

Abstract
Amino acids 17-35 of the thrombospondin1 (TSP1) N-terminal domain (NTD) bind cell surface calreticulin to signal focal adhesion disassembly, cell migration, and anoikis resistance in vitro. However, the in vivo relevance of this signaling pathway has not been previously determined. We engineered local in vivo expression of the TSP1 calreticulin-binding sequence to determine the role of TSP1 in tissue remodeling. Surgical sponges impregnated with a plasmid encoding the secreted calreticulin-binding sequence [NTD (1-35)-EGFP] or a control sequence [mod NTD (1-35)-EGFP] tagged with enhanced green fluorescent protein were implanted subcutaneously in mice. Sponges expressing NTD (1-35)-EFGP formed a highly organized capsule despite no differences in cellular composition, suggesting stimulation of collagen deposition by the calreticulin-binding sequence of TSP1. TSP1, recombinant NTD, or a peptide of the TSP1 calreticulin-binding sequence (hep I) increased both collagen expression and matrix deposition by fibroblasts in vitro. TSP1 stimulation of collagen was inhibited by a peptide that blocks TSP1 binding to calreticulin, demonstrating the requirement for cell surface calreticulin. Collagen stimulation was independent of TGF-β activity and Smad phosphorylation but was blocked by an Akt inhibitor, suggesting that signaling through the Akt pathway is important for regulation of collagen through TSP1 binding to calreticulin. These studies identify a novel function for the NTD of TSP1 as a mediator of collagen expression and deposition during tissue remodeling.
Tissue remodeling is a highly orchestrated process that requires coordinated regulation of cell migration, proliferation, extracellular matrix deposition and remodeling, and eventual cell regression. The extracellular matrix provides both biochemical and mechanical cues to regulate these complex cellular responses to injury and repair. A family of extracellular matrix proteins, the matricellular proteins, has been shown to regulate cell behavior and extracellular matrix deposition during tissue remodeling and wound repair.1–3
Thrombospondin 1 (TSP1) is a multifunctional, matricellular protein that constitutes 25% of the protein released from the α-granules of activated platelets.4,5 TSP1 is present in wounds and expressed by cells involved in wound healing, including macrophages, fibroblasts, endothelial cells, and vascular smooth muscle cells.6–9 TSP1 knockout mice display compromised wound healing, characterized by reduced macrophage infiltration and a delay in capillary angiogenesis, but persistence of granulation tissue.9 It induces focal adhesion disassembly, stimulates cell motility, activates latent transforming growth factor-β (TGF-β), and inhibits nitric oxide signaling.10–12 Depending on whether the N- or C-terminal domain of TSP1 is engaged, it is either anti- or proangiogenic.12–14 TSP1 can be proapoptotic to endothelial cells, but it also stimulates cell survival by signaling resistance to anoikis.15 These diverse and sometimes paradoxical activities can be ascribed to its interactions with multiple receptors, including integrins, syndecans,13,16 CD47,17 CD36,18 low-density lipoprotein receptor-related protein 1 (LRP1),19 and calreticulin (CRT).20
To date, the role of TSP1 in tissue remodeling has largely been studied through injury models in TSP1-null mice.9,21,22 These models have been useful for identifying many functions of TSP1 but are limited in their ability to mimic tissue remodeling in a normal organism, in which TSP1 expression, proteolysis, and interactions with multiple receptors will be modulated both temporally and spatially. The susceptibility of TSP1 to proteolytic cleavage by a wide spectrum of proteases suggests that cells are likely to be exposed to fragments of TSP1 during tissue remodeling.23 Both the N- and C-terminal domains can be detected separately from the full-length TSP1 molecule in vivo.23,24 Therefore, ongoing questions include whether TSP1 can signal simultaneously through multiple receptors and whether isolated domains elicit responses distinct from the intact molecule. For these reasons, in vivo models expressing isolated TSP1 domains on a wild-type genetic background are relevant to the physiological conditions of TSP1 in wound healing.
Previously, we showed that amino acids 17-35 of the N-terminal domain (NTD) of TSP1 signal focal adhesion disassembly and increased cell migration in vitro.25 Furthermore, signaling through this sequence prevents anoikis.19 This sequence in the NTD binds to a cell surface cocomplex of CRT and LRP1 and stimulates signaling through focal adhesion kinase (FAK), extracellular signal related kinase (ERK), and phosphoinositide 3-kinase (PI3 kinase), which results in transient phosphorylation of Akt and down-regulation of Rho kinase.26,27 Signaling downstream from TSP1 engagement of the CRT/LRP1 cocomplex induces an intermediate state of adhesion in endothelial cells, fibroblasts, and vascular smooth muscle cells.25,26 Intermediate adhesion is characterized by a reduced number of focal adhesions and actin stress fibers without the loss of cell attachment or spreading.28 This intermediate adhesive state precedes migration in response to the TSP1 CRT-binding sequence.28,29 Induction of intermediate adhesion, cell migration, and anoikis resistance are similarly regulated by TSP1, a recombinant trimeric form of the NTD (NoC1), and by a synthetic peptide comprising the CRT-binding sequence (aa 17-35, hep I peptide). Furthermore, TSP1 binding to aa19-36 in the NTD of CRT is necessary for TSP-CRT binding and induction of signaling, and cells lacking this site in CRT do not respond to TSP1.15,30,31
The role of TSP1 binding to the CRT-LRP1 complex in vivo is unknown. Based on previous studies, we hypothesized that local expression of the secreted CRT-binding sequence of TSP1 at sites of injury in vivo would signal intermediate cell adhesion and migration of CRT-expressing cells to increase cellularity of wounds. To test this hypothesis, we used an in vivo mouse model of the foreign body response to drive local expression of a secreted enhanced green fluorescent protein (EGFP)-tagged fusion protein of the TSP1 CRT-binding sequence. Unexpectedly, our results showed that the CRT-binding sequence of TSP1 stimulates the formation of a highly organized collagen capsule, which reduced cellular infiltration into the sponges. In vitro studies confirmed that TSP1 stimulates fibrillar collagen expression by fibroblasts and increased incorporation of collagen into the extracellular matrix in a CRT-dependent manner. Although TSP1 can activate latent TGF-β, the recombinant TSP1 NTD protein NoC1 stimulated collagen independently of both TGF-β activity and Smad2 phosphorylation. Rather, the CRT-binding sequence requires Akt activity to stimulate collagen. These studies identify a previously unknown role for the NTD of TSP1 in tissue remodeling through a CRT-dependent TGF-β–independent stimulation of collagen matrix formation.
Materials and Methods
Antibodies
The following antibodies were purchased: rat anti-mouse F4/80 antibody, clone CI:A3-1 (AbD Serotec, Raleigh, NC); rat anti-CD31 antibody (BD Biosciences Pharmingen, San Diego, CA); rabbit anti-human Ki-67 (Abcam Inc., Cambridge, MA); mouse anti-α-smooth muscle actin (Vector Labs, Burlingame, CA); rabbit anti-human collagen I or rabbit anti-mouse collagen I (MD Biosciences, St. Paul, MN) for immunoblots; rabbit anti-collagen I for immunocytochemistry (Abcam Inc); phosphorylated Smad-2, rabbit polyclonal (ser 465/467) (Cell Signaling Technology, Danvers, MA); Smad 2/3, mouse monoclonal (clone 18/Smad2/3) (BD Transduction Laboratories, San Jose, CA). Mouse anti-EGFP was a gift of Dr. Mary Ann Accavitti-Loper of the UAB Epitope Recognition and ImmunoReagent Core and was biotinylated with a ChromaLink Biotin Labeling Kit (SoluLink, San Diego, CA).
Proteins and Peptides
Platelet TSP1 stripped of associated TGF-β was purified as previously described using gel filtration chromatography equilibrated with Tris buffered saline, pH 11.0.32 TSP1 preparations had less than 0.3 pmol/L active TGF-β/10 nmol/L TSP1 (data not shown). Recombinant EGFP was purchased from BD Biosciences. Peptides were purchased from AnaSpec, Inc.: hep I (aa17-35 TSP1): ELTGAARKGSGRRLVKGPD; control peptide modified hep I: ELTGAARAGSGRRLVAGPD; scrambled hep I: RSKAGTLGERDLKPGARVG; TSP1 binding sequence from CRT, CRT19.36: RWIESKHKSDFGKFVLSS; control for CRT19.36, CRT20A.30A: RWIESAAASDKFGLAAASS.31 All peptides are >95% pure by HPLC and mass spectrometry. Recombinant trimeric N-terminal domain of TSP1 (NoC1) was a kind gift of Dr. Deane Mosher (University of Wisconsin, Madison).
