

Abstract
In renal fibrosis, interstitial fibroblasts have an increased proliferative phenotype, and the numbers of interstitial fibroblasts closely correlate with the extent of kidney damage. The mechanisms underlying proliferation and resulting expansion of the interstitium remain largely unknown. Here we define the intracellular signaling events by which tissue plasminogen activator (tPA) promotes renal interstitial fibroblast proliferation. tPA promoted the proliferation of renal interstitial fibroblasts independent of its protease activity. The mitogenic effect of tPA required Tyr4507 phosphorylation of the cytoplasmic tail of its receptor LDL receptor–related protein 1. tPA triggered sequential proliferative signaling events involving Erk1/2, p90RSK, GSK3β phosphorylation, and cyclin D1 induction. Blockade of Erk1/2 activation or knockdown of p90RSK suppressed tPA-induced GSK3β phosphorylation, cyclin D1 expression, and fibroblast proliferation. In contrast, expression of constitutively active Mek1 mimicked tPA in inducing GSK3β phosphorylation and cyclin D1 expression. Ectopic overexpression of an uninhibitable GSK3β mutant eliminated tPA-induced cyclin D1 expression. In the murine obstruction model, tPA deficiency reduced renal GSK3β phosphorylation and induction of PCNA and FSP-1. These findings show that tPA induces Tyr4507 phosphorylation of LDL receptor–related protein 1, which in turn leads to the downstream phosphorylation of Erk1/2, p90RSK, and GSK3β, followed by the induction of cyclin D1 in murine interstitial fibroblasts. This study implicates tPA as a mitogen that promotes interstitial fibroblast proliferation, leading to expansion of these cells.
The hallmark of chronic kidney disease is renal interstitial fibrosis, which is characterized by avid inflammation, proliferation of interstitial cells, extensive deposition of extracellular matrix components, and the eventual destruction of normal kidney structure.1–3 In general, the extent of tubulointerstitial fibrosis largely predicts the prognosis of patients with chronic kidney disease.1,2,4 Interstitial fibroblasts are considered to be the primary matrix-producing cells and principal mediators of renal fibrosis associated with progressive renal failure.2,5,6 The size of the interstitial fibroblast population closely correlates with the extent of interstitial injuries.6–8 In the fibrotic kidney, fibroblasts display an increased proliferative phenotype and expand in the interstitial region.7,9,10 However, the underlying mechanisms and the regulation of the fate of these cells remain largely unknown.
Recent studies demonstrate that tPA is actually a molecule with dual functions.11–13 As a member of the serine protease family, tPA participates in the activation of various zymogens and certain growth factors and plays a pivotal role in the homeostasis of blood coagulation/fibrinolysis and matrix regulation.14–17 As a cytokine, tPA executes multiple biological functions by binding to its membrane receptor, the LDL receptor–related protein-1 (LRP-1) and triggering profound intracellular signaling events.11–14,18 In the unilateral ureteral obstruction (UUO) model, the expression of tPA and LRP-1 in the obstructed kidney are significantly increased compared with control kidney, suggesting that tPA signaling is up-regulated during kidney injury.12,13 In addition, tPA-deficient mice are protected from obstruction induced fibrotic injury and demonstrate significantly fewer activated fibroblasts than wild-type mice.12,19 Thus, we hypothesized that tPA may be an endogenous factor governing the fate of interstitial fibroblasts and controlling the size of these cells population.
LRP-1 functions as a tPA receptor and mediates most of the cytokine actions of tPA.11–14,20 Mature LRP-1 consists of an extracellular 515-kDa α subunit and an 85-kDa β subunit with a transmembrane segment and cytoplasmic tail. The cytoplasmic tail of the β subunit contains numerous tyrosine residues in the vicinity of two NPXY motifs.20,21 Phosphorylation of the tyrosine residue(s) is believed to be required for the binding of signaling adaptor proteins that mediate the signal transduction of its ligands,22,23 although the exact role of tyrosine phosphorylation in tPA signaling remains undefined.
In the present study, we demonstrate that tPA acts as a mitogen to promote the proliferation of renal interstitial fibroblasts. The mitogenic effect of tPA is mediated by the phosphorylation of Tyr4507 within the distal NPXY motif of LRP-1, initiating a cascade of proliferative signaling events involving phosphorylation of Erk1/2, p90RSK, GSK3β, and induction of cyclin D1.
Materials and Methods
Antibodies and Reagents
The antibodies against phospho-specific and total GSK3β, cyclin D1, phospho-specific and total Erk1/2, phospho-specific p90RSK, RSK1/2/3, GAPDH, and HA-tag were purchased from Cell Signaling Technology (Beverly, MA). Mouse anti–α-tubulin, anti-bromodeoxyuridine (BrdU) antibodies, normal mouse IgG, and MTT were obtained from Sigma (St. Louis, MO). Specific phospho-tyrosine antibody PY20 was purchased from BD Biosciences (Franklin Lakes, NJ). Mouse monoclonal anti–LRP-1 (11H4) antibody was prepared as described previously.13 The secondary HRP-conjugated antibodies were obtained from Thermo Fisher Scientific (Rockford, IL). Recombinant human single-chain tPA was purchased from American Diagnostica Inc. (Stamford, CT). The nonenzymatic tPA was supplied by Molecular Innovations Inc. (Southfield, MI). Genistein, PD98059, wortmannin, and SC68376 were obtained from Calbiochem-Novabiochem Corp. (La Jolla, CA). PCNA antibody and protein A/G plus agarose bead were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-S100A4 (FSP-1) antibody was provided by Dako (Glostrup, Denmark). Cell culture media, fetal bovine serum (FBS), and supplements were purchased from Invitrogen (Carlsbad, CA). All other chemicals were of analytic grade and were obtained from Sigma or Fisher Scientific (Pittsburgh, PA) unless otherwise indicated.
Cell Culture
Normal rat kidney interstitial fibroblasts (NRK-49F), mouse homozygous LRP-deficient embryo fibroblasts (PEA-13), and its wild-type counterpart MEF-1 were maintained as previously described.11–13
Cell Proliferation Assay and Cytotoxicity Assay
After starvation in serum-free medium for 24 hours, cells were incubated with tPA in serum-free medium for various periods of time. Proliferation of cells was determined by cell number count, 3-(4,5-dimethylthiazol-2-yl)−2,5-diphenyltetrazolium bromide (MTT) assay, BrdU incorporation assay, or quick cell proliferation assay kit (Biovision Inc.) as previously described.24,25 The quick cell proliferation assay kit is a (2-(2-methoxy-4-nitrophenyl)−3-(4-nitrophenyl)−5-(2,4-disulfophenyl)-2H-tetrazolium) (WST-8)–based colorimetric assay which correlates well with [3H]-thymidine incorporation assay.25 Cytotoxicity of specific reagents was evaluated by lactate dehydrogenase (LDH)-cytotoxicity assay kit (Biovision Inc) according to manufacturer's instructions.
Site-Directed Mutagenesis
The LRP-1 minireceptor expression vector (pHA-mLRP2), consisting of the ligand-binding domain II and the β subunit of LRP-1, was prepared as described previously.26 The four tyrosine residues (Tyr4473, Tyr4476, Tyr4507, and Tyr4511) on the β subunit of LRP-1 (pHA-mLRP) were mutated to alanine using a site-directed mutagenesis kit (Stratagene). The resultant mutants (Y4473A, Y4476A, Y4507A, and Y4511A) were sequenced to ensure fidelity.
