

Abstract
The sigma-1 receptor is widely distributed in the central nervous system and periphery. Originally mischaracterized as an opioid receptor, the sigma-1 receptor binds a vast number of synthetic compounds but does not bind opioid peptides; it is currently considered an orphan receptor. The sigma-1 receptor pharmacophore includes an alkylamine core, also found in the endogenous compound N,N-dimethyltryptamine (DMT). DMT acts as a hallucinogen, but its receptor target has been unclear. DMT bound to sigma-1 receptors and inhibited voltage-gated sodium ion (Na+) channels in both native cardiac myocytes and heterologous cells that express sigma-1 receptors. DMT induced hypermobility in wild-type mice but not in sigma-1 receptor knockout mice. These biochemical, physiological, and behavioral experiments indicate that DMT is an endogenous agonist for the sigma-1 receptor.
The sigma-1 receptor binds a broad range of synthetic compounds (1). It has long been suspected that the sigma-1 receptor is targeted by endogenous ligands, and several candidates have been proposed (2, 3). Although progesterone and other neuroactive steroids are known to bind sigma-1 receptors and regulate some of their functions (1, 4), they do not exhibit agonist properties on sigma-1–regulated ion channels in electrophysiological experiments (5).
Our search for a sigma receptor endogenous ligand (or ligands) was based on a variant of the canonical sigma-1 receptor ligand pharmacophore (6), but with a more basic structure (Fig. 1A). Otherwise dissimilar sigma-1 receptor ligands possess a common N-substituted pharmacophore (Fig. 1A): an N,N-dialkyl or N-alkyl-N-aralkyl product, most easily recognized in the high-affinity sigma-1 receptor ligand, fenpropimorph (7). Similar chemical backbones can be derived from other sigma-1 receptor ligands such as haloperidol and cocaine (Fig. 1A). N-substituted trace amines harbor this sigma-1 receptor ligand pharmacophore, but their interactions with sigma receptors have not been determined. Of particular interest is the only known endogenous mammalian N,N-dimethylated trace amine, N,N-dimethyltryptamine (DMT) (8–10). In addition to being one of the active compounds in psychoactive snuffs (yopo, epená) and sacramental teas (ayahuasca, yagé) used in native shamanic rituals in South America, DMT can be produced by enzymes in mammalian lung (11) and in rodent brain (12). DMT has been found in human urine, blood, and cerebrospinal fluid (9, 13). Although there are no conclusive quantitative studies measuring the abundance of endogenous DMT because of its rapid metabolism (14), DMT concentrations can be localized and elevated in certain instances. Evidence suggests that DMT can be locally sequestered into brain neurotransmitter storage vesicles and that DMT production increases in rodent brain under environmental stress (8). Although a family of heterotrimeric GTP-binding protein (G protein)–coupled receptors (GPCRs) known as the trace amine receptors (TARs) was discovered in 2001 (15), only two members of this family respond to trace amines and have been renamed trace amine-associated receptors (TAARs) (16). Because other binding targets for trace amines and DMT are likely (8), we first examined the sigma-1 receptor binding affinities of the trace amines and their N-methylated and N,N-dimethylated counterparts.
Fig. 1.

Sigma-1 receptor ligand pharmacophore and binding affinities. (A) A basic sigma-1 receptor ligand pharmacophore variant of Glennon et al. (6) was derived by removal of the red bonds from the sigma-1 receptor ligands fenpropimorph, haloperidol, and cocaine. (B) Competitive binding curves of tryptamine,N-methyltryptamine, and DMT, against the radioactive sigma-1 receptor ligand [3H]-(+)-pentazocine. Curves are shown as percent specific binding (5 μM haloperidol). (C) Sigma-1 and sigma-2 receptor Kd values of trace amines and their N-methylated and N,N-dimethylated derivatives (scheme S2). Included are SEM values (n = 3 binding experiments) and R2 values for a nonlinear regression curve fit. Solid arrows denote the direction of increasing affinity.
