in a nonlinear manner to determine the level of a given metabolite. To investigate this possibility, we performed support vector machine regression using a nonlinear kernel (Fan et al., 2005). We found only one metabolite whose level was significantly predicted by a group of enzymes (glutamate; R = 0.42, P value corrected = 3E-05). Multiple repetitions of the analysis, leaving out one enzyme at a time, indicated that GluDH was the only enzyme with a major effect on Glu levels.
Relationships between Enzyme Activation State and Metabolite Levels
Optimized in vitro measurements provide information about the maximum catalytic capacity. In vivo activity also depends on the concentrations of substrates and regulatory effectors and, in some cases, posttranslational modification. Two of the enzymes in our platform are subject to a posttranslational modification that can be monitored by appropriate sample handling and assay procedures. Rubisco is posttranslationally activated by carbamylation of Lys-201. The inactive and active form can be distinguished by assaying activity immediately or after incubation with high bicarbonate concentrations in alkaline conditions to convert the decarbamylated into the carbamylated form (Portis, 2003). NADP-MDH is reversibly reduced and activated by thioredoxin-m. Assay of maximum activity in vitro requires pretreatment with DTT, whereas rapid assay in the absence of DTT detects only the reduced form (Scheibe, 2004). These two enzymes were assayed without pretreatment to monitor the amount of posttranslationally activated enzyme (RubisCOin and NADP-MDHin). This revealed further biologically relevant correlations: NADP-MDHin was negatively correlated with malate (Rs = −0.38, pBH = ×0.005), and RubisCOin correlated positively with NR activity (Rs = +0.35, pBH = 0.01) (see Supplemental Data Set 2A online).
Relationships between Enzyme Activities and Rosette Biomass
Several enzyme activities correlate weakly with rosette FW, especially when they are expressed on a protein basis (see Supplemental Figure 1B online). Using an analogous approach to that taken previously for metabolites (Meyer et al., 2007; Sulpice et al., 2009), we investigated whether a stronger regression can be obtained using the 35 enzyme activities as predictors in a PLS regression on biomass. In contrast with metabolites, this did not improve the regression (Rpls = 0.04; P = 0.07). This was so, irrespective of whether the enzyme activities were expressed on a FW or a total protein basis. This lack of additivity is probably a consequence of the highly coordinated changes of enzyme activities.
We therefore searched for a way to integrate information about the variance in enzyme activity that did not depend on variation between the activities of individual enzymes. The highly coordinated changes of maximum enzyme activities are likely to be due to coordinated changes in the amount of protein invested in enzymes, rather than parallel changes in their Kcat. We used literature values for enzyme specific activities (Table 2) to compute the amount of protein invested in each enzyme in each accession (see Supplemental Data Set 1F online). As shown by Piques et al. (2009), the enzyme amount estimated in this way correlates well with protein abundance measured using mass spectrometry. To provide an overview of how the protein abundance varies between enzymes, we estimated the average value across all the accessions (Table 2). As expected, Rubisco represents a large proportion of total leaf protein (43.7%). Other enzymes contribute between 0.01 and 3%. Grouped on a pathway basis, Calvin-Benson cycle enzymes contribute almost 50% of the total protein, Calvin-Benson cycle enzymes without Rubisco 5%, sucrose and starch synthesis <1%, respiration 3%, amino acid metabolism 2.6%, and sucrose breakdown 0.2% of total leaf protein.
The estimated protein in all of the enzymes was summed for each accession and expressed as a percentage of the total protein in that accession. This term will be referred to as relative investment of protein in metabolism (RPM). There was a significant positive correlation (Rs = 0.33, pBH = 0.0001) between RPM and FW (Figure 6A). As Rubisco represents the vast majority of the rosette protein, this relationship might be very sensitive to changes in Rubisco. The same calculation was therefore repeated for all enzymes except Rubisco. A similar correlation was retained (Rs = 0.36; pBH = 0.00002; Figure 6B). An analogous plot in which the fraction of total rosette protein found in Rubisco was plotted against FW yielded an Rs of 0.29 (pBH = 0.0008) (see Supplemental Data Set 2A online). This stable relationship reflects the fact that there is a fairly conserved relationship (R = 0.51) between the fraction of the total protein that is invested in Rubisco and the fraction that is invested in the other 34 enzymes (Figure 6C).
Figure 6.

Correlation between Biomass and the Fraction of Total Rosette Protein Invested in Enzymes of Central Metabolism.
The amount of protein invested in a given enzyme was calculated from the measured activity using literature values for the specific activity (see Table 2 and Supplemental Data Set 1F online). This was summed for all enzymes in a given accession and divided by the total rosette protein in that accession to calculate the relative investment of protein in metabolism (RPM).
(A) Summed protein in enzymes as a percentage of total protein (=RPM) versus rosette biomass. Each data point is a separate accession.
(B) Summed protein in enzymes minus Rubisco as a percentage of total protein versus rosette biomass.
(C) Relationship between summed protein in enzymes-Rubisco as a percentage of total protein and summed protein in Rubisco as a percentage of total protein.
(D) Total rosette protein versus rosette biomass.
(E) Summed protein in enzymes as a percentage of total protein (=RPM) versus total rosette protein. Data for all accessions are given in Supplemental Data Set 1F online.
(F) R2 for the correlation between parameters and biomass. The corresponding Rs and P values were for starch versus FW, −0.46, 4E-08; RPM versus FW +0.34, 1E-0.04; starch + RPM versus FW, 0.54, 3E-11; starch versus RPM, −0.061, 0.49).
(G) Relationship between starch content, RPM, and biomass in 129 accessions. Each data point represents a single accession. Starch content and RPM values are taken from Supplemental Data Sets 1E and 1F online. The accessions were divided depending on their biomass into six classes (<150, 151 to 200, 201 to 250, 251 to 300, 301 to 350, and >351 mg/plant), which are distinguished by color coding, with the darkness of the shading indicating an increasingly large biomass (see legend in the panel). Asterisk denotes some of the smallest accessions (Lov5, Bla11, Nok2, and Tamm2) with a low value of RPM and a high starch content; “x” denotes small accessions (Pyl1 and Van0) with a high starch content but a high RPM; “+” denotes small accessions (Be0 and Nok1) with a low RPM but a low starch content.
[See online article for color version of this figure.]
To identify enzymes with a particularly strong predictive value for RPM, the amount of protein in a given enzyme was regressed on the summed protein in all enzymes. An analogous calculation was also performed in which Rubisco was omitted from the summed enzyme protein. Especially high Rs values were obtained for NADP-GAPDH, TPI, TK, NAD-GAPDH, and AlaAT (Table 2, rightmost columns). As already noted, total leaf protein shows an inverse relationship to rosette FW (Figure 6D; see also Sulpice et al., 2009). Consequently, accessions with a high RPM tend to have low total leaf protein (Figure 6E).
Sulpice et al. (2009) used PLS regression to show that the set of 62 metabolites analyzed in this study have predictive power for starch, as well as rosette FW, and that the VIP values (variance importance in the prediction; this gives the weighting of individual metabolites for the prediction of biomass) of the predictor metabolites were similar in both regressions. This implies that starch integrates metabolic status and that the regulatory network that determines starch levels contributes to the regulation of biomass. We investigated whether the prediction of biomass provided by RPM is additive to that provided by starch (Figure 6F). This was done by performing multiple linear regression with cross validation. In this data set, the R2s of starch with FW was 0.21, which is slightly lower than in the smaller data set in Sulpice et al. (2009). The R2s of RPM with biomass was 0.11, and the combined R2s provided by starch and RPM was 0.29. An F-test was applied to evaluate the statistical significance of the increase of explained variance when RPM is added to starch as predictor. The corresponding P value of 6 × 10−5 demonstrates that this increase is highly significant. Regression analysis indicated that starch and RPM are independent parameters (R2s = 0.003; Figure 6F). This was further tested by including an interaction term between RPM and starch. This did not improve the prediction of biomass but instead resulted in a lower value of the adjusted R2, which was not significant in an ANOVA test. Evaluation of Akaike’s Information criterion for the two alternative models also showed that the model without the interaction term was to be preferred.
