

Abstract
Protective effects of adiponectin (APN; an adipocytokine) were shown against various oxidative challenges; however, its therapeutic implications and the mechanisms underlying hepatic iron overload remain unclear. Herein, we show that the deleterious effects of iron dextran on liver function and iron deposition were significantly reversed by adiponectin gene therapy, which was accompanied by AMP-activated protein kinase (AMPK) phosphorylation and heme oxygenase (HO)-1 induction. Furthermore, AMPK-mediated peroxisome proliferator-activated receptor-α (PPARα) activation by APN was ascribable to HO-1 induction. Additionally, we revealed direct transcriptional regulation of HO-1 by the binding of PPARα to a PPAR-responsive element (PPRE) by various experimental assessments. Interestingly, overexpression of HO-1 in hepatocytes mimicked the protective effect of APN in attenuating iron-mediated injury, whereas it was abolished by SnPP and small interfering HO-1. Furthermore, bilirubin, the end-product of the HO-1 reaction, but not CO, protected hepatocytes from iron dextran-mediated caspase activation. Herein, we demonstrate a novel functional PPRE in the promoter regions of HO-1, and APN-mediated HO-1 induction elicited an antiapoptotic effect and a decrease in iron deposition in hepatocytes subjected to iron challenge.
Adiponectin (APN), an adipocytokine first described as the most abundant protein produced by adipocytes, appears to serve as a central regulatory protein in many of the physiological pathways controlling lipid and carbohydrate metabolism and to mediate various vascular processes.1 APN displays both antiinflammatory and antiatherogenic properties,2,3 and its levels are paradoxically decreased in obesity and insulin-resistance states including metabolic syndrome and diabetes, as well as hypertension and coronary artery disease.4
APN interacts with two types of receptors, adipoR1 and adipoR2.5 In general, the binding of APN to AdipoR1 activates p38 mitogen-activated protein kinase (MAPK), AMP-activated kinase (AMPK), and peroxisome proliferator-activated receptor-α (PPARα), which regulate the inhibition of gluconeogenesis and fatty acid oxidation, whereas APN binding to AdipoR2 mainly activates the AMPK and PPARα pathways, which stimulate energy dissipation and inhibit inflammation and oxidative stress.
PPARα modulates target gene expressions in response to ligand activation after heterodimerization with the retinoid X receptor and binding to peroxisome proliferator-responsive elements (PPREs) of target genes.6,7 In addition to ligand-dependent activation, PPARs, including PPARα and PPARγ, have been shown to be activated by phosphorylation.8,9 Therefore, PPAR-mediated modulation of gene transcription by APN may form the basis for its novel role as a regulator of gene expression.10 Additionally, PPAR activation was shown to play a beneficial role in preventing various disease states. For instance, treatment with synthetic PPARα and PPARγ ligands by lipid lowering fibrates and insulin-sensitizing thiazolidinediones, respectively, inhibits vascular inflammation, atherosclerosis, and restenosis through induction of the vasculoprotective and antiinflammatory enzyme, heme oxygenase (HO)-1, in vascular cells.11 Additionally, the PPARγ response was also found to increase expression of APN in human vascular cells.12
HO is a rate-limiting enzyme in the degradation of heme to produce equimolar amounts of CO, iron, and biliverdin, which is further converted to the antioxidant, bilirubin, by biliverdin reductase.13,14 Two HO isozymes were identified as having distinct genes.15 Among them, HO-1, a stress–response protein, can be induced by various oxidative-inducing agents, including heme, heavy metals, UV radiation, cytokines, and endotoxin.16,17 Recently, numerous in vitro and in vivo studies showed that the induction of HO-1 is an important cellular protective mechanism against oxidative injury.15 Both isoforms might be largely responsible for the recycling of iron through its liberation from heme and hemoproteins, although their contribution to total iron homeostasis has not been carefully examined. The destination of iron has not been clearly addressed, although the hypothesis that iron is safely stored in the iron storage protein, ferritin, is favored. In contrast to expectations, evidence has recently accumulated suggesting that HO-1 is required for mammalian iron reutilization.18 The first human case of HO-1 deficiency and mice with a targeted HO-1 null mutation both developed serum iron deficiency, but also pathological iron overload, indicating that HO-1 is crucial for the expulsion of iron from tissue stores.18–20 Furthermore, our lab previously showed that overexpression of HO-1 in vascular smooth muscle cells produced a lesser extent of iron deposition caused by additional hemin treatment compared with control cells. We also showed a lesser extent of iron accumulation in aortic tissues of mice with Adv-HO-1 gene therapy than in control mice.21
Humans are susceptible to iron metabolism disorders. For example, dietary iron deficiency causes millions of cases of anemia yearly, while functional hypoferremia contributes to the anemia that is frequently observed in chronic inflammatory disease.22 While these conditions result from iron insufficiency, other human disorders are caused by excessive iron storage such as hemochromatosis and thalassemia. Frequent blood transfusions often result in iron overloading, which requires iron chelation. Additionally, hereditary hemochromatosis is a disorder of increased iron absorption and storage yielding multiorgan pathology, which affects approximately 1 in 200 individuals within white populations.
The interplay of APN and HO-1 in protecting against various disease states has not been clearly elucidated. Although it was shown that up-regulation of HO-1 causes adipose remodeling and increases APN secretion, both of them play synergistic actions in modulating the metabolic syndrome phenotype.23 Whether APN conversely regulates HO-1 expression and its potential implications against iron-mediated injury in liver has not been addressed.
Excess iron was shown to cause oxidative stress, as well as iron deposition, multiple organ failure, and anemia in clinical cases. We attempted to unravel the molecular mechanism of adiponectin-mediated HO-1 induction and its beneficial actions in preventing deleterious effects from hepatic iron overload both in vivo and in vitro.
Materials and Methods
Cell Culture and Reagents
We purchased mouse BALB/c embryonic liver cells from the Bioresource Collection and Research Center (Hsinchu, Taiwan) and cultured them in Dulbecco's modified Eagle's medium (DMEM) supplemented with an antibiotic/antifungal solution and 10% fetal bovine serum (FBS) (pH 7.2). DMEM, FBS, and tissue culture reagents were obtained from Life Technologies (Gaithersburg, MD). We purchased SB 202190 from Tocris (Ellisville, MO), SnPP from Porphyrin Products (Logan, UT), and all other chemicals were of reagent grade and were obtained from Sigma (St Louis, MO).
Production of Recombinant Adeno-Associated Virus Carrying APN
Full-length APN was obtained by polymerase chain reaction (PCR) amplification from a human complementary (c)DNA library, flanking the BamHI and XbaI restriction cutting sites, and was cloned into the pAAV-MCS vector. To produce the AAV virus, a three-plasmid cotransfection method was used.24 The plasmids used in transfection were as follows: (i) the AAV-CMV-APN plasmid with the gene driven by the CMV promoter, which carried the promoter-driven transgene flanked by AAV inverted terminal repeats; (ii) the pHelper plasmid, which contained helper genes from the adenovirus; and (iii) the pseudotyped AAV packaging plasmid containing the AAV8 serotype capsid gene coupled with the AAV2 rep gene. The wild-type of AAV (the control) or AAV-APN (experimental group) was purified two times by CsCl gradient ultracentrifugation, and the titer of vector genome particles was determined using a previously described method.25 The recombinant viruses with 1 × 1012 viral particles in 50 μl of PBS were injected into a mouse tail vein 2 weeks ahead of the iron insults.
Animal Model of Iron Overload and Biochemical Assays
Animal care and treatment were performed according to protocols approved by the Institutional Animal Care and Utilization Committee, Academia Sinica in conformity with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996). Male C57BL/6J (8 weeks; 20–25 g) were used in this study. These mice were fed a regular chow diet and maintained under conventional housing conditions in our animal facility. Five groups of C57BL/6J mice were used: 1, AAV; 2, AAV + iron; 3, AAV-APN; 4, AAV-APN + iron; and 5, AAV-APN + SnPP + iron. After administration of AAV or AAV-APN for 2 weeks, mice were intraperitoneally injected with iron-dextran five times per week at a dose of 1.25 mg/25 g for 2 weeks to cause iron overload in the liver, whereas control mice were injected with 0.1 ml of 10% dextrose (n = 15 per group). SnPP was given 2 hours ahead of the iron challenge and at a dose of 15 mg/kg three times a week, by an i.p. injection for 2 weeks. Levels of aspartate transaminase (AST; GOT), alanine transaminase (ALT; GPT) activity, creatinine, and blood urea nitrogen were measured in the serum using FUJI DRI-CHEM SLIDE (Fujifilm, Kanagawa-ken, Japan). The method for determining bilirubin in the culture medium in hepatocytes with APN treatment was described previously.26 Serial paraffin-embedded sections (10 μm) were produced to determine tissue iron deposition using an iron staining kit (Sigma), and frozen sections (20 μm) were prepared for the TUNEL assay.