Plasmid Transfection, siRNA Inhibition, and Adenovirus Infection
The eukaryotic expression vectors encoding the HA-tagged wild-type (pGSK3β-WT, Addgene Inc.) and uninhibitable mutant (pGSK3β-S9A) GSK3β were kindly provided by Dr. Jim Woodgett (Toronto, Canada) and Dr. Gail V. W. Johnson Voll (Birmingham, AL). Transient plasmids transfection, siRNA inhibition, and adenovirus infection were performed as previously described.11,13,27
Western Blot Analysis
Whole cell lysates were prepared as previously described and separated on 10% SDS polyacrylamide gels.11–13 The PVDF membrane with transferred proteins was incubated overnight at 4°C with various primary antibodies in TBST buffer (20 mmol/L Tris-HCl, 150 mmol/L NaCl, and 0.1% Tween 20) containing 5% nonfat milk, then with HRP-conjugated secondary antibodies for 1 hour. After extensive washes in TBST buffer, the signals on the membrane were visualized using the SuperSignal West Pico Chemiluminescent Substrate kit (Thermo Fisher Scientific).
Coimmunoprecipitation
Cells were treated with 10 nmol/L tPA for various periods of time then subjected to immunoprecipitation as described previously.12,13 In brief, cell lysates were incubated with the monoclonal antibody against LRP-1 (11H4) or IgG at 4°C overnight. Then, protein A/G PLUS agarose beads were added and incubated at 4°C for 3 hours. After three washes with lysis buffer, the proteins binding to the beads were extracted in reducing sample buffer, separated on 10% SDS polyacrylamide gel, and analyzed by Western blotting using anti-phosphotyrosine and anti-HA antibodies, respectively.
Animal Model
Homozygous tPA knockout (tPA−/−) and wild-type (tPA+/+) mice were generated from the original breeding pairs purchased from the Jackson Laboratory (Bar Harbor, ME) and maintained as previously described.11,12 The background strains of these mice were C57BL/6. Animal studies were performed by using an approved protocol by the Institutional Animal Care and Use Committee at the Penn State University College of Medicine. The left kidneys of sex-matched mice weighing 20–22 g (five animals per group) were subjected to UUO using established procedures described elsewhere.11,12 The right unobstructed kidneys served as controls. At day 7 and 14 after UUO, mice were sacrificed and kidney samples were used for analysis.
Indirect Immunofluorescence Staining
Snap-frozen kidney was cryosectioned at 5-μm thickness and subjected to double staining as previously described.12 Stained tissues were mounted with Vectashield anti-fade mounting media with DAPI (4′, 6-diamidino-2-phenylindole, HCl) (Vector Laboratories Inc., Burlingame, CA) and viewed under an Olympus BX60 fluorescence microscope equipped with a digital camera (Olympus America Inc., Center Valley, PA). Sections stained without primary antibodies were served as negative controls.
Statistical Analysis
All experiments were performed at least three times. Data are presented as means ± SEM. Statistical analysis of the data were performed using SigmaStat software (Jandel Scientific Software). Comparison between multiple groups was performed by using one-way analysis of variance followed by the Student-Newman-Keuls test or Student's t-test between two groups. A P value of less than 0.05 was considered statistically significant.
Results
tPA Promotes Renal Interstitial Fibroblast Proliferation
To investigate the effect of tPA on interstitial fibroblast proliferation, normal rat kidney interstitial fibroblasts (NRK-49F) were incubated with tPA for 48 hours after 24 hours of serum starvation and then examined for cell proliferation by BrdU incorporation, MTT assay, cell number count, and WST-8 assay which correlates well with [3H]-thymidine incorporation assay.25 Immunofluorescence staining revealed that tPA promoted nuclear incorporation of BrdU (Figure 1A). Likewise, tPA increased cell growth as reflected by WST-8 assay (Figure 1B), MTT assay (Figure 1C), and cell number count (Figure 1D). Of note, tPA also induced fibroblast growth in a time- (Figure 1E) and concentration-dependent manner (Figure 1, F and G). These results indicate that tPA increases fibroblast proliferation.
Figure 1.
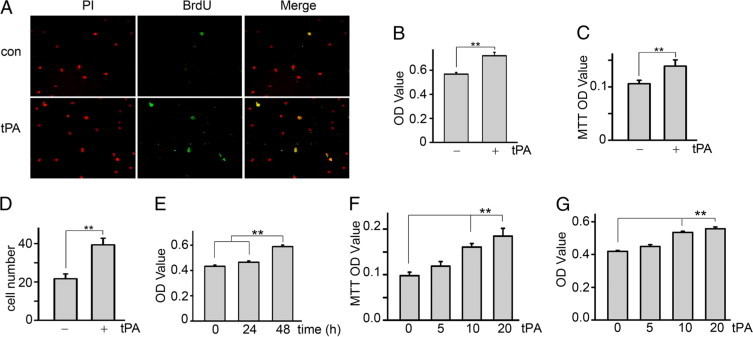
tPA time- and dose-dependently promotes interstitial fibroblast proliferation. NRK-49F cells were incubated with serum-free medium for 24 hours and then left untreated (control, con) or treated with 10 nmol/L tPA for an additional 48 hours. Cell proliferation was evaluated by BrdU incorporation (A), WST-8 based colorimetric assay (B), MTT assay (C), and cell number count (D, ×104). In A, nuclei are red (PI, propidium iodide), and proliferating cells are green (BrdU). For time-response experiments, NRK-49F cells were treated with 10 nmol/L tPA for various times as indicated, and cell number was evaluated by the WST-8 assay (E). For dose-dependent effects, NRK-49F cells were treated with 0, 5, 10, and 20 nmol/L tPA for 48 hours, and cell number was assessed by MTT (F) and WST-8 (G) assays. **P < 0.01 (n = 4–6).
tPA Activates a Cascade of Proliferative Signaling Involving the Phosphorylation of Erk1/2, p90RSK, GSK3β, and Induction of Cyclin D1
We next investigated cell proliferation–related signaling events in interstitial fibroblasts after tPA treatment. It was found in previous studies that tPA did not activate Akt or p38 Mitogen-activated protein kinase (MAPK) in NRK-49F fibroblasts.11,13 tPA rapidly induced Erk1/2 phosphorylation as early as 2 minutes (Figure 2A) followed by p90RSK phosphorylation after 4 minutes (Figure 2B). Intriguingly, tPA also triggered phosphorylation of GSK3β from 10 minutes to 1 hour (Figure 2C). The time course of GSK3β phosphorylation after the activation of p90RSK suggests that GSK3β may be a downstream effector of p90RSK kinase in the tPA signaling pathway. Subsequently, tPA induced the sustained expression of cyclin D1 between 2 hours to 18 hours (Figure 2D). Thus, it is likely that tPA activates proliferative signaling involving phosphorylation of Erk1/2, p90RSK, GSK3β, and induction of cyclin D1 in a temporally coordinated manner.
Figure 2.