These results imply that almost one-third of the variance in biomass in this large population of accessions is related to these two integrative metabolic parameters. Their contribution is visualized in Figure 6G. For each accession, the starch content is plotted against the RPM value. The accessions were divided into six classes based on their biomass. These classes are identified by color coding, with increasing darkness signifying increasing biomass. Some of the smallest accessions (Lov5, Bla11, Nok2, and Tamm2 marked with an asterisk in the diagram) have a low value of RPM and a high starch content. However, when accessions are compared in the moderately sized or large classes, low RPM tends to be accompanied by high starch and vice versa. The largest accessions are characterized by relatively low starch and high RPM values but do not have extreme values for either parameter. In several cases, outliers for the relationship between biomass and one parameter can be explained by an extreme value for the other parameter. For example, Pyl1 and Van0, (marked as “x” in the figure) are small accessions with a high starch content but have a high RPM, whereas Be0 and Nok1 (marked with “+” in the figure) are small accessions with a low RPM but a low starch content.
Van0 contains a mutation in the ERECTA locus (van Zanten et al., 2009). Mining the currently available genome sequence data for the accessions used in this study (Est-1, Fei-0, Bay-1, Bur-0, and Cvi-1) did not reveal any other likely erecta nonsilent alleles.
DISCUSSION
Metabolism is usually studied by perturbing the activities of single enzymes. However, metabolic networks are generated by interactions between enzymes. Arabidopsis accessions have allelic diversity for hundreds or thousands of genes, allowing a systems-level study of how a metabolic network responds to simultaneous changes of many enzyme activities. This response was analyzed by measuring a large set of enzyme activities, metabolites, and biomass in 129 accessions. The resulting data matrix reveals a very high frequency of positive correlations between enzyme activities, a moderate frequency of correlations between related metabolites, and remarkably few correlations between enzyme activities and metabolite levels. It provides insights into the nature, and consequences, of genetic variation at these two levels of metabolic function and its relationship with biomass formation.
Absolute Enzyme Activities Differ between Pathways in a Manner That Broadly Reflects Pathway Stoichiometry
Enzymes from the same pathway have more closely related absolute activities than do enzymes from different pathways (Figure 1). The range of activities measured here broadly reflects differences in fluxes through the different pathways. Enzyme activities in the Calvin-Benson cycle that we measured were, on average, ~20-fold higher than those in anabolic pathways like sucrose and starch synthesis, nitrogen assimilation, and amino acid synthesis. Based on known pathway stoichiometry (Heldt, 1997), it can be calculated that synthesis of one molecule of sucrose requires the net fixation of 12 molecules of CO2. Depending on the position of the enzyme in the Calvin-Benson cycle, 12 to 24 catalytic cycles will be needed to regenerate 12 molecules of the acceptor Ru1,5bisP and allow fixation of 12 molecules of CO2. This number will be 50 to 60% higher in ambient conditions due to photorespiration (Pettersson and Ryde-Pettersson, 1988; Poolman et al., 2001; Reumann and Weber, 2006; Zhu et al., 2007). The activities of most enzymes for sucrose degradation and respiration were even lower. This reflects the fact that the rate of respiration is typically 10- to 100-fold lower than the rate of photosynthesis (Heldt, 1997; Ivanova et al., 2008). The qualitative match between absolute enzyme activities and the fluxes that they carry indicates that there has been selection to optimize the efficiency of allocation of C and nitrogen between different enzymes and pathways.
For some respiratory enzymes (e.g., NAD-MDH and fumarase), we measured activities similar to those of the Calvin-Benson cycle enzymes. In addition to their function in the TCA cycle, these enzymes have an additional role in catalyzing the synthesis of organic acids. Malate accumulates to high levels in the vacuole of many plants (Heldt, 1997), and fumarate also accumulates in Arabidopsis (Chia et al., 2000). Organic acids are synthesized as a counteranion when nitrate is assimilated and may also have an osmotic function and provide a temporary carbon store (Zell et al., 2010). This may explain why the activities of NAD-MDH and fumarase are much higher than those of other respiratory enzymes.
Coordinated Changes of Enzymes That Are Required for Photosynthesis
Enzymes from a given pathway show coordinated changes of activity in this large population of Arabidopsis accessions. Examples include enzymes in the Calvin-Benson cycle and, to a lesser extent, enzymes for starch and sucrose synthesis and amino acid metabolism. These coordinated changes are retained when enzyme activities are examined in relation to total rosette protein content. They result in changes in the total rosette protein because the enzymes in central metabolism, especially Rubisco, represent a considerable part of the total rosette protein.
The coordinated nature of the variation in enzyme activities in this panel of accessions has two important functional implications. First, a coordinated change in the activities of several enzymes is an effective strategy to alter pathway flux (Kacser and Burns, 1973; Kacser and Acerenza, 1993; Fell and Thomas, 1995). Second, the changes in the relative levels of individual enzymes in this panel of accessions are so small that they are unlikely to have a detrimental effect on efficiency with which the other enzymes are used. For example, there are almost no instances where the activity of one Calvin-Benson cycle enzyme changes >2-fold relative to the activity of another Calvin-Benson cycle enzyme. As discussed in the Introduction, a 2-fold decrease in the activity of individual enzymes in mutants and transgenic plants leads to only minor changes in flux for some enzymes and has no effect for other enzymes (see also Stitt et al., 2010b). Zhu et al. (2007) developed a model of the Calvin-Benson cycle, photorespiration and photosynthetic end product synthesis, and used it to predict that a small reallocation of protein toward Rubisco, Ald, SBPase, and plastid FBPase and away from other Calvin-Benson cycle enzymes could potentially increase the rate of photosynthesis. We found no evidence for more variation in the ratio between Rubisco and PGK, NADP-GAPDH; TPI and TK than would be expected on the basis of the variation in the total data set.
In some cases, enzymes in different pathways show coordinated changes of activity. For example, enzymes from the pathways of sucrose and starch synthesis and amino acid synthesis correlate with Calvin-Benson cycle enzymes. Although nominally classified as separate pathways, they are actually mutually interdependent because the synthesis of sucrose, starch, and amino acids depends on the supply of triose phosphates from the Calvin-Benson cycle. A recent study of an RIL population found that AGP and pPGI correlated with Rubisco (R > 0.3), whereas enzymes for sucrose degradation and respiration did not (R < 0.17) (Keurentjes et al., 2008). Another example of a coordinated response that spans several nominally different pathways is provided by NADP-MDH, NAD-MDH, NADP-ICDH, TPI, and NAD-GAPDH. In addition to their roles in the Calvin-Benson cycle, glycolysis, and the TCA cycle, these enzymes catalyze metabolite shuttles that transfer ATP and redox groups between subcellular compartments. During photosynthesis, the light reactions generate NADPH and ATP in the chloroplast stroma. While much of this is used in the chloroplast, some is transferred to the remainder of the cell. Mature chloroplasts have no mechanism for direct export of NAD(P)H and only a limited capacity for ATP export (Heldt, 1997). Instead, ATP and reducing groups are exported via a metabolite shuttle involving the chloroplastic NADP-GAPD, the triose phosphate translocator, and the cytosolic NAD-GAPDH, and excess reducing equivalents are exported by the malate valve involving the plastidic NADP-MDH and extraplastidic NAD-MDH (Scheibe, 2004).
Enzymes in other pathways, especially sucrose degradation, show divergent responses. Plants possess alternative and potentially redundant pathways for sucrose degradation via SuSy and FK or via INV, GK, and FK. No evidence was found for a correlation between enzymes that are required in one or the other of these pathways. These enzymes, in particular INV, also varied in an uncoordinated manner in a Cvi × Ler RIL population (Keurentjes et al., 2008). This implies that Arabidopsis tolerates large changes in the relative levels of enzymes in C breakdown pathways.
NR provides another interesting example of an enzyme whose maximum activity varies independently of other enzymes. NR is a key site for the regulation of nitrate assimilation. In addition to transcriptional and translational regulation (Nussaume et al., 1995; Kaiser and Huber, 2001; MacKintosh and Meek, 2001; Lea et al., 2006), NR is subject to posttranslational regulation involving phosphorylation followed by the Mg2+-dependent binding of a 14-3-3-protein (Nussaume et al., 1995; Kaiser and Huber, 2001; MacKintosh and Meek, 2001; Lea et al., 2006). One possible explanation for the divergent responses may be that NR activity is mainly regulated by posttranslational mechanisms. Nitrogen metabolism in tobacco (Nicotiana tabacum) was not markedly affected by constitutive overexpression of native NIA (encoding NR) but was strongly modified by introduction of a modified and constitutively active form of the enzyme (Lea et al., 2006).