HO-1 Enzymatic Activity
The generation of bilirubin was used to estimate the activity of HO-1 enzyme.27 Livers were pooled and homogenized on ice in a Tris-HCl lysis buffer (pH 7.4, containing 0.5% Triton X-100 and protease inhibitors). Homogenates (100 μl) were further mixed with 0.8 mmol/L NADPH, 0.8 mmol/L glucose-6-phosphate, 1.0 unit of glucose-6-phosphate dehydrogenase, 1 mmol/L MgCl2, hemin (0.25 mmol/L), and mouse liver cytosol containing biliverdin reductase at 4°C. The reaction was conducted at 37°C for 15 minutes in the dark and terminated by the addition of chloroform. Insoluble material was removed by centrifugation, and supernatants were analyzed for bilirubin concentration by comparing their absorbance at 464 and 530 nm. Controls included samples prepared without the NADPH generating system.
Recombinant APN Production
Full-length cDNA of human APN was subcloned into the pET-20b(+) vector at the restriction cutting sites of BamHI and NdeI. The resulting construct was transformed into BL-21 Escherichia coli followed by 1 mmol/L IPTG induction for 20 hours when the OD600 of the E. coli broth reached 0.5. APN was harvested from E. coli by a standard protocol with the addition of Ni-NTA column and FPLC gel filtration column (HiLoad 16/60 superdex 2000) purification, which was resolved on sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels to ensure the purity and identity.
Constructs of Plasmid Variants and the Luciferase Activity Assay of the PPARα Enhancer and HO-1 Promoter
The pBV-luc plasmid containing the prototypic PPAR response element (5-AGGTCAAAGGTCA-3) from the acyl-CoA oxidase gene promoter was a gift from Dr. Vogelstein (Johns Hopkins University, Baltimore, MD).28 The PPARα and RXRα cDNAs in pcDNA3-Flag vectors were kind gifts from Dr. Song-Kun Shyue (Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan). The method to obtain the pGL3/hHO-1 reporter plasmid, which contains a 3293-bp fragment located −3106 to +186 bp relative to the transcription start site of the human HO-1 gene, and the method for the reporter activity assay was described previously.29
PPAR Small Interfering (si)RNA Preparation and Transient Transfection
PPARα siRNAs (5′-GAACAUCGAGUGUCGAAUATT-3′ and 5′-GACUACCAGUACUUAGGAATT-3′) duplexes were chemically synthesized by Ambion (Austin, TX). Hepatocytes were seeded in a 6-well plate and transfected with either 100 pmole of PPARα siRNA (#s72004 and #s72005 Ambion), scrambled control siRNA (#4611, Ambion), or GAPDH siRNA (#4624, Ambion) in a 100-μL volume with siPORTNeoFX. The efficiency of siRNA silencing was analyzed by Western blotting after transfection for 24 hours, followed by APN treatment for the indicated time periods.
Analysis of Gene Expression by Reverse-Transcription (RT)-PCR and Western Blot Analysis
The method to obtain total RNA for the RT-PCR analysis was as described previously30 with minor modifications. Sequences of the primer pairs for amplification of each gene were 5′-ATGCCAGTACTGCCGTTTTC-3′and 5′-GGCCTTGACCTTGTTCATGT-3′ for the PPARα gene (220 bp); 5′-CACGCATATACCCGCTACCT-3′and 5′-CCAGAGTGGTCATTCGAGCA-3′ for the HO-1 gene (175 bp); and 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′ for the GAPDH gene (451 bp). Total RNA, at 5 μg, of extracts from hepatocytes was used. The level of the housekeeping gene GAPDH was analyzed and used to demonstrate the presence of the same amount of total cDNA in each RNA sample. The expression level of HO-1 was detected from the same samples using appropriate primers. Bands separated on 2% agarose gels were visualized and quantified with an electrophoresis image analysis system (Eastman Kodak, Rochester, NY).
Antibodies for HO-1 (1: 2000, Assay Designs, Ann Arbor, MI), PPARα, Bcl-XL, BAX, and Lamin A/C (1: 500, Santa Cruz, CA), pPPARα (1: 500, Ser21, ABR Affinity Bioreagents, Rockford, IL), cleaved caspase 3 (1: 500, Cayman Chemical, Ann Arbor, Michigan), GAPDH (1: 2000, Ab Frontier, Seoul, Korea), and total and phosphorylated AMPK (1: 500, Millipore, Burlington, MA) and p38 (1: 500, Cell Signaling Technology, Dancers, MA) were included in the assay. Hepatocytes in 10-cm2 dishes after treatment with 30 μg/ml of APN for the indicated time points were harvested and partitioned into cytosol and nuclear fractions by NE-PER nuclear extraction reagents (Pierce, Rockford, IL) with the addition of protease inhibitors according to the manufacturer's instructions. The indicated cellular fractions (50 μg) were electrophoresed on a 10% SDS-polyacrylamide gel and then transblotted onto a Hybond-P membrane (GE Health care, Hong Kong). The subsequent procedures were described elsewhere.31
Establishment of HO-1 Transfectants
pcDNA-HO-1, a constitutive expression vector, carries full-length human HO-1 cDNA under the control of a cytomegalovirus (CMV) promoter/enhancer sequence. Short hairpin (sh)RNA against rat HO-1 was generated in the pSM2 vector (Open Biosystems, Huntsville, AL) and amplified in an E. coli system. Confirmation was verified by restriction site analysis and sequencing. We transfected pcDNA-HO-1, pcDNA, or pSM2-shHO-1 (4 μg/3.5-cm Petri dish) into hepatocytes using the jetPEI (Polyplus-transfection, San Marcos, CA). After transfection, cells were plated in DMEM with 10% FBS and 400 μg/ml of G418 for pcDNA variants, or 2 μg/ml of puromycin for pSM2-shHO-1 as selective pressures. G418- or puromycin-resistant cells were selected and expanded. The level of HO-1 was analyzed by Western blotting.
Electrophoretic Mobility Shift Assay (EMSA) and Chromatin Immunoprecipitation (ChIP) Assay
The EMSA was performed as described previously32 with minor modifications. To prepare the nuclear protein extracts, hepatocytes in 10-cm2 dishes after treatment with 30 μg/ml of APN for 1 hour were subjected to NE-PER nuclear extraction reagents (Pierce) with the addition of protease inhibitors. The subsequent procedures for the nuclear protein extraction followed the manufacturer's instructions. With in silico analysis using MatInspector Professional software, there was a putative PPRE in the mouse HO-1 promoter region. The PPRE fragment spanning −682 to −701 bp of the HO-1 promoter is ATAAACCATGGAAAAGTTAA (the putative PPRE is boxed), which was synthesized and end-labeled with biotin according to the manufacturer's protocol (Pierce Biotechnology, Rockford, IL), as a probe for the EMSA. Briefly, unlabeled oligonucleotides (1 μmol/L) were incubated in TdT reaction buffer containing biotin-11-dUTP (0.5 μmol/L) and TdT (0.2 U/μl) at 37°C for 30 minutes, followed by the addition of 2.5 μl EDTA (0.2 mol/L, pH 8.0) to stop each reaction and 50 μl chloroform/isoamyl alcohol to extract the TdT. Extracted nuclear proteins (10 μg) were incubated with biotin-labeled (1 pmol) probes at 15°C for 30 minutes in binding buffer containing 1 μg of poly deoxyinosinedeoxycytidine (dI-dC) (Panomics, Redwood City, CA). For competition with unlabeled oligonucleotides, a 100-fold molar excess of unlabeled oligonucleotides relative to the biotin-labeled probes was added to the binding assay. The mixture was separated on a 6% nondenaturing polyacrylamide gel at 4°C in 1× TBE (90 mmol/L Tris borate and 2 mmol/L EDTA; pH 8.3) and then transblotted onto a Hybond N+ membrane (Amersham Pharmacia Biotech, Freiburg, Germany). Blots were incubated with blocking buffer, followed by the addition of streptavidin-horseradish peroxidase (HRP) conjugates. Blots were imaged by means of an enhanced chemiluminescence system.
A ChIP assay was performed according to the instructions of Upstate Biotechnology (Lake Placid, NY) with minor modifications. Briefly, 6 × 105 cells were cultured in 100-mm dishes and pretreated with or without PPARα antagonists for 1 hour before APN administration for another 1 hour. The resulting supernatant was subjected to overnight co-immunoprecipitation (IP) using an anti-PPARα antibody, or the same amount of a nonspecific antibody (GAPDH) as a negative control, followed by incubation with a salmon sperm DNA/protein G agarose slurry to immobilize the DNA-protein-antibody complex. DNA-protein complexes were then eluted with 200 μl of elution buffer (Tris-EDTA buffer containing 1% SDS) for 30 minutes, and the cross-links were reversed by overnight incubation at 65°C. DNA was purified with a PCR purification kit (Qiagen, Hilden, Germany). The DNA filtrates were amplified by PCR with primers flanking the promoter of the HO-1 gene containing the putative PPRE: HO-1 forward (5′-GCTCAGATTCCCCACCTGTA-3′) and reverse primers (5′-ACCTTCCCGGAACTCTTAGG-3′). Additionally, the template was replaced with double-distilled (dd)H2O as a negative internal control. The PCR products were electrophoresed on a 2% agarose gel, and PCR products of the expected size of 162 bp were visualized, quantified using the Image analysis system, and eluted from the agarose gel for sequencing to verify the site of amplification.