Competition assays against the sigma-1 receptor–specific ligand, (+)-[3H]-pentazocine (10 nM), determined that the nonmethylated trace amines tryptamine, phenethylamine, and tyramine bound the sigma-1 receptor poorly (Fig. 1C), with dissociation constant (Kd) values of 431, 97.4, and >30,000 μM, respectively. By contrast, the N-methylated and N,N-dimethylated derivatives of these compounds bound sigma-1 receptors more tightly, with a clear increase in affinity as the ligands approached the sigma-1 receptor ligand pharmacophore (Fig. 1, A and B). With the exception of the N-methylated tyramines, this trend did not apply to the sigma-2 receptor, which differs pharmacologically and functionally from the sigma-1 receptor (Fig. 1C). Tryptamine, phenethylamine, and N-methyltyramine had the highest sigma-2 receptor affinities, with Kd values of 4.91, 7.31, and 6.61 μM, respectively. In contrast to sigma-1 receptors, N-methylation and N,N-dimethylation of tryptamine and phenethylamine decreased sigma-2 receptor affinity (Fig. 1C).
We tested the ability of tryptamine, N-methyl-tryptamine, and DMT to block sigma receptor photolabeling in rat liver homogenates by two radioactive photoaffinity labels, the sigma-1 receptor–specific cocaine derivative 3-[125I]iodo-4-azidococaine ([125I]-IACoc) (17) (Fig. 2A) and the sigma-1 and sigma-2 receptor fenpropimorph derivative 1-N-(2′,6′-dimethyl-morpholino)-3-(4-azido-3-[125I]iodo-phenyl) propane ([125I]IAF) (18) (Fig. 2B). Both of these compounds have been used to identify the drug binding region of the sigma-1 receptor (18, 19). As anticipated, [125I]-IACoc [sigma-1 Kd = 0.126 nM (17)] photolabeling of the 26-kD sigma-1 receptor (Fig. 2A) was protected best by DMT, with 61% protection by 50 μM DMT and almost 100% protection by 100 μM DMT. By contrast, tryptamine and N-methyltryptamine protected minimally against sigma-1 receptor [125I]-IACoc photolabeling, even at these high concentrations (Fig. 2A). Similarly, [125I]IAF photolabeling of the sigma-1 [Kd = 194 nM (18)] receptor showed that DMT was the most potent protector. Ten micromolar DMT provided 31% protection, whereas 50 and 100 μM DMT provided 43 and 69% protection, respectively (Fig. 2B). With the exception of N-methyltryptamine, protection of [125I]IAF sigma-2 [Kd = 2780 nM (18)] receptor photolabeling paralleled the sigma-2 binding data. Tryptamine afforded the greatest protection of sigma-2 receptor photolabeling, with values of 47, 78, and 79% for 10, 50, and 100 μM, respectively (Fig. 2B).
Fig. 2.

Tryptamine, N-methyltryptamine, and DMT inhibition of photolabeling. Rat liver membranes (100 μg per lane) were suspended in the presence or absence of the protecting drugs. Samples were photolyzed with (A) 1 nM carrier-free [125I]-IACoc or (B) 1 nM carrier-free [125I]IAF. Ten micromolar (+)-pentazocine (P) protected sigma-1 receptor photolabeling, whereas 10 μM haloperidol (H) protected both sigma-1 and sigma-2 receptors. Percent band intensities are shown as compared to controls performed in the absence of protecting ligand (−).
An important biological activity of sigma receptor activation is the inhibition of ion channels, which operates through protein-protein interactions without mediation by G proteins and protein kinases (20–22). In addition to modulating various types of voltage-activated K+ channels (21, 23, 24), the sigma-1 receptor associates with the Kv1.4 K+ channel in posterior pituitary nerve terminals, as well as in Xenopus oocytes (22). Sigma receptor ligands also modulate N-, L-, P/Q-, and R-type Ca2+ channels in rat sympathetic and parasympathetic neurons (25). Sigmareceptor ligands modulate cardiac voltage-gated Na+ channels (hNav1.5) in human embryonic kidney 293 (HEK293) cells, COS-7 cells, and neonatal mouse cardiac myocytes (26). To evaluate the capacity of DMT to induce physiological responses by binding to sigma receptors, we examined the action of DMT on voltage-activated Na+ current. Patch-clamp recordings from HEK293 cells stably expressing the human cardiac Na+ channel hNav1.5 revealed voltage-activated Na+ currents (INa) in response to voltage steps from −80 to −10 mV (Fig. 3B). Application of 100 μM DMT inhibited INa by 62 ± 3% (n = 3 HEK293 cells), which reversed upon DMT removal. With hNav1.5 transiently transfected into COS-7 cells, 100 μM DMT inhibited INa by only 22 ± 4% (n = 3 COS-7 cells), but photolabeling has shown that these cells have much lower concentrations of endogenous sigma-1 receptors compared to HEK293 cells (fig. S1 and Fig. 3B). The difference between DMT inhibition of INa in HEK293 and COS-7 cells (Fig. 3B, P < 0.03) thus demonstrates the dependence of INa inhibition on sigma-1 receptors. Experiments in cardiac myocytes demonstrated the same DMT action in a native preparation (Fig. 3C) and enabled further demonstration of sigma-1 receptor dependence by using a sigma-1 receptor knockout mouse (27). [125I]IAF photolabeling of liver homogenates from wild-type (WT) and sigma-1 receptor knockout (KO) mice indeed showed the absence of sigma-1 receptor (26 kD) in the KO samples (Fig. 3A). In WT neonatal cardiac myocytes, 100 μM DMT reversibly inhibited INa by 29 ± 3% (n = 7 WT myocytes), whereas INa was reduced by only 7 ± 2% (n = 7 KO myocytes) in KO myocytes (Fig. 3C, P < 0.002).