Coordinated Changes of Sets of Related Metabolites
Metabolites were moderately correlated with each other at a global level. This resembles previous studies with Arabidopsis rosettes (Meyer et al., 2007; Sulpice et al., 2009) and tomato fruit (Schauer et al., 2006, 2008). There were strong correlations between metabolites in specific sectors of metabolism. This included the amino acids, as seen previously (Galili and Hofgen, 2002; Foyer et al., 2003; Lu et al., 2008). There was also a strong correlation between total amino acids and sucrose and starch, the two main carbohydrates formed during photosynthesis. Similar correlations have been reported in physiological studies of single or near-isogenic genotypes (see Introduction). However, starch and sucrose did not correlate with individual amino acids. Instead, 2-oxoglutarate correlated with Gln, Glu, Asp, and several minor amino acids, including Arg and Phe. 2-Oxoglutarate is the immediate C-acceptor during nitrogen assimilation in plants, where it reacts with Gln to form two molecules of Glu in the reaction catalyzed by GOGAT. These results indicate genetic conservation of a regulatory network linking 2-oxoglutarate and amino acid biosynthesis. Physiological studies in tobacco and Arabidopsis have shown that the levels of 2-oxoglutarate and Glu are often correlated (Muller et al., 2001; Fritz et al., 2006), and there is growing evidence that Glu plays a key role in the regulation of nitrogen metabolism (Fritz et al., 2006; Forde and Lea, 2007).
Metabolites Change Independently of Maximal Enzyme Activities
There were very few correlations between enzyme activities and metabolites. We also failed to uncover more links when we used multivariate statistics to search for combinations of enzymes that might determine the level of a metabolite, or permitted nonlinear interactions. Several explanations can be advanced for this lack of connectivity between enzyme activities and metabolites.
One explanation is that enzymes catalyze fluxes and that there is no simple relationship between flux and the concentrations of metabolic intermediates. The coordinated changes of enzyme activities that occur in this panel of accessions may, paradoxically, result in there being even fewer correlations of enzyme activities with metabolic intermediates. As pointed out (Kacser and Acerenza, 1993), a coordinated increase of enzyme activities will increase flux while leaving the levels of the metabolic intermediates essentially unaltered. This does not account for the absence of correlations between enzymes and metabolites that are pathway products, like starch, sucrose, many of the amino acids, and organic acids that accumulate to high levels in the vacuole, like malate and fumarate. However, the levels of pathway products may still represent a balance between the rates of synthesis and utilization. For example, sucrose is exported from the leaf, and amino acids are exported or used for protein synthesis in the leaf. In one case, our data set does allow a direct assessment of flux. Starch is synthesized in the light and almost completely remobilized at night in the conditions used in our experiment (Gibon et al., 2004a; Usadel et al., 2008), so the level at the end of the light period therefore reflects the rate of synthesis in the preceding light period. The starch content did not correlate with the activity of two enzymes from the pathway of starch synthesis, AGP or pPGI (Rs = –0.04 and 0.17, respectively).
A second explanation is that metabolite levels are not determined by maximum enzyme activities. When the maximum activity of an enzyme is decreased, there is usually no effect on flux until a threshold is reached (Kacser and Burns, 1973; Fell and Thomas, 1995; Stitt et al., 2010b). As discussed in the introduction, for four (NADP-GAPDH, PGK, TPI, and TK) of the six Calvin-Benson cycle enzymes in our data set, it has been empirically determined that a 2-fold decrease of the maximum activity has no detectable effect on the rate of photosynthesis (Stitt and Schulze, 1994; Stitt and Sonnewald, 1995). Our measurements now show that the relative activities of these enzymes vary by <2-fold in this large population of accessions. This explains why they do not correlate with metabolites that are produced by photosynthesis. Rubisco and Ald do have measureable flux control coefficients for photosynthesis in ambient conditions (i.e., a relatively small decrease in their maximum activity leads to a detectable inhibition of CO2 fixation). Rubisco activity correlated with starch and sucrose, which are the two main end-products of photosynthesis, whereas Ald showed little variation in its activity in this panel of accessions. The correlation matrix between maximum Calvin-Benson cycle enzyme activities and metabolites in this panel of accessions is therefore consistent with the results of earlier studies, which used reverse genetics to alter the maximum activity of individual enzymes. Crucially, the matrix reveals which enzymes actually show changes of their maximum activity in different Arabidopsis accessions and could therefore contribute to the genetic control of metabolism. The coordinated changes of maximum enzyme activities in the Calvin-Benson cycle and related pathways that occur in this panel of accessions will provide a very effective means to change fluxes through central photosynthetic metabolism.
In vivo enzyme activity is usually lower than the maximum activity. There are various reasons for this (Rolleston, 1972; Newsholme and Start, 1973; Junker et al., 2007). One is that many reactions are close to their thermodynamic equilibrium in vivo. In this case, the net flux is the difference between the rates of the forward and the reverse reactions and is much smaller than total rate of catalysis. The second is that in vivo activity is typically restricted by inhibitors, low levels of substrates and activators, and/or posttranslational regulation. In the terminology of Newsholme and Start (1973), coarse regulation refers to changes in the maximal activity brought about by expression and protein turnover, and fine regulation refers to modulation of the activity by substrate availability, levels of activators and inhibitors, and posttranslational activation. Fine regulation makes a far larger contribution than does gene expression to the regulation of glycolysis when yeast is grown in conditions that alter the ATP yield (Daran-Lapujade et al., 2007). This implies that additional information about fine regulation will be required to link changes in maximum enzyme activity to the responses of metabolites.
It is difficult to assess the impact of fine regulation when it is caused by changes in substrate saturation and the levels of metabolite effectors because this requires detailed information about the kinetic properties of an enzyme and the in vivo levels of all its major effectors. By measuring the posttranslational regulation of Rubisco and NADP-MDH, we were able to uncover two biologically relevant correlations that were not apparent from measurements of maximum enzyme activities.
The lack of connectivity between enzymes and metabolites highlights the complexity of the interactions between different enzymes in pathways in central metabolism and the importance of regulation of enzyme activities by metabolite ligands and posttranslational modification (fine regulation). Further integration of global data on metabolism will require determinations of fluxes, the acquisition and integration of information concerning allosteric regulators and posttranslational regulation, and the analysis of these results in the context of quantitative metabolic models (Pettersson and Ryde-Pettersson, 1988; Poolman et al., 2001; Uys et al., 2007; Zhu et al., 2007).
Biomass Correlates with the Relative Investment of Protein in Enzymes
Our study provides a large matrix of over 100 metabolic traits, which can be analyzed to identify metabolic traits that are linked with increased biomass. Multivariate statistics has already been applied to predict rosette biomass from a large set of metabolites (Meyer et al., 2007) and to show that starch is an integrative biomarker that captures much of the information present in the metabolite profile and has a highly significant negative correlation with biomass (Sulpice et al., 2009). Starch accumulates in the light period and is degraded to support metabolism and growth at night (Smith and Stitt, 2007). When Col-0 is grown in different photoperiods, there is a strong correlation (R > 0.99) between the relative growth rate and the rate of starch degradation (Gibon et al., 2009). In this study, starch was measured at the end of the light period. It has been shown previously that starch is almost completely degraded by the end of the night in a subset of 20 accessions (Cross et al., 2006). The amount of starch accumulated at the end of the light period therefore provides an indicator for the amount of C available for metabolism and growth at night. The negative correlation between starch and biomass indicates that accessions that produce a large biomass are able to use their C more efficiently for growth. This biomarker therefore presumably reflects the efficiency with which C is allocated for biomass production.
Many individual enzyme activities correlate weakly and positively with growth, especially when they are expressed on a total protein basis. However, multivariate analysis on the 35 enzyme activities did not provide a prediction of biomass. The reason is probably that little additional information can be extracted by a combined analysis because the enzyme activities are already strongly correlated with each other. This prompted us to use the measured enzyme activities to compute the fraction of total rosette protein that is invested in the enzymatic machinery for central metabolism (RPM). We found a significant positive correlation between RPM and rosette biomass. We interpret this parameter as measure of the efficiency with which protein is invested for the purpose of carbon acquisition. Many of the most strongly correlating enzymes are involved in the Calvin-Benson cycle or related pathways. This relationship did not depend on any single enzyme. Indeed, our results show that information about one or a small number of enzymes suffice to generate a proxy value for this term. The robustness is presumably a consequence of the coordinated nature of the responses of enzyme activities. The importance of optimizing investment in the photosynthetic machinery is highlighted by the fact that half or more of the total protein in leaves is invested in the photosynthetic machinery (Farquhar et al., 2001; Zhu et al., 2007).