Cell Culture
Mouse embryonic fibroblasts (MEFs) from wild-type and CRT-null mice were a gift of Dr. Marek Michalak, University of Alberta. Human foreskin fibroblasts were a gift of Dr. Laura Timares, University of Alabama at Birmingham and were used with the approval of the UAB Institutional Review Board. Bovine aortic endothelial cells (BAECs) and MEFs were maintained in DMEM with 4.5 g/L glucose (Gibco, Invitrogen, Carlsbad, CA), supplemented with 2 mmol/L l-glutamine and 10% fetal bovine serum (FBS). Human foreskin fibroblasts were cultured in fibroblast growth media (FGM) Bullet Kit (with insulin and recombinant human fibroblast growth factor supplements) (Lonza, Walkersville, MD) and 10% FBS. Ascorbic acid was purchased from Sigma (St. Louis, MO).
EGFP Constructs
All plasmids were prepared from individual colonies (QIAGEN, Valencia, CA) and sequenced to confirm correct fragment insertion and/or mutagenesis. The TSP1 signal sequence and the first 35 amino acids of the NTD (−55/+105) were isolated from pGEM4 vector containing human TSP1 cDNA (gift of Deane F. Mosher, University of Wisconsin). The fragment was extracted by PCR and engineered with restriction sites NheI (5′-TGGGCGCTAGCAGCTCCACCATGGGGC-3′) and HindIII (5′-TGGGCTAAGCTTGTCGGGGCCCTTCAC-3′) (IDT, Coralville, IA). The purified fragment was ligated into a commercially available vector, pEGFP-N1 (BD Biosciences, Palo Alto, CA) using restriction sites NheI and HindIII. The resulting construct was designated “NTD (1-35)-EGFP.” Two site mutations of NTD (1-35)-EGFP were performed to replace two lysines [TSP1 + 42 (aa 24) and TSP1 + 50 (aa 32)] with alanine using a site-directed mutagenesis kit (Stratagene, La Jolla, CA). Lysine + 42 was mutated using 5′-GGCCGCCCGCGCGGGGTCTGGGC-3′ (IDT, Coralville, IA). The resulting vector was sequenced to ensure sequence replacement and then mutated at lysine + 50 using 5′-CGCCGACTGGTGGCGGGCCCCGACAAG-3′ (IDT, Coralville, IA). The resulting vector with two amino acid substitutions was designated “mod NTD (1-35)-EGFP.”
Interference Reflection Microscopy
Focal adhesion disassembly assays were performed as previously described and quantified using a Zeiss Axiovert 10 microscope equipped for interference reflection microscopy.25 At least 300 cells were scored per coverslip, and all treatments were performed in triplicate. Cells with at least five focal adhesions were scored as positive.
In Vitro Cell Transfections
Endotoxin-free plasmid DNA was extracted using either EndoFree Plasmid Maxi Kit or EndoFree Plasmid Giga Kit (QIAGEN) per kit instructions. MEFs were transfected via nucleofection using MEF 1 Nucleofector Kit from Amaxa Biosystems (Amaxa GmbH, Lonza) in an Amaxa Nucleofector II using program A-23. Transfected cells were cultured in DMEM + 10% FBS. EGFP expression was confirmed by fluorescence microcopy (Nikon Eclipse TE200). Expression of EGFP in transfected cells was seen as early as 6 hours and up to 96 hours postnucleofection. Transfection efficiencies, calculated at 24 hours, averaged between 60 to 80%.
Gene-Activated Matrix (GAM) Sponge Preparation
The GAM sponge implant method was modified from Bonadio et al.33 Briefly, polyvinyl alcohol sponges (First Aide Bandage Company, New London, CT) were cut to 0.9-cm disks with a cork borer, rinsed in sterile Dulbecco's PBSCa2+/Mg2+ (DPBS) (Mediatech Inc., Manassas, VA), exposed to UV light for 30 minutes per side. Sponges were dried overnight in a laminar flow hood. GAM was prepared on ice with 0.6 mg of plasmid DNA mixed with 0.6 mg of bovine type I collagen (BD Biosciences) and brought to a final volume of 0.8 ml with DPBS. The pH of each GAM was measured and adjusted to pH 7.4 with NaOH. Sponges were then filled with 0.8 ml GAM, placed under vacuum for 3 hours to ensure even GAM distribution throughout the sponge, and then incubated overnight at 37°C to polymerize the GAM. Polymerized GAM-filled sponges were frozen at −80°C for one hour and then lyophilized before implantation.
Sponge Implantation and Retrieval
All in vivo experimental procedures were performed with approval from the University of Alabama at Birmingham Institutional Animal Care and Use Committee. Two sponges were implanted on the mid-back subcutaneously and bilaterally with at least 0.5 cm between sponges in male C57BLl/6 mice, age 6–8 weeks (Jackson Laboratories, Bar Harbor, ME). Sponges were harvested at 1, 3, 7, 14, and 21 days post implantation. One half of each sponge was processed for morphological and immunohistochemical analysis. The other half was used to harvest the wound fluid.
EGFP Live Animal Imaging
At five days postimplantation, mice (one per group) were anesthetized with isofluorane and fur was removed with depilatory cream. GFP 515 nm emission was imaged using a Xenogen imaging system in the Laboratory for Multi-Modality Imaging Assessment and Small Animal Imaging Core at UAB. Results are depicted by false color of fluorescence intensity.
Histology and Immunohistochemistry
Harvested sponges were cut in half laterally to expose the central midline of the sponge. Sponges were fixed for 4 hours in 10% formaldehyde and then 70% ethanol for subsequent staining with hematoxylin and eosin or Masson's trichrome. Sponges used for immunohistochemistry for CD31 and EGFP were fixed in 10% zinc-buffered formalin. All sponges were paraffin embedded and sectioned to 6 μm thickness.
Sections stained with Masson's trichrome were used to quantify collagen accumulation in the pericapsular region by MetaMorph analysis. Briefly, a region of normal dermis was used to set the selection threshold. Hue, light, and intensity were selected to include only the blue-stained, basket weave patterned dermis and to exclude other structures in the skin and dermal muscle (the panniculus carnosus). These parameters were then applied to the sponge implant. The inclusion of collagen and exclusion of other materials and structures was confirmed by eye and calibrated individually for each slide. Collagen accumulation was calculated as the percentage of the total pericapsular area highlighted by the selection threshold criteria.
Immunohistochemistry for Ki-67 and α-smooth muscle actin (α-SMA) were performed on formaldehyde-fixed sections. Sections for Ki-67 staining were boiled in 0.01 mol/L sodium citrate, pH 6.0, for 10 minutes and then blocked in 4% BSA, 2.5% goat serum for 30 minutes. Sections were incubated overnight at 4°C in primary antibody at the following dilutions: 1:200 for anti-CD31, 1:50 for biotinylated anti-EGFP, 1:200 for anti-Ki-67, and 1:25 for anti-α-SMA. Secondary antibody was applied for 1 hour at room temperature (omitted for biotinylated anti-EGFP), followed by neutravidin-HRP and developed with DAB reagent (Vector Laboratories).
Soluble Collagen (Sircol) Assay
Human foreskin fibroblasts were grown in six-well plates with FGM with 10% FBS for 48 hours. Cultures were switched to 0.8 ml of medium with 0.5% FBS with 2 μmol/L ascorbic acid and treated daily for another 48 hours. Conditioned media were collected on ice with a protease inhibitor cocktail (Sigma) and centrifuged 3000 × g 4°C to remove cell debris. Conditioned media (200 μl) was added to 900 μl picric acid Sirius Red Sircol reagent (Biocolor Ltd., Accurate Chemical Scientific, Westbury, NY). MEFs were plated at 6 × 104 cells per well in six-well plates and grown in DMEM with 10% FBS for 24 hours and then switched to media with 0.5% FBS with 2 μmol/L ascorbic acid and treatments; treatments and media were refreshed daily over 72 hours. Conditioned media were harvested as above, and 300 μl media was incubated with 1 ml of the picric acid Sirius Red Sircol reagent. Samples were rotated for 30 minutes and then centrifuged at 13,500 × g for 15 minutes at room temperature. Excess Sircol reagent was removed, pellets were resuspended in an alkali reagent (provided by manufacturer), and absorbance was read at 540 nm. Collagen concentrations were determined from a standard curve of rat-tail collagen I (provided by manufacturer).