Fig. 3.

Sodium channel inhibition by DMT. (A) In the presence or absence of 10 μM haloperidol, wild type (WT) orsigma-1receptorknock-out (KO) mouse liver homogenates (200 μg/lane) were photolabeled with 1 nM [125I]IAF. (B) Examples of INa evoked by steps from −80 to −10 mV in HEK293 or COS-7 cells expressing hNav1.5 channel in the absence (control, black), presence (DMT, red), and after wash out (recovery, blue) of 100 μM DMT. Average inhibition by DMT was determined by measuring peak INa. Bars represent mean ± SEM (n = 3 cells). INa inhibition in HEK293 cells differed significantly from that in COS-7 cells (*P < 0.03). (C) Examples of INa evoked as described in (B) in neonatal cardiac myocytes from WT and KO mice in the absence (control, black), presence (DMT, red), and after wash out (recovery, blue) of 100 μM DMT. Current inhibition in WT was significantly different from that in KO (*P < 0.002, n = 7 neonatal cardiac myocytes).
Both DMT and sigma receptor ligands influence animal behavior. DMT injection induces hypermobility in rodents concurrently treated with the monoamine oxidase inhibitor pargyline (28), and this action is not antagonized by blockers of dopamine or serotonin receptors, but is potently inhibited by haloperidol (28). Although haloperidol is thought to act in part through the dopamine D2 receptor system, it is also a potent sigma-1 receptor agonist [sigma-1 inhibition constant (Ki) = 3 nM (29); sigma-2 Ki = 54 nM (29)] when inhibiting voltage-gated ion channels (5, 25). Haloperidol reduces brain concentrations of DMT (8) and DMT inhibits haloperidol binding in brain tissue more robustly than the dopamine agonist apomorphine (8). On the basis of these findings, which were discovered before sigma receptor identification, DMT has been hypothesized to act through an unknown “hallucinogen” receptor (8). We confirmed results (28) that intraperitoneal (ip) administration of DMT (2 mg per kilogram of body weight) 2 hours after pargyline (75 mg/kg, ip) injection induced hypermobility in WT mice (7025 ± 524.1 cm, n = 12 WT mice) in an open-field assay. Identical drug treatments in sigma-1 receptor KO mice had no hypermobility action (2328 ± 322.9 cm, n = 12 KO mice, P < 0.0001; Fig. 4, A and B). This result is particularly important to our understanding of sigma-1 receptor biological function because the KO mice are viable and fertile (27). The sigma-1 receptor dependence of DMT-induced hypermobility parallels that induced by the sigma-1 receptor ligand (+)-SKF10047 in WT but not in KO mice (27). As a positive control, methamphetamine, which is thought to act through catecholaminergic systems, induced hypermobility in both WT and KO mice (3 mg/kg, ip, n = 6 mice; Fig. 4, B and C) with a reduced onset rate compared with that seen for DMT (Fig. 4, A and C). This indicates that behavioral actions of DMT depend on the sigma-1 receptor, which may provide an alternative research area for psychiatric disorders that have not been linked to dopamine or N-methyl-D-aspartate systems.
Fig. 4.