The prediction of biomass provided by the relative investment of protein in metabolism (RPM) is independent of the prediction provided by starch, and the summed R2 for these two integrative parameters accounts for almost one-third of the total variance in rosette biomass in this large population of Arabidopsis accessions. Formally, this additivity is a consequence of the lack of connectivity between enzyme activities and metabolite levels. In practical terms, this means that a relatively small number of robotized analytic measurements can be used to identify two major metabolic determinants of biomass. Functionally, it indicates that investment in photosynthetic machinery and optimization of carbon use are largely separate processes. While both are likely to be subject to multigenic regulation, our results indicate that these may be largely nonoverlapping networks. This may allow these two parameters to be used in the future to identify complementary genotypes that score highly for one and weakly for the other to investigate how they contribute to productivity in segregating populations.
Finally, our approach is based on the idea that the variation for metabolic traits in accessions is generated by large numbers of small perturbations. It is possible that part of the variance is generated by a small number of major polymorphisms that have a large impact on one or many metabolic traits. For example, ERECTA is a pleiotropic regulator of developmental and physiological processes, as well as a modulator of responses to environmental stimuli (van Zanten et al., 2009). In an analysis of a Ler × Cvi RIL population, Keurentjes et al. (2008) noted that the ERECTA locus, which is polymorphic between the parents, colocates with strong QTL for several metabolic traits studies and suggested that ERECTA may be responsible for a subtle simultaneous regulation of primary metabolism in parallel with, or as a consequence of, its effects on development. Our accession panel contains at least two (accessions Ler and Van0) with a polymorphism at the ERECTA locus (van Zanten et al., 2009). As more genome resequencing data become available, it will become possible to use candidate gene association mapping to assess the contribution of such major locus polymorphism to genetic variation in metabolism at a species level.
METHODS
Selection of Accessions
Arabidopsis thaliana accessions used in this study were obtained from various sources: Col-0 from G. Rédei (University of Missouri-Columbia); C24 from J.P. Hernalsteens (Vrije Universiteit Brussels, Belgium); Ler from M. Koornneef (Wageningen University, The Netherlands); Eil0, Lip0, Rsch0, Te, and Yo0 from S. Misera (Institut für Pflanzengenetik und Kulturpflanzenforschung, Gatersleben, Germany); Bor4, Est1, Fei0, Lov5, NFA8, RRS7, RRS10, TAMM2, and Tsu1 from D. Weigel (Max Planck Institute, Tuebingen); Ak1, Akita, Bur0, Ct1, Enkheim, Edi0, Jea, Kn0, Lip0, Mh1, Mt0, Nok1, Oy0, Petergof, Pyl1, Ru1, Shakdara, Stw0, Ta0, Te0, Tsu0, and Yo0 from the Versailles stock center. All others were retrieved from the Nottingham Arabidopsis Stock Centre, through which all accessions are now available. Accessions were homogenized by single-seed propagation and were bulk amplified prior to the analysis (Toerjek et al., 2003). Geographical origin of the accessions is available at Vnat (http://dbsgap.versailles.inra.fr/vnat/).
For the selection of a diverse collection of Arabidopsis accessions, 406 Arabidopsis accessions were analyzed using 115 single nucleotide polymorphism (SNP) markers (Toerjek et al., 2003; Schmid et al., 2005). Heterozygous genotypes of SNPs were excluded from further analysis (<2%). All 406 accessions as well as 115 SNPs were used in the Mstrat software (Gouesnard et al., 2001) to generate a core collection of accessions with maximal allelic richness. One hundred core collections were generated independently, and the core set with the highest Nei index was retained. Selected accessions were genotyped with an additional set of 149 SNP markers, which are a subset of the 289 SNP markers available at http://naturalsystems.uchicago.edu/naturalsystems/lab/index_files/page0007.html. Genotyping was done at Sequenom. Finally, a cutoff of >15 accessions in the minor allele class was used to exclude 81 markers where a highly asymmetric distribution would lead to poor statistical resolution.
Growth Conditions and Experimental Design
Growth conditions were exactly as previously described by Cross et al. (2006). Briefly, seeds were germinated and grown for the first 7 d with a daylength of 16 h, temperature 6°C at night and 20°C during day, humidity 75%, and luminosity 145 μmol m−2 s−1. After 7 d, seedlings were transferred to a phytotron. Growth was continued in an 8-h-light/16-h-dark regime at temperatures and humidities of 16°C and 75% at night and of 20°C and 60% during the day. Illumination was 145 μmol m−2 s−1. At the age of 2 weeks, plants of average sizes were transferred to pots of 6 cm in diameter (five plants per pot). After 1 further week in the short-day conditions outlined above, plants were moved to a controlled small growth chamber for 2 more weeks. Daylength was then 8 h, temperature a constant 20°C, and illumination an average 125 μmol m−2 s−1. Plants were watered daily. Harvests of 15 plant rosettes (five samples containing each three plants) were performed at the end of the light period. Each sample typically contained three rosettes, equivalent to ~500 mg FW, depending on the accessions. The entire sample was powdered under liquid nitrogen and stored at –80°C until its use.
Single accessions were grown in at least two independent experiments and in total eight independent experiments were performed, each including between 20 and 108 accessions. Within each experiment, the position of the pots containing individual accessions was randomized.
Enzyme and Metabolite Assays
Chemicals were purchased as described (Gibon et al., 2004b). Total protein, starch, glucose, fructose, sucrose, and total amino acids were assayed as described (Cross et al., 2006). Malate and fumarate were assayed as described (Nunes-Nesi et al., 2007).
For enzyme measurements, aliquots of 20 mg frozen FW were extracted by vigorous mixing with extraction buffer (Nunes-Nesi et al., 2007). AGP, NAD-GAPDH, NADP-GAPDH, PFP, PK, SPS, TK, cFBPase, GK, FK, G6PDH, NADP-ICDH, shikimate dehydrogenase, AlaAT, AspAT, fumarase, GLDH, PEPC, INV, GOGAT, NR, and GS were assayed as described (Gibon et al., 2004b). PFK, CS, NAD-IDH, NAD-MDH, and NADP-MDH were assayed as described by Nunes-Nesi et al. (2007). PGK and glycerate kinase were assayed as described by Huege et al. (2007). Rubisco was assayed as described by Sulpice et al. (2007). TPI was assayed as described by Burrell et al. (1994). cPGI and pPGI were assayed as described by Weeden and Gottlieb (1982). PGM was assayed as described by Manjunath et al. (1998). SuSy and UGP were assayed as described (Keurentjes et al., 2008). Ald was assayed by incubating for 20 min crude extract or dihydroxyacetone phosphate standards in a freshly prepared medium containing 0 or 5 mM fructose-1,6-bisphosphate, 1 U·mL−1 triose-P isomerase, 2 U·mL−1 glycerol-3P dehydrogenase, 0.3 mM NAD+, 5 mM MgCl2, 1 mM EDTA, 0.05% Triton X-100, and 100 mM Tricine buffer, pH 8.5. The reaction was stopped by addition of an equal volume of 0.5 M HCl. After incubation for 10 min at room temperature and neutralization with 0.5 M NaOH, the glycerol-3P produced was then determined using the glycerol-3-P/DHAP–based cycling protocol described by Gibon et al. (2004b).
GC Analysis
Metabolite extraction for GC-MS was performed on the exact same samples as used for enzymes and metabolites determined by spectrophotometric methods as described previously (Schauer et al., 2006). Fifty milligrams of Arabidopsis shoots were homogenized using a ball mill precooled with liquid nitrogen. Derivatization and GC-MS analysis were performed as described previously (Lisec et al., 2006).
ANOVA
Statistical analyses were performed with SAS-JMP (SAS Institute). A two-way factorial ANOVA was used to partition the variance into genetic and experimental variance separately for each trait using the following model:
where Yijk are the experimental responses, μ is the general mean, Ai is the effect of the ith accession genotype, Ej is the effect of the jth experiment, and εjik is the error of Yijk.
