

Abstract
The mechanisms underlying the exaggerated distal airway inflammation and hyperresponsiveness that characterize acute exacerbations of asthma are largely unknown. Using BALB/c mouse experimental models, we demonstrated a potentially important role for alveolar macrophages (AM) in the development of an allergen-induced exacerbation of asthma. To induce features of airway inflammation and remodeling characteristic of mild chronic asthma, animals were systemically sensitized and exposed to low mass concentrations (≈3 mg/m3) of aerosolized ovalbumin for 30 minutes per day, 3 days per week, for 4 weeks. A subsequent single moderate-level challenge (≈30 mg/m3) was used to trigger an acute exacerbation. In chronically challenged animals, cytokine expression by AM was not increased, whereas after an acute exacerbation, AM exhibited significantly enhanced expression of proinflammatory cytokines, including interleukin (IL) 1β, IL-6, CXCL-1, and tumor necrosis factor α. In parallel, there was a marked increase in the expression of several cytokines by CD4+ T-lymphocytes, notably the Th2 cytokines IL-4 and IL-13. Importantly, AM from an acute exacerbation stimulated the expression of Th2 cytokines when cocultured with CD4+ cells from chronically challenged animals, and their ability to do so was significantly greater than AM from either chronically challenged or naïve controls. Stimulation was partly dependent on interactions involving CD80/86. We conclude that in an acute exacerbation of asthma, enhanced cytokine expression by AM may play a critical role in triggering increased expression of cytokines by pulmonary CD4+ T-lymphocytes.
Asthma is an acute-on-chronic inflammatory disease of the airways, typified by reversible airflow obstruction, inflammation, and airway hyperresponsiveness (AHR). Acute episodes, or exacerbations of asthma, account for the majority of health care resource utilization events.1 Exacerbations are characterized by inflammation in the distal airways, increased recruitment of eosinophils and neutrophils, diminished airflow, and exaggerated AHR.2 Although respiratory viral infections are the most common trigger of exacerbations, they may also be initiated by exposure to high levels of allergens, and both these factors interact to increase the risk and severity of acute exacerbations.3–4
In chronic asthma, there is significant accumulation of CD4+ T lymphocytes in the airways.5 These cells typically exhibit a so-called Th2 cytokine profile, characterized by production of interleukin (IL) 4, IL-5, and IL-13, which contribute to the recruitment of eosinophils and to the development of AHR.6–8 In addition, accumulating evidence from both human and animal studies indicates a role for Th1 cytokines such as interferon (IFN)-γ in asthma.9 More recently, it has been suggested that Th17 cells, which secrete IL-17A, may also contribute to the pathogenesis of the disease.10–11
Alveolar macrophages (AM) have long been recognized to be activated in asthmatic inflammation, with evidence of increased secretion of cytokines such as tumor necrosis factor (TNF) α and IL-6.12 Activation of AM can be triggered by allergen challenge.13–14 AM from atopic asthmatics, but not from atopic nonasthmatics or nonatopic individuals, have the capacity to stimulate peripheral blood CD4+ T-lymphocytes to secrete Th2 cytokines such as IL-5.15 This could be triggered by cytokines secreted by AM or by antigen presentation in combination with co-stimulatory molecules such as CD80 and CD86. However, whether pulmonary CD4+ T-lymphocytes can similarly be activated by AM during acute exacerbations of asthma is unknown. Human studies are difficult to undertake, while animal experiments have hitherto been limited by the lack of suitable models of acute exacerbations of chronic asthma.
We have previously described a murine model of mild chronic asthma in which sensitized animals are challenged with a low mass concentration of aerosolized ovalbumin for 6 weeks. This elicits airway-specific inflammation, with eosinophil recruitment on a background of chronic inflammation, accompanied by changes of structural remodeling of the airways and development of hyperresponsiveness.16 The model is now widely recognized as uniquely replicating key features of chronic asthma in patients.17–18 More recently, we have developed a model of an allergen-induced acute exacerbation of chronic asthma19–20 based on the chronic model. After 4 weeks of low-level challenge, mice are exposed on a single occasion to a 10-fold higher concentration of aerosolized ovalbumin. This moderate-level challenge, which still uses a much lower mass concentration than in conventional animal models of allergic airway inflammation, triggers rapid and enhanced accumulation of inflammatory cells, including eosinophils and neutrophils, around intrapulmonary airways. Furthermore, it is associated with a distinct pattern of AHR arising from the small distal airways,19 similar to that seen in clinical acute exacerbations. The overall severity of inflammation is much less marked than in conventional short-term models. While all experimental models of asthma have significant limitations, this has been recognized as a valid murine model that more closely resembles acute asthma in patients.18
In the present study, we investigated the profile of expression of cytokines by AM and pulmonary CD4+ T-lymphocytes in our model of an acute exacerbation and compared it to the model of mild chronic asthma. We found that in the model of mild asthma, cytokine expression by AM was not increased. Induction of an acute exacerbation by moderate-level challenge elicited greatly enhanced expression of the proinflammatory cytokines IL-1β, IL-6, CXCL-1, and TNF-α. In parallel, there was a marked increase in expression and secretion of the Th2 cytokines IL-4, IL-5, and IL-13 by CD4+ T cells. Importantly, in coculture experiments we showed that AM from an acute exacerbation could selectively induce Th2 cytokine production by primed T cells, and that this was in part mediated by interactions involving CD80/86.