Preparation of Deoxycholate (DOC) Insoluble Matrices
DOC extractions of the detergent insoluble ECM were performed as a modification of Midwood et al.34 MEFs were plated (4.5–6 × 104 cells) and then grown in DMEM with 10% FBS for 24 hours. Human foreskin fibroblasts were plated (4.5–6 × 104 cells) and then grown in FGM with 10% FBS for 24 hours. Cells were then switched to the appropriate media with 2 μmol/L ascorbic acid containing 0.5% FBS and treated daily for 72 hours. Wells were rinsed twice in DPBS and then harvested by scraping with 300 μl of 4% DOC (4% DOC in 20 mmol/L Tris-HCl, pH 8.8, with 200 μU/ml DNase and protease inhibitors) and then homogenized with a 27½ gauge needle and tumbled gently at 4°C overnight.35 Precipitates were pelleted for 30 minutes at 13,500 × g The soluble cell fraction was collected in the supernatant. The detergent insoluble ECM in the pellet was washed in 200 μl 4% DOC and the pellets resuspended in 30 μl 1% SDS in 25 mmol/L Tris-HCl. The total insoluble pellet (ECM) and 30 μl of the soluble fraction were diluted in 4× Laemmli buffer with 10% β-mercaptoethanol and heated at 95°C for 10 minutes before loading on SDS-PAGE. In additional studies to examine ECM collagen, we used a modified 1% DOC-SDS extraction protocol. Cellular and pericellular material were extracted by solubilization with 250 μl 1.0% DOC for 15 minutes. Extracts were collected, sheared, tumbled, and centrifuged as above and the DOC soluble material collected. The DOC insoluble ECM which remained bound to the wells was then extracted in 50 μl 2× Laemmli buffer (2% SDS with 5% β-mercaptoethanol) and protease inhibitors.36,37
Immunoblotting
DOC soluble and insoluble ECM fractions or cell lysates extracted with 4× Laemmli buffer were separated on by SDS-PAGE on 4 to 15% gradient Tris-HCl gels under reducing conditions (Bio-Rad Laboratories, Hercules, CA). Cell lysates immunoblotted for phospho-Smad 2 detection were separated on 10% SDS-PAGE Tris-HCl gels. Proteins were transferred onto nitrocellulose membranes, and then membranes were stained with Ponceau S to detect protein. Membranes were blocked in 1% casein-PBS and then probed with primary antibody for 1 hour at room temperature with the exception of anti-phospho Smad 2, which was incubated at 4°C overnight. Blots were washed with Tris buffered saline, 0.1% Tween-20. All secondary HRP-labeled antibodies were used at a dilution of 1:10,000 (Jackson ImmunoResearch, West Grove, PA). Blots were developed with Western Lighting Plus reagent (Perkin Elmer, Waltham, MA). Blots were stripped for 10 minutes with either ReBlot Strong for collagen blots or ReBlot Mild for phospho-Smad blots (Millipore-Chemicon, Billerica, MA) and then reprobed as described above.
Immunocytochemistry of Collagen Fibrils
Human foreskin fibroblasts were plated on washed coverslips coated with FGM with10% FBS at 15,000 cells per well. Cells were cultured in FGM with 10% FBS for 36 hours and then switched to media with 0.5% FBS for 24 hours. Treatments were administered daily for 72 hours. Cells were fixed in 3% formalin, permeabilized with 0.05% Tween-20, and then blocked in 2.5% goat serum + 4% bovine serum albumin for 20 minutes at room temperature. Cells were incubated with rabbit anti-type I collagen (1:200) for 2 hours at room temperature and goat anti-rabbit Texas-Red (1:300) for 1 hour at room temperature. Nuclei were stained using Hoechst 33342. Coverslips were mounted with Vectashield. Cells were imaged using a Leica SP1 confocal laser-scanning microscope at the UAB High Resolution Imaging Facility. Contrast was adjusted postimaging equally for all images.
Cell Signaling Inhibitors
PI3K inhibitor, LY294002, was purchased from Promega (Madison, WI) and was used at 5 μmol/L, a concentration which inhibits Akt phosphorylation in response to the TSP1 CRT-binding domain.15 Akt inhibitor II, which inhibits Akt binding to phosphatidylinositol3,4,5-trisphosphate (PIP3), was purchased from Calbiochem (Gibbstown, NJ) and was used at a concentration of 2 μmol/L.38 MEFs (4.5 × 104 cells per well) were plated in six-well plates at in DMEM with 10% FBS for 48 hours and then switched to media with 0.5% FBS and 2 μmol/L ascorbic acid. Cells were then treated with inhibitors or vehicle (DMSO, Sigma) for one hour and then with 30 nmol/L NoC1 or media control. Treatments, including inhibitors, were refreshed daily over a 72-hour period. Cell layers were harvested in 4× Laemmli buffer with 10% β-mercaptoethanol (Sigma) at 72 hours. SDS-PAGE immunoblots were then performed as described above.
PAI-1 Reporter Luciferase Activity Assay for Detection of Active TGF-β in Conditioned Media
Active TGF-ß in the conditioned medium was quantified by the mink lung epithelial cell (MLEC) PAI-1promoter luciferase reporter assay using MLECs stably transfected with the TGF-β response element of the PAI-1 promoter-luciferase reporter construct as described previously.39,40 All three mammalian TGF-β isoforms are detected by this assay. The MLEC reporter cells were obtained from Dr. Daniel Rifkin (New York University Medical Center).
Statistics
Data were analyzed using a two-parameter Student's t-test (paired or unpaired as indicated) or one way analysis of variance with P < 0.05 considered significant.
Results
Secreted CRT-Binding Fusion Proteins Replicate TSP1 and Hep I Functions in Vitro
A mouse model of the foreign body response was used to assess the potential biological significance of TSP1 signaling through the CRT-LRP1 complex. In this method, first described by Bonadio et al,33 polyvinyl alcohol sponges are filled with a GAM comprised of partially solubilized collagen I and plasmid DNA expressing the gene of interest. The GAM-filled sponges are implanted subcutaneously to elicit a foreign body response. Invading macrophages and fibroblasts perceive the exogenous collagen I as a provisional matrix, phagocytose the GAM, and thus become locally transfected.33
To express the secreted CRT-binding sequence of TSP1 (aa 17-35) in the GAM, a plasmid encoding the TSP1 signal peptide and the first 35 amino acids of TSP1 with downstream EGFP was constructed [NTD (1-35)-EGFP]. In prior studies, we established that substitution of lysines at positions 24 and 32 renders the sequence inactive.31 Therefore, the NTD (1-35) sequence was modified by site-directed mutagenesis to substitute alanine for lysine at amino acids 24 and 32 and used as a negative control [mod NTD (1-35)-EGFP] (Figure 1).
Figure 1.
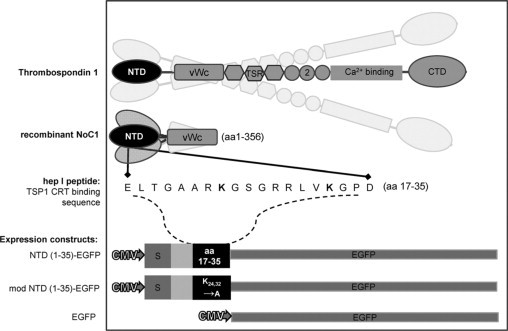
Illustration of TSP1-derived molecules used in vivo and in vitro. Platelet-derived trimeric human thrombospondin-1 (TSP1) (aa 1-1152); recombinant TSP1 N-terminal domain trimer (NoC1; aa 1-356); synthetic CRT-binding TSP1 peptide (hep I; aa 17-35); secreted CRT-binding domain EGFP construct [NTD (1-35)-EGFP; TSP 1 aa1-35] and secreted nonfunctional mutated construct [mod NTD (1-35)-EGFP]; and nonsecreted EGFP parent vector. S indicates signal sequence; EGFP, enhanced green fluorescent protein; TSR, type 1 thrombospondin repeat; 2, type 2 thrombospondin repeat.