The explained genetic variances of a genotype main effect (η2A) and of an experiment effect η2(E) were calculated as follows:
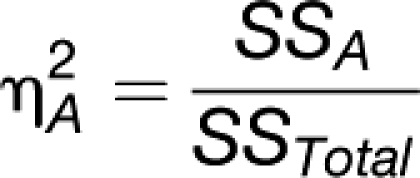 |
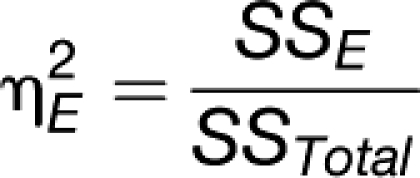 |
where SSA, SSE, and SSTotal are the sum of squares of the factor accession, experiment, and the total sum of squares, respectively, which were obtained from the ANOVA procedure.
Partial F-Test for Model Selection and Coefficient of Variation
The F statistic we used is defined by F = RSS(βk) − RSS(βk+1)/(RSS(βk+1))/(n − k − 2)), with RSS denoting the residual sum of squares for the models expressed by the coefficient vectors βk and βk+1, and k and k+1 representing the number of variables already selected and n the number of samples.
The CV was defined as the ratio of the standard deviation to the mean of all mean values obtained for a trait/accession pair.
Support Vector Machines
To evaluate nonlinear relationships between enzyme activities and metabolites, support vector machines regression was applied using the R function with a radial kernel “svm” (Chang and Lin, 2001). To detect the important predictors from p input variables, the svm prediction in cross-validation was p times repeated each time leaving out one predictor. The predictor for which the elimination yields the lowest correlation between predicted and actual response in cross-validation is then regarded as the most important one.
PLS Regression and Cross-Validation
PLS identifies the combinations of the original predictor variables with maximum covariance with the response (Wold, 1975; Barker and Rayens, 2003). These orthogonal combinations replace the original data matrix and are used in a multivariate ordinary least squares regression to predict the response. The optimal number of components is determined by the maximum proportion of explained variance obtained in 5-fold cross-validation. The observations are divided into five subsets, a training set (four subsets) is used to build a model that is applied to predict the response of the remaining subset, and this procedure is repeated five times to estimate the response for all observations. Cross-validation was also applied each time to the training set. The estimated vector is correlated with the measured response to obtain a measure of the predictive power of the predictor variables.
The weight of a predictor j in the linear combination resulting in PLS component i is denoted as wij. The VIP of each predictor j gives an estimate of the importance of that predictor for the PLS prediction using the h most important orthogonal components and is calculated as the sum of the wij (i=1, … h) multiplied by the correlation of PLS component i with the response (Chong and Jun, 2005).
Significance of Coordinated Changes between Enzyme Activities
For each enzyme, the activity in a given accession was normalized on the mean activity of that enzyme in all of the accessions. This allowed activity ratios in the same range to be obtained for each enzyme pair. The activity ratios for each enzyme-enzyme pair belonging to the pathways tested were calculated in each accession. The values obtained for all accessions were then depicted as a frequency plot. To test for significance of the plots obtained, the normalized activities were shuffled 1000 times for the accessions, ratios calculated and combined, and their standard deviation calculated. The frequency distribution of the standard deviations obtained from 1000 such shufflings was tested against the standard deviation of the real values (see Supplemental Figure 3 online).
Cartographic Representation of the Network
A matrix of correlation between all trait pairs was generated. Significance levels were set at pBH < 0.01. The 412 significant pairs (>5886) that resulted were considered as a network in which a vertex corresponds to a trait and a link between two vertices corresponds to significant correlations between these two traits. This network was then subjected to the cartography algorithm (Guimera and Amaral, 2005). Essentially, this algorithm divides the network into modules or groups of vertices that are more connected between themselves than to nodes from other modules, yielding a cartographic representation of a complex network. In implementing this algorithm, negative correlations were considered to be equal to positive correlations.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. Supporting Information for the Network Analysis of the Entire Data Set.
Supplemental Figure 2. Correlations between Enzymes Involved in Metabolic Shuttles That Allow Transfer of Redox Groups between Different Subcellular Compartments.
Supplemental Figure 3. Frequency Histogram of the Enzyme-Enzyme Ratios for Normalized Enzyme Activities for Different Pathways.
Supplemental Data Set 1A. Geographical Origin of the Accessions Studied.
Supplemental Data Set 1B. List of Metabolites Analyzed, with Abbreviations and the Assignments to Metabolic Sectors.
Supplemental Data Set 1C. List of the Enzymes Analyzed, with Abbreviations and Assignments of Pathways.
Supplemental Data Set 1D. Genetic Variation in Metabolic Parameters Relating to Central C and N Metabolism in the Arabidopsis Accessions Used for the Study.
Supplemental Data Set 1E. Normalized Average Data for All Traits Determined in the Accessions.
Supplemental Data Set 1F. Calculation of RPM (Relative Investment of Protein in Metabolism).
Supplemental Data Set 2A. Spearman Rs Values and Associated P Values for All Pairwise Correlations, Based on Fresh Weight.
Supplemental Data Set 2B. Spearman Rs Values and Associated P Values for All Pairwise Correlations, Based on Protein Content.
Acknowledgments
This research was supported by the German Ministry for Education and Research (GABI-EVAST, GABI GNADE, GoFORSYS, and SEPSAPE), the European Community (FP7 IP AGRON-OMICS), and the Max Planck Society. J.M.R. was supported by an Alexander von Humboldt Fellowship.
References
- Adam N.R., Wall G.W., Kimball B.A., Pinter P.J., LaMorte R.L., Hunsaker D.J., Adamsen F.J., Thompson T., Matthias A.D., Leavitt S.W., Webber A.N. (2000). Acclimation response of spring wheat in a free-air CO2 enrichment (FACE) atmosphere with variable soil nitrogen regimes. 1. Leaf position and phenology determine acclimation response. Photosynth. Res. 66: 65–77 [DOI] [PubMed] [Google Scholar]
- Aranzana M.J., et al. (2005). Genome-wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PLoS Genet. 1: e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerenfaller K., Grossmann J., Grobei M.A., Hull R., Hirsch-Hoffmann M., Yalovsky S., Zimmermann P., Grossniklaus U., Gruissem W., Baginsky S. (2008). Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science 320: 938–941 [DOI] [PubMed] [Google Scholar]
- Barker M., Rayens W. (2003). Partial least squares for discrimination. J. Chemometr. 17: 166–173 [Google Scholar]
- Baxter I., Muthukumar B., Park H.C., Buchner P., Lahner B., Danku J., Zhao K., Lee J., Hawkesford M.J., Guerinot M.L., Salt D.E. (2008). Variation in molybdenum content across broadly distributed populations of Arabidopsis thaliana is controlled by a mitochondrial molybdenum transporter (MOT1). PLoS Genet. 4: e1000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler E.S., Thornsberry J.M. (2002). Plant molecular diversity and applications to genomics. Curr. Opin. Plant Biol. 5: 107–111 [DOI] [PubMed] [Google Scholar]
- Burrell M.M., Mooney P.J., Blundy M., Carter D., Wilson F., Green J., Blundy K.S., Aprees T. (1994). Genetic manipulation of 6-phosphofructokinase in potato tubers. Planta 194: 95–101 [Google Scholar]
- Calenge F., Saliba-Colombani V., Mahieu S., Loudet O., Daniel-Vedele F., Krapp A. (2006). Natural variation for carbohydrate content in Arabidopsis. Interaction with complex traits dissected by quantitative genetics. Plant Physiol. 141: 1630–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causse M., Rocher J.P., Henry A.M., Charcosset A., Prioul J.L., Devienne D. (1995). Genetic dissection of the relationship between carbon metabolism and early growth in maize, with emphasis on key enzyme loci. Mol. Breed. 1: 259–272 [Google Scholar]