Materials and Methods
Mice, Sensitization, and Challenge
The protocols used for sensitization and inhalational challenge have previously been described.19 Briefly, specific pathogen-free female BALB/c mice aged 7–8 weeks (Animal Resources Centre, Perth, Western Australia) were systemically sensitized by intraperitoneal injection of 50 μg of alum-precipitated chicken egg ovalbumin (OVA) (Grade V, ≥98% pure, Sigma, Sydney, Australia) 21 and 7 days before inhalational challenge, then exposed to aerosolized OVA in a whole body inhalation exposure chamber (Unifab Corporation, Kalamazoo, MI).16 Chronic low-level challenge involved exposure to ≈3 mg/m3 aerosolized OVA for 30 minutes per day on 3 days per week for 4 weeks. At the end of this period, a single-moderate level challenge (≈30 mg/m3) was used to induce an acute exacerbation. Particle concentration within the chamber was continuously monitored using a DustTrak 8520 instrument (TSI, St Paul, MN). All experimental procedures complied with the requirements of the Animal Care and Ethics Committee of the University of New South Wales (reference numbers: 06/119B and 08/09B). Experimental groups, each comprising 6–8 animals, included mice that received chronic challenge with OVA aerosol for 4 weeks and mice in which an acute exacerbation was induced by an additional single moderate-level challenge with aerosolized OVA. Control groups included mice which were given a single moderate-level exposure to aerosolized OVA without prior chronic challenge and a group of naïve animals. For most readouts, results were replicated in at least 3 separate experiments.
To assess whether endotoxin (LPS) contamination of OVA might have contributed to the induction of airway inflammation and cellular response in an acute exacerbation, results were compared with an experiment in which the final moderate-level challenge was performed using low-endotoxin OVA (EndoGrade ovalbumin, endotoxin concentration <1 EU/mg, Hyglos GmbH, Regensburg, Germany).
Cell Isolation
Mice were killed by exsanguination after an overdose of sodium pentobarbital 4 hours after the final airway challenge (this time point was selected on the basis of our earlier studies using this model),19 and lungs were perfused with saline to remove blood from the pulmonary capillary bed. Bronchoalveolar lavage (BAL) was performed by inflating the lungs with 2 × 1 ml of PBS. Recovered fluid was centrifuged and the supernatant collected and stored at −20°C for immunoassays. In the moderate-challenge group, induction of an acute exacerbation was confirmed by a subsequent leukocyte differential count (lymphocytes 6.8 ± 1.1% vs 0.9 ± 0.2% in naïve animals, eosinophils 1.5 ± 0.5% versus 0.0 ± 0.0%, neutrophils 11.2 ± 0.4% vs 1.1 ± 0.5%, P < 0.001 for all comparisons). To purify AM, BAL cells were resuspended in RPMI-1640 and incubated at 37°C in 6- or 24-well plates for 30 minutes. Plates were washed at least four times to remove nonadherent cells, after which AM were used for coculture experiments or resuspended in TriReagent (Sigma) for extraction of RNA. Adherent cells were >90% AM by morphological criteria and immunostaining for F4/80.
For isolation of CD4+ T-lymphocytes, two pairs of lungs were pooled for each sample and diced into fine fragments (<0.3 mm3). Tissue was dispersed in ≈3 ml of a mixture of Type IV collagenase with minimal nonspecific protease activity (Worthington Biochemical, Lakewood, NJ) and DNase (Roche Diagnostics, Sydney, Australia). After incubation at 37°C on a roller mixer for 40 minutes, samples were vortexed and passed through a 70-μm cell strainer to release cells. Cells were resuspended in erythrocyte lysis buffer and washed with PBS. This yielded a mixed population of cells, the majority of which were identified as lymphocytes and macrophages on a Leishman's-stained smear. Cell viability was >90% for all preparations.
CD4+ T-lymphocytes were purified using a FlowComp magnetic bead isolation kit (Invitrogen, Carlsbad, CA) according to the manufacturer's protocols. Cell purity was assessed by flow cytometry, staining with conjugated antibodies to CD3 (FITC), CD4 (PerCP), and CD8 (PE) or with appropriate isotype controls (BD Pharmingen, San Jose, CA). Lymphocytes were selected by gating the population on the basis of forward and side scatter and were confirmed by CD3+ staining. Analysis was on a FACSort (Becton Dickinson, Mountain View, CA) using CellQuest Pro software (BD Pharmingen). The isolated lymphocyte population was determined to be ≈98.0% CD3+/CD4+.
Coincubation of AM and CD4+ T-Lymphocytes
Coincubation of adherent AM with nonadherent CD4+ T-lymphocytes was performed at a ratio of 1:2.5 (4 × 105: 1 × 106 cells per well) in 24-well tissue culture plates in RPMI 1640 (containing 10% FBS, 1% penicillin/streptomycin) at 37°C in an atmosphere containing 5% CO2. After 4 hours, nonadherent CD4+ T-lymphocytes were collected and lysed in TriReagent (Sigma) for extraction of RNA. In other coculture experiments, purified macrophages were incubated with or without a mouse CTLA-4 Fc chimera (2.5 μg/ml; R&D Systems, Sydney, Australia) for 30 minutes before the addition of CD4+ T-lymphocytes.
ELISpot Analysis
Assessment of cytokine-secreting cells was performed using appropriately coated plates (R&D Systems) according to the manufacturer's instructions. CD4+ T-lymphocytes were cultured at 2 × 105 cells per well in RPMI 1640 (containing 10% FBS, 1% penicillin/streptomycin) at 37°C in an atmosphere containing 5% CO2 for 18 hours. Cytokine-producing cells were detected using streptavidin-alkaline phosphotase (MabTech, Sydney, Australia) followed by BCIP/NBT substrate (Sigma) and counted using an ELISpot reader (Autoimmun Diagnostika GmbH, Strassberg, Germany).