Expression and functionality of the constructs were confirmed in vitro by transiently transfecting the plasmids into three cell types which have been previously used to demonstrate focal adhesion disassembly stimulated by both TSP1 and hep I.10,15 Localization of EGFP to the Golgi apparatus in both NTD (1-35)-EGFP and mod NTD (1-35)-EFP transfected cells was confirmed by fluorescent microscopy (data not shown). Secretion of the proteins of the predicted size was confirmed by EGFP pull-down and immunoblotting in conditioned medium from NTD (1-35)-EGFP and mod NTD (1-35)-EGFP transfected cells (Figure 2A). To determine whether the CRT-binding functions of TSP1, the recombinant NTD (NoC1), and the hep I peptide are replicated by the secreted NTD (1-35)-EGFP fusion protein, conditioned media from transfected cells were assayed for the ability to stimulate focal adhesion disassembly. Conditioned media containing secreted NTD (1-35)-EGFP stimulated focal adhesion disassembly of BAECs to levels similar to that of cells treated with 10 nmol/L hep I peptide. In addition, conditioned medium from NTD (1-35)-EGFP transfected cells diluted to 1:1000 and applied to adherent BAECs had focal adhesion disassembly activity equivalent to 10 nmol/L hep I peptide (Figure 2B).
Figure 2.
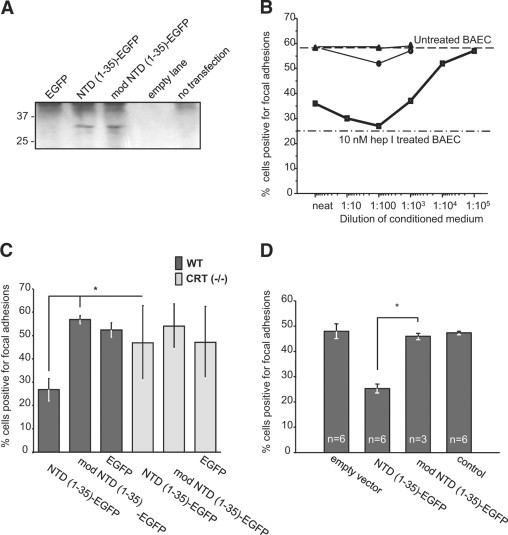
In vitro, NTD (1-35)-EGFP and mod NTD (1-35)-EGFP are functional substitutes for the synthetic peptides. A: EGFP fusion proteins were detected in the conditioned media of MEFs cells transfected with NTD (1-35)-EGFP or mod NTD (1-35)-EGFP. EGFP-containing proteins were isolated from conditioned media of transfected [EGFP, NTD (1-35)-EGFP or mod NTD (1-35)-EGFP] or control cells (not transfected) 72 hours after transfection by nucleoporation. EGFP containing proteins were isolated by pull-down using mouse anti-GFP coupled beads. Bound proteins were eluted in Laemmli buffer, separated by SDS-PAGE, transferred to nitrocellulose, and then immunoblotted for GFP. B–D: The presence of focal adhesions was assessed by interference reflection microscopy. At least 300 cells were counted per coverslip. Cells with more than five focal adhesions per cell were scored as positive. B: BAECs responded to NTD (1-35)-EGFP in a dose-dependent manner. Conditioned medium from NTD (1-35)-EGFP–transfected cells was diluted 1:10 with conditioned medium from mock-transfected cells. NTD (1-35)-EGFP–conditioned medium (squares), pEGFP-N1-transfected cell conditioned media (circles), and mock-transfected medium (triangles). hep I peptide (10 nmol/L) was used as a positive control for focal adhesion disassembly (dash-dot line). C: Wild-type and CRT (−/−) MEFs were transfected with NTD (1-35)-EGFP constructs. Twenty-four hours after plating, cells were analyzed by interference reflection microscopy for the presence of focal adhesions. Results are expressed as the mean number of focal adhesion positive cells ± SD; n = 3 replicates with P values calculated by two-parameter unpaired Student's t-test. D: NTD (1-35)-EGFP-secreted protein was active in the presence of serum. MEFs were transfected and grown for 24 hours in medium containing 10% FBS. Transfection efficiencies were measured and then transfected cell populations were mixed with mock-transfected cells to normalize the percentage of construct-expressing cells to 10% of each coculture population. Results are the mean percentage of focal adhesion positive cells ± SD; n for each group is indicated on figure. *P < 0.05.
To establish that the NTD (1-35)-EGFP protein modified focal adhesions through binding to cell surface CRT, wild-type, and CRT (−/−) MEFs were transfected with the constructs and examined for the presence of focal adhesions by interference reflection microscopy after 48 hours. Wild-type MEFs transfected with NTD (1-35)-EGFP had reduced numbers of cells positive for focal adhesions as compared to NTD (1-35)-EGFP transfected CRT (−/−) MEFs, indicating that the NTD (1-35)-EGFP protein requires cell surface CRT to signal focal adhesion labilizing activity (Figure 2C).
To be effective in vivo, secreted fusion proteins must be functional in a serum-rich environment such as a wound. Therefore, MEFs were transfected with the EGFP constructs and grown for 24 hours in medium containing 10% FBS. Transfection efficiencies were measured, and then the transfected cell populations were mixed with mock-transfected cells to normalize the number of expressing cells in each culture and thus normalize exogenous protein expression. In cocultures with 10% of the cells expressing the NTD (1-35)-EGFP construct, MEFs plated in the presence of 10% FBS had reduced levels of strongly adherent cells (more than six focal adhesions per cell), which persisted for at least 24 hours (Figure 2D). Despite reducing construct expression to only 10% of the cells, more than 60% of cells had reduced focal adhesions, indicating paracrine signaling to nontransfected cells in the coculture. Cocultures containing either 10% mod NTD (1-35)-EGFP or EGFP transfected cells showed no reduction in focal adhesion-positive cells.
Together these studies establish that the secreted CRT-binding fusion protein, NTD (1-35)-EGFP, requires cell surface CRT to signal focal adhesion disassembly similar to TSP1, NoC1, and the hep I peptide and that it can signal in both an autocrine and paracrine fashion. Importantly, this CRT-binding fusion protein maintains function in the presence of serum, similar to the milieu present in wounds.
The CRT-Binding EGFP Fusion Protein Is Expressed in Vivo
Initial studies were performed to determine whether the EGFP fusion proteins are expressed in vivo through GAM-mediated delivery. In vivo imaging, immunohistochemistry for GFP, and measurement of GFP in fluid collected from the sponges were used to evaluate fusion protein expression. Expression of the EGFP fusion proteins in vivo was confirmed by detection of the EGFP signal in the sponge implant region using an IVIS-100 imaging system for live animal visualization of fluorescence. At day 5 postimplantation, expression was localized solely to the region of the sponge implants for all three constructs (Figure 3A). EGFP fusion proteins were also detected in day-7 wound fluid collected from sponges by measuring fluorescence at 530 nm. Levels of EGFP fusion protein expression were quantified by comparing fluorescence to a standard curve of recombinant EGFP. At day 7, both mod NTD (1-35)-EGFP and NTD (1-35)-EGFP were detected in the fluid at concentrations ranging from 28–131 nmol/L (Figure 3B). There was no significant difference in expression levels between modified and NTD (1-35)-EGFP–expressing sponges. Importantly, no fluorescence was detected in the wound fluids from sponges impregnated with plasmid for expression of the nonsecreted EGFP protein (data not shown). This indicates that detection of the EGFP fusion proteins in the sponge fluid reflects active cellular secretion of fusion protein rather than an artifact of processing. EGFP expression and localization within the sponges were also assessed by immunohistochemical localization of GFP in sections of sponges harvested on days 14 and 21 (day 14 shown, Figure 3C). GFP expression was detected along the interface between the dermal panniculus carnosus muscle and sponge and in cells within the sponge body. EGFP was detected only intracellularly in the EGFP-GAM sponges, whereas EGFP expression was both intracellular and extracellular in the NTD (1-35)-EGFP and mod NTD (1-35)-EGFP GAM sponges, consistent with secretion of the fusion proteins. Staining intensity was similar between the NTD (1-35)-EGFP and the mod NTD (1-35)-EGFP plasmid loaded sponges. These results confirm local expression of the secreted EGFP fusion proteins from day 5 to day 21. Importantly, the concentration of NTD (1-35)-EGFP fusion protein measured in the wound fluid (average = 37 nmol/L) is comparable to concentrations of the synthetic hep I peptide (10 nmol/L – 1 μmol/L) used in in vitro assays for focal adhesion disassembly, cell migration, and anoikis prevention.15,26,29
Figure 3.