- Chia D.W., Yoder T.J., Reiter W.D., Gibson S.I. (2000). Fumaric acid: An overlooked form of fixed carbon in Arabidopsis and other plant species. Planta 211: 743–751 [DOI] [PubMed] [Google Scholar]
- Chong I.G., Jun C.H. (2005). Performance of some variable selection methods when multicollinearity is present. Chemom. Intell. Lab. Syst. 78: 103–112 [Google Scholar]
- Cross J.M., von Korff M., Altmann T., Bartzetko L., Sulpice R., Gibon Y., Palacios N., Stitt M. (2006). Variation of enzyme activities and metabolite levels in 24 Arabidopsis accessions growing in carbon-limited conditions. Plant Physiol. 142: 1574–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curien G., Ravanel S., Dumas R. (2003). A kinetic model of the branch-point between the methionine and threonine biosynthesis pathways in Arabidopsis thaliana. Eur. J. Biochem. 270: 4615–4627 [DOI] [PubMed] [Google Scholar]
- Daran-Lapujade P., Rossell S., van Gulik W.M., Luttik M.A.H., de Groot M.J.L., Slijper M., Heck A.J.R., Daran J.M., de Winde J.H., Westerhoff H.V. (2007). The fluxes through glycolytic enzymes in Saccharomyces cerevisiae are predominantly regulated at posttranscriptional levels. Proc. Natl. Acad. Sci. USA 104: 15753–15758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debolt S., Melino V., Ford C.M. (2007). Ascorbate as a biosynthetic precursor in plants. Ann. Bot. (Lond.) 99: 3–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan R.E., Chen P.H., Lin C.J. (2005). Working set selection using the second order information for training SVM. J. Mach. Learn. Res. 6: 1889–1918 [Google Scholar]
- Farquhar G.D., von Caemmerer S., Berry J.A. (2001). Models of photosynthesis. Plant Physiol. 125: 42–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell D.A., Thomas S. (1995). Physiological control of metabolic flux - The requirement for multisite modulation. Biochem. J. 311: 35–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiehn O., Kopka J., Dormann P., Altmann T., Trethewey R.N., Willmitzer L. (2000). Metabolite profiling for plant functional genomics. Nat. Biotechnol. 18: 1157–1161 [DOI] [PubMed] [Google Scholar]
- Forde B.G., Lea P.J. (2007). Glutamate in plants: Metabolism, regulation, and signalling. J. Exp. Bot. 58: 2339–2358 [DOI] [PubMed] [Google Scholar]
- Foyer C.H., Parry M., Noctor G. (2003). Markers and signals associated with nitrogen assimilation in higher plants. J. Exp. Bot. 54: 585–593 [DOI] [PubMed] [Google Scholar]
- Fridman E., Pleban T., Zamir D. (2000). A recombination hotspot delimits a wild-species quantitative trait locus for tomato sugar content to 484 bp within an invertase gene. Proc. Natl. Acad. Sci. USA 97: 4718–4723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz C., Mueller C., Matt P., Feil R., Stitt M. (2006). Impact of the C-N status on the amino acid profile in tobacco source leaves. Plant Cell Environ. 29: 2055–2076 [DOI] [PubMed] [Google Scholar]
- Galili G., Hofgen R. (2002). Metabolic engineering of amino acids and storage proteins in plants. Metab. Eng. 4: 3–11 [DOI] [PubMed] [Google Scholar]
- Geiger D.R., Servaites J.C. (1994). Diurnal regulation of photosynthetic carbon metabolism in C-3 plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 45: 235–256 [Google Scholar]
- Gibon Y., Bläsing O.E., Palacios-Rojas N., Pankovic D., Hendriks J.H.M., Fisahn J., Hohne M., Gunther M., Stitt M. (2004a). Adjustment of diurnal starch turnover to short days: Depletion of sugar during the night leads to a temporary inhibition of carbohydrate utilization, accumulation of sugars and post-translational activation of ADP-glucose pyrophosphorylase in the following light period. Plant J. 39: 847–862 [DOI] [PubMed] [Google Scholar]
- Gibon Y., Blaesing O.E., Hannemann J., Carillo P., Hohne M., Hendriks J.H.M., Palacios N., Cross J., Selbig J., Stitt M. (2004b). A robot-based platform to measure multiple enzyme activities in Arabidopsis using a set of cycling assays: Comparison of changes of enzyme activities and transcript levels during diurnal cycles and in prolonged darkness. Plant Cell 16: 3304–3325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibon Y., Pyl E.-T., Sulpice R., Höhne M., Stitt M. (2009). Adjustment of growth, starch turnover, protein content and central metabolism to a decrease of the carbon supply when Arabidopsis is grown in very short photoperiods. Plant Cell Environ. 32: 859–874 [DOI] [PubMed] [Google Scholar]
- Gibon Y., Usadel B., Blaesing O.E., Kamlage B., Hoehne M., Trethewey R., Stitt M. (2006). Integration of metabolite with transcript and enzyme activity profiling during diurnal cycles in Arabidopsis rosettes. Genome Biol. 7: R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford R.M., Evans L.T. (1981). Photosynthesis, carbon partitioning, and yield. Annu. Rev. Plant Physiol. Plant Mol. Biol. 32: 485–509 [Google Scholar]
- Gouesnard B., Bataillon T.M., Decoux G., Rozale C., Schoen D.J., David J.L. (2001). MSTRAT: An algorithm for building germ plasm core collections by maximizing allelic or phenotypic richness. J. Hered. 92: 93–94 [DOI] [PubMed] [Google Scholar]
- Guimera R., Amaral L.A.N. (2005). Functional cartography of complex metabolic networks. Nature 433: 895–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimera R., Mossa S., Turtschi A., Amaral L.A.N. (2005). The worldwide air transportation network: Anomalous centrality, community structure, and cities' global roles. Proc. Natl. Acad. Sci. USA 102: 7794–7799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haake V., Geiger M., Walch-Liu P., Engels C., Zrenner R., Stitt M. (1999). Changes in aldolase activity in wild-type potato plants are important for acclimation to growth irradiance and carbon dioxide concentration, because plastid aldolase exerts control over the ambient rate of photosynthesis across a range of growth conditions. Plant J. 17: 479–489 [Google Scholar]
- Haake V., Zrenner R., Sonnewald U., Stitt M. (1998). A moderate decrease of plastid aldolase activity inhibits photosynthesis, alters the levels of sugars and starch, and inhibits growth of potato plants. Plant J. 14: 147–157 [DOI] [PubMed] [Google Scholar]
- Hansen B.G., Halkier B.A., Kliebenstein D.J. (2008). Identifying the molecular basis of QTLs: eQTLs add a new dimension. Trends Plant Sci. 13: 72–77 [DOI] [PubMed] [Google Scholar]
- Harrison E.P., Willingham N.M., Lloyd J.C., Raines C.A. (1998). Reduced sedoheptulose-1,7-bisphosphatase levels in transgenic tobacco lead to decreased photosynthetic capacity and altered carbohydrate accumulation. Planta 204: 27–36 [Google Scholar]
- Heldt H.W. (1997). Plant Biochemistry and Molecular Biology. (Oxford, UK: Oxford University Press; ). [Google Scholar]
- Henkes S., Sonnewald U., Badur R., Flachmann R., Stitt M. (2001). A small decrease of plastid transketolase activity in antisense tobacco transformants has dramatic effects on photosynthesis and phenylpropanoid metabolism. Plant Cell 13: 535–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmeyr J.H.S., Cornish-Bowden A. (2000). Regulating the cellular economy of supply and demand. FEBS Lett. 476: 47–51 [DOI] [PubMed] [Google Scholar]
- Huege J., Sulpice R., Gibon Y., Lisec J., Koehl K., Kopka J. (2007). GC-EI-TOF-MS analysis of in vivo carbon-partitioning into soluble metabolite pools of higher plants by monitoring isotope dilution after (CO2)-C-13 labelling. Phytochemistry 68: 2258–2272 [DOI] [PubMed] [Google Scholar]
- Ivanova H., Keerberg H., Parnik T., Keerberg O. (2008). Components of CO2 exchange in leaves of C-3 species with different ability of starch accumulation. Photosynthetica 46: 84–90 [Google Scholar]
- Junker B.H., Lonien J., Heady L.E., Rogers A., Schwender J. (2007). Parallel determination of enzyme activities and in vivo fluxes in Brassica napus embryos grown on organic or inorganic nitrogen source. Phytochemistry 68: 2232–2242 [DOI] [PubMed] [Google Scholar]