Cytokine Assays
The concentration of cytokines in undiluted BAL fluid was measured using a Bioplex assay (Mouse 23-Plex panel, Biorad Laboratories, Hercules, CA) according to the manufacturer's instructions.
RNA Isolation and PCR Analysis
RNA was isolated from CD4+ T-lymphocytes and AM using TriReagent (Sigma) according to the manufacturer's instructions. After DNase treatment (Turbo DNase, Ambion, Scoresby, Australia), samples were reverse transcribed into cDNA using Superscript III (Invitrogen). Semiquantitative real-time PCR was used to assess expression of cytokines with detection of amplified products using SYBR green. Primers were custom-designed in house to allow the use of identical thermocyler conditions, thus permitting simultaneous assessment of multiple cytokines. Reactions were performed using an ABI Prism 7700 Sequence Detector (Applied Biosystems, Melbourne, Australia) and expression was normalized to HPRT.
Statistical Analysis
In general, data are presented as arithmetic means ± SEM. Bioplex data are shown as log-transformed mean relative expression in a heat map generated using the R statistical/graphics language (http://www.r-project.org/, last accessed December 15, 2009). Data were analyzed by a Kruskal–Wallis test or one-way analysis of variance followed by a Dunn's or Bonferroni posttest as appropriate. A P value of <0.05 was considered significant. The software package GraphPad Prism 5.01 (GraphPad Software, San Diego, CA) was used for data analysis and preparation of graphs.
Results
Cytokine Expression by AM
After chronic low-level challenge alone, or after a single moderate-level challenge, AM isolated by adherence for 30 minutes did not exhibit significantly elevated expression of mRNA for TNF-α, IL-1β, IL-6, or CXCL-1/KC. However, after induction of an acute exacerbation, expression of mRNA for these proinflammatory cytokines was markedly elevated, relative to both naïve and chronically challenged animals (Figure 1, A–D). Because longer periods of adherent culture nonspecifically activated naïve AM (data not shown) it was not possible to compare cytokine secretion by AM from the various groups of OVA-challenged mice.
Figure 1.
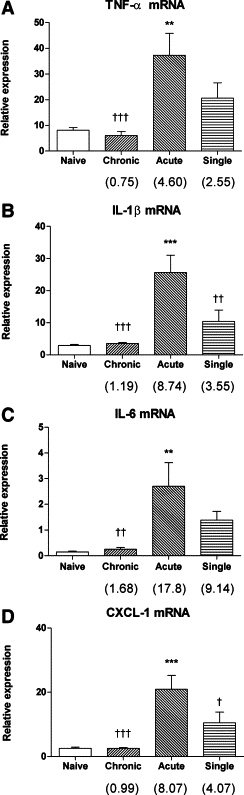
Enhanced expression of mRNA for major proinflammatory cytokines by AM from the acute exacerbation group: TNF-α (A), IL-1β (B), IL-6 (C), and CXCL-1/KC (D). Data are the mean ± SEM (n = 6 samples per group) relative to HPRT. Fold change relative to the naïve group is shown in parentheses. Significant differences relative to the naïve group are as shown as **P < 0.01 and ***P < 0.001; relative to the acute exacerbation group are shown as †P < 0.05, ††P < 0.01, and †††P < 0.001.
Although the mass concentrations of aerosolized OVA delivered in our model are much lower than in conventional mouse models of allergic airway inflammation, it was possible that endotoxin, which frequently contaminates OVA preparations,21 might have contributed to the induction of airway inflammation and cellular responses in an acute exacerbation. Therefore, we also assessed macrophage responses in an experiment in which the final moderate-level challenge was performed using low-endotoxin OVA. As shown in Table 1, the effect of low-endotoxin OVA in stimulating expression of mRNA for proinflammatory cytokines by AM was indistinguishable from that of conventional OVA.
Table 1.
Stimulation of AM after Induction of an Acute Exacerbation by Conventional or Low-Endotoxin OVA
| Cytokine | Conventional OVA (Sigma) | Low-Endotoxin OVA (Hyglos) |
|---|---|---|
| TNF-α | 4.6 ± 1.1 | 5.2 ± 1.7 |
| IL-1β | 8.7 ± 1.8 | 11.3 ± 3.5 |
| IL-6 | 16.3 ± 5.2 | 13.2 ± 2.8 |
| CXCL-1 | 6.9 ± 1.8 | 7.9 ± 1.5 |
Stimulation of expression of cytokine mRNA relative to the corresponding naïve control AM (n = 6 samples per group).
Cytokine Expression by CD4+ T-Lymphocytes
The numbers of CD4+ T-lymphocytes obtained from all groups of challenged animals were increased approximately twofold compared to naïve mice. Relative expression of mRNA for the Th2 cytokines IL-4, IL-5, and IL-13, as well as for IL-10 and IL-17A, was modestly increased in the chronic challenge group compared to naïve mice, while expression of mRNA for IFN-γ was significantly increased (P < 0.05) (Figure 2A). In the acute exacerbation group, there was a further significant increase in expression of mRNA for IL-4, IL-10, IL-13, and MIP-1α (fold change respectively 11.53, 2.77, 2.93, and 4.09; P < 0.05 for all comparisons, see Supplemental Table S1 at http://ajp.amjpathol.org) compared to the chronic challenge group, together with moderately increased expression of IL-5 (Figure 2A). However, there was no further increase in expression of mRNA for other cytokines. Levels of expression in the acute exacerbation group were in general significantly higher than in the single moderate-level challenge group, except for the chemokines MIP-1α and RANTES.