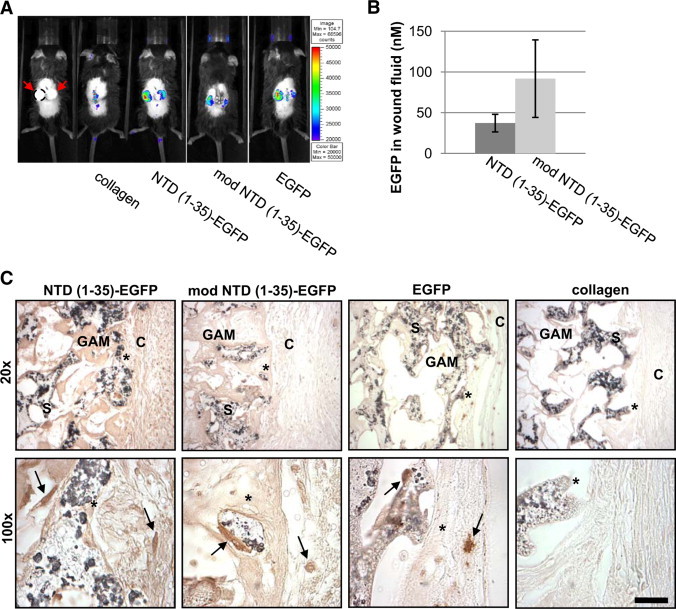
EGFP expression is detected in sponges from days 5–14. A: At day 5, EGFP was detected in vivo at sponge implantation sites by fluorescent imaging. Far left panel shows sponges without fluorescent image overlay. Red arrows: implanted sponges, perimeter of left sponge denoted by a dashed line. Autofluorescence was observed where tissue glue still adheres to the healing incision (collagen and EGFP). Pseudocolor indicates relative intensity of EGFP fluorescence at 515 nm. B: EGFP was detected in the wound fluid from sponges containing NTD (1-35)-EGFP and mod NTD (1-35)-EGFP constructs at day seven. Results are mean molar concentration of EGFP in fluid calculated from an EGFP standard curve of fluorescence with excitation at 485 nm and emission at 530 nm ± SD; n = 6 NTD (1-35)-EGFP sponges and n = 4 modNTD (1-35)-EGFP sponges C: Immunohistochemistry of EGFP expression in sponges at day 14. Paraffin sections of sponges show cells expressing EGFP (arrows). C indicates capsule; S, sponge; GAM, gene-activated matrix infused into sponges. Asterisk indicates the same location at different magnifications. Scale bar = 25 μm.
Histological Analysis of Sponge Implants
The CRT-binding sequence of TSP1 increases cell migration of adherent cells in vitro.29 Therefore, we hypothesized that the expression of the CRT-binding sequence of TSP1 via the NTD (1-35)-EGFP construct would increase granuloma tissue formation through stimulation of increased cell migration into the sponge implant. Unexpectedly, sponges expressing NTD (1-35)-EGFP harvested at 7 days postimplantation showed a reduction in cellularity within the sponge body and an increase in collagen capsule formation as compared to the mod NTD (1-35)-EGFP sponges (Figure 4). Furthermore, the differences in capsule formation persisted over 14 days. Both mod NTD (1-35)-EGFP and EGFP control sponges had surrounding pericapsular areas with dense cellular infiltrates but only sparse and loosely organized collagen fibrils. The organization of the GAM collagen was clearly distinguishable from the organization of the collagen deposited in the pericapsular space: this was confirmed with images obtained using polarized light to detect collagen fibril orientation (data not shown). The intensity and location of the blue color in the Masson's Trichrome-stained sponge sections were used to quantify the collagen deposition and ECM density in the pericapsular sponge regions (Figure 5A). Capsule area in Masson's trichrome-stained sections did not differ between treatment groups at days 7 or 14 (Figure 5B). However, the density of the capsular collagen in NTD (1-35)-EGFP sponges was significantly increased as compared to controls at day 14 (Figure 5C).
Figure 4.
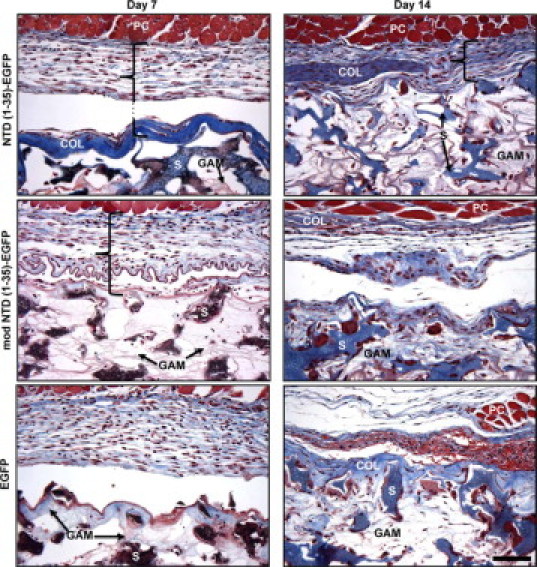
Collagen capsule formation is increased in NTD (1-35)-EGFP–expressing sponges at days 7 and 14. Sections of representative sponges harvested at days 7 and 14 were stained with Masson's trichrome. Collagen stained blue, muscle and red blood cells stained red, and cell nuclei stained brown. PVA sponge material stained brown/blue at day 7 and blue at day 14. Bracket denotes pericapsular region of the sponge. Plasmid infused collagen (GAM) could be seen between the sponge material (S). Thick collagen capsules (COL) were observed at the perimeter of the sponges beneath the panniculus carnosus dermal muscle layer (PC) in NTD (1-35)-EGFP but not in control plasmid-expressing sponges. Scale bar = 50 μm.
Figure 5.
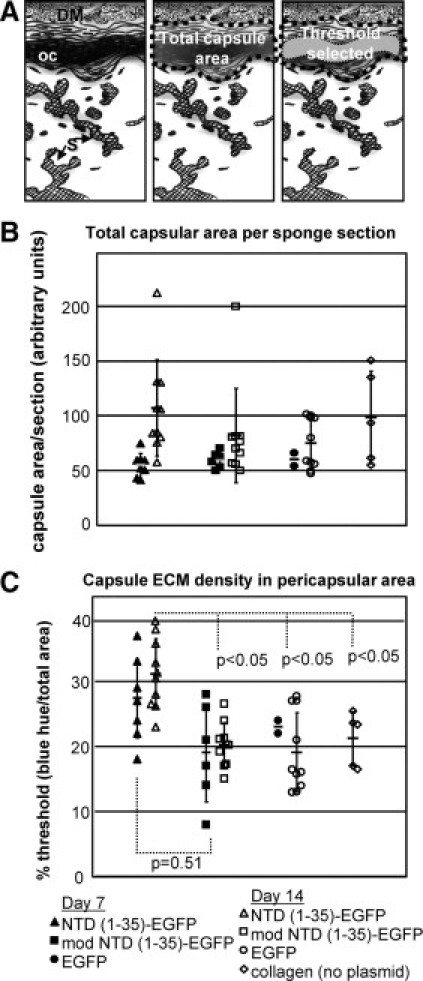
Collagenous ECM capsule density is increased in NTD (1-35)-EGFP sponges. A: Diagram of capsule quantification methodology. Images were captured along the entire pericapsular area of the sponge (left). DM indicates dermal muscle; oc, organized capsule; S, sponge material. The entire capsule region was selected (center) with the area containing organized collagen quantified based on the intensity of blue staining in Masson's trichrome–stained sections (right) using MetaMorph software and expressed as the percentage of the total capsular region that was designated as thresholded by these criteria. B: Total capsular area from sponges harvested at day 7 (filled symbols) and day 14 (open symbols). EGFP (circle), mod NTD (1-35)-EGFP (squares), NTD (1-35)-EGFP (triangles), and collagen only (diamonds). C: Collagen organization is expressed as the percentage of the total area defined by the threshold parameters. P values were calculated using the unpaired two-parameter Student's t-test.