- Kacser H., Acerenza L. (1993). A universal method for achieving increases in metabolite production. Eur. J. Biochem. 216: 361–367 [DOI] [PubMed] [Google Scholar]
- Kacser H., Burns J.A. (1973). The control of flux. In Rate Control of Biological Processes, Davies D.D., (Cambridge, UK: Cambridge University Press; ), pp. 65–104 [Google Scholar]
- Kaiser W.M., Huber S.C. (2001). Post-translational regulation of nitrate reductase: mechanism, physiological relevance and environmental triggers. J. Exp. Bot. 52: 1981–1989 [DOI] [PubMed] [Google Scholar]
- Keurentjes J.J.B., Fu J.Y., de Vos C.H.R., Lommen A., Hall R.D., Bino R.J., van der Plas L.H.W., Jansen R.C., Vreugdenhil D., Koornneef M. (2006). The genetics of plant metabolism. Nat. Genet. 38: 842–849 [DOI] [PubMed] [Google Scholar]
- Keurentjes J.J.B., Sulpice R., Gibon Y., Fu J., Koornneef M., Stitt M., Vreugdenhil D. (2008). Integrative analyses of genetic variation in enzyme activities of primary carbohydrate metabolism reveal distinct modes of regulation in Arabidopsis thaliana. Genome Biol. 9: R129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliebenstein D., Pedersen D., Barker B., Mitchell-Olds T. (2002). Comparative analysis of quantitative trait loci controlling glucosinolates, myrosinase and insect resistance in Arabidopsis thaliana. Genetics 161: 325–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliebenstein D.J., West M.A.L., van Leeuwen H., Loudet O., Doerge R.W., St Clair D.A. (2006). Identification of QTLs controlling gene expression networks defined a priori. BMC Bioinformatics 7: 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopka J. (2006). Current challenges and developments in GC-MS based metabolite profiling technology. J. Biotechnol. 124: 312–322 [DOI] [PubMed] [Google Scholar]
- Lea U.S., Leydecker M.T., Quillere I., Meyer C., Lillo C. (2006). Posttranslational regulation of nitrate reductase strongly affects the levels of free amino acids and nitrate, whereas transcriptional regulation has only minor influence. Plant Physiol. 140: 1085–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leakey A.D., Ainsworth E.A., Bernacchi C.J., Rogers A., Long S.P., Ort D.R. (2009). Elevated CO2 effects on plant carbon, nitrogen, and water relations: Six important lessons from FACE. J. Exp. Bot. 60: 2859–2876 [DOI] [PubMed] [Google Scholar]
- Lisec J., Meyer R.C., Steinfath M., Redestig H., Becher M., Witucka-Wall H., Fiehn O., Torjek O., Selbig J., Altmann T., Willmitzer L. (2008). Identification of metabolic and biomass QTL in Arabidopsis thaliana in a parallel analysis of RIL and IL populations. Plant J. 53: 960–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisec J., Schauer N., Kopka J., Willmitzer L., Fernie A.R. (2006). Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat. Protoc. 1: 387–396 [DOI] [PubMed] [Google Scholar]
- Long S.P., Ainsworth E.A., Leakey A.D.B., Nosberger J., Ort D.R. (2006). Food for thought: Lower-than-expected crop yield stimulation with rising CO2 concentrations. Science 312: 1918–1921 [DOI] [PubMed] [Google Scholar]
- Loudet O., Chaillou S., Camilleri C., Bouchez D., Daniel-Vedele F. (2002). Bay-0 x Shahdara recombinant inbred line population: a powerful tool for the genetic dissection of complex traits in Arabidopsis. Theor. Appl. Genet. 104: 1173–1184 [DOI] [PubMed] [Google Scholar]
- Loudet O., Chaillou S., Merigout P., Talbotec J., Daniel-Vedele F. (2003). Quantitative trait loci analysis of nitrogen use efficiency in Arabidopsis. Plant Physiol. 131: 345–358 Erratum. Plant Physiol. 131: 1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Savage L.J., Ajjawi I., Imre K.M., Yoder D.W., Benning C., DellaPenna D., Ohlrogge J.B., Osteryoung K.W., Weber A.P., Wilkerson C.G., Last R.L. (2008). New connections across pathways and cellular processes: Industrialized mutant screening reveals novel associations between diverse phenotypes in Arabidopsis. Plant Physiol. 146: 1482–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKintosh C., Meek S.E.M. (2001). Regulation of plant NR activity by reversible phosphorylation, 14-3-3 proteins and proteolysis. Cell. Mol. Life Sci. 58: 205–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunath S., Lee C.H.K., VanWinkle P., Bailey-Serres J. (1998). Molecular and biochemical characterization of cytosolic phosphoglucomutase in maize - Expression during development and in response to oxygen deprivation. Plant Physiol. 117: 997–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mate C.J., Hudson G.S., Voncaemmerer S., Evans J.R., Andrews T.J. (1993). Reduction of ribulose-bisphophate carboxylase activase levels in tobacco (Nicotiana tabacum) by antisense RNA reduces ribulose-bisphosphate carboxylase carbamylation and impairs photosynthesis. Plant Physiol. 102: 1119–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer R.C., Steinfath M., Lisec J., Becher M., Witucka-Wall H., Torjek O., Fiehn O., Eckardt A., Willmitzer L., Selbig J., Altmann T. (2007). The metabolic signature related to high plant growth rate in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 104: 4759–4764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell-Olds T., Pedersen D. (1998). The molecular basis of quantitative genetic variation in central and secondary metabolism in Arabidopsis. Genetics 149: 739–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller C., Scheible W.R., Stitt M., Krapp A. (2001). Influence of malate and 2-oxoglutarate on the NIA transcript level and nitrate reductase activity in tobacco leaves. Plant Cell Environ. 24: 191–203 [Google Scholar]
- Newsholme E.A., Start C. (1973). Regulation in Metabolism. (New York: John Wiley & Sons; ). [Google Scholar]
- Nordborg M., et al. (2005). The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol. 3: 1289–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes-Nesi A., Carrari F., Gibon Y., Sulpice R., Lytovchenko A., Fisahn J., Graham J., Ratcliffe R.G., Sweetlove L.J., Fernie A.R. (2007). Deficiency of mitochondrial fumarase activity in tomato plants impairs photosynthesis via an effect on stomatal function. Plant J. 50: 1093–1106 [DOI] [PubMed] [Google Scholar]
- Nussaume L., Vincentz M., Meyer C., Boutin J.P., Caboche M. (1995). Posttranscriptional regulation of nitrate reductase by light is abolished by an N-terminal deletion. Plant Cell 7: 611–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul M.J., Foyer C.H. (2001). Sink regulation of photosynthesis. J. Exp. Bot. 52: 1383–1400 [DOI] [PubMed] [Google Scholar]
- Paul M.J., Knight J.S., Habash D., Parry M.A.J., Lawlor D.W., Barnes S.A., Loynes A., Gray J.C. (1995). Reduction in phosphoribulokinase activity by antisense RNA in transgenic tobacco - Effect on CO2 assimilation and growth in low irradiance. Plant J. 7: 535–542 [Google Scholar]
- Paul M.J., Pellny T.K. (2003). Carbon metabolite feedback regulation of leaf photosynthesis and development. J. Exp. Bot. 54: 539–547 [DOI] [PubMed] [Google Scholar]
- Pettersson G., Ryde-Pettersson U. (1988). A mathematical model of the Calvin photosynthesis cycle. Eur. J. Biochem. 175: 661–672 [DOI] [PubMed] [Google Scholar]
- Piques M., Schulze W.X., Höhne M., Usadel B., Gibon Y., Rohwer J., Stitt M. (2009). Ribosome and transcript copy numbers, polysome occupancy and enzyme dynamics in Arabidopsis. Mol. Syst. Biol. 5: 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolman M.G., Olcer H., Lloyd J.C., Raines C.A., Fell D.A. (2001). Computer modelling and experimental evidence for two steady states in the photosynthetic Calvin cycle. Eur. J. Biochem. 268: 2810–2816 [DOI] [PubMed] [Google Scholar]
- Portis A.R. (2003). Rubisco activase - Rubisco's catalytic chaperone. Photosynth. Res. 75: 11–27 [DOI] [PubMed] [Google Scholar]
- Price G.D., Evans J.R., Voncaemmerer S., Yu J.W., Badger M.R. (1995). Specific reduction of glyceraldehyde-3-phosphate dehydrogenase activity by antisense RNA reduces CO2 assimilation via a reduction in ribulose-bisphosphate regeneration in transgenic tobacco plants. Planta 195: 369–378 [DOI] [PubMed] [Google Scholar]