Determination of Reactive Oxygen Species (ROS) Generation and Apoptosis in Hepatocytes
Production of cellular ROS was evaluated by analyzing changes in the fluorescence intensity resulting from oxidation of the intracellular fluoroprobe, 5-(6)-chloromethyl-20,70-dichlorodihydrofluorescein diacetate (CM-H2DCFDA). In brief, hepatocytes grown on a coverglass from each treatment group were loaded with 10 μmol/L of the nonfluorescent dye 20,70-dichlorodihydrofluorescein diacetate (H2DCFDA, Molecular Probes, Eugene, OR) at 37°C for 30 minutes in the dark followed by 20 μmol/L of iron dextran challenge for 1 hour. Wells were washed twice with Hank's balanced salt solution (HBSS) containing calcium and magnesium. Apoptosis in hepatocytes with iron dextran challenge was identified by a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay with an in situ Cell Death Detection kit (Roche, Mannheim, Germany) according to the manufacturer's instructions. Fluorescence was viewed with a CCD camera (DP72, Olympus, Melville, NY) attached to a microscope system (BX51, Olympus) at ×100 magnification.
Immunofluorescence Staining
Hepatocytes were cultured on poly-L-lysine-coated 0.17-mm coverslips, and numbers of cells, which had been transfected with pcDNA3 or pcDNA3-Flag-PPARα for 48 hours followed by 30 μg/ml APN administration for 1 hour, were counted at 50–80% confluence. Then cells were fixed in 4% formaldehyde for 15 minutes. Cells were permeabilized with 0.2% Triton and 0.1% Tween 20 in blocking buffer (3% BSA in PBS) for 2 hours. Antibodies used for staining included mouse anti-Flag (1:1000, Sigma) followed by Texas red-conjugated goat anti-mouse IgG (IgG) (1:200, Jackson Immunoresearch, West Grove, PA). Coverslips were mounted on slides with Vectashield anti-fade (Vector Laboratories, Inc., Burlingame, CA) diluted 1:1 with PBS, and images were obtained with a DMI 6000B CS laser confocal microscope (Leica, Heidelberg, Germany) using an HCX PL APO l-blue ×63/1.40–0.60 NA oil-immersion objective lens for hepatocytes. Images were acquired with a CM350 CCD camera (Applied Precision, Issaquah, WA) using TCS SP5 confocal spectral microscope imaging system software (Leica) and processed with Photoshop 7.0 software (Adobe System, San Diego, CA). Four coverslips in each experimental group were examined.
Statistical Analysis
Values are expressed as the mean ± SEM of at least three experiments. The significance of the difference from the control groups was analyzed by Student's t-test or one-way analysis of variance (analysis of variance). A value of P < 0.05 was considered statistically significant.
Results
Therapeutic Effects and Mechanisms of APN in Mice Subjected to Iron Dextran Overload
To examine the effect of APN gene transfer in liver on iron dextran-mediated damage, we administered adeno-associated virus (AAV) or AAV-APN intravenously to 8-week-old C57BL/6J mice for 2 week before intraperitoneal iron dextran challenge for another 2 weeks. At 4 weeks, these animals were sacrificed, and their blood samples and livers were collected for further analysis. As shown in the Table 1, there was no significant difference in body weight, food intake, or levels of creatinine and blood urea nitrogen among groups of mice. However, mice given iron dextran have severe liver dysfunction including increased GOT and GPT levels (Figure 1A) and apoptosis (Figure 1B), which were statistically alleviated by AAV-APN therapy. Additionally, Western blot analysis showed that mice with APN therapy had significantly increased AMPK phosphorylation, HO-1 overexpression (Figure 1C), and HO-1 activity (Figure 1D), but not those with iron dextran or AAV treatment alone. Notably, iron accumulation in the liver subjected to iron overload was eliminated by AAV-APN therapy, indicative of the correlation between HO-1 induction and iron reutilization (Figure 1E). Nevertheless, the therapeutic effect of AAV-APN in protecting liver from iron-mediated damage was reversed by blockage of HO-1 activity by SnPP in the assessment of the levels of GOT/GPT, apoptotic cell death, HO-1 activity, etc (Figure 1, A–E).
Table 1.
Effects of APN or the Combined Treatment of APN and Iron Dextran or SnPP on Mice Body Weights, Food Intake, and Serum Levels of Creatinine and BUN
| AAV |
AAV-APN |
||||
|---|---|---|---|---|---|
| + | Iron | + | Iron | SnPP + Iron | |
| Weight, g | 23.57 ± 0.33 | 22.50 ± 2.27 | 23.23 ± 0.05 | 22.50 ± 2.27 | 22.87 ± 1.23 |
| Food intake, g/d/5ea | 13.2 ± 2.1 | 11.2 ± 1.5 | 13.5 ± 1.1 | 12.5 ± 1.6 | 11.5 ± 1.2 |
| CRE, mg/dl | 0.47 ± 0.05 | 0.33 ± 0.12 | 0.35 ± 0.05 | 0.40 ± 0.08 | 0.43 ± 0.05 |
| BUN, mg/dl | 31.9 ± 3.8 | 28.4 ± 1.6 | 26.5 ± 5.3 | 23.0 ± 4.4 | 31.7 ± 7.1 |
Body weight, food intake, and levels of creatinine and BUN were determined at 4 weeks after viral administration, or the combination of iron dextran challenge and SnPP treatment. Data are mean ± SD (n = 15 per each group).
Figure 1.
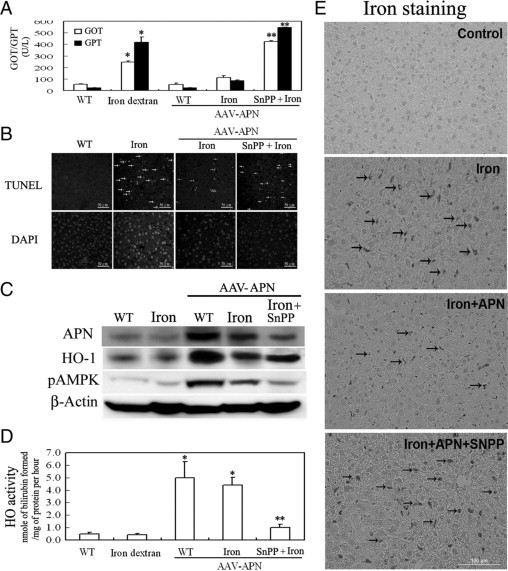
Therapeutic effects and mechanisms of adiponectin (APN) associated with HO-1 induction in murine hepatic iron overload. A: Mice were intravenously injected with an AAV-APN gene for two weeks before SnPP and iron dextran challenge for another two weeks. The method for determining serum levels of GOT and GPT followed the manufacturer's instructions. GOT and GPT levels are expressed as ΔOD460/min/mg protein. B: Representative photomicrographs of iron-mediated apoptosis in hepatocytes stained with TUNEL, as indicated by arrows, and the nuclei marker, DAPI. C: Western blot analysis of AMPK phosphorylation and HO-1 induction by APN in mice with iron overload for one week. Liver tissues (50 μg) from each group were assessed for the induction of pAMPK and heme oxygenase (HO)-1. A representative result of three separate experiments is shown. D: Assessment of hepatic HO-1 activity after mice were exposed to iron dextran challenge for two weeks. E: Elimination of iron accumulation with hepatic iron overload by APN. Liver sections from various treatments were stained to determine the extent of iron deposition for one week. The green staining of deposited iron is indicated by arrows, and a representative result of three separate experiments is shown. Results are expressed as the mean ± SD (n = 15). *P < 0.005, vs. the given empty AAV alone and **P < 0.01 vs. AAV-APN with additional iron dextran challenge.
APN Concentration- and Time-Dependently Induced HO-1 Expression at the Transcriptional Level
To examine the interplay between APN and HO-1, hepatocytes were given an increasing concentration of APN from 0–50 μg/ml for HO-1 induction and then analyzed by RT-PCR and Western blotting. As demonstrated in Figure 1A, HO-1 concentration-dependently increased from 0 to 30 μg/ml of APN administration for 6 hours, but there was no further increase at 50 μg/ml. Furthermore, cells were transiently transfected with the pGL3-basic luciferase-reporter vector containing the human HO-1 promoter (−3106/+186) for 24 hours, as described previously.30 Adding a concentration of 30 μg/ml of APN for 6 hours resulted in a significant increase in HO-1 luciferase activity by approximately 2.0-fold (Figure 2A). Additionally, cells treated with APN showed significant induction of HO-1 in a time-dependent fashion at both the messenger (m)RNA and protein levels. Significant up-regulation of HO-1 occurred at 2 hours of APN treatment, had increased by approximately 3.0-fold at 6 hours, and significant induction of HO-1 was sustained for up to 24 hours of treatment, the longest time period examined (Figure 2B). Likewise, the activity of HO-1 was significantly increased by measuring the byproducts of the HO-1 reaction, bilirubin (Figure 2C). The pattern of the time-dependent increase of bilirubin in hepatocytes with APN administration was an approximately 6-hour delayed response relative to the time course of mRNA and protein induction of HO-1 by APN in Figure 2B. The phenomenon of HO-1 induction by recombinant APN was further verified in hepatocytes infected with Adv-APN on days 1, 3, and 5, as demonstrated in Figure 2D.
Figure 2.