Cellular Populations of Sponge Implants
To determine whether the increased capsular organization might be the consequence of increased cellularity, total cell number and specific cell types within the sponges were quantified. For these analyses, cells in two regions were examined: the “sponge body” as defined by all of the area containing sponge material, and the “pericapsular area” defined as the region between the cutaneous muscle and the sponge material. Cellularity throughout the sponge did not differ between groups through day 3 of treatment (data not shown). At day 7 postimplantation, the pericapsular area around the sponges in all treatment groups was densely populated with cells and, despite differences in capsule density, there was no significant difference in pericapsular cellularity between treatment groups (Table 1). However, at day 7 the NTD (1-35)-EGFP–expressing sponges had reduced infiltration of cells into the sponge body as compared to control sponges (Table 1). Because the NTD (1-35)-EGFP–expressing sponges had increased capsular organization by day 7, it is possible that the capsule acts as a physical barrier to cellular infiltration. In fact, across all treatments there is an inverse relationship between collagen capsule organization and cell number within the sponge on day 7 (data not shown). In all treatment groups, the number of cells within the sponge body increased between days 7 and 14, whereas the cell number in the pericapsular region between days 7 and 14 remained similar.
Table 1.
Average Number of Cells per Sponge Region
| Sponge region | NTD (1-35)-EGFP | mod NTD (1-35)-EGFP | P value |
|---|---|---|---|
| Day 7 pericapsular | 128 ± 36 | 182 ± 108 | 0.287 |
| Day 7 sponge body | 12 ± 2 | 20 ± 9 | 0.021 |
| Day 14 pericapsular | 152 ± 102 | 166 ± 103 | 0.772 |
| Day 14 sponge body | 69 ± 43 | 65 ± 67 | 0.882 |
Tranverse hematoxylin-stained sections of each sponge were analyzed for total cell number in the pericapsular (between striated muscle and sponge) and sponge body (entire sponge area) regions. At least ten ×40 fields per region were quantified in each sponge. P values were calculated using the two parameter paired Student's t-test, n = 6–8 sponges per group.
To assess whether the increased capsular density in the NTD (1-35)-EGFP–expressing sponges might be due to differences in cell types, myofibroblast and endothelial cell populations were evaluated. Immunostaining for α-SMA, a marker for myofibroblasts, was observed within the sponge body and the pericapsular area (Figure 6), but staining did not differ between treatment groups. Numbers of endothelial cells, as identified by CD31 staining, also were not significantly different between treatment groups (Table 2). In addition, there was no correlation between cell proliferation as measured by Ki-67 and sponge capsule organization (Table 2). Similarly, there were no differences in numbers of foreign body giant cells observed within the sponge bodies between treatment groups (Table 2). Together these data suggest that the differences in collagen capsule organization in the NTD (1-35)-EGFP–expressing sponges are not due to differences in cell proliferation, myofibroblast, macrophage/foreign body giant cell, or endothelial cell number in the pericapsular space.
Figure 6.
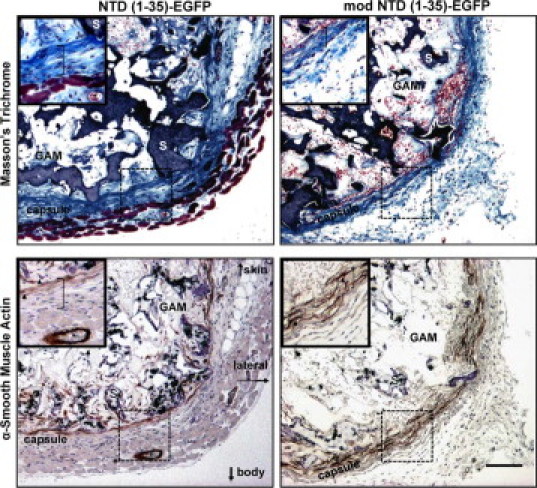
Myofibroblasts are associated with the collagen capsule and granulation tissue in NTD (1-35) and mod NTD (1-35) treatment groups at day seven. Top two panels: Masson's trichrome stain. Bottom two panels: Immunohistochemical staining for α-SMA. Images are from representative serial sections. Insets show magnified sections of the pericapsular region (hatched boxes) from the ventral side of the sponge (S). Muscle from the panniculus carnosus is visible in the NTD (1-35)-EGFP sponge (reddish tissue). Arrowheads in insets mark α-SMA–stained cells. Asterisk denotes a blood vessel in the muscle layer with positive α-SMA staining of the vessel wall. S indicates sponge material; GAM, gene-activated matrix; brackets in insets denote the capsular layer. Scale bar = 100 μm.
Table 2.
Cell Populations in Day-14 Sponges
| Sponge Type | CD31-positive pericapsular endothelial cells (mean percent area positive for CD31) | Foreign body giant cells (mean number of cells with ≥5 nuclei per area) | Ki67-positive pericapsular cells (mean percent positive cells per area) |
|---|---|---|---|
| NTD (1-35)-EGFP | 2.4 ± 1.1 | 20 ± 8 | 73 ± 32 |
| mod NTD (1-35)-EGFP | 2.2 ± 0.7 | 35 ± 18 | 43 ± 7 |
Cell populations in the pericapsular area were quantified by the following criteria: endothelial cells by measuring the area positive for CD31; foreign body giant cells by counting the number of cells with ≥5 nuclei per cell; and proliferating cells by counting the number of cells positive for Ki67. Ten random ×40 fields were counted in each sponge section, n = 6–10 sponges per group. Results are expressed as the mean percent of total cells scored as positive ± SD. There were no statistically significant differences between the experimental and control groups.
The CRT-Binding Sequence of TSP1 Stimulates Soluble Collagen Expression in Vitro
The absence of differences in either cell number or population with NTD (1-35)-EGFP expression suggests that the TSP1 CRT-binding domain directly stimulates collagen expression and fibril formation. To address this hypothesis, human foreskin fibroblasts were treated with the CRT-binding hep I peptide or the control modified hep I peptide and assayed for collagen secretion by the Sircol assay. At 48 hours of treatment, levels of soluble collagen in conditioned media of hep I–treated cells were significantly increased as compared to cells treated with modified hep I or media alone (Figure 7A). Platelet TSP1 has similar effects on stimulating fibroblast expression of soluble collagens (Figure 7B). Furthermore, this action of TSP1 occurs through binding to CRT, because collagen stimulation is blocked by a peptide of the TSP1-binding sequence of CRT (CRT19.36), which blocks TSP1-CRT interactions, but not by the control peptide (CRT20.30A), which does not bind TSP1.15 In further experiments, we also tested the ability of recombinant trimeric NTD, NoC1, to stimulate collagen expression in MEFs. NoC1 has been shown to recapitulate other functions (anoikis survival, focal adhesion disassembly) attributed to TSP1 binding to the CRT/LRP1 cocomplex and more closely represents the form of the NTD proteolytically released from TSP1 at wound sites.15,24,41 These studies showed that NoC1, similar to the CRT-binding peptide hep I and to TSP1, also increased soluble collagen expression as measured by the Sircol assay (Figure 7C). Furthermore, the ability of NoC1 to stimulate collagen expression was inhibited by the CRT peptide 19.36, but not by the non-binding control peptide (Figure 7C).
Figure 7.
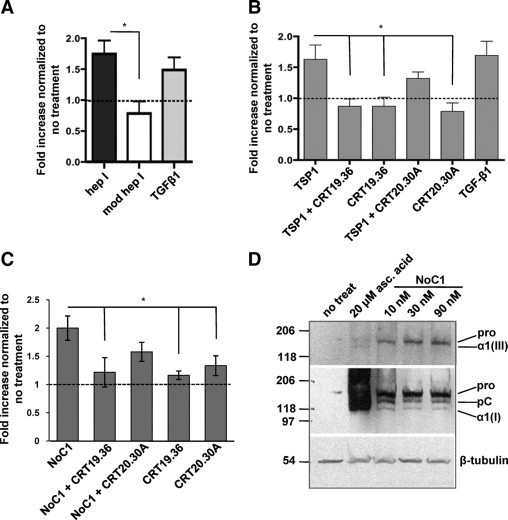
Collagen secretion and deposition by fibroblasts in vitro are increased by the CRT-binding sequence of TSP1. A: Sircol assay of human foreskin fibroblasts treated once daily for two days in media with 2 μmol/L ascorbic acid with 10 pmol/L TGF-β1, 10 μmol/L hep I peptide, 10 μmol/L modified hep I peptide, or media with 2 μmol/L ascorbic acid (no treatment). Results are expressed as the mean fold increase normalized to cells receiving no treatment ± SD; n = 3. Dashed lines in A–C indicate normalized value for no treatment. B: Sircol assay of human foreskin fibroblasts treated daily for two days with media with 2 μmol/L ascorbic acid and 10 nmol/L TSP1 or TSP1 incubated with either 20-fold molar excess of peptide CRT 19.36, or control peptide CRT 20.30A. Results are expressed as the mean fold increase normalized to cells receiving no treatment ± SD; n = 4. C: MEFs were treated daily for three days in media with 0.5% FBS, 2 μmol/L ascorbic acid with 30 nmol/L NoC1 with either 25-fold molar excess of peptide CRT 19.36, or control peptide CRT 20.30A. Soluble collagens were quantified by the Sircol assay. Results are expressed as the mean fold increase in soluble collagen normalized to cells receiving no treatment ± SD as analyzed by one-way analysis of variance; n = 4 separate experiments. D: MEFs were treated daily for four days in media with 2 μmol/L ascorbic acid with 10, 30, or 90 nmol/L NoC1 or with 20 μmol/L ascorbic acid. Cell layers were harvested with Laemmli buffer, separated by SDS-PAGE, and then immunoblotted with antibody to collagen III (top). Membranes were stripped and re-probed with anti-collagen I antibody (middle). Blots were stripped and reprobed with anti–β-tubulin as a loading control (bottom). *P < 0.05.