- Prioul J.L., Pelleschi S., Sene M., Thevenot C., Causse M., de Vienne D., Leonardi A. (1999). From QTLs for enzyme activity to candidate genes in maize. J. Exp. Bot. 50: 1281–1288 [Google Scholar]
- Quick W.P., Schurr U., Fichtner K., Schulze E.D., Rodermel S.R., Bogorad L., Stitt M. (1991). The impact of decreased Rubisco on photosynthesis, growth, allocation and storage in tobacco plants which have been transformed with antisense Rbcs. Plant J. 1: 51–58 [Google Scholar]
- Reumann S., Weber A.P.M. (2006). Plant peroxisomes respire in the light: Some gaps of the photorespiratory C-2 cycle have become filled - others remain. Biochim. Biophys Acta 1763: 1496–1510 [DOI] [PubMed] [Google Scholar]
- Rogers A., Ainsworth E.A. (2006). The response of foliar carbohydrates to elevated carbon dioxide concentration. In Managed Ecosystems and CO2: Case Studies, Processes and Perspectives, Nösberger J., Long S.P., Norby R.J., Stitt M., Hendrey G.R., Blum H., (Berlin: Springer Verlag; ), pp. 293–305 [Google Scholar]
- Rolleston F.S. (1972). A theoretical background to the use of measured intermediates in the study of the control of intermediary metabolism. Curr. Top. Cell. Regul. 5: 47–54 [Google Scholar]
- Rowe H.C., Hansen B.G., Halkier B.A., Kliebenstein D.J. (2008). Biochemical networks and epistasis shape the Arabidopsis thaliana metabolome. Plant Cell 20: 1199–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer N., Semel Y., Balbo I., Steinfath M., Repsilber D., Selbig J., Pleban T., Zamir D., Fernie A.R. (2008). Mode of inheritance of primary metabolic traits in tomato. Plant Cell 20: 509–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer N., et al. (2006). Comprehensive metabolic profiling and phenotyping of interspecific introgression lines for tomato improvement. Nat. Biotechnol. 24: 447–454 [DOI] [PubMed] [Google Scholar]
- Scheibe R. (2004). Malate valves to balance cellular energy supply. Physiol. Plant. 120: 21–26 [DOI] [PubMed] [Google Scholar]
- Schmid K.J., Ramos-Onsins S., Ringys-Beckstein H., Weisshaar B., Mitchell-Olds T. (2005). A multilocus sequence survey in Arabidopsis thaliana reveals a genome-wide departure from a neutral model of DNA sequence polymorphism. Genetics 169: 1601–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A.M., Stitt M. (2007). Coordination of carbon supply and plant growth. Plant Cell Environ. 30: 1126–1149 [DOI] [PubMed] [Google Scholar]
- Sønderby I.E., Hansen B.G., Bjarnholt N., Ticconi C., Halkier B.A., Kliebenstein D.J. (2007). A systems biology approach identifies a R2R3 MYB gene subfamily with distinct and overlapping functions in regulation of aliphatic glucosinolates. PLoS ONE 2: e1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks C.A., et al. (2001). Potential for manipulating carbon metabolism in wheat. Ann. Appl. Biol. 138: 33–45 [Google Scholar]
- Stitt M. (1995). The use of transgenic plants to study the regulation of plant carbohydrate-metabolism. Aust. J. Plant Physiol. 22: 635–646 [Google Scholar]
- Stitt M., Huber S.C., Kerr P. (1987). Control of photosynthetic sucrose synthesis. In Biochemistry of Plants, Hatch M.D., Boardman N.K., (London: Academic Press; ), pp. 327–409 [Google Scholar]
- Stitt M., Lunn J., Usadel B. (2010b). Arabidopsis and primary photosynthetic metabolism - More than the icing on the cake. Plant J. 61: 1067–1091 [DOI] [PubMed] [Google Scholar]
- Stitt M., Schulze D. (1994). Does Rubisco control the rate of photosynthesis and plant growth - An exercise in molecular ecophysiology. Plant Cell Environ. 17: 465–487 [Google Scholar]
- Stitt M., Sonnewald U. (1995). Regulation of metabolism in transgenic plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 46: 341–368 [Google Scholar]
- Stitt M., Sulpice R., Keurentjes J. (2010a). Metabolic networks: How to identify key components in the regulation of metabolism and growth. Plant Physiol. 152: 428–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulpice R., et al. (2009). Starch as a major integrator in the regulation of plant growth. Proc. Natl. Acad. Sci. USA 106: 10348–10353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulpice R., Tschoep H., Von Korff M., Bussis D., Usadel B., Hohne M., Witucka-Wall H., Altmann T., Stitt M., Gibon Y. (2007). Description and applications of a rapid and sensitive non-radioactive microplate-based assay for maximum and initial activity of D-ribulose-1,5-bisphosphate carboxylase/oxygenase. Plant Cell Environ. 30: 1163–1175 [DOI] [PubMed] [Google Scholar]
- Sumner L.W., Mendes P., Dixon R.A. (2003). Plant metabolomics: Large-scale phytochemistry in the functional genomics era. Phytochemistry 62: 817–836 [DOI] [PubMed] [Google Scholar]
- Thevenot C., Simond-Cote E., Reyss A., Manicacci D., Trouverie J., Le Guilloux M., Ginhoux V., Sidicina F., Prioul J.L. (2005). QTLs for enzyme activities and soluble carbohydrates involved in starch accumulation during grain filling in maize. J. Exp. Bot. 56: 945–958 [DOI] [PubMed] [Google Scholar]
- Toerjek O., Berger D., Meyer R.C., Muessig C., Schmid K.J., Soerensen T.R., Weisshaar B., Mitchell-Olds T., Altmann T. (2003). Establishment of a high-efficiency SNP-based framework marker set for Arabidopsis. Plant J. 36: 122–140 [DOI] [PubMed] [Google Scholar]
- Usadel B., Bläsing O.E., Gibon Y., Retzlaff K., Höhne M., Günter M., Stitt M. (2008). Global transcript levels respond to small changes of the carbon status during a progressive exhaustion of carbohydrates in Arabidopsis rosettes. Plant Physiol. 146: 1834–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uys L., Botha F.C., Hofmeyr J.H.S., Rohwer J.M. (2007). Kinetic model of sucrose accumulation in maturing sugarcane culm tissue. Phytochemistry 68: 2375–2392 [DOI] [PubMed] [Google Scholar]
- van Zanten M., Snoek L.B., Proveniers M.C., Peeters A.J. (2009). The many functions of ERECTA. Trends Plant Sci. 14: 214–218 [DOI] [PubMed] [Google Scholar]
- von Roepenack-Lahaye E., Degenkolb T., Zerjeski M., Franz M., Roth U., Wessjohann L., Schmidt J., Scheel D., Clemens S. (2004). Profiling of Arabidopsis secondary metabolites by capillary liquid chromatography coupled to electrospray ionization quadrupole time-of-flight mass spectrometry. Plant Physiol. 134: 548–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeden N.F., Gottlieb L.D. (1982). Dissociation, reassociation, and purification of plastid and cytosolic phosphoglucose isomerase isozymes. Plant Physiol. 69: 717–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzell A.M., Rowe H.C., Hansen B.G., Ticconi C., Halkier B.A., Kliebenstein D.J. (2007). Linking metabolic QTLs with network and cis-eQTLs controlling biosynthetic pathways. PLoS Genet. 3: 1687–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter H., Huber S.C. (2000). Regulation of sucrose metabolism in higher plants: Localization and regulation of activity of key enzymes. Crit. Rev. Plant Sci. 19: 31–67 [DOI] [PubMed] [Google Scholar]
- Wold H. (1975). Soft Modelling by Latent Variables. (London: Academic; ). [Google Scholar]
- Zell M.B., Fahnenstich H., Maier A., Saigo M., Voznesenskaya E.V., Edwards G.E., Andreo C., Schleifenbaum F., Zell C., Drincovich M.F., Maurino V.G. (2010). Analysis of Arabidopsis with highly reduced levels of malate and fumarate sheds light on the role of these organic acids as storage carbon molecules. Plant Physiol. 152: 1251–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K.Y., Aranzana M.J., Kim S., Lister C., Shindo C., Tang C.L., Toomajian C., Zheng H.G., Dean C., Marjoram P., Nordborg M. (2007). An Arabidopsis example of association mapping in structured samples. PLoS Genet. 3: e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.G., de Sturler E., Long S.P. (2007). Optimizing the distribution of resources between enzymes of carbon metabolism can dramatically increase photosynthetic rate: A numerical simulation using an evolutionary algorithm. Plant Physiol. 145: 513–526 [DOI] [PMC free article] [PubMed] [Google Scholar]