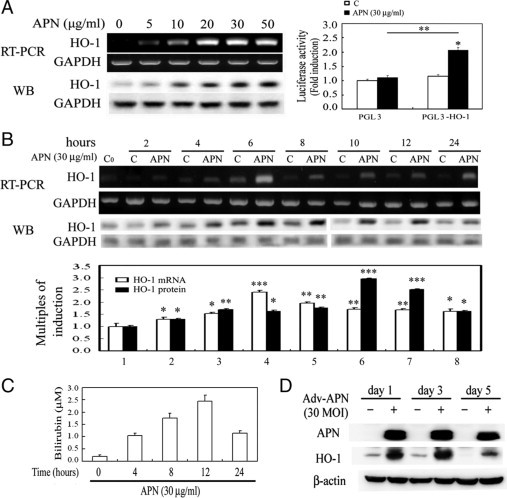
Regulation of heme oxygenase (HO)-1 induction by adiponectin (APN) in concentration- and time-dependent fashions. A: Concentration-dependent induction of HO-1 by APN. Left: The effect of increasing concentrations (0–50 μg/ml) of APN for six hours on the induction of the HO-1 protein level was analyzed by Western blotting. Right: Mouse hepatocytes were transiently transfected with pGL3/HO-1 and pRL-TK for 24 hours, followed by 30 μg/ml of APN treatment for six hours. The methods are described in Materials and Methods. Results are presented as the mean ± SEM of four independent experiments. *P < 0.05 vs. pGL3 alone and **P < 0.05 vs. pGL3 with additional APN treatment. Time-course induction of (B) HO-1 mRNA/protein and (C) HO-1 activity in hepatocytes by APN treatment. Cells were treated with adiponectin (30 μg) for the indicated time points and analyzed by RT-PCR and Western blotting. Equal loading in each lane or transfer was confirmed using GAPDH mRNA or by incubation with an anti-GAPDH antibody. Representative results of three separate experiments are shown. Additionally, the bilirubin concentration in 0.5 ml of cell medium from hepatocytes at various time points of 30 μg/ml of APN treatment was assessed, as described in Materials and Methods. Data were obtained from six independent experiments. Results are expressed as the mean ± SEM. Significantly different (*P < 0.05, **P < 0.01, and ***P < 0.005 vs. the control group). D: Verification of APN-induced HO-1 overexpression by an adenovirus carrying the APN gene in hepatocytes. After cells were infected with 30 MOI of Adv variants for one, three, and five days, cell lysates were prepared, and the expression of HO-1 protein was determined by Western blot analysis.
Signal Transduction Pathways Involved in APN-Mediated HO-1 Induction
Given that the molecular actions of APN are through AMPK, p38MAPK, or PPARα,5,33 the effects of APN on these molecules were examined by Western blots. Hepatocytes with 20 minutes of APN treatment significantly increased phosphorylation of AMPK, followed by PPARα activation at 1 hour. This phenomenon was correlated with HO-1 induction at 6 hours of APN administration because blockage of activation of AMPK by an AMPK inhibitor, compound C, reversed APN-mediated PPARα and subsequent HO-1 induction, as shown in Figure 3A. On the contrary, altering the phosphorylation of neither p38MAPK at 20 minutes of APN administration in hepatocytes nor a p38MAPK inhibitor, SB202190, was shown to eliminate PPARα activation at 1 hour and consequent induction of HO-1 at 6 hours of APN administration (Figure 3B).
Figure 3.
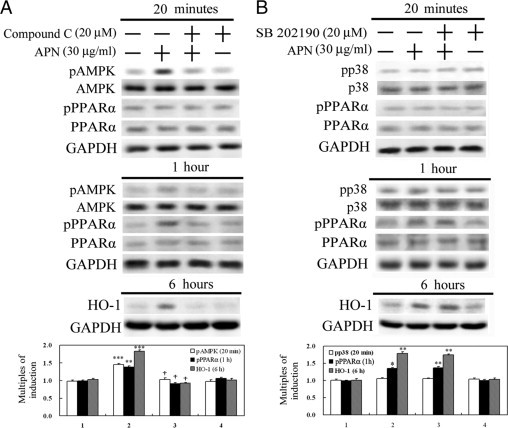
Determination of signal transduction pathways involved in adiponectin (APN)-mediated heme oxygenase (HO)-1 induction. Hepatocytes were pretreated with a pAMPK inhibitor, compound C (A), or a p38MAPK inhibitor, SB202190 (B), for 30 minutes before treatment with APN for 20 minutes or one or six hours to determine the respective active forms or protein levels by Western blot analysis. Phosphorylated and total forms of AMPK and p38MAPK were harvested at 20 minutes of APN treatment, while the activation of PPARα and consequent HO-1 protein level were determined at one and six hours of treatment, respectively. Three samples were analyzed in each group, and values are presented as the mean ± SEM. Representative results of three separate experiments are shown. *P < 0.05, **P < 0.01, and ***P < 0.005 vs. the control group; †P < 0.01 vs. the APN-treated group.
Involvement of PPARα in APN-Mediated HO-1 Induction
Next, to examine whether PPARα serves as a downstream effector of pAMPK, PPRE enhancer activity and nuclear translocation of PPARα were assayed in hepatocytes treated with APN at the indicated time points. Luciferase activity driven by the PPARα enhancer was markedly increased by approximately 84-fold in HEK293 cells transiently simultaneously transfected with vectors of pRL-TK, pBV-Luc-PPRE, pcDNA-PPARα, and pcDNA-RXR for 24 hours followed by 6 hours of APN administration (Figure 4A). Additionally, similar to the effect of Wy-14643 (a PPARα agonist), 1 hour of APN administration caused an increase in phosphorylation of PPARα in the nuclear fraction, which was effectively reversed by compound C in Figure 4B, suggesting the likelihood that PPARα is the downstream effector of pAMPK activation in this event. Furthermore, in addition to compound C (an AMPK inhibitor), GW6471 (a selective PPARα antagonist) also effectively reversed HO-1 induction with APN administration for 6 hours (Figure 4C). Furthermore, knockdown of PPARα by siRNA was used to examine its roles in the increased APN-mediated HO-1 induction by RT-PCR and Western blot analyses. Results in Figure 4D show that cells with PPARα knockdown had significantly decreased APN-mediated HO-1 induction relative to the control group or cells transfected with scrambled siRNA.
Figure 4.
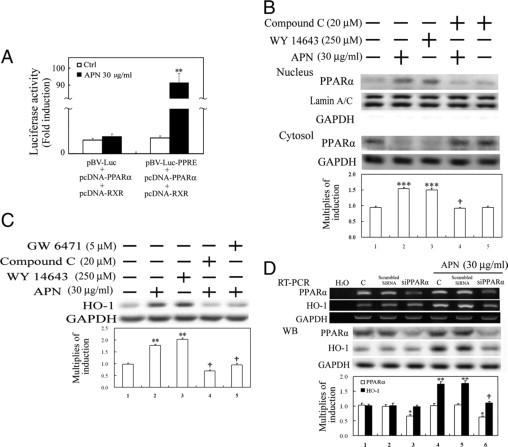
Involvement of PPARα activation in adiponectin (APN)-mediated heme oxygenase (HO)-1 induction. APN increased PPRE enhancer-driven luciferase activity (A) and nuclear translocation of PPARα activation (B) and subsequent HO-1 induction (C). A: HEK 293 cells were transiently simultaneously transfected with pBV-Luc-PPARα enhancer and Renilla control vectors or with additional transfection of pcDNA-PPARα, and pcDNA-RXR (0.5 μg/well) for five hours, after which fresh medium was added for overnight inoculation. Cell lysates were harvested after cells were treated with APN for six hours. B: Cells were respectively pretreated with either AMPK or PPARα antagonist, or a PPARα agonist for one hour before the administration of APN for another one hour for nuclear translocation of PPARα by Western blot analysis and (C) subsequent HO-1 induction at six hours of APN treatment. D: Elimination by PPARα knockdown of APN-mediated HO-1 induction. The method of cells with PPARα knockdown was described in Materials and Methods. Equal loading or transfer was confirmed by incubation with an anti-GAPDH or anti-lamin A/C antibody. Representative results of three separate experiments are shown, and data are presented as the mean ± SEM (*P < 0.05, **P < 0.01, and ***P < 0.001 vs. the control; †P < 0.05 vs. APN alone).
Effects of APN and PPARα Overexpression in HO-1 Induction
To further confirm the essential role of PPARα in the induction of HO-1 by APN, hepatocytes were transiently transfected with the pcDNA3-Flag vector carrying full-length PPARα cDNA for overexpression. The results in Figure 5A demonstrate that overexpression of exogenous PPARα increased nuclear translocation of PPARα in the nuclear fraction and then HO-1 induction by approximately 2.0-fold in the Western blot analysis of total cell lysates, which were further enhanced to approximately 6.0-fold by APN administration. This suggests that PPARα activation by APN is responsible for HO-1 induction. Additionally, APN-mediated PPARα nuclear translocation was examined by immunofluorescence staining. In control cells, expression of Flag-PPARα was mainly detected in the cytosol (Figure 5B). However, the Flag-PPARα protein was significantly translocated into the nucleus on APN stimulation. No exogenous overexpression of PPARα was detected in the cytosolic or nucleus fractions when cells were transfected with the pcDNA3 vector alone (data not shown).
Figure 5.