The CRT-Binding Sequence of TSP1 Stimulates ECM Deposition of Fibrillar Collagens in Vitro
The dense organization of the collagenous capsule in the NTD (1-35)-expressing sponges suggests that this sequence of TSP1 stimulates increased deposition of collagen into the ECM, in addition to stimulation of soluble collagen expression. SDS lysates of the cell layer (ECM and cellular collagen) from MEFs treated with increasing concentrations of NoC1 trimer showed an increase in both collagens type I and III, with a dose response observed for collagen III (Figure 7D). Although NoC1 treatment increased collagen protein expression as compared to untreated cells, it did not increase the processing of the N- and C-terminal propeptides of procollagen α1(I) as did treatment with 20 μmol/L ascorbic acid. To further distinguish intra- and pericellular collagen from ECM bound collagen, a differential ECM extraction protocol was used.36 Indeed, MEFs stimulated with hep I peptide, TSP1, or NoC1 showed increased type I collagen in the DOC insoluble, SDS soluble ECM as compared to ECM extracted from either media, mod hep I peptide, or scrambled hep I peptide-treated cells (Figure 8A). The action of the recombinant NTD in stimulating increased collagen deposition in the ECM is due to its binding to cell surface CRT, because NoC1 stimulation of ECM collagen is blocked in the presence of the TSP-binding CRT19.36 peptide but not by the non-TSP binding CRT20.30A peptide (Figure 8B). Treatment of human foreskin fibroblasts with the CRT-binding hep I peptide also increased organization of type I collagen fibrils as compared to mod hep I peptide or medium controls (Figure 8C). In fact, collagen I fibrils observed in hep I–treated cells were similar in organization and abundance to cells treated with 20 μmol/L ascorbic acid. Further analyses of 4% DOC insoluble ECM fractions showed an increase in type I collagen in this fraction in human foreskin fibroblasts treated with hep I peptide in the presence of 20 μmol/L ascorbic acid (Figure 8D).
Figure 8.
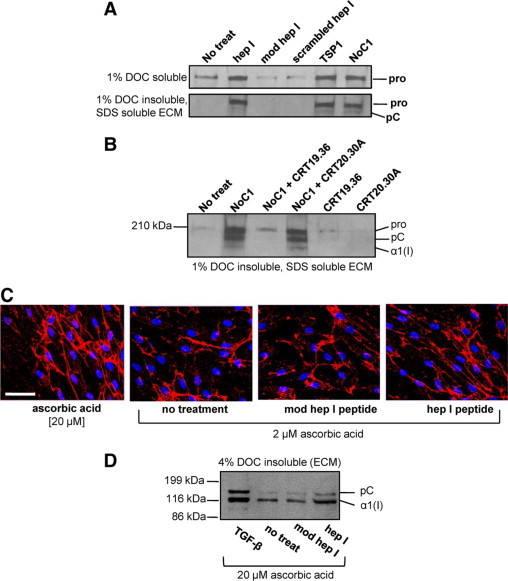
TSP1 binding to CRT increases collagen deposition into the insoluble extracellular matrix fraction of fibroblasts. A: MEFs were treated daily for three days in media with 0.5% FBS, 2 μmol/L ascorbic acid, and with 10 μmol/L hep I, 10 μmol/L modified hep I, 10 μmol/L scrambled hep I, 67 nmol/L TSP1, or 30 nmol/L NoC1. Cellular and pericellular protein was harvested with 1% DOC extraction (top), and the DOC-insoluble ECM was harvested with 2× SDS Laemmli buffer (bottom), separated by SDS-PAGE, and transferred to nitrocellulose membranes for immunoblotting with rabbit anti-mouse collagen I. Pro indicates procollagen; pC, procollagen with N-terminal propeptide removed, α1 (I), collagen I with both C- and N-terminal propeptides removed. B: MEFs were treated daily for three days as described above and with 30 nmol/L NoC1 in the presence or absence of 25-fold molar excess of CRT19.36 or control CRT20.30A peptide (750 nmol/L). The 1% DOC insoluble, SDS-soluble ECM was harvested, separated by SDS-PAGE, and immunoblotted for type I collagen. C: Type I collagen fibril formation in human foreskin fibroblasts was detected by immunofluorescence with rabbit anti-collagen I antibody and a Texas Red-labeled secondary antibody (red). Cell nuclei were stained with Hoechst 33342 (blue). Cells were treated daily for three days in the presence of 20 μmol/L ascorbic acid with no additional treatment or in the presence of 2 μmol/L ascorbic acid with either 5 μmol/L hep I peptide, 5 μmol/L modified hep I peptide, or media only. Scale bar = 60 μm. D: Human foreskin fibroblasts were treated daily for three days in FGM containing 0.5% FBS, 20 μmol/L ascorbic acid, and 5 μmol/L hep I peptide, 5 μmol/L modified hep I peptide, or 10 pmol/L TGF-β1. Cell matrices were separated by extraction in 4% DOC, and DOC-insoluble ECM was separated by SDS-PAGE and transferred to nitrocellulose membranes for immunoblotting with rabbit anti-human collagen I. Bands were labeled as described above.
Together, these data indicate that the CRT-binding sequence of TSP1 increases collagen secretion and importantly, deposition and organization into ECM.
TSP1/CRT Stimulation of Collagen I Is Not the Result of Increased Active TGF-β Secretion
It is well established that TSP1 activates latent TGF-β,42 which is a potent stimulator of collagen production and deposition.43 Although we demonstrated that the NoC1 domain of TSP1, which lacks the TGF-β–binding and activation sites, stimulates collagen expression (Figure 7, C and D) and that TSP1 and NoC1 stimulation of collagen can be inhibited by incubation with peptides that block TSP1 binding to CRT (Figures 7, B and C and 8B), it has not been previously established whether signaling through the NoC1 domain might alter TGF-β signaling through unknown mechanisms. Despite stimulation of type I collagen in the DOC insoluble ECM by both NoC1 and TSP1, there was no stimulation of TGF-β activity in the conditioned media of treated cells as compared to untreated cultures (Figure 9A). Similarly, NoC1 and TSP1 did not stimulate Smad 2 phosphorylation by fibroblasts over a 24-hour time course, although cells responded to TGF-β1 treatment with increased Smad 2 phosphorylation at 1 hour (Figure 9B).
Figure 9.