Figure 2.
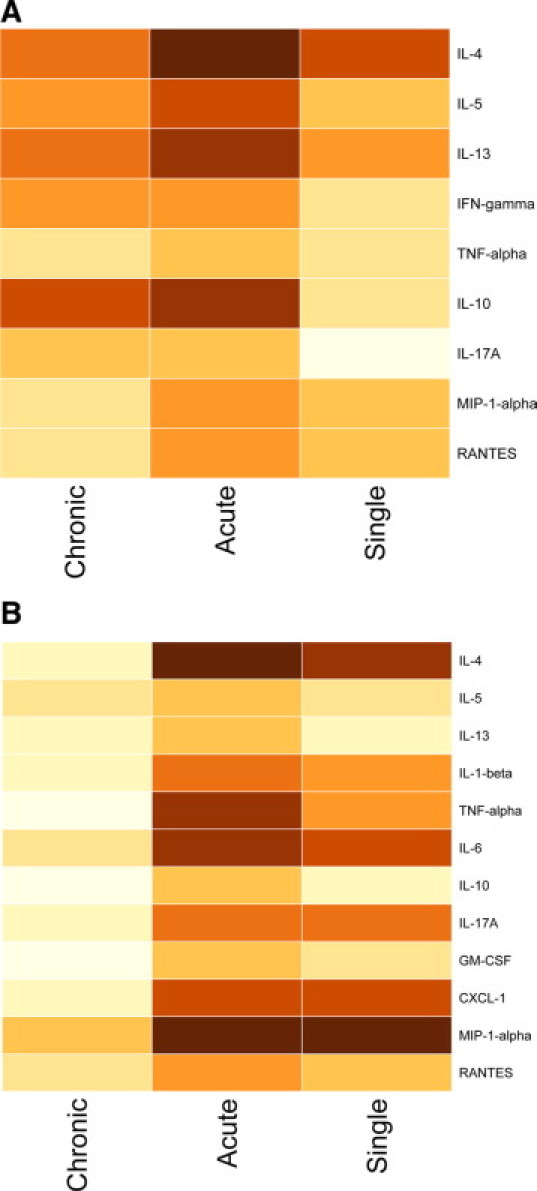
Heat maps illustrating expression of mRNA for various cytokines by CD4+ T-lymphocytes (A) and concentrations of various cytokines in BAL fluid (B). Levels in the acute exacerbation group, compared to the chronic challenge and single moderate challenge groups, are shown as log-transformed mean fold change relative to the naïve group, converted to increasingly darker colors.
For selected cytokines, secretion by CD4+ T-cells was confirmed using ELISpot assays. The observed changes in the proportions of cells secreting IL-4 and IL-5 were consistent with the data from the mRNA studies. Thus, relative to cells from naïve animals, the proportion of IL-4–secreting cells was slightly elevated in cells from chronically challenged animals and significantly increased in cells from animals in the acute exacerbation group (P < 0.05) (Table 2). The proportion of cells secreting IL-5 was significantly increased after chronic challenge (P < 0.05) but was not increased further after an acute exacerbation.
Table 2.
ELISpot Assessment of Cytokine Secretion by Pulmonary CD4+ T Cells
Numbers of pulmonary CD4+ cells secreting IL-4 (n = 3 samples per group) and IL-5 (n = 4) expressed as fold change relative to the naïve group. Significant differences compared to the naïve group are shown as
P <0.05.
Cytokine Concentration in BAL Fluid
To confirm that elevated expression of cytokine mRNA by AM and CD4+ T-lymphocytes was paralleled by increased protein production, we assessed cytokine concentration in BAL fluid. In samples from animals that received chronic challenge alone, there was no significant increase in the concentration of any cytokines tested. At 4 hours after induction of an acute exacerbation there was a significant increase in the concentration of IL-1β, IL-4, IL-6, IL-10, and IL-13, as well as of TNF-α, GM-CSF, CXCL-1, and the eosinophil chemoattractants MIP-1α and RANTES (P < 0.001 for all, see Supplemental Table 2 at http://ajp.amjpathol.org) (Figure 2B). In animals that only received a single moderate-level challenge, there was also a significant increase in the concentration of several of these cytokines, notably IL-1β, IL-4, IL-6, CXCL-1, MIP-1α, and RANTES (Figure 2B).
Cytokine Expression by CD4+ T-Lymphocytes after Coculture with AM
To define the capacity of activated AM to stimulate CD4+ T-lymphocytes, AM recovered from naïve or chronically challenged animals, or from mice 4 hours after an acute exacerbation, were cocultured with CD4+ T-lymphocytes from chronically challenged animals (ie, primed T cells). CD4+ T cells cocultured with AM from an acute exacerbation exhibited significantly increased expression of mRNA for IL-4, −5, −6, and −13, compared to T cells cultured with AM from naïve animals (Figure 3, A–D). In contrast, expression of mRNA for these cytokines was not increased in CD4+ T cells incubated with AM from chronically challenged mice.
Figure 3.