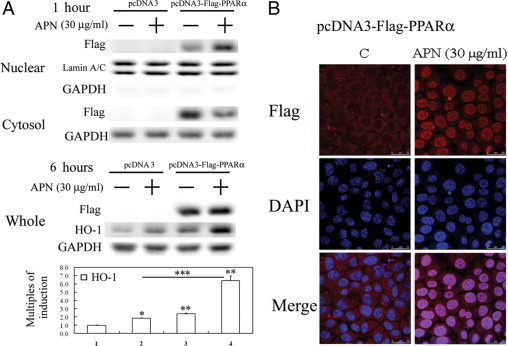
Adiponectin (APN) increased nuclear translocation of exogenous PPARα and subsequent heme oxygenase (HO)-1 induction in hepatocytes with PPARα overexpression by (A) Western blot analysis of cytosol and nuclear fractions of PPARα and total cell lysates of HO-1 protein level and (B) fluorescent confocal microscopy. A: Cells were treated with APN for one or six hours and harvested for cytosolic-nuclear partitioning of PPARα. Weights of the cell lysates were 50 μg for the cytosolic and nuclear fractions analyzed, and GAPDH and lamin A/C were used as internal controls for these fractions. Data are presented as the mean ± SEM (*P < 0.01, and **P < 0.005 vs. the control; ***P < 0.01 vs. cells transfected with pcDNA3 with additional APN treatment). B: Cells were treated with APN (30 μg/ml) for one hour, then immunostained with an anti-Flag antibody followed by incubation with a second antibody conjugated with Texas red. Red color represents Flag-positive staining in the cytosol or nuclei. Identical fields stained with Flag were also stained using DAPI to reveal the positions of cell nuclei. Micrographs of representative fields were recorded. Representative results of three separate experiments are shown.
Effects of APN and PPARα Agonist on Binding of PPARα to the Putative PPRE of the HO-1 Promoter Region
Furthermore, to precisely prove the binding of PPARα to the putative PPRE derived from the HO-1 promoter region, an EMSA was performed in hepatocytes with APN administration or additional pretreatment with an antagonist of AMPK or PPARα (ie, compound C or GW6471, respectively). Results of the EMSA in Figure 6A show the increase in the DNA binding activities of PPARα in cells treated with APN and Wy14643 (as a positive control). The increased binding activity of PPARα to PPRE in cells treated with APN was abolished by the addition of compound C and GW6471, and the competition of a 100-fold molar excess of unlabeled oligonucleotides relative to the biotin-labeled probe. In addition, the association of PPARα with the PPRE region of the HO-1 promoter was further confirmed using a ChIP assay. We created APN-induced association with the PPARα-DNA complex by pulling down the PPRE fragment of the HO-1 gene promoter using an anti-PPARα antibody and using an anti-GAPDH antibody as a negative control. The immunoprecipitated PPRE fragments were amplified by PCR to examine the binding of PPARα to the fragment. Figure 6B shows that APN and Wy14643 increased the association of PPARα recruitment to PPRE at 1 hour of treatment by approximately 1.6-fold, whereas additional pretreatment with compound C and GW6471 reversed this effect by APN, in agreement with the findings in Figure 6A. Together, these results suggest that APN increases PPRE-mediated up-regulation of HO-1 by increasing the binding activity of PPARα to PPRE.
Figure 6.
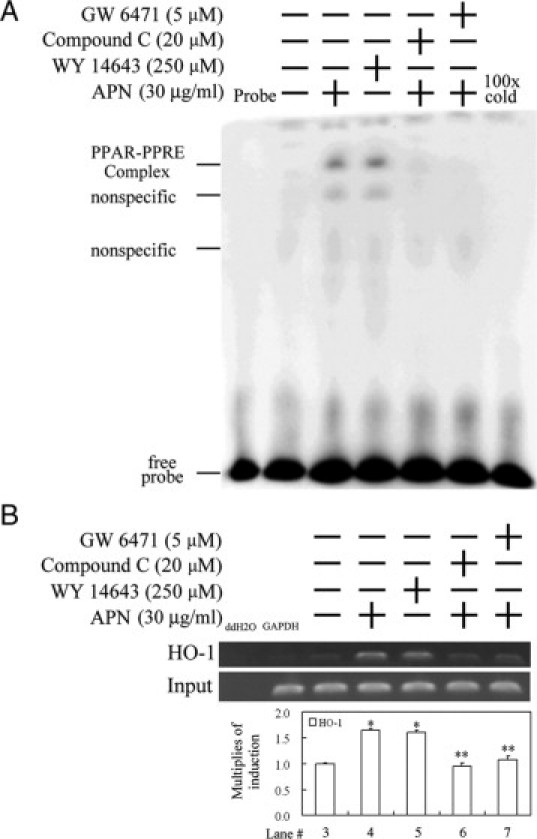
Adiponectin (APN)-mediated increase in the binding activity of PPARα to the PPRE of the heme oxygenase (HO)-1 promoter region by an electrophoretic mobility shift assay (EMSA) and chromatin immunoprecipitation (ChIP). A: Cells were cultured and pretreated with a PPARα or AMPK antagonist for 1 hour before the addition of 30 μg/ml APN for one hour, with Wy14643 used as a positive control. The putative PPRE-binding activity derived from the HO-1 promoter region of nuclear proteins was assayed by EMSA in cells with the indicated treatments. 100× cold denotes a 100-fold molar excess of unlabeled oligonucleotides relative to the biotin-labeled probe; this was added to the binding assay for competition with the unlabeled oligonucleotide. The mobility of specific PPARα complexes is indicated. B: Cell lysate was subjected to a ChIP assay. The DNA associated with the PPRE was immunoprecipitated with an anti-PPARα antibody, and PCR amplification was used to determine the extent of PPARα association with the functional PPRE in an HO-1 promoter fragment of 162 bp. Distilled water (ddH2O) and anti-GAPDH were used, respectively, as negative controls for the PCR and ChIP assays. Representative results of three separate experiments are shown, and data are presented as the mean ± SEM (*P < 0.01 vs. the control; **P < 0.01 vs. APN alone).
Therapeutic Effect and Mechanism of APN in Protecting Against Iron-Mediated Hepatic Apoptosis
We previously demonstrated that APN gene therapy effectively rescued iron dextran-mediated liver function and iron deposition as shown in Figure 1. The underlying molecular mechanisms of APN were further examined in vitro using hepatocyte cell culture. To optimize the therapeutic effect of APN on the various concentrations of iron dextran, hepatocytes were treated with increasing concentrations of 10–40 μmol/L of iron dextran. Results in Figure 7A show that the physiological concentration of 30 μg/ml of APN significantly alleviated caspase 3 activation induced by iron dextran at the concentrations of 10 and 20 μmol/L, but not 40 μmol/L. Therefore, 20 μmol/L of iron dextran was used for the following experiments. Iron dextran-mediated ROS generation in hepatocytes is shown in Figure 7B using CM-H2DCFDA fluorescence, which was significantly eliminated by APN. Additionally, hepatocytes were pretreated with APN for 1 hour before 20 μmol/L iron dextran challenge for 18 hours to assess the therapeutic intervention of APN in preventing hepatic iron overload. Apoptotic-related molecules such as Bcl-XL (anti-apoptosis), Bax (pro-apoptosis), and caspase-3 were examined by Western blot analysis. Increasing protein levels of Bcl-XL by APN and Wy-14643 were accompanied by elimination of iron-mediated caspase 3 activation. Contrarily, the induction of Bcl-XL by APN was abolished by pretreatment with compound C and GW6471 for 1 hour, which produced a concomitant increase in cleaved caspase 3 (Figure 8A). However, there was no apparent alteration in Bax across all treatments. Next, to further explore whether HO-1 is responsible for Bcl-XL induction as we reported previously in another setting,26 an HO-1 inhibitor, SnPP, was added to block HO-1 activity to assess its impacts on the antiapoptotic effects of APN. Results in Figure 8B demonstrate that the addition of SnPP treatment decreased Bcl-XL protein levels, which consequently abolished APN-mediated protection against iron-mediated caspase 3 activation. Whether caspase 3 activation was correlated with cell apoptosis was examined by a TUNEL assay in Figure 8C, which demonstrates that APN-mediated protection against iron-mediated cell apoptosis was alleviated by the addition of compound C, GW6471, or SnPP, suggesting that the induction of HO-1 by APN is via a pAMPK- and PPARα-dependent pathway, through which HO-1 exerts the protective effect of APN on hepatocytes with iron overload.
Figure 7.