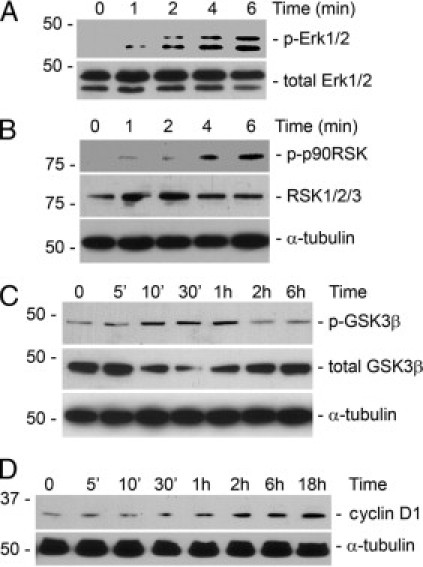
tPA activates mitogenic signaling via sequential phosphorylation of Erk1/2, p90RSK, GSK3β, and induction of cyclin D1. NRK-49F cells were incubated with 10 nmol/L tPA for various periods of time as indicated and then subjected to Western blotting for phospho-Erk1/2 (A), phospho-p90RSK (B), phospho-GSK3β (C), and cyclin D1 (D). The samples were also probed with antibodies against total Erk1/2, GSK3β, RSK1/2/3, and α-tubulin, respectively.
The Mitogenic Effect of tPA Is Independent of its Protease Activity
tPA is a hybrid molecule with dual functions: protease and cytokine. As a protease, tPA converts plasminogen into biologically active plasmin and participates in the posttranslational activation of several growth factors including PDGF-CC.15–17 Accordingly, it is possible that tPA increases fibroblast proliferation through its protease-mediated activation of those growth factors. To test this, NRK-49F cells were incubated with a mutant, catalytically inactive tPA in which the serine within the active site of the enzyme was replaced with alanine.28 As shown in Figure 3, nonenzymatic tPA also induced phosphorylation of GSK3β (Figure 3A) and fibroblast proliferation (Figure 3B) comparable to wild-type tPA, indicating that the mitogenic effect of tPA is independent of its protease activity. Although previous studies showed that tPA induces matrix metalloproteinase-9 (MMP-9) expression in NRK-49F fibroblasts,13 MMP-9 is not likely to mediate the mitogenic effect of tPA as MMP-9 per se did not induce NRK-49F fibroblast proliferation (Figure 3C).
Figure 3.
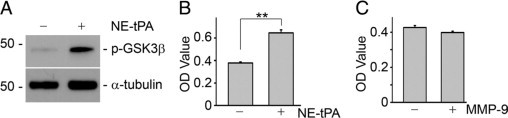
Mitogenic effect of tPA is independent of its protease activity. A: After treatment with nonenzymatic mutant tPA (NE-tPA, 10 nmol/L) for one hour, NRK-49F cells were subjected to Western blot with anti-phospho GSK3β and α-tubulin antibodies. B: NRK-49F cells were treated with NE-tPA (10 nmol/L) for 48 hours, and cell growth was assessed by WST-8 assay. **P < 0.01 (n = 4). C: MMP-9 did not affect fibroblast proliferation. NRK-49F cells were incubated with MMP-9 (10 nmol/L) for 48 hours and then subjected to WST-8 assay.
LRP-1 Mediates the Proliferative Effect of tPA
As a cell membrane receptor for tPA, LRP-1 mediates multiple cytokine functions of tPA in NRK-49F cells.11–13 To investigate whether LRP-1 also mediates the mitogenic effect of tPA, we compared the ability of tPA to induce cell growth in mouse embryonic fibroblasts with (MEF-1) or without (PEA-13) LRP-1 expression. As shown in Figure 4A, tPA induced proliferation in MEF-1 (LRP-1+/+) cells but failed to do so in the LRP-1–deficient PEA-13 fibroblasts. LRP-1 deficiency in PEA-13 was confirmed by Western blot (Figure 4A, inset). Moreover, knockdown of LRP-1 expression in NRK-49F cells by small interfering RNA (siRNA) clearly decreased tPA-induced GSK3β phosphorylation (Figure 4B) and subsequently fibroblast proliferation (Figure 4C).
Figure 4.
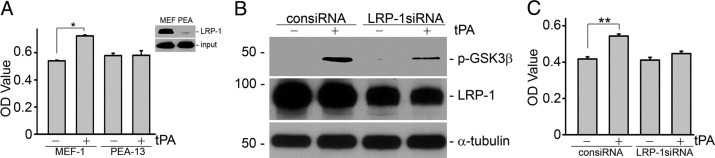
LRP-1 mediates the proliferative effect of tPA. A: tPA failed to induce cell growth in LRP-1–deficient fibroblasts. Mouse fibroblasts MEF-1 (LRP-1+/+) and PEA-13 (LRP-1−/−) were treated with 10 nmol/L tPA for 48 hours, and cell growth was assessed by WST-8 assay. *P < 0.05, n = 4. Deficiency of LRP-1 in PEA-13 cells was confirmed by Western blot (A, inset). NRK-49F cells were transfected with either control siRNA (consiRNA) or LRP-1–specific siRNA (LRP-1siRNA), followed by treatment with tPA (10 nmol/L) as indicated. Knockdown of LRP-1 in NRK-49F cells by siRNA inhibition abrogated tPA-induced GSK3β phosphorylation (B) and cell proliferation (C). **P < 0.01, n = 4.
Tyrosine Phosphorylation of LRP-1 NPXY4507 Is Indispensable for the Mitogenic Effect of tPA
Our previous investigations found that tPA induces rapid tyrosine phosphorylation of LRP-1 intracellular β subunit13 and blockade of the tyrosine phosphorylation by a specific tyrosine kinase inhibitor, genistein, eliminated the subsequent signaling events and biological effects of tPA.12 To determine whether tyrosine phosphorylation of LRP-1 is required for tPA-induced cell growth, we evaluated the effect of genistein pretreatment on NRK-49F fibroblast proliferation. As shown in Figure 5, pretreatment of NRK-49F fibroblasts with genistein at the concentration previously shown to abolish the tyrosine phosphorylation of LRP-112 completely suppressed the mitogenic effect of tPA (Figure 5A), and its inhibitory effect was independent of cytotoxicity (Figure 5B).
Figure 5.
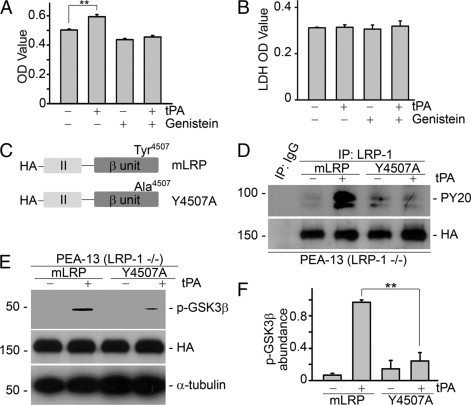
Phosphorylation of tyrosine residue Tyr4507 on LRP-1 is essential for tPA activated proliferative signaling and cell growth. A: Pretreatment with the specific tyrosine kinase inhibitor genistein (50 μmol/L) for 30 minutes abolished tPA-induced proliferation in NRK-49F fibroblasts. **P < 0.01, n = 4. B: No evident cytotoxicity of 50 μmol/L genistein treatment was detected by lactate dehydrogenase (LDH)-cytotoxicity assay (units are arbitrary). C: Graphic illustration of the structure of LRP-1 minireceptor (mLRP) and its mutant. Tyrosine residue Tyr4507 was mutated to alanine (Y4507A) by site-directed mutagenesis. D: tPA failed to induce tyrosine phosphorylation of LRP-1 in PEA-13 (LRP-1−/−) cells transfected with mutant Y4507A plasmid. PEA-13 fibroblasts were transfected with mLRP and mutant Y4507A plasmids followed by treatment with vehicle or tPA (10 nmol/L) for 0.5 minutes. Cell lysates were then immunoprecipitated by mouse IgG or mouse anti–LRP-1 antibody followed by immunoblotting with specific anti-tyrosine phosphorylation antibody PY20 and anti-HA antibody. E and F: Ectopic expression of mutant Y4507A plasmid abolished tPA-induced GSK3β phosphorylation. PEA-13 fibroblasts were transfected with mLRP and mutant Y4507A plasmids followed by treatment with vehicle or tPA (10 nmol/L) for 1 hour. Cell lysates were subjected to Western blot with anti–phospho-GSK3β, anti-HA, and anti–α-tubulin antibodies. Representative Western blot (E) and graphic demonstration of the relative abundance of phospho-GSK3β (F). **P < 0.01, n = 3.
We next identified the tyrosine phosphorylation site(s) responsible for tPA-induced proliferative signaling. To this end, each of four possible tyrosine phosphorylation sites (Tyr4473, Tyr4476, Tyr4507, and Tyr4511) in the LRP-1 minireceptor plasmid was mutated into alanine and designated as Y4473A, Y4476A, Y4507A, and Y4511A, respectively. The LRP-1 minireceptor (mLRP) consists of the ligand-binding domain II and the intact β subunit (Figure 5C) and is sufficient to restore the biological functions of tPA in LRP-1–deficient fibroblasts (PEA-13).11 To minimize the competition from endogenous LRP-1 tyrosine phosphorylation, LRP-1–deficient PEA-13 fibroblasts were transfected with the mLRP mutants. We found that the ability of tPA to induce GSK3β phosphorylation in PEA-13 cells transfected with Y4473A, Y4476A, and Y4511A mutants was comparable to that of wild-type mLRP (data not shown). However, tPA failed to induce tyrosine phosphorylation of LRP-1 (Figure 5D) and subsequent GSK3β phosphorylation (Figure 5, E and F) in PEA-13 fibroblasts transfected with the Y4507A mutant. Thus, phosphorylation of tyrosine Tyr4507 is essential for tPA signaling in murine fibroblasts.
Activation of Erk1/2 Is Necessary and Sufficient for Interstitial Fibroblast Proliferation
In line with the previous finding that tPA did not activate the PI3K/Akt pathway in NRK-49F fibroblasts,11,13 we found that the PI3K inhibitor, wortmannin, failed to block tPA-induced GSK3β phosphorylation (Figure 6A). We next examined the role of Erk1/2 in the mitogenic pathway induced by tPA. NRK-49F fibroblasts were incubated with tPA in the presence or absence of PD98059, a specific inhibitor of Erk1/2 upstream kinase mitogen-activated protein kinase/Erk kinase-1 (Mek1). PD98059 dramatically decreased tPA-mediated GSK3β phosphorylation (Figure 6, B and C), eliminated tPA-induced cyclin D1 expression (Figure 6D), and eventually abolished the mitogenic effect of tPA in interstitial fibroblasts (Figure 6, E and F) at a concentration previously shown to completely suppress Erk1/2 activation in NRK-49F cells.13 Infection of NRK-49F fibroblasts with constitutively active Mek1 adenovirus mimicked the effects of tPA on GSK3β phosphorylation (Figure 6G) and cyclin D1 induction (Figure 6H). Thus, Erk1/2 activation by tPA is indispensable for the proliferation of interstitial fibroblasts.
Figure 6.
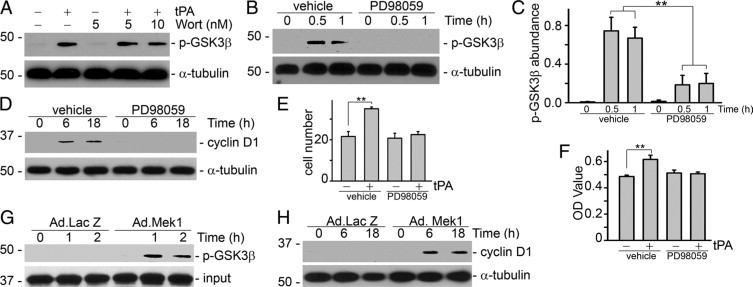
Erk1/2 activation is necessary and sufficient for the mitogenic effect of tPA. NRK-49F cells were pretreated with PI3K inhibitor wortmannin (Wort, 5 and 10 nmol/L) or Mek1 inhibitor PD98059 (20 μmol/L) for 30 minutes and then incubated with tPA (10 nmol/L) for various time periods as indicated. Cell lysates were probed with phospho-GSK3β, cyclin D1, and α-tubulin. A: Pretreatment with the PI3K inhibitor did not affect tPA-induced GSK3β phosphorylation. B and C: PD98059 abrogated tPA-induced GSK3β phosphorylation. B: Representative Western blot. C: Quantitative representation of the relative abundance of phospho-GSK3β. **P < 0.01, n = 3. PD98059 also suppressed tPA-induced cyclin D1 expression (D) and cell proliferation (E and F). Cell growth was evaluated by cell number count (E, ×104) and WST-8 assay (F). **P < 0.01, n = 4–6. Infection of constitutively active Mek1 adenovirus (Ad.MEK1) was sufficient to induce GSK3β phosphorylation (G) and cyclin D1 expression (H). NRK-49F cells were infected with adenovirus for periods of time as indicated. An adenoviral vector containing the β-galactosidase gene (Ad. LacZ) was used as a control.
p90RSK Mediates tPA Mitogenic Signaling and Effect
The temporal sequence of p90RSK phosphorylation followed immediately by GSK3β phosphorylation (Figure 2, B and C) is consistent with the possibility that GSK3β may be a downstream effector of p90RSK kinase in the tPA signaling pathway. To test this, p90RSK was knocked down using siRNA. As shown Figure 7, knockdown of RSK expression by both p90RSK siRNA1 and 2 abrogated tPA-induced phosphorylation of GSK3β (Figure 7, A and B), cyclin D1 expression (Figure 7C), and proliferation of interstitial fibroblasts (Figure 7D). Hence, p90RSK is an upstream kinase responsible for GSK3β/cyclin D1 signaling and the mitogenic effect of tPA.
Figure 7.
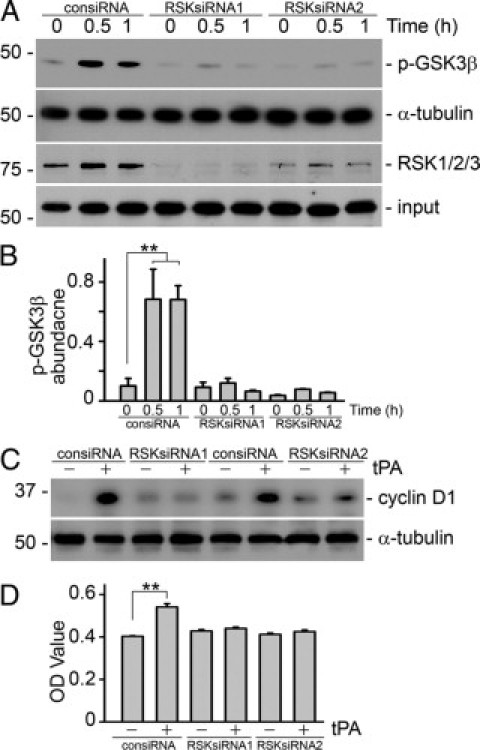
p90RSK is essential for tPA-induced proliferation. Knockdown of p90RSK in NRK-49F cells by both siRNA1 and 2 abolished tPA-induced GSK3β phosphorylation (A and B), cyclin D1 induction (C), and fibroblast proliferation (D). NRK-49F cells were transfected with either control siRNA (consiRNA) or p90RSK-specific siRNA (RSKsiRNA), followed by treatment with tPA (10 nmol/L) as indicated. Cell lysates were immunoblotted with antibodies against RSK1/2/3, phospho-GSK3β, cyclin D1, and α-tubulin. (B) Quantitative representation of the relative abundance of phospho-GSK3β. **P < 0.01, n = 3. Cell growth was assessed by WST-8 assay, **P < 0.01, n = 4.
GSK3β Phosphorylation Is Required for the Proliferative Signaling of tPA
GSK3β is constitutively active in mammalian cells. Unlike most other protein kinases, GSK3β is primarily regulated by inactivation. Many GSK3β downstream targets including cyclin D1 are inhibited by GSK3β-mediated phosphorylation. Phosphorylation of Ser9 on GSK3β inactivates its kinase activity, leading to derepression of its downstream targets.29 Because p90RSK is the upstream kinase responsible for the phosphorylation of GSK3β (Figure 7), we next investigated the role of GSK3β in tPA mitogenic signaling. As shown in Figure 8A, mutation of Ser9 to Ala9 in the regulatory domain of GSK3β (GSK3β-S9A) renders it unable to be inactivated by phosphorylation. Because GSK3β suppresses cyclin D1 expression, forced overexpression of wild-type GSK3β in mouse fibroblasts partially inhibited cyclin D1 induction by tPA. Overexpression of the uninhibitable mutant GSK3β-S9A completely eliminated the induction of cyclin D1, even though expression of GSK3β-S9A appeared to be less than that of wild-type GSK3β (Figure 8, B and C). This indicates that phosphorylation of Ser9 and inactivation of GSK3β by tPA is indispensable to its proliferative signaling.
Figure 8.
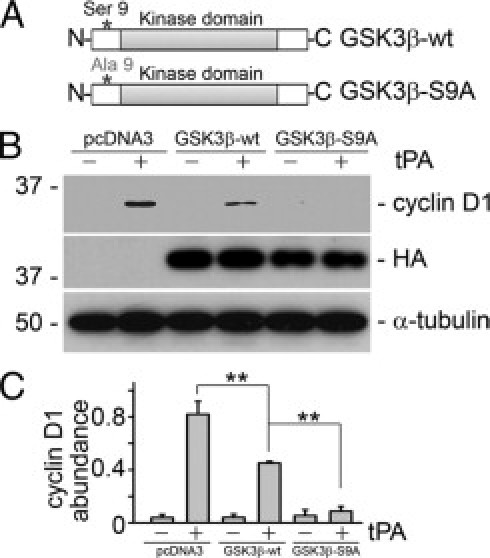
Phosphorylation of GSK3β is necessary for the effect of tPA. A: Graphic illustration of the structure of wild-type (GSK3β-wt) and mutant GSK3β (GSK3β-S9A) plasmids. B and C: Overexpression of the uninhibitable mutant GSK3β-S9A abolished tPA-induced cyclin D1 expression. MEF-1 fibroblasts were transfected with pcDNA3, GSK3β-wt, and GSK3β-S9A plasmids followed by tPA (10 nmol/L) treatment for 8 hours. Cell lysates were probed with anti-cyclin D1, anti-HA, and anti–α-tubulin antibodies. Representative Western blot (B) and graphic demonstration of the relative abundance of phospho-GSK3β (C). **P < 0.01, n = 3.
tPA Activates Proliferative Signaling in Vivo
To examine the mitogenic effect of tPA in vivo, we studied the proliferative signaling events in the obstruction-induced fibrosis model in tPA wild-type and knockout mice. It was found that GSK3β phosphorylation (Figure 9, A, B, D, and E), PCNA expression (Figure 9, F–I), and FSP-1 abundance (Figure 9, K–N) were dramatically induced in the fibrotic kidneys from the wild-type mice at 7 days (Figure 9, A, D, F, H, K, and M) and 14 days (Figure 9, B, E, G, I, L, and N) after UUO, whereas phosphorylation of GSK3β and expression of PCNA and FSP-1 were significantly reduced in tPA knockout mice compared with that from tPA wild-type controls (Figure 9), suggesting that mitogenic signaling and fibroblast proliferation were attenuated in tPA-deficient mice. In addition, double immunofluorescence staining of phospho-GSK3β (red, cytoplasm), PCNA (red, nuclei), and αSMA (green, cytoplasm) confirmed that in the fibrotic kidneys, the interstitial proliferating cells were largely αSMA-positive fibroblasts (left, Figure 9, C and J). tPA ablation suppressed the proliferation of these fibroblasts (right, Figure 9, C and J). Therefore, tPA also activates mitogenic signaling and promotes renal interstitial fibroblasts proliferation in vivo.
Figure 9.
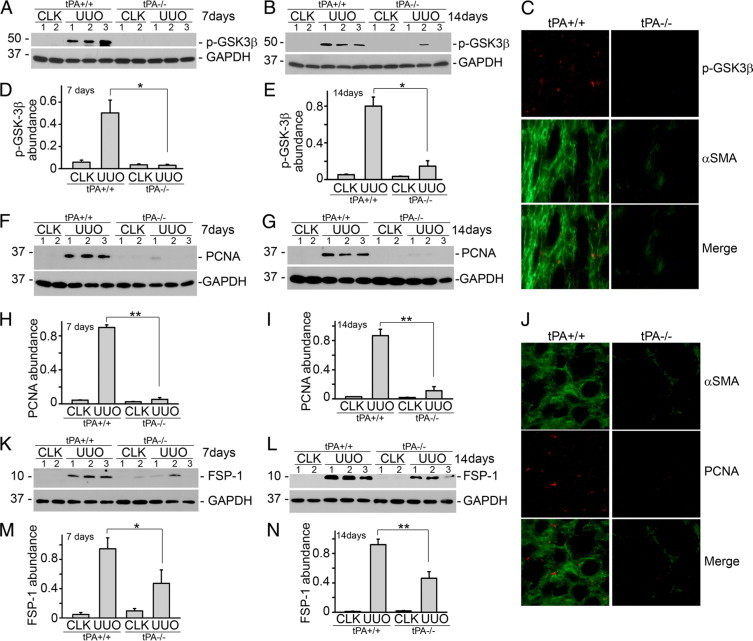
tPA activates proliferative signaling in vivo. tPA knockout mice exhibit reduced induction of phospho-GSK3β (A, B, D, and E), PCNA (F–I), and FSP-1 (K–N) in fibrotic kidneys. At seven days (A, D, F, H, K, and M) and 14 days (B, E, G, I, L, and N) after unilateral ureteral obstruction (UUO), kidney homogenates were probed with antibodies against phospho-GSK3β, PCNA, FSP-1, and GAPDH. Lane numbers indicate individual mice. Quantitative analysis of the relative abundance is shown in (D, H, and M) for seven days and (E, I, and N) for 14 days. *P < 0.05 and **P < 0.01, n = 5. CLK, contralateral unobstructed kidney. C: Double immunostaining of phospho-GSK3β (red) and αSMA (green) in the kidney after seven days obstruction. J: Double staining of PCNA (red, nuclei) and αSMA (green, cytoplasm) in the kidney after seven days obstruction.
Discussion
As the major matrix-producing cells, the size of the interstitial fibroblast population is a critical determinant of outcomes in chronic kidney injury.1,2,4,6–8 In the normal adult kidney, interstitial fibroblasts represent a small group of quiescent cells with a low turnover rate. In response to chronic injuries, fibroblasts undergo a phenotypic change, become functionally activated, acquire higher mitogenic potential, and produce excessive matrix.2,6–10 These events coincide with the concomitant inductions of tPA and its receptor, LRP-1, within the renal interstitium,12,13 suggesting that tPA may be a key endogenous factor regulating the fate of interstitial fibroblasts. In the present study, we demonstrated that tPA activates a cascade of proliferative signaling events via its membrane receptor LRP-1 and promotes interstitial fibroblast expansion. This finding, together with our recently discovered cytoprotective activities of tPA,11 establishes a previously unrecognized function of tPA in controlling the fate of interstitial fibroblasts. It is clear that tPA accelerates the excessive accumulation of interstitial fibroblasts and promotes renal fibrosis by both inducing proliferation and reducing the apoptosis of these cells.
tPA was originally discovered as a member of the serine protease family, whose main function is to activate numerous zymogens into active enzymes involved in fibrinolysis and extracellular matrix degradation. Recently, studies from us and other groups have found that tPA also elicits many biological activities through protease-independent mechanisms.11–13,30–38 These observations eventually established that tPA is hybrid molecule having both protease and cytokine functions.13 We determined in the present studies that the mitogenic actions of tPA are independent of its protease activity (Figure 3) and mediated via LRP-1 (Figure 4). Notably, tPA also activated proliferative signaling in vivo (Figure 9), suggesting that tPA may be an important endogenous factor modulating the phenotypic change of interstitial fibroblasts from quiescence to higher mitogenic potential.
Although it is not a dedicated receptor for tPA, LRP-1 mediates most of the cytokine functions of tPA.11–13,18,30–32,35–38 LRP-1 also mediates the mitogenic effects of tPA as both knockout and siRNA-mediated knockdown of LRP-1 abolished tPA-induced proliferative signaling and cell growth (Figure 4). Our earlier study demonstrated that tPA triggers a rapid tyrosine phosphorylation of LRP-1 which takes place before other signaling events.13 We were able to show that phosphorylation of these tyrosine residues on LRP-1 β subunit are critical for tPA signal transduction and biological functions as indicated using genistein, a specific tyrosine kinase inhibitor (Figure 5, A and B). However, until now, the precise site of phosphorylation and the role of signaling in tPA-induced proliferation were unknown. Previous structural analyses had determined that there are four tyrosine residues on the cytoplasmic domain of LRP-1, two of which (Tyr4473 and Tyr4507) are present in the context of NPXY motifs. The two NPXY motifs on the cytoplasmic domain are thought to act as docking sites for signaling adaptor proteins.21 To identify which, if any, of these tyrosine residue site(s) are responsible for the mitogenic signaling of tPA, we generated four mutants with individual tyrosine residue mutated to alanine (Y4473A, Y4476A, Y4507A, and Y4511A) using LRP-1 minireceptor (mLRP) as a template. To avoid possible interference of endogenous LRP-1 tyrosine phosphorylation, wild-type mLRP and individual mutants were transfected into LRP-1–deficient fibroblasts (PEA-13). We found that only Y4507A suppressed tPA-induced tyrosine phosphorylation (Figure 5D) and downstream signaling (Figure 5, E and F) whereas the other 3 mutants had no effect (data not shown). This result clearly demonstrates that tyrosine residue Tyr4507 is essential for tPA signaling.
LRP-1–mediated Erk activation by tPA has been reported in various cellular systems13,33,35; however, the exact mechanism has not been fully understood. Tyrosine residues on the β subunit of LRP-1 provide docking sites for signaling adaptor protein including Shc,22,39,40 which on phosphorylation will then recruit Grb2-Sos, potentiating activation of Ras.40 In addition, it was recently discovered that binding of tPA to LRP-1 transactivates Trk receptor–dependent Erk activation in neurons.18 Our current finding is in line with other studies which showed that Tyr4507 can be phosphorylated by Src kinase in vitro22,41 and that tPA activates Src family kinase.18 Because phosphorylation of Tyr4507 by v-Src induces association of LRP-1 with the adaptor protein Shc,22 it seems likely that Shc may mediate Ras-Erk1/2 signal transduction of tPA and subsequent proliferative signaling. Further investigation will be needed to confirm the role of Shc in tPA-activated Erk signaling.
In this study we also determined that tPA-induced p90RSK phosphorylation mediates GSK3β phosphorylation and inactivation. As shown in Figure 7, A and B, knockdown of p90RSK by RNA silencing abolished tPA-induced phosphorylation and inactivation of GSK3β. This result is supported by previous work which found that GSK3β is a substrate of and can be phosphorylated by p90RSK in vitro.42 The p90 ribosomal s6 kinases (RSKs) are a group of serine/threonine kinases that regulate diverse cellular process, such as cell growth, cell motility, and cell survival.43 There are four RSK isoforms (RSK1–4), of which RSK1 is also designated as p90RSK. RSK isoforms share 75%−80% sequence identity and are expressed ubiquitously in human tissue including kidney44; they are known to play an important role in the MAPK signaling cascade and are downstream effectors of the Ras-Erk1/2 signaling. Erk1/2 activation phosphorylates and activates RSKs.43,45 RSKs, in turn, activate various signaling events through selection of different phosphorylation substrates including GSK3β and Bad.43 GSK3β has been implicated in the phosphorylation of many proteins and in the regulation of several cellular events. In contrast to most other protein kinases which induce the activation of a substrate, the action of GSK3β inhibits its downstream targets including cyclin D1 by ubiquination and proteosomal degradation.46 GSK3β is constitutively active in mammalian cells. Phosphorylation of GSK3β leads to its inactivation. So, by phosphorylating and inhibiting GSK3β, tPA promotes the dephosphorylation, stabilization, and induction of cyclin D1, which facilitates fibroblast entry into the S phase of cell division cycle and induces cell growth. Intriguingly, our recent study found that the cytoprotective effect of tPA is mediated by Bad phosphorylation induced by p90RSK activation.11 This result, together with our current finding that p90RSK mediates the mitogenic effect of tPA, suggests that p90RSK is likely the pivotal control point in the signaling cascade of tPA, regulating the fate and destiny of interstitial fibroblasts in the diseased kidney. Therefore, p90RSK may be a promising therapeutic target for blocking the fibrogenic effects of tPA under pathological conditions.
As summarized in Figure 10, tPA binds to the membrane receptor LRP-1, triggers tyrosine phosphorylation of Tyr4507 on LRP-1, and activates Erk1/2 and p90RSK; p90RSK in turn phosphorylates and inhibits its downstream substrate GSK3β; inhibition of GSK3β promotes the stabilization and induction of cyclin D1, resulting in cell-cycle progression and cell growth. Blockade of any step within this signaling pathway eliminates the mitogenic effect of tPA. Thus, tPA contributes to the accumulation of interstitial fibroblasts in the diseased kidney through dual mechanisms: i) via phosphorylation of p90RSK/GSK3β and induction of cyclin D1 to induce cell proliferation; ii) via activation of p90RSK/Bad to inhibit apoptotic cell death.11
Figure 10.
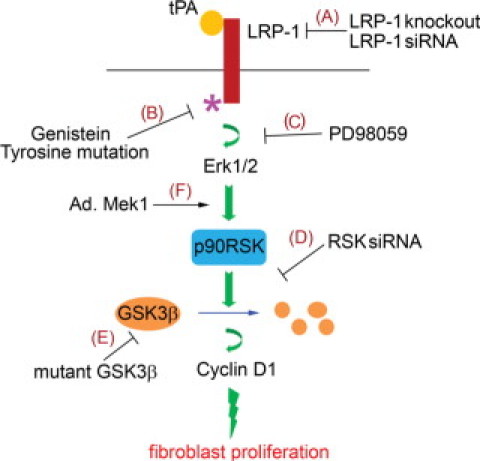
Schematic illustration of the signaling transduction pathway leading to tPA induced fibroblast proliferation. tPA binds to membrane receptor LRP-1, inducing LRP-1 phosphorylation at Tyr4507 residue, which in turn triggers the phosphorylation of Erk 1/2, p90RSK, GSK3β, and induction of cyclin D1. Blockade of any step within this signaling cascade by different strategies (A–E) abolishes the mitogenic effect of tPA, whereas overexpression of Mek1 mimics tPA and promotes interstitial fibroblast/myofibroblast proliferation (F).
Acknowledgements
We thank Drs. Jim Woodgett (Toronto, Canada) and Gail V. W. Johnson Voll (Birmingham, AL) for providing the necessary plasmids and Joe Bednarczyk (Molecular Genetics Core at Penn State College of Medicine) for technical assistance.
Footnotes
Supported by grants from American Heart Association and Kidney Foundation of Central Pennsylvania (K.H.).
References
- 1.Grande MT, Lopez-Novoa JM. Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat Rev Nephrol. 2009;5:319–328. doi: 10.1038/nrneph.2009.74. [DOI] [PubMed] [Google Scholar]
- 2.Strutz F, Zeisberg M. Renal fibroblasts and myofibroblasts in chronic kidney disease. J Am Soc Nephrol. 2006;17:2992–2998. doi: 10.1681/ASN.2006050420. [DOI] [PubMed] [Google Scholar]
- 3.Kaissling B, Le Hir M. The renal cortical interstitium: morphological and functional aspects. Histochem Cell Biol. 2008;130:247–262. doi: 10.1007/s00418-008-0452-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bohle A, Strutz F, Muller GA. On the pathogenesis of chronic renal failure in primary glomerulopathies: a view from the interstitium. Exp Nephrol. 1994;2:205–210. [PubMed] [Google Scholar]
- 5.Neilson EG. Mechanisms of disease: fibroblasts–a new look at an old problem. Nat Clin Pract Nephrol. 2006;2:101–108. doi: 10.1038/ncpneph0093. [DOI] [PubMed] [Google Scholar]
- 6.Qi W, Chen X, Poronnik P, Pollock CA. The renal cortical fibroblast in renal tubulointerstitial fibrosis. Int J Biochem Cell Biol. 2006;38:1–5. doi: 10.1016/j.biocel.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 7.Muller GA, Zeisberg M, Strutz F. The importance of tubulointerstitial damage in progressive renal disease. Nephrol Dial Transplant. 2000;15(Suppl 6):76–77. doi: 10.1093/ndt/15.suppl_6.76. [DOI] [PubMed] [Google Scholar]
- 8.Zeisberg M, Strutz F, Muller GA. Role of fibroblast activation in inducing interstitial fibrosis. J Nephrol. 2000;13(Suppl 3):S111–S120. [PubMed] [Google Scholar]
- 9.Muller GA, Rodemann HP. Characterization of human renal fibroblasts in health and disease: i. Immunophenotyping of cultured tubular epithelial cells and fibroblasts derived from kidneys with histologically proven interstitial fibrosis. Am J Kidney Dis. 1991;17:680–683. doi: 10.1016/s0272-6386(12)80351-9. [DOI] [PubMed] [Google Scholar]
- 10.Rodemann HP, Muller GA, Knecht A, Norman JT, Fine LG. Fibroblasts of rabbit kidney in culture. I Characterization and identification of cell-specific markers. Am J Physiol. 1991;261:F283–F291. doi: 10.1152/ajprenal.1991.261.2.F283. [DOI] [PubMed] [Google Scholar]
- 11.Hu K, Lin L, Tan X, Yang J, Bu G, Mars WM, Liu Y. tPA protects renal interstitial fibroblasts and myofibroblasts from apoptosis. J Am Soc Nephrol. 2008;19:503–514. doi: 10.1681/ASN.2007030300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu K, Wu C, Mars WM, Liu Y. Tissue-type plasminogen activator promotes murine myofibroblast activation through LDL receptor-related protein 1-mediated integrin signaling. J Clin Invest. 2007;117:3821–3832. doi: 10.1172/JCI32301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu K, Yang J, Tanaka S, Gonias SL, Mars WM, Liu Y. Tissue-type plasminogen activator acts as a cytokine that triggers intracellular signal transduction and induces matrix metalloproteinase-9 gene expression. J Biol Chem. 2006;281:2120–2127. doi: 10.1074/jbc.M504988200. [DOI] [PubMed] [Google Scholar]
- 14.Hu K, Mars WM, Liu Y. Novel actions of tissue-type plasminogen activator in chronic kidney disease. Front Biosci. 2008;13:5174–5186. doi: 10.2741/3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mars WM, Zarnegar R, Michalopoulos GK. Activation of hepatocyte growth factor by the plasminogen activators uPA and tPA. Am J Pathol. 1993;143:949–958. [PMC free article] [PubMed] [Google Scholar]
- 16.Yee JA, Yan L, Dominguez JC, Allan EH, Martin TJ. Plasminogen-dependent activation of latent transforming growth factor beta (TGF beta) by growing cultures of osteoblast-like cells. J Cell Physiol. 1993;157:528–534. doi: 10.1002/jcp.1041570312. [DOI] [PubMed] [Google Scholar]
- 17.Fredriksson L, Li H, Fieber C, Li X, Eriksson U. Tissue plasminogen activator is a potent activator of PDGF-CC. EMBO J. 2004;23:3793–3802. doi: 10.1038/sj.emboj.7600397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi Y, Mantuano E, Inoue G, Campana WM, Gonias SL. Ligand binding to LRP1 transactivates Trk receptors by a Src family kinase-dependent pathway. Sci Signal. 2009;2:ra18. doi: 10.1126/scisignal.2000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang J, Shultz RW, Mars WM, Wegner RE, Li Y, Dai C, Nejak K, Liu Y. Disruption of tissue-type plasminogen activator gene in mice reduces renal interstitial fibrosis in obstructive nephropathy. J Clin Invest. 2002;110:1525–1538. doi: 10.1172/JCI16219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lillis AP, Van Duyn LB, Murphy-Ullrich JE, Strickland DK. LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol Rev. 2008;88:887–918. doi: 10.1152/physrev.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J Clin Invest. 2001;108:779–784. doi: 10.1172/JCI13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barnes H, Ackermann EJ, van der Geer P. v-Src induces Shc binding to tyrosine 63 in the cytoplasmic domain of the LDL receptor-related protein 1. Oncogene. 2003;22:3589–3597. doi: 10.1038/sj.onc.1206504. [DOI] [PubMed] [Google Scholar]
- 23.Gotthardt M, Trommsdorff M, Nevitt MF, Shelton J, Richardson JA, Stockinger W, Nimpf J, Herz J. Interactions of the low density lipoprotein receptor gene family with cytosolic adaptor and scaffold proteins suggest diverse biological functions in cellular communication and signal transduction. J Biol Chem. 2000;275:25616–25624. doi: 10.1074/jbc.M000955200. [DOI] [PubMed] [Google Scholar]
- 24.Matsuo N, Shiraha H, Fujikawa T, Takaoka N, Ueda N, Tanaka S, Nishina S, Nakanishi Y, Uemura M, Takaki A, Nakamura S, Kobayashi Y, Nouso K, Yagi T, Yamamoto K. Twist expression promotes migration and invasion in hepatocellular carcinoma. BMC Cancer. 2009;9:240. doi: 10.1186/1471-2407-9-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanemura Y, Mori H, Kobayashi S, Islam O, Kodama E, Yamamoto A, Nakanishi Y, Arita N, Yamasaki M, Okano H, Hara M, Miyake J. Evaluation of in vitro proliferative activity of human fetal neural stem/progenitor cells using indirect measurements of viable cells based on cellular metabolic activity. J Neurosci Res. 2002;69:869–879. doi: 10.1002/jnr.10377. [DOI] [PubMed] [Google Scholar]
- 26.Obermoeller-McCormick LM, Li Y, Osaka H, FitzGerald DJ, Schwartz AL, Bu G. Dissection of receptor folding and ligand-binding property with functional minireceptors of LDL receptor-related protein. J Cell Sci. 2001;114:899–908. doi: 10.1242/jcs.114.5.899. [DOI] [PubMed] [Google Scholar]
- 27.Miyazaki T, Katagiri H, Kanegae Y, Takayanagi H, Sawada Y, Yamamoto A, Pando MP, Asano T, Verma IM, Oda H, Nakamura K, Tanaka S. Reciprocal role of ERK and NF-kappaB pathways in survival and activation of osteoclasts. J Cell Biol. 2000;148:333–342. doi: 10.1083/jcb.148.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olson ST, Swanson R, Day D, Verhamme I, Kvassman J, Shore JD. Resolution of Michaelis complex, acylation, and conformational change steps in the reactions of the serpin, plasminogen activator inhibitor-1, with tissue plasminogen activator and trypsin. Biochemistry. 2001;40:11742–11756. doi: 10.1021/bi0107290. [DOI] [PubMed] [Google Scholar]
- 29.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akkawi S, Nassar T, Tarshis M, Cines DB, Higazi AA. LRP and alphavbeta3 mediate tPA activation of smooth muscle cells. Am J Physiol Heart Circ Physiol. 2006;291:H1351–H1359. doi: 10.1152/ajpheart.01042.2005. [DOI] [PubMed] [Google Scholar]
- 31.An J, Zhang C, Polavarapu R, Zhang X, Zhang X, Yepes M. Tissue-type plasminogen activator and the low-density lipoprotein receptor-related protein induce Akt phosphorylation in the ischemic brain. Blood. 2008;112:2787–2794. doi: 10.1182/blood-2008-02-141630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao C, Lawrence DA, Li Y, Von Arnim CA, Herz J, Su EJ, Makarova A, Hyman BT, Strickland DK, Zhang L. Endocytic receptor LRP together with tPA and PAI-1 coordinates Mac-1-dependent macrophage migration. EMBO J. 2006;25:1860–1870. doi: 10.1038/sj.emboj.7601082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee HY, Hwang IY, Im H, Koh JY, Kim YH. Non-proteolytic neurotrophic effects of tissue plasminogen activator on cultured mouse cerebrocortical neurons. J Neurochem. 2007;101:1236–1247. doi: 10.1111/j.1471-4159.2007.04417.x. [DOI] [PubMed] [Google Scholar]
- 34.Liot G, Roussel BD, Lebeurrier N, Benchenane K, Lopez-Atalaya JP, Vivien D, Ali C. Tissue-type plasminogen activator rescues neurones from serum deprivation-induced apoptosis through a mechanism independent of its proteolytic activity. J Neurochem. 2006;98:1458–1464. doi: 10.1111/j.1471-4159.2006.03982.x. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Lee SR, Arai K, Lee SR, Tsuji K, Rebeck GW, Lo EH. Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nat Med. 2003;9:1313–1317. doi: 10.1038/nm926. [DOI] [PubMed] [Google Scholar]
- 36.Yepes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, Lawrence DA. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J Clin Invest. 2003;112:1533–1540. doi: 10.1172/JCI19212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X, Polavarapu R, She H, Mao Z, Yepes M. Tissue-type plasminogen activator and the low-density lipoprotein receptor-related protein mediate cerebral ischemia-induced nuclear factor-kappaB pathway activation. Am J Pathol. 2007;171:1281–1290. doi: 10.2353/ajpath.2007.070472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhuo M, Holtzman DM, Li Y, Osaka H, DeMaro J, Jacquin M, Bu G. Role of tissue plasminogen activator receptor LRP in hippocampal long-term potentiation. J Neurosci. 2000;20:542–549. doi: 10.1523/JNEUROSCI.20-02-00542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barnes H, Larsen B, Tyers M, van Der Geer P. Tyrosine-phosphorylated low density lipoprotein receptor-related protein 1 (Lrp1) associates with the adaptor protein SHC in SRC-transformed cells. J Biol Chem. 2001;276:19119–19125. doi: 10.1074/jbc.M011437200. [DOI] [PubMed] [Google Scholar]
- 40.Strickland DK, Gonias SL, Argraves WS. Diverse roles for the LDL receptor family. Trends Endocrinol Metab. 2002;13:66–74. doi: 10.1016/s1043-2760(01)00526-4. [DOI] [PubMed] [Google Scholar]
- 41.Loukinova E, Ranganathan S, Kuznetsov S, Gorlatova N, Migliorini MM, Loukinov D, Ulery PG, Mikhailenko I, Lawrence DA, Strickland DK. Platelet-derived growth factor (PDGF)-induced tyrosine phosphorylation of the low density lipoprotein receptor-related protein (LRP). Evidence for integrated co-receptor function betwenn LRP and the PDGF. J Biol Chem. 2002;277:15499–15506. doi: 10.1074/jbc.M200427200. [DOI] [PubMed] [Google Scholar]
- 42.Stambolic V, Woodgett JR. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem J. 1994;303(Pt 3):701–704. doi: 10.1042/bj3030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol. 2008;9:747–758. doi: 10.1038/nrm2509. [DOI] [PubMed] [Google Scholar]
- 44.Nguyen TL. Targeting RSK: an overview of small molecule inhibitors. Anticancer Agents Med Chem. 2008;8:710–716. doi: 10.2174/187152008785914770. [DOI] [PubMed] [Google Scholar]
- 45.Carriere A, Ray H, Blenis J, Roux PP. The RSK factors of activating the Ras/MAPK signaling cascade. Front Biosci. 2008;13:4258–4275. doi: 10.2741/3003. [DOI] [PubMed] [Google Scholar]
- 46.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]