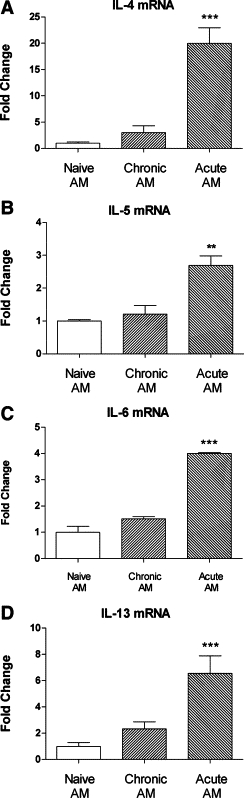
Enhanced expression of mRNA for various cytokines by primed CD4+ T cells cocultured with AM from naïve animals, chronically challenged animals, or animals in which an acute exacerbation had been induced: A: IL-4, B: IL-5, C: IL-6, and D: IL-13. Data are the mean ± SEM (n = 3 samples per group) fold change relative to naïve AM. Significant differences relative to naïve AM are shown as **P < 0.01 and ***P < 0.001.
The stimulatory effect of AM from an acute exacerbation in coculture was selective for primed CD4+ T cells. Relative to naïve T cells, there was consistent evidence of elevated expression of mRNA for IL-4 (≈10-fold), IL-5 (≈3-fold), IL-6 (≈10-fold), IL-10 (≈10-fold), and IL-13 (≈20-fold) (P < 0.05 for all comparisons). However, expression of mRNA for IFN-γ was not increased in primed as compared to naïve T cells.
To define the contribution of the CD80/86 costimulatory molecules to the activation of primed CD4+ T cells by AM, the coincubation experiments were repeated using AM from an acute exacerbation that had been preincubated with a CTLA-4 Fc chimera, to prevent signaling via CD80/86. As previously demonstrated, stimulation of primed CD4+ T cells by acutely challenged AM was observed, as measured by significantly enhanced expression of mRNA for IL-4, −5, −6, and −13 (Figure 4, A–D). In cocultures preincubated with CTLA-4 Fc chimera, there was a significant reduction in expression of mRNA for IL-5, −6, and −13, as well as a modest reduction in mRNA for IL-4 which was not statistically significant.
Figure 4.
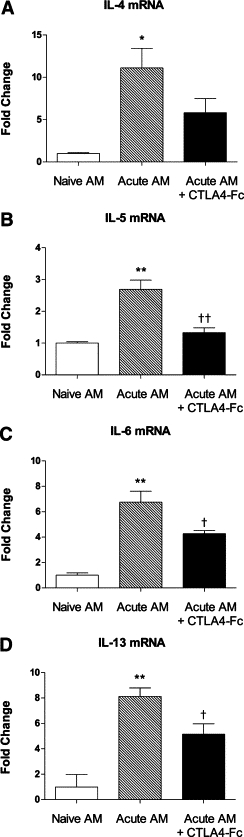
Inhibition of the enhanced expression of cytokine mRNA by primed CD4+ T cells, as a result of preincubation of cocultured AM from an acute exacerbation with CTLA-4 Fc chimera to prevent costimulation via CD80/86: A: IL-4, B: IL-5, C: IL-6, and D: IL-13. Data are the mean ± SEM (n = 6 samples per group) fold change relative to naïve AM. Significant differences relative to naïve AM are shown as *P < 0.05 and **P < 0.01; relative to non–CTLA-4-treated cultures are shown as †P < 0.05 and ††P < 0.01.
Discussion
Acute exacerbations of asthma are characterized by increased airway inflammation, which extends further distally2 and is associated with recruitment of both eosinophils and significant numbers of neutrophils.22–23 In parallel, patients develop worsening airflow obstruction and its consequences, which may be difficult to manage and can be life-threatening.1,24 The cellular and molecular mechanisms underlying these changes remain undefined. In this study, we have examined the role of alveolar macrophage- and pulmonary T cell–derived cytokines in an experimental model of an acute exacerbation of chronic asthma19 and have identified potentially important mediators by comparison with our model of mild chronic asthma.16
The only experimental difference between our model of an acute exacerbation of asthma and the model of mild chronic asthma on which it is based is the mass concentration of the final aerosol challenge with ovalbumin. Whereas the chronic asthma model involves repeated low-level exposure to ≈3 mg/m3 of ovalbumin aerosol, induction of an acute exacerbation involves a single additional moderate-level challenge with ≈30 mg/m3 of aerosolized OVA (a concentration which is still between 10- and 100-fold lower than in conventional short-term models of allergic airway inflammation). Within 4 hours, the moderate-level challenge induces more marked airway inflammation which extends further distally, together with a pattern of AHR distinct from that seen in the chronic challenge model.19 As shown in this study, these changes are associated with a significant increase in expression of a variety of cytokines. A reasonable explanation for this response is that the higher mass concentration of particles allows OVA to reach the distal airways, leading to enhanced antigen uptake and presentation. In this setting, primed CD4+ T-lymphocytes might be able to rapidly interact with antigen-presenting cells.
While dendritic cells are well recognized as playing a critical role in antigen presentation to naïve T cells and the induction of a Th2-biased response,25 other antigen-presenting cells are likely to have important roles in activating primed T cells.26 Macrophages in the alveolar space are “front line” cells in terms of host defense, can interact with dendritic cells in the uptake and presentation of particulate antigen,27 and can also independently present antigen.28 Therefore, we investigated the expression of cytokines by AM in our models. We found that there was no significant increase in the expression of mRNA for proinflammatory cytokines by AM in the chronic challenge model. However, AM expressed significantly enhanced levels of mRNA for TNF-α, IL-1β, IL-6, and CXCL-1/KC in the setting of an experimentally induced acute exacerbation. These increases were much greater than in mice that received a single moderate-level challenge, indicating that AM in the inflammatory environment of chronic asthma had an altered capacity to express cytokines after allergen challenge.