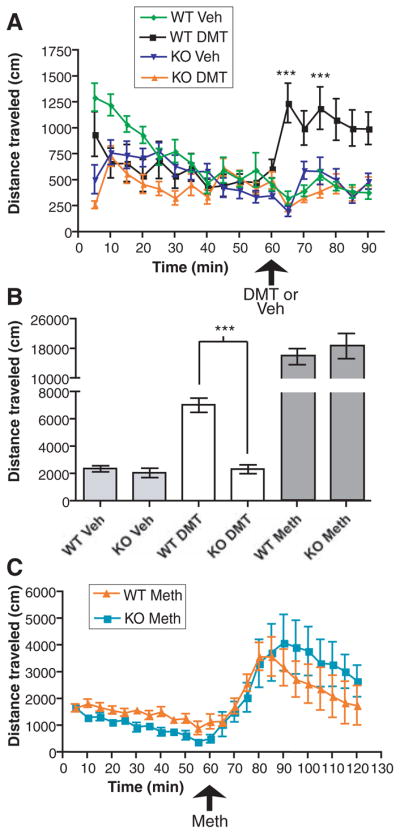
DMT-induced hypermobility abrogated in the sigma-1 KO mouse. (A) Distances traveled by WT and KO mice were measured in an open-field assay in 5-min increments. Pargyline was injected 2 hours before DMT or vehicle (Veh) ip injection. Bars represent mean ± SEM (n = 8 to 14 mice). WT mice showed a significant (***P < 0.0001) increase in mobility in response to DMT as compared to KO mice. (B) Total distance traveled over 30 min after DMT, vehicle (Veh), or methamphetamine (Meth, n = 6 mice) injection in WT and KO mice. (C) Methamphetamine serves as a positive control for hypermobility in KO mice.
The binding, biochemical, physiological, and behavioral studies reported here all support the hypothesis that DMT acts as a ligand for the sigma-1 receptor. On the basis of our binding results and the sigma-1 receptor pharmacophore, endogenous trace amines and their N-methyl and N,N-dimethyl derivatives are likely to serve as endogenous sigma receptor regulators. Moreover, DMT, the only known mammalian N,N-dimethylated trace amine, can activate the sigma-1 receptor to modulate Na+ channels. The recent discovery that the sigma-1 receptor functions as a molecular chaperone (30) may be relevant, because sigma-1 receptors, which are observed in the endoplasmic reticulum, associate with plasma membrane Kv 1.4 channels (22) and may serve as a molecular chaperone for ion channels. Furthermore, the behavioral effect of DMT may be due to activation or inhibition of sigma-1 receptor chaperone activity instead of, or in addition to, DMT/sigma-1 receptor modulation of ion channels. These studies thus suggest that this natural hallucinogen could exert its action by binding to sigma-1 receptors, which are abundant in the brain (1, 27). This discovery may also extend to N,N-dimethylated neurotransmitters such as the psychoactive serotonin derivative N,N-dimethylserotonin (bufotenine), which has been found at elevated concentrations in the urine of schizophrenic patients (10). The finding that DMT and sigma-1 receptors act as a ligand-receptor pair provides a long-awaited connection that will enable researchers to elucidate the biological functions of both of these molecules.
Supplementary Material
Acknowledgments
We thank the Corinna Burger laboratory for use of their mouse behavior equipment, and A. Paul and T. Mavlyutov for providing [125I]IAF and [125I]-IACoc, respectively. Supported by the Molecular and Cellular Pharmacology (MCP) Graduate Program training grant from NIH T32 GM08688 and by the NIH Ruth L. Kirschstein National Research Service Award (NRSA) (F31 DA022932) from the National Institute on Drug Abuse (to D.F.). This work was funded by NIH grants R01 MH065503 (to A.E.R.) and NS30016 (to M.B.J.).