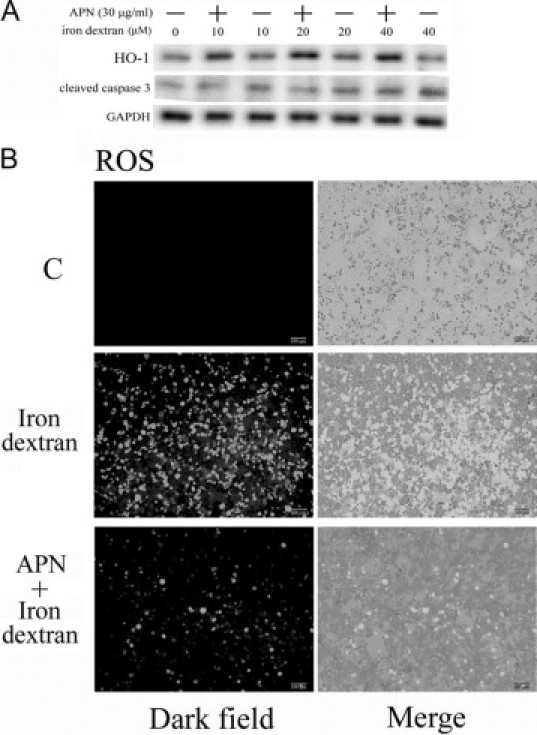
Adiponectin (APN) attenuates iron dextran-induced caspase 3 activation and reactive oxygen species (ROS) production. A: Therapeutic effect of APN in caspase 3 activation induced by varying concentrations of iron dextran. Cells were pretreated with 30 μg/ml of APN for one hour, followed by challenge with various concentrations of iron (0–40 μmol/L) B: Representative CM-H2DCFDA fluorescent photomicrographs of hepatocytes challenged with 20 μmol/L of iron dextran. Results are representative data from three separate experiments.
Figure 8.
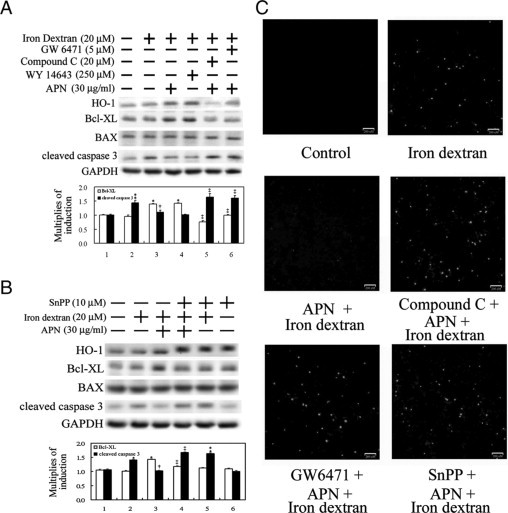
Elimination of iron dextran-mediated caspase 3 activation and apoptosis by adiponectin (APN) through AMPK-mediated PPARα activation and subsequent heme oxygenase (HO)−1 induction. A: Cells were pretreated with compound C, GW6471, or DMSO as the control for one hour before another one hour of APN or phosphate-buffered saline administration, followed by iron dextran challenge for 18 hours. Additionally, cells were treated with WY-14643 as a positive control for HO-1 induction. B: Effect of HO-1 on APN-mediated protection against iron-mediated hepatic injury. Cells were pretreated with SnPP for one hour to block HO-1 activity before APN administration. Fifty micrograms of total cell lysate was analyzed for the protein level of apoptotic-related molecules by Western blotting. Membranes were probed with an anti-GAPDH antibody to verify equivalent loading. Bar charts in the lower panel show the band intensities of indicated molecules by densitometry. Data were derived from three independent experiments and are presented as the mean ± SEM. Significantly different (*P < 0.01, and **P < 0.005 vs. the control; †P < 0.05 vs. iron dextran alone; ‡P < 0.01 vs. APN and iron dextran-treated group). C: Cells grown on coverslips with the above-mentioned various pretreatments, then challenged with 20 μmol/L of iron dextran for two days. Nuclei with positive stains are indicated as having undergone cell apoptosis by the TUNEL assay. Representative results of three separate experiments are shown.
To verify further the protective effect of APN by HO-1 induction, permanent hepatocyte clones with HO-1 overexpression and HO-1 knockdown were included. As presented in Figure 9A, hepatocytes overexpressing HO-1 showed an increase in the Bcl-XL expression level and then caspase 3 inactivation, whereas the opposite effects were observed in cells with HO-1 knockdown. The actual mechanism of the antiapoptotic effect of HO-1 in hepatocytes challenged with iron dextran was examined by including the end-products of HO-1 reaction, CO (CORMII) and bilirubin. The result in Figure 9B show that bilirubin significantly suppressed iron-mediated caspase-3 activation with a concomitant increase in Bcl-xL, compared with what CORM(II) did. This suggests that the antiapoptotic effect of HO-1 results from bilirubin. Furthermore, hepatocytes overexpressing HO-1 showed significantly decreased iron deposition relative to the control group, and the greatest extent of iron deposition was demonstrated in cells with HO-1 knockdown (Figure 9C), suggesting that HO-1 not only has an antiapoptotic effect, but also increases iron metabolism in hepatocytes.
Figure 9.
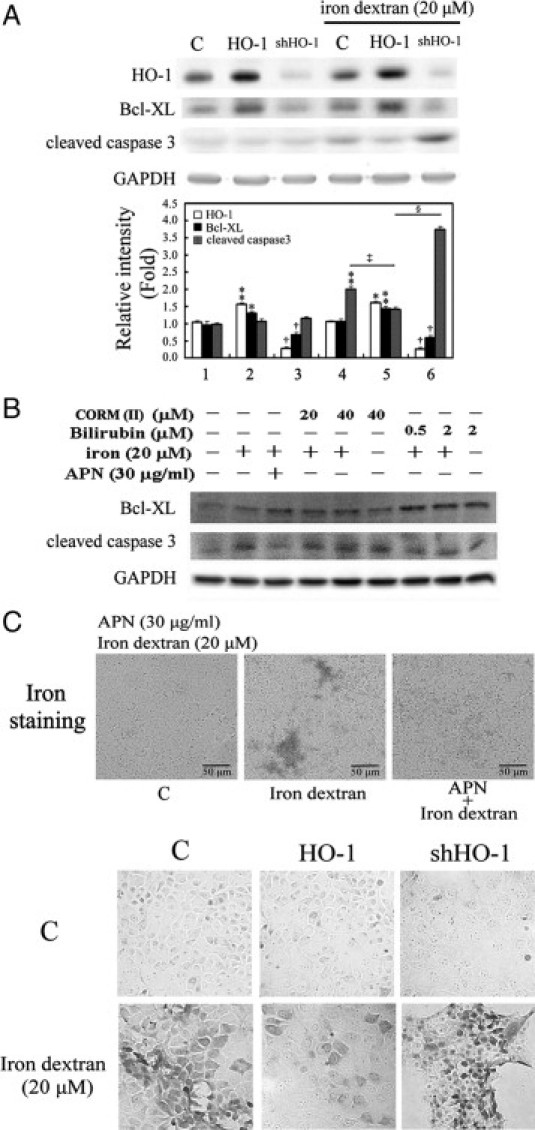
Effects of adiponectin (APN)-mediated heme oxygenase (HO)-1 induction in apoptosis and iron accumulation in hepatocytes by iron dextran challenge. A: Hepatocytes permanently transformed with HO-1 or small hairpin (sh)HO-1 were challenged with 20 μmol/L iron dextran for two days. Data are presented as the mean ± SEM (*P < 0.05, and **P < 0.01 vs. the control; †P < 0.01 vs. cells overexpressing HO-1; ‡P < 0.005 vs. iron alone; §P < 0.001 vs. cells overexpressing HO-1 with the additional iron insult). B: The mechanism of HO-1 in iron dextran-mediated apoptosis by the administration of CO or bilirubin, end-products of HO-1 reaction. Cells were treated with CO (CORMII) or bilirubin to mimic the antiapoptotic effect of HO-1, followed by iron challenge for 18 hours for the Western blot analysis of Bcl-XL or cleaved caspase 3, respectively. Equal loading or transfer was confirmed by incubation with an anti-GAPDH antibody. C: Effect of APN (upper) and hepatocytes overexpressing or knocking down HO-1 (lower) on 20 μmol/L of iron dextran insult in vitro. Intracellular iron deposition was examined by Perls' iron staining in iron challenge for two days. Original magnification: ×200. Representative results of three separate experiments are shown.
Discussion
APN is generally considered to act as a regulatory protein in controlling lipid and carbohydrate metabolism. Its role in the antiapoptotic effect has not been clearly illustrated. Herein, we demonstrate the therapeutic effects of APN in protecting hepatocytes from iron-mediated cell apoptosis and excess iron deposition both in vivo and in vitro. Interestingly, the biological concentration of APN in blood is approximate 5–30 μg/ml, thus accounting for approximately 0.01% of the total plasma protein.34 The concentration of APN used in this study was compatible with its physiological levels in human plasma, suggesting its clinical implications, especially for treating hepatic iron overload, which was not previously reported.
Herein, the protective effect of APN in hepatic iron overload occurs through HO-1–mediated antiapoptosis and iron reutilization. The molecular mechanism of HO-1 induction by APN is AMPK-mediated PPARα activation, although activation of PPARα/γ and HO-1 induction has been shown to increase APN expression.11,12 The phosphorylation of AMPK increases nuclear translocation of PPARα and subsequently HO-1 induction. The evidence for PPARα-mediated HO-1 in hepatocytes with APN treatment was verified using a pharmaceutical inhibitor of PPARα (GW6471) as well as PPARα overexpression and knockdown systems. Hepatocytes overexpressing PPARα mimicked the effect of APN in HO-1 induction, whereas siPPARα and a PPARα antagonist blocked APN-mediated HO-1 induction. Additionally, we pinpointed a PPARα-binding site in the mouse HO-1 promoter region, through which the binding activity was increased by APN-mediated PPARα activation according to the ChIP assay and EMSA. Furthermore, the antiapoptotic and anti-iron deposition effects of APN in hepatocytes with excess iron insult were abolished when using a pharmaceutical inhibitor of HO-1, SnPP, or in cells with HO-1 knockdown, suggesting that the protective effect of APN might be at least, in part, through HO-1 induction.