After induction of an acute exacerbation, we also found that expression by pulmonary CD4+ T-lymphocytes of a number of cytokines was strikingly increased. Notably, we found that relative expression of mRNA for the Th2 cytokines IL-4 and IL-13, already increased in the chronic challenge model, was markedly up-regulated. This was accompanied by an increase in the proportion of cells actively secreting these cytokines. While such findings are not altogether surprising in the Th2-biased immunological environment of this experimental model,29 the demonstration that expression of Th2 cytokines is elevated after induction of an acute exacerbation of asthma is novel. Currently, no information is available regarding expression of pulmonary T cell–derived cytokines during an acute exacerbation of clinical asthma. However, Katsunuma and colleagues have demonstrated enhanced expression of IL-4 by peripheral blood T-cells from children suffering an exacerbation.30 Our findings are consistent with those data.
Corresponding to the enhanced expression of mRNA for cytokines by AM and T cells, we found that levels of various proinflammatory and chemoattractant cytokines were markedly increased in BAL fluid from animals in which an acute exacerbation had been induced. Notably, these included all of the AM-derived cytokines and the Th2 cytokines for which relative expression of mRNA was increased. Of particular interest in this context are the marked elevation of concentrations of TNF-α, IL-4, IL-6, and IL-17A. The enhanced expression of TNF-α is likely to be especially relevant to the pathogenesis of asthmatic exacerbations, because inhibition of this mediator has been demonstrated to be of therapeutic value.31
A key finding of this study was that AM from an acute exacerbation exhibited an increased capacity to stimulate the production of cytokines by primed CD4+ T cells. Importantly, this enhanced expression of Th2 cytokines was absent when AM from chronically challenged or naïve animals were cultured with primed T cells. In this model of an acute exacerbation of asthma, induction of enhanced expression of Th2 cytokines in coculture was at least partially dependent on costimulation via CD80 and CD86, because the response was significantly, although not completely, abrogated when AM were preincubated with a soluble CTLA-4 Fc molecule. An important mechanism by which macrophages activate T cells is via the costimulatory molecules CD80 and 86, through the T cell coreceptor CTLA-4. Inhibition of this signaling pathway by the introduction of soluble CTLA-4-Ig has previously been shown to inhibit CD80/86 induced T cell proliferation and IL-2 production.32 Moreover, in a mouse model of allergic airway inflammation, blockade of CD86 was associated with reduced AHR and accumulation of eosinophils after ovalbumin challenge.33
Collectively, these results identify a proinflammatory role for AM in an acute exacerbation of asthma. Although AM have been shown to be able to stimulate expression of Th2 cytokines by peripheral blood T-lymphocytes,15 this is the first study to demonstrate that activated AM can promote the expression of Th2 cytokines by pulmonary CD4+ T-lymphocytes. Our findings contrast with previous reports suggesting that AM suppress Th2 responses.34–35 However, other investigators have shown that the immunosuppressive activity of AM is negated in an inflammatory environment,36 and this is undoubtedly relevant to AM in the asthmatic lung.
Although most acute exacerbations of clinical asthma are triggered by viral infection rather than by allergen exposure, our results are of particular importance in the context of identifying a common pathogenetic mechanism that links allergen- and virus-induced exacerbations. In a murine model of rhinovirus-induced exacerbation of acute allergic airway inflammation, Bartlett et al have shown that rhinovirus infection increased allergen-induced production of IL-4 and IL-13, while allergen challenge increased rhinovirus infection-induced production of IFN-γ.37 Thus enhanced expression of IL-4 and IL-13, possibly driven by AM, may be part of a “final common pathway” of the pathogenesis of acute exacerbations of asthma. The associated enhanced expression of the regulatory cytokine IL-10, and the eosinophil-selective chemoattractant cytokines MIP-1α and RANTES, may also be part of this process.
In conclusion, we have demonstrated that in a mouse model of an acute exacerbation of chronic asthma associated with worsening inflammation and AHR, expression by alveolar macrophages of a variety of proinflammatory and chemoattractant cytokines is significantly elevated. In parallel, we have shown that pulmonary CD4+ T-lymphocytes exhibit markedly enhanced expression of numerous cytokines, notably the Th2 cytokines IL-4 and IL-13. Elevated levels of all of these cytokines are present in BAL fluid. Collectively, these results suggest the possibility that after exposure to a trigger for an acute exacerbation, AM in the inflammatory environment of chronic asthma might contribute to activation of CD4+ T-lymphocytes and thus elicit cytokine-driven distal airway inflammation and its consequences. Furthermore, these results suggest that activated alveolar macrophages might be a potential target for therapeutic intervention in asthma.
Acknowledgements
We thank Dr. Taline Hampartzoumian for assistance with flow cytometry.
Footnotes
Supported by grants from the National Health and Medical Research Council of Australia.