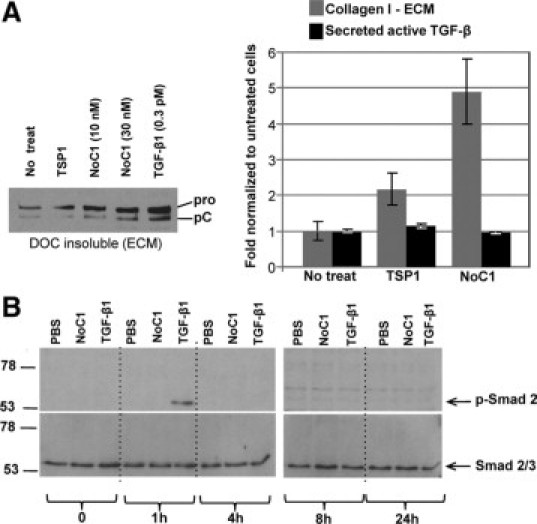
NoC1 does not increase active TGF-β or Smad2 phosphorylation. A: Levels of active TGF-β in the conditioned media of MEFs treated with 10 nmol/L of NoC1 or 10 nmol/L TSP1 in the presence of 2 μmol/L ascorbic acid for 48 hours were measured by PAI-1 luciferase assay (black bars). Collagen I ECM levels were calculated by densitometry of immunoblots for collagen I in the 4% DOC insoluble fraction of treated cells (gray bars). Results are reported as the fold change normalized to the mean ± SD. A representative collagen blot from this experiment is shown. B: Immunoblot for phosphorylated Smad 2 and total Smad 2/3 in MEF cell lysates from cells treated with media containing 2 μmol/L ascorbic acid and PBS, 10 nmol/L NoC1, or 8 pmol/L active TGF-β1 for 0, 1, 4, 8, and 24 hours. Cell lysates were harvested, separated by SDS-PAGE, and then immunoblotted for phosphorylated Smad 2. Blots were stripped and reprobed for total Smad 2/3.
TSP1/CRT Stimulation of Collagen I Is Akt-Dependent
Engagement of the cell surface CRT/LRP1 cocomplex by TSP1 initiates cellular signaling that induces transient association of Giα2 with LRP1, activation of PI3 kinase, and phosphorylation of Akt.15,44 PI3 kinase-dependent Akt activation is important for signaling of anoikis resistance by TSP1.15 Therefore, pharmacological inhibitors were used to determine whether the PI3 kinase/Akt cascade is also required for regulation of collagen through the TSP1 CRT-binding sequence. Cells were treated with NoC1 in presence or absence of either a PI3 kinase inhibitor, LY294002, or an Akt inhibitor, Akt inhibitor II, which blocks PIP3 binding to Akt.38 Levels of type I collagen protein in SDS lysates of the cell layer were determined by immunoblotting. These concentrations of PI3 kinase and Akt inhibitors were sufficient to block NoC1 stimulation of Akt phosphorylation (data not shown). Treatment of cells with the Akt inhibitor II blocked NoC1 stimulation of collagen type I protein to that of untreated control cells, and the Akt inhibitor had no effect on basal levels of collagen protein (Figure 10). Collagen levels in cells stimulated with NoC1 and treated with the PI3K inhibitor were comparable to NoC1-stimulated cells treated with Akt inhibitor II; however, baseline collagen levels were reduced by 80% in the presence of LY294002. These data suggest that NoC1 stimulates collagen I protein expression through an Akt-dependent mechanism.
Figure 10.
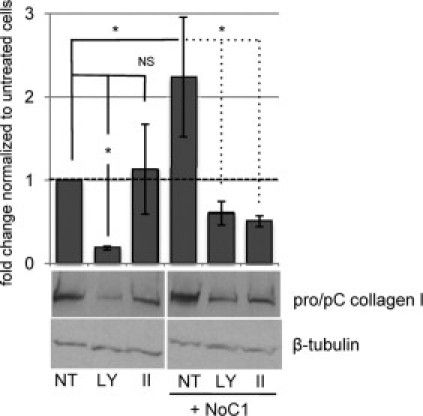
An Akt inhibitor blocks collagen I protein expression in response to NoC1. MEFs cultured in media with 2 μmol/L ascorbic acid were treated daily for three days with either PI3-kinase inhibitor, 5 μmol/L LY294002 (LY), 2 μmol/L Akt inhibitor II (II), or DMSO vehicle (NT) in the presence or absence of 30 nmol/L NoC1. Cell layers harvested in Laemmli buffer were separated by SDS-PAGE and immunoblotted for type I collagen. Note that the processed and unprocessed procollagen bands were not resolved in the Laemmli extracts of the total cell lysates. Blots were stripped and reprobed for β-tubulin as a loading control. Blots were analyzed by densitometric analysis, and collagen levels were normalized to the loading control for each sample. Results are expressed as the mean fold change as compared to cells treated with DMSO ± SD (n = 3).
Discussion
These studies are the first to address the specific role of TSP1 binding to the CRT/LRP1 coreceptor complex in vivo. To ensure that only CRT/LRP1 signaling would be engaged, we locally expressed the first 35 amino acids of TSP1, which include the CRT-binding sequence. Initially, we hypothesized that overexpression of the CRT-binding sequence of TSP1 in vivo during injury would increase cell migration and thus increase granulation tissue formation. Surprisingly, expression of the NTD (1-35)-EGFP construct resulted in the formation of a dense collagenous/ECM capsule, despite similar cell number and cellular composition in the pericapsular region. In vitro studies showed that TSP1, the recombinant NTD, or the CRT-binding sequence of TSP1 stimulates collagen protein expression and deposition into the ECM by mouse embryonic and human neonatal fibroblasts in a TGF-β–independent Akt-dependent manner. Collagen I protein expression was also increased in hep I–treated primary adult human fibroblasts (data not shown). The increased collagen deposition in the adult mice in vivo and the in vitro responses by mouse embryonic fibroblasts and human neonatal fibroblasts suggest that this function of the TSP1 NTD is conserved in both embryonic/neonatal and adult collagen regulation. Interestingly, although we observed differences in collagen protein expression and ECM deposition, we failed to observe stimulation of COL1A2 mRNA by the TSP1 CRT-binding sequence or by NoC1 (data not shown), suggesting that primary regulation by TSP1 is posttranscriptional. Together, these studies identify a novel role for TSP1 in tissue remodeling by stimulation of collagen expression and organization through its CRT-binding sequence.
The role of TSP1 in collagen regulation has been studied indirectly in dermal excisional wound healing models using either TSP1 or TSP1 and TSP2 double knockout mice.9,45,46 In these mice, as well as in TSP2 knockout mice, a reduction in collagen deposition in the granulation tissues of dermal wounds was reported, although a significant defect in collagen fibril formation was reported for only the TSP2 null mice.45,46 The reduced granulation tissue observed in TSP1-null mice has been attributed to attenuation of macrophage recruitment to wound sites but has also been shown to be at least partly due to reduced activation of TGF-β.9,47 In our studies, we used a model of the foreign body response, which is a specialized form of repair that entails prolonged inflammation with a characteristic remodeling phase that involves robust collagen deposition and encapsulation of the foreign material. Our results using this foreign body sponge implant model suggest that the altered granulation tissue in dermal excisional wounds of the TSP1-null mice might also be attributed in part to insufficient collagen deposition in the absence of signaling from the TSP1 NTD.
In contrast to these findings suggesting that TSP1 stimulates collagen deposition in wounds, others have reported that TSP1 suppresses collagen. Zhou et al demonstrated in an ex vivo muscle explant model that type I collagen α1 and α2 mRNAs are up-regulated in TSP1(−/−) endothelial cells and that this activity is suppressed by the addition of exogenous soluble TSP1.48 Moura et al used an arterial ligation model to induce medial thickening of the carotid artery in TSP1-null mice and showed that these mice exhibit increased collagen deposition within the neointima.49 These findings suggest that TSP1 negatively regulates collagen expression by mouse endothelial and vascular smooth muscle cells. None of these studies in the TSP1-null models addressed which receptor(s) mediate TSP1 regulation of collagen I. It is not clear why these findings differ from the results of our present studies using sponge implants in wild-type mice and in vitro studies in normal human and murine fibroblasts or from the excisional dermal wound studies performed by other groups in the TSP1-null mice9,50; it is possible that these differences might reflect cell type–specific regulation of collagen.9 Moreover, these paradoxical findings illustrate the utility of both knockout and local overexpression models for determining tissue and disease specific functions of TSPs.
The increased capsule organization in sponges expressing NTD (1-35)-EGFP is consistent with the functions of related matricellular proteins in sponge implant models. Expression of TSP2 antisense cDNA reduced collagen encapsulation of sponge implants in wild-type mice.51 Sponge implant models using matricellular knockout mice indicate that plasminogen activator inhibitor-1 (PAI-1)52 and SPARC (secreted protein acidic and rich in cysteine)53 knockout mice, respectively, have reduced collagen deposition or collagen encapsulation of the foreign body as compared with wild-type mice. Expression of the matricellular protein, tenascin-C, positively correlated with increased foreign body collagen encapsulation of prosthetic hip implants.54