We demonstrated in vivo or in vitro that iron dextran causes free radical production (Figure 7B), but does not obviously induce HO-1 in mouse hepatocytes (Figures 1C and 7A). This agrees with the finding that iron dextran up-regulates ferritin, but not heme oxygenase-1, in the rat kidney with iron overload.35 However, these results differ from the results by Ibrahim et al,36 showing that the increase in HO-1 as a response to oxidative stress from iron dextran causes a cascade of events that leads to cellular protection in the rat liver. This might have been due a discrepancy of animal species, or the schedule or doses of iron dextran being given to these animals. However, our data raised an interesting possibility that the protective effect of APN on iron hepatotoxicity might be attributable to an HO-1–mediated antiapoptotic action, but the mechanism by which APN itself exerts antiapoptotic effects warrants further investigation. Moreover, the results presented in this study provide evidence that the protective effect of HO-1 against iron dextran-induced apoptosis is through the antiapoptotic effect of bilirubin (Figure 9B). Although bilirubin and CO are important byproducts of HO-1 reaction,37 the biological actions of bilirubin has been shown to scavenge ROS in vitro38 and inhibit NADPH oxidase,39 thereby reducing oxidant-mediated cellular damage and attenuating oxidant stress in vivo.40 However, CO has been demonstrated to significantly contribute to the antiinflammatory properties of HO-1 by suppressing inflammatory cytokines through activation of both sGC and p38 MAPK.41,42
APN not only decreased iron-mediated apoptosis, but also eliminated the extent of iron deposition in hepatocytes. Likewise, HO-1 overexpression mimicked the protective effect of APN in hepatic iron overload, whereas the protection by APN was eliminated in cells with HO-1 knockdown, which agrees with our previous publication that overexpression of HO-1 decreases iron deposition in atherosclerotic lesions of ApoE-deficient mice.21 Therefore, APN might be a potential therapeutic target for iron-induced hepatic injury. The underlying mechanisms of antiapoptosis by HO-1 were extensively elucidated37,39,40; however, little information has shed light on the molecular mechanism of HO-1 in iron reutilization, which warrants further investigation.
Given that adiponectin induces HO-1, it is tempting to postulate that the antioxidant and antiapoptotic properties of adiponectin may, at least in part, be related to HO-1 induction. However, there is increasing interest in the prooxidant activity of HO in diabetes, which is possibly due to increased redox-active iron in disease states. It was shown that great enhancement of HO-1 expression by diabetes mellitus, hypercholesterolemia, and smoking may be related to the severity of human atherosclerosis.43 Additionally, diabetes-induced oxidative damage to the heart is eliminated by SnPP, an HO-1 activity inhibitor, suggesting that the injury is intimately associated with up-regulation of HO-1 expression and activity.44 However, they could not rule out the possibility that the induction of HO-1 is the consequence other than the cause of diabetes mellitus mediated oxidative damage to the heart and that tissues in pathological condition might not properly respond to the increasing free iron on the increase in HO-1 activity. Furthermore, significant positive correlations were reported between the degree of renal failure and HO-1 levels in diabetic uremic patients; nevertheless, those authors speculated that HO-1 is induced to counteract the intracellular prooxidant status in diabetic nephropathy.45 Together with those findings, the timing and levels of HO-1 induction, in concert with properly sequestering redox-active iron into ferritin, might be critical for HO-1 to exert its therapeutic protection effects, although the cumulative evidence favors HO-1 as a therapeutic target for oxidative-related diseases.21,37,39,40
This is the first report to demonstrate that APN eliminates iron-induced hepatic apoptosis through PPARα-mediated HO-1 induction. It was previously shown that HO-1 overexpression increases the APN level by the synergistic modulation of metabolic syndrome. Additionally, the beneficial effects of HO-1 expression on myocardial ischemia-reperfusion involve increases in nitric oxide synthase expression and activity and serum APN in mildly diabetic rats.23 Conversely, herein we proved that APN increased PPARα-mediated HO-1 induction in hepatocytes, which consequently decreased excess iron-induced hepatic apoptosis and iron deposition. Interestingly, the biological concentration of APN (30 μg/ml) used in vitro might suggest that in addition to the metabolic importance of APN, it might have antiapoptotic and antioxidant effects via HO-1 induction in the liver. Thus, APN, via induction of HO-1, synergistically exerts protective effects of antioxidation and iron reutilization in the model of hepatic iron overload in mice. However, our experimental setting differs from that of Kim et al,46 who showed that HO-1 with ensuing APN secretion decreased the levels of proinflammatory cytokines and triglyceride and caused weight loss in fat mice with excessive ROS and increased cytokines. In line with that finding, recent studies demonstrated that HO-1 inducers (ie, CoPP and hemin) increase APN levels and improve insulin sensitivity and glucose tolerance in obese and streptozotocin-induced diabetic mice,47,48 which might be due to the likelihood of increased HO-1 expression acting as a chaperone protein to protect APN from oxidative destruction. In combination with our present finding showing that APN increased HO-1 levels, APN and HO-1 might influence each other's induction, although whether the induction of HO-1 by APN in turn up-regulates APN remains to be determined. Herein, we provide evidence that APN in concert with HO-1 might cause synergistic protection against iron overload, iron-mediated apoptosis, and other oxidative or metabolic-related disorders.
The results presented in this study showed the interwoven relationship of adiponectin, PPARα, and HO-1, which suggests their crucial protective roles in modulating antiapoptotic and metabolic processes of APN in hepatic iron overload. Notably, determining whether APN can serve as an alternative gene therapy for patients with HO-1 deficiency or with iron overload requires further clinical investigation.
Footnotes
Supported by grants from Tzu-Chi University (TCIRP95007-02), Hualien, Taiwan and from the National Science Council, (NSC97-2320-B-038-017-MY3). Taiwan.
H.L. and C.-H.Y. contributed equally to this work.
Current address of H.L.: Department of Physiology, School of Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan.
References
- 1.Rabin KR, Kamari Y, Avni I, Grossman E, Sharabi Y. Adiponectin: linking the metabolic syndrome to its cardiovascular consequences. Expert Rev Cardiovasc Ther. 2005;3:465–471. doi: 10.1586/14779072.3.3.465. [DOI] [PubMed] [Google Scholar]
- 2.Wulster-Radcliffe MC, Ajuwon KM, Wang J, Christian JA, Spurlock ME. Adiponectin differentially regulates cytokines in porcine macrophages. Biochem Biophys Res Commun. 2004;316:924–929. doi: 10.1016/j.bbrc.2004.02.130. [DOI] [PubMed] [Google Scholar]
- 3.Yokota T, Oritani K, Takahashi I, Ishikawa J, Matsuyama A, Ouchi N, Kihara S, Funahashi T, Tenner AJ, Tomiyama Y, Matsuzawa Y. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood. 2000;96:1723–1732. [PubMed] [Google Scholar]
- 4.Yuhki K, Kawabe J, Ushikubi F. Fat, keeping the heart healthy? Nat Med. 2005;11:1048–1049. doi: 10.1038/nm1005-1048. [DOI] [PubMed] [Google Scholar]
- 5.Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006;116:1784–1792. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 7.Kim DJ, Murray IA, Burns AM, Gonzalez FJ, Perdew GH, Peters JM. Peroxisome proliferator-activated receptor-beta/delta inhibits epidermal cell proliferation by down-regulation of kinase activity. J Biol Chem. 2005;280:9519–9527. doi: 10.1074/jbc.M413808200. [DOI] [PubMed] [Google Scholar]
- 8.Juge-Aubry CE, Hammar E, Siegrist-Kaiser C, Pernin A, Takeshita A, Chin WW, Burger AG, Meier CA. Regulation of the transcriptional activity of the peroxisome proliferator-activated receptor alpha by phosphorylation of a ligand-independent trans-activating domain. J Biol Chem. 1999;274:10505–10510. doi: 10.1074/jbc.274.15.10505. [DOI] [PubMed] [Google Scholar]
- 9.Shalev A, Siegrist-Kaiser CA, Yen PM, Wahli W, Burger AG, Chin WW, Meier CA. The peroxisome proliferator-activated receptor alpha is a phosphoprotein: regulation by insulin. Endocrinology. 1996;137:4499–4502. doi: 10.1210/endo.137.10.8828512. [DOI] [PubMed] [Google Scholar]