C.H. and M.M.S. contributed equally to this study.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Web Extra Material
Expression of mRNA for various cytokines by CD4+ T cells (supplemental data for Figure 2A)
Cytokine concentrations in BAL fluid (supplemental data for Figure 2B)
References
- 1.Holgate ST. Exacerbations: the asthma paradox. Am J Respir Crit Care Med. 2005;172:941–943. doi: 10.1164/rccm.2507007. [DOI] [PubMed] [Google Scholar]
- 2.Martin RJ. Therapeutic significance of distal airway inflammation in asthma. J Allergy Clin Immunol. 2002;109:S447–S460. doi: 10.1067/mai.2002.121409. [DOI] [PubMed] [Google Scholar]
- 3.Green RM, Custovic A, Sanderson G, Hunter J, Johnston SL, Woodcock A. Synergism between allergens and viruses and risk of hospital admission with asthma: case-control study. BMJ. 2002;324:763. doi: 10.1136/bmj.324.7340.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murray CS, Poletti G, Kebadze T, Morris J, Woodcock A, Johnston SL, Custovic A. Study of modifiable risk factors for asthma exacerbations: virus infection and allergen exposure increase the risk of asthma hospital admissions in children. Thorax. 2006;61:376–382. doi: 10.1136/thx.2005.042523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 6.Foster PS, Martinez-Moczygemba M, Huston DP, Corry DB. Interleukins-4, -5, and -13: emerging therapeutic targets in allergic disease. Pharmacol Ther. 2002;94:253–264. doi: 10.1016/s0163-7258(02)00220-6. [DOI] [PubMed] [Google Scholar]
- 7.Hamelmann E, Cieslewicz G, Schwarze J, Ishizuka T, Joetham A, Heusser C, Gelfand EW. Anti-interleukin 5 but not anti-IgE prevents airway inflammation and airway hyperresponsiveness. Am J Respir Crit Care Med. 1999;160:934–941. doi: 10.1164/ajrccm.160.3.9806029. [DOI] [PubMed] [Google Scholar]
- 8.Kumar RK, Herbert C, Yang M, Koskinen AM, McKenzie AN, Foster PS. Role of interleukin-13 in eosinophil accumulation and airway remodelling in a mouse model of chronic asthma. Clin Exp Allergy. 2002;32:1104–1111. doi: 10.1046/j.1365-2222.2002.01420.x. [DOI] [PubMed] [Google Scholar]
- 9.Kumar RK, Webb DC, Herbert C, Foster PS. Interferon-gamma as a possible target in chronic asthma. Inflamm Allergy Drug Targets. 2006;5:253–256. doi: 10.2174/187152806779010909. [DOI] [PubMed] [Google Scholar]
- 10.Louten J, Boniface K, de Waal Malefyt R. Development and function of TH17 cells in health and disease. J Allergy Clin Immunol. 2009;123:1004–1011. doi: 10.1016/j.jaci.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 11.Oboki K, Ohno T, Saito H, Nakae S. Th17 and allergy. Allergol Int. 2008;57:121–134. doi: 10.2332/allergolint.R-07-160. [DOI] [PubMed] [Google Scholar]
- 12.Gosset P, Tsicopoulos A, Wallaert B, Vannimenus C, Joseph M, Tonnel AB, Capron A. Increased secretion of tumor necrosis factor alpha and interleukin-6 by alveolar macrophages consecutive to the development of the late asthmatic reaction. J Allergy Clin Immunol. 1991;88:561–571. doi: 10.1016/0091-6749(91)90149-i. [DOI] [PubMed] [Google Scholar]
- 13.Viksman MY, Bochner BS, Peebles RS, Schleimer RP, Liu MC. Expression of activation markers on alveolar macrophages in allergic asthmatics after endobronchial or whole-lung allergen challenge. Clin Immunol. 2002;104:77–85. doi: 10.1006/clim.2002.5233. [DOI] [PubMed] [Google Scholar]
- 14.Lensmar C, Katchar K, Eklund A, Grunewald J, Wahlstrom J. Phenotypic analysis of alveolar macrophages and lymphocytes following allergen inhalation by atopic subjects with mild asthma. Respir Med. 2006;100:918–925. doi: 10.1016/j.rmed.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 15.Tang C, Rolland JM, Li X, Ward C, Bish R, Walters EH. Alveolar macrophages from atopic asthmatics, but not atopic nonasthmatics, enhance interleukin-5 production by CD4+ T cells. Am J Respir Crit Care Med. 1998;157:1120–1126. doi: 10.1164/ajrccm.157.4.9706118. [DOI] [PubMed] [Google Scholar]
- 16.Temelkovski J, Hogan SP, Shepherd DP, Foster PS, Kumar RK. An improved murine model of asthma: selective airway inflammation, epithelial lesions and increased methacholine responsiveness following chronic exposure to aerosolised allergen. Thorax. 1998;53:849–856. doi: 10.1136/thx.53.10.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fulkerson PC, Rothenberg ME, Hogan SP. Building a better mouse model: experimental models of chronic asthma. Clin Exp Allergy. 2005;35:1251–1253. doi: 10.1111/j.1365-2222.2005.02354.x. [DOI] [PubMed] [Google Scholar]
- 18.Nials AT, Uddin S. Mouse models of allergic asthma: acute and chronic allergen challenge. Dis Model Mech. 2008;1:213–220. doi: 10.1242/dmm.000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siegle JS, Hansbro N, Herbert C, Yang M, Foster PS, Kumar RK. Airway hyperreactivity in exacerbation of chronic asthma is independent of eosinophilic inflammation. Am J Respir Cell Mol Biol. 2006;35:565–570. doi: 10.1165/rcmb.2006-0135OC. [DOI] [PubMed] [Google Scholar]
- 20.Ito K, Herbert C, Siegle JS, Vuppusetty C, Hansbro N, Thomas PS, Foster PS, Barnes PJ, Kumar RK. Steroid-resistant neutrophilic inflammation in a mouse model of an acute exacerbation of asthma. Am J Respir Cell Mol Biol. 2008;39:543–550. doi: 10.1165/rcmb.2008-0028OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watanabe J, Miyazaki Y, Zimmerman GA, Albertine KH, McIntyre TM. Endotoxin contamination of ovalbumin suppresses murine immunologic responses and development of airway hyper-reactivity. J Biol Chem. 2003;278:42361–42368. doi: 10.1074/jbc.M307752200. [DOI] [PubMed] [Google Scholar]
- 22.Wenzel SE, Schwartz LB, Langmack EL, Halliday JL, Trudeau JB, Gibbs RL, Chu HW. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med. 1999;160:1001–1008. doi: 10.1164/ajrccm.160.3.9812110. [DOI] [PubMed] [Google Scholar]
- 23.in't Veen JC, Smits HH, Hiemstra PS, Zwinderman AE, Sterk PJ, Bel EH. Lung function and sputum characteristics of patients with severe asthma during an induced exacerbation by double-blind steroid withdrawal. Am J Respir Crit Care Med. 1999;160:93–99. doi: 10.1164/ajrccm.160.1.9809104. [DOI] [PubMed] [Google Scholar]
- 24.McFadden ER., Jr Acute severe asthma. Am J Respir Crit Care Med. 2003;168:740–759. doi: 10.1164/rccm.200208-902SO. [DOI] [PubMed] [Google Scholar]
- 25.Lambrecht BN, Hammad H. Biology of lung dendritic cells at the origin of asthma. Immunity. 2009;31:412–424. doi: 10.1016/j.immuni.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 26.McLachlan JB, Jenkins MK. Migration and accumulation of effector CD4+ T cells in nonlymphoid tissues. Proc Am Thorac Soc. 2007;4:439–442. doi: 10.1513/pats.200606-137MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blank F, Rothen-Rutishauser B, Gehr P. Dendritic cells and macrophages form a transepithelial network against foreign particulate antigens. Am J Respir Cell Mol Biol. 2007;36:669–677. doi: 10.1165/rcmb.2006-0234OC. [DOI] [PubMed] [Google Scholar]
- 28.Hume DA. Macrophages as APC and the dendritic cell myth. J Immunol. 2008;181:5829–5835. doi: 10.4049/jimmunol.181.9.5829. [DOI] [PubMed] [Google Scholar]
- 29.Foster PS, Webb DC, Yang M, Herbert C, Kumar RK. Dissociation of T helper type 2 cytokine-dependent airway lesions from signal transducer and activator of transcription 6 signalling in experimental chronic asthma. Clin Exp Allergy. 2003;33:688–695. doi: 10.1046/j.1365-2222.2003.01647.x. [DOI] [PubMed] [Google Scholar]
- 30.Katsunuma T, Kawahara H, Suda T, Ishii T, Ohya Y, Akasawa A, Saito H, Oshida T, Sugita Y. Analysis of gene expressions of T cells from children with acute exacerbations of asthma. Int Arch Allergy Immunol. 2004;134:29–33. doi: 10.1159/000077530. [DOI] [PubMed] [Google Scholar]
- 31.Berry M, Brightling C, Pavord I, Wardlaw A. TNF-alpha in asthma. Curr Opin Pharmacol. 2007;7:279–282. doi: 10.1016/j.coph.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 32.Freeman GJ, Borriello F, Hodes RJ, Reiser H, Gribben JG, Ng JW, Kim J, Goldberg JM, Hathcock K, Laszlo G. Murine B7–2, an alternative CTLA4 counter-receptor that costimulates T cell proliferation and interleukin 2 production. J Exp Med. 1993;178:2185–2192. doi: 10.1084/jem.178.6.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crosby JR, Guha M, Tung D, Miller DA, Bender B, Condon TP, York-DeFalco C, Geary RS, Monia BP, Karras JG, Gregory SA. Inhaled CD86 antisense oligonucleotide suppresses pulmonary inflammation and airway hyper-responsiveness in allergic mice. J Pharmacol Exp Ther. 2007;321:938–946. doi: 10.1124/jpet.106.119214. [DOI] [PubMed] [Google Scholar]
- 34.Fireman E, Onn A, Levo Y, Bugolovov E, Kivity S. Suppressive activity of bronchial macrophages recovered by induced sputum. Allergy. 1999;54:111–118. doi: 10.1034/j.1398-9995.1999.00864.x. [DOI] [PubMed] [Google Scholar]
- 35.Tang C, Inman MD, van Rooijen N, Yang P, Shen H, Matsumoto K, O'Byrne PM. Th type 1-stimulating activity of lung macrophages inhibits Th2-mediated allergic airway inflammation by an IFN-gamma-dependent mechanism. J Immunol. 2001;166:1471–1481. doi: 10.4049/jimmunol.166.3.1471. [DOI] [PubMed] [Google Scholar]
- 36.Bilyk N, Holt PG. Cytokine modulation of the immunosuppressive phenotype of pulmonary alveolar macrophage populations. Immunology. 1995;86:231–237. [PMC free article] [PubMed] [Google Scholar]
- 37.Bartlett NW, Walton RP, Edwards MR, Aniscenko J, Caramori G, Zhu J, Glanville N, Choy KJ, Jourdan P, Burnet J, Tuthill TJ, Pedrick MS, Hurle MJ, Plumpton C, Sharp NA, Bussell JN, Swallow DM, Schwarze J, Guy B, Almond JW, Jeffery PK, Lloyd CM, Papi A, Killington RA, Rowlands DJ, Blair ED, Clarke NJ, Johnston SL. Mouse models of rhinovirus-induced disease and exacerbation of allergic airway inflammation. Nat Med. 2008;14:199–204. doi: 10.1038/nm1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expression of mRNA for various cytokines by CD4+ T cells (supplemental data for Figure 2A)
Cytokine concentrations in BAL fluid (supplemental data for Figure 2B)