Footnotes
References and Notes
- 1.Hayashi T, Su TP. CNS Drugs. 2004;18:269. doi: 10.2165/00023210-200418050-00001. [DOI] [PubMed] [Google Scholar]
- 2.Bouchard P, et al. Eur J Neurosci. 1995;7:1952. doi: 10.1111/j.1460-9568.1995.tb00718.x. [DOI] [PubMed] [Google Scholar]
- 3.Su TP, Weissman AD, Yeh SY. Life Sci. 1986;38:2199. doi: 10.1016/0024-3205(86)90572-2. [DOI] [PubMed] [Google Scholar]
- 4.Su TP, London ED, Jaffe JH. Science. 1988;240:219. doi: 10.1126/science.2832949. [DOI] [PubMed] [Google Scholar]
- 5.Wilke RA, et al. J Physiol. 1999;517:391. doi: 10.1111/j.1469-7793.1999.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glennon RA, et al. J Med Chem. 1994;37:1214. doi: 10.1021/jm00034a020. [DOI] [PubMed] [Google Scholar]
- 7.Moebius FF, Reiter RJ, Hanner M, Glossmann H. Br J Pharmacol. 1997;121:1. doi: 10.1038/sj.bjp.0701079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barker SA, Monti JA, Christian ST. Int Rev Neurobiol. 1981;22:83. doi: 10.1016/s0074-7742(08)60291-3. [DOI] [PubMed] [Google Scholar]
- 9.Franzen F, Gross H. Nature. 1965;206:1052. doi: 10.1038/2061052a0. [DOI] [PubMed] [Google Scholar]
- 10.Jacob MS, Presti DE. Med Hypotheses. 2005;64:930. doi: 10.1016/j.mehy.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Axelrod J. Science. 1961;134:343. doi: 10.1126/science.134.3475.343. [DOI] [PubMed] [Google Scholar]
- 12.Saavedra JM, Axelrod J. Science. 1972;175:1365. doi: 10.1126/science.175.4028.1365. [DOI] [PubMed] [Google Scholar]
- 13.Beaton JM, Morris PE. Mech Ageing Dev. 1984;25:343. doi: 10.1016/0047-6374(84)90007-1. [DOI] [PubMed] [Google Scholar]
- 14.Burchett SA, Hicks TP. Prog Neurobiol. 2006;79:223. doi: 10.1016/j.pneurobio.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Borowsky B, et al. Proc Natl Acad Sci USA. 2001;98:8966. doi: 10.1073/pnas.151105198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindemann L, et al. Genomics. 2005;85:372. doi: 10.1016/j.ygeno.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 17.Kahoun JR, Ruoho AE. Proc Natl Acad Sci USA. 1992;89:1393. doi: 10.1073/pnas.89.4.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pal A, et al. Mol Pharmacol. 2007;72:921. doi: 10.1124/mol.107.038307. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y, Hajipour AR, Sievert MK, Arbabian M, Ruoho AE. Biochemistry. 2007;46:3532. doi: 10.1021/bi061727o. [DOI] [PubMed] [Google Scholar]
- 20.Lupardus PJ, et al. J Physiol. 2000;526:527. doi: 10.1111/j.1469-7793.2000.00527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang H, Cuevas J. J Pharmacol Exp Ther. 2005;313:1387. doi: 10.1124/jpet.105.084152. [DOI] [PubMed] [Google Scholar]
- 22.Aydar E, Palmer CP, Klyachko VA, Jackson MB. Neuron. 2002;34:399. doi: 10.1016/s0896-6273(02)00677-3. [DOI] [PubMed] [Google Scholar]
- 23.Wilke RA, et al. J Biol Chem. 1999;274:18387. doi: 10.1074/jbc.274.26.18387. [DOI] [PubMed] [Google Scholar]
- 24.Kennedy C, Henderson G. Neuroscience. 1990;35:725. doi: 10.1016/0306-4522(90)90343-3. [DOI] [PubMed] [Google Scholar]
- 25.Zhang H, Cuevas J. J Neurophysiol. 2002;87:2867. doi: 10.1152/jn.2002.87.6.2867. [DOI] [PubMed] [Google Scholar]
- 26.Johannessen MA, Ramos-Serrano A, Ramachandran S, Ruoho AE, Jackson MB. Sigma receptor modulation of voltage-dependent sodium channels. Program No. 466.22, Annual Neuroscience Meeting; San Diego, CA. 5 November 2007. [Google Scholar]
- 27.Langa F, et al. Eur J Neurosci. 2003;18:2188. doi: 10.1046/j.1460-9568.2003.02950.x. [DOI] [PubMed] [Google Scholar]
- 28.Jenner P, Marsden CD, Thanki CM. Br J Pharmacol. 1980;69:69. doi: 10.1111/j.1476-5381.1980.tb10884.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsumoto RR, Pouw B. Eur J Pharmacol. 2000;401:155. doi: 10.1016/s0014-2999(00)00430-1. [DOI] [PubMed] [Google Scholar]
- 30.Hayashi T, Su TP. Cell. 2007;131:596. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.