- 10.Hertz R, Berman I, Keppler D, Bar-Tana J. Activation of gene transcription by prostacyclin analogues is mediated by the peroxisome-proliferators-activated receptor (PPAR) Eur J Biochem. 1996;235:242–247. doi: 10.1111/j.1432-1033.1996.00242.x. [DOI] [PubMed] [Google Scholar]
- 11.Kronke G, Kadl A, Ikonomu E, Bluml S, Furnkranz A, Sarembock IJ, Bochkov VN, Exner M, Binder BR, Leitinger N. Expression of heme oxygenase-1 in human vascular cells is regulated by peroxisome proliferator-activated receptors. Arterioscler Thromb Vasc Biol. 2007;27:1276–1282. doi: 10.1161/ATVBAHA.107.142638. [DOI] [PubMed] [Google Scholar]
- 12.Iwaki M, Matsuda M, Maeda N, Funahashi T, Matsuzawa Y, Makishima M, Shimomura I. Induction of adiponectin, a fat-derived antidiabetic and antiatherogenic factor, by nuclear receptors. Diabetes. 2003;52:1655–1663. doi: 10.2337/diabetes.52.7.1655. [DOI] [PubMed] [Google Scholar]
- 13.Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2:2557–2568. [PubMed] [Google Scholar]
- 14.Ponka P. Cell biology of heme. Am J Med Sci. 1999;318:241–256. doi: 10.1097/00000441-199910000-00004. [DOI] [PubMed] [Google Scholar]
- 15.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 16.Platt JL, Nath KA. Heme oxygenase: protective gene or Trojan horse. Nat Med. 1998;4:1364–1365. doi: 10.1038/3947. [DOI] [PubMed] [Google Scholar]
- 17.Otterbein LE, Choi AM. Heme oxygenase: colors of defense against cellular stress. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1029–L1037. doi: 10.1152/ajplung.2000.279.6.L1029. [DOI] [PubMed] [Google Scholar]
- 18.Poss KD, Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci USA. 1997;94:10919–10924. doi: 10.1073/pnas.94.20.10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, Ohta K, Kasahara Y, Koizumi S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest. 1999;103:129–135. doi: 10.1172/JCI4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noyes WD, Bothwell TH, Finch CA. The role of the reticulo-endothelial cell in iron metabolism. Br J Haematol. 1960;6:43–55. doi: 10.1111/j.1365-2141.1960.tb06216.x. [DOI] [PubMed] [Google Scholar]
- 21.Juan SH, Lee TS, Tseng KW, Liou JY, Shyue SK, Wu KK, Chau LY. Adenovirus-mediated heme oxygenase-1 gene transfer inhibits the development of atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2001;104:1519–1525. doi: 10.1161/hc3801.095663. [DOI] [PubMed] [Google Scholar]
- 22.Means RT, Jr, Krantz SB. Progress in understanding the pathogenesis of the anemia of chronic disease. Blood. 1992;80:1639–1647. [PubMed] [Google Scholar]
- 23.L'Abbate A, Neglia D, Vecoli C, Novelli M, Ottaviano V, Baldi S, Barsacchi R, Paolicchi A, Masiello P, Drummond GS, McClung JA, Abraham NG. Beneficial effect of heme oxygenase-1 expression on myocardial ischemia-reperfusion involves an increase in adiponectin in mildly diabetic rats. Am J Physiol Heart Circ Physiol. 2007;293:H3532–H3541. doi: 10.1152/ajpheart.00826.2007. [DOI] [PubMed] [Google Scholar]
- 24.Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rabinowitz JE, Rolling F, Li C, Conrath H, Xiao W, Xiao X, Samulski RJ. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J Virol. 2002;76:791–801. doi: 10.1128/JVI.76.2.791-801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sue YM, Cheng CF, Chang CC, Chou Y, Chen CH, Juan SH. Antioxidation and anti-inflammation by haem oxygenase-1 contribute to protection by tetramethylpyrazine against gentamicin-induced apoptosis in murine renal tubular cells. Nephrol Dial Transplant. 2009;24:769–777. doi: 10.1093/ndt/gfn545. [DOI] [PubMed] [Google Scholar]
- 27.Abraham NG, da Silva JL, Lavrovsky Y, Stoltz RA, Kappas A, Dunn MW, Schwartzman ML. Adenovirus-mediated heme oxygenase-1 gene transfer into rabbit ocular tissues. Invest Ophthalmol Vis Sci. 1995;36:2202–2210. [PubMed] [Google Scholar]
- 28.He TC, Chan TA, Vogelstein B, Kinzler KW. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99:335–345. doi: 10.1016/s0092-8674(00)81664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Juan SH, Cheng TH, Lin HC, Chu YL, Lee WS. Mechanism of concentration-dependent induction of heme oxygenase-1 by resveratrol in human aortic smooth muscle cells. Biochem Pharmacol. 2005;69:41–48. doi: 10.1016/j.bcp.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 30.Pang PH, Lin YH, Lee YH, Hou HH, Hsu SP, Juan SH. Molecular mechanisms of p21 and p27 induction by 3-methylcholanthrene, an aryl-hydrocarbon receptor agonist, involved in antiproliferation of human umbilical vascular endothelial cells. J Cell Physiol. 2008;215:161–171. doi: 10.1002/jcp.21299. [DOI] [PubMed] [Google Scholar]
- 31.Lin H, Lee JL, Hou HH, Chung CP, Hsu SP, Juan SH. Molecular mechanisms of the antiproliferative effect of beraprost, a prostacyclin agonist, in murine vascular smooth muscle cells. J Cell Physiol. 2008;214:434–441. doi: 10.1002/jcp.21214. [DOI] [PubMed] [Google Scholar]
- 32.Shih CM, Lin H, Liang YC, Lee WS, Bi WF, Juan SH. Concentration-dependent differential effects of quercetin on rat aortic smooth muscle cells. Eur J Pharmacol. 2004;496:41–48. doi: 10.1016/j.ejphar.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 33.Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta. 2007;1771:952–960. doi: 10.1016/j.bbalip.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257:79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 35.Ishizaka N, Saito K, Noiri E, Sata M, Mori I, Ohno M, Nagai R. Iron dextran causes renal iron deposition but not renal dysfunction in angiotensin II-treated and untreated rats. Nephron Physiol. 2004;98:107–113. doi: 10.1159/000081559. [DOI] [PubMed] [Google Scholar]
- 36.Ibrahim NG, Hoffstein ST, Freedman ML. Induction of liver cell haem oxygenase in iron-overloaded rats. Biochem J. 1979;180:257–263. doi: 10.1042/bj1800257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abraham NG, Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev. 2008;60:79–127. doi: 10.1124/pr.107.07104. [DOI] [PubMed] [Google Scholar]
- 38.Kushida T, Quan S, Yang L, Ikehara S, Kappas A, Abraham NG. A significant role for the heme oxygenase-1 gene in endothelial cell cycle progression. Biochem Biophys Res Commun. 2002;291:68–75. doi: 10.1006/bbrc.2002.6403. [DOI] [PubMed] [Google Scholar]
- 39.Kwak JY, Takeshige K, Cheung BS, Minakami S. Bilirubin inhibits the activation of superoxide-producing NADPH oxidase in a neutrophil cell-free system. Biochim Biophys Acta. 1991;1076:369–373. doi: 10.1016/0167-4838(91)90478-i. [DOI] [PubMed] [Google Scholar]
- 40.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 41.Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA, Choi AM. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6:422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 42.Zhan Y, Kim S, Izumi Y, Izumiya Y, Nakao T, Miyazaki H, Iwao H. Role of JNK, p38, and ERK in platelet-derived growth factor-induced vascular proliferation, migration, and gene expression. Arterioscler Thromb Vasc Biol. 2003;23:795–801. doi: 10.1161/01.ATV.0000066132.32063.F2. [DOI] [PubMed] [Google Scholar]
- 43.Song J, Sumiyoshi S, Nakashima Y, Doi Y, Iida M, Kiyohara Y, Sueishi K. Overexpression of heme oxygenase-1 in coronary atherosclerosis of Japanese autopsies with diabetes mellitus: Hisayama study. Atherosclerosis. 2009;202:573–581. doi: 10.1016/j.atherosclerosis.2008.05.057. [DOI] [PubMed] [Google Scholar]
- 44.Farhangkhoee H, Khan ZA, Mukherjee S, Cukiernik M, Barbin YP, Karmazyn M, Chakrabarti S. Heme oxygenase in diabetes-induced oxidative stress in the heart. J Mol Cell Cardiol. 2003;35:1439–1448. doi: 10.1016/j.yjmcc.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 45.Calabrese V, Mancuso C, Sapienza M, Puleo E, Calafato S, Cornelius C, Finocchiaro M, Mangiameli A, Di Mauro M, Stella AM, Castellino P. Oxidative stress and cellular stress response in diabetic nephropathy. Cell Stress Chaperones. 2007;12:299–306. doi: 10.1379/CSC-270.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim DH, Burgess AP, Li M, Tsenovoy PL, Addabbo F, McClung JA, Puri N, Abraham NG. Heme oxygenase-mediated increases in adiponectin decrease fat content and inflammatory cytokines tumor necrosis factor-alpha and interleukin-6 in Zucker rats and reduce adipogenesis in human mesenchymal stem cells. J Pharmacol Exp Ther. 2008;325:833–840. doi: 10.1124/jpet.107.135285. [DOI] [PubMed] [Google Scholar]
- 47.Li M, Kim DH, Tsenovoy PL, Peterson SJ, Rezzani R, Rodella LF, Aronow WS, Ikehara S, Abraham NG. Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes. 2008;57:1526–1535. doi: 10.2337/db07-1764. [DOI] [PubMed] [Google Scholar]
- 48.Ndisang JF, Jadhav A. Heme oxygenase system enhances insulin sensitivity and glucose metabolism in streptozotocin-induced diabetes. Am J Physiol Endocrinol Metab. 2009;296:E829–E841. doi: 10.1152/ajpendo.90783.2008. [DOI] [PubMed] [Google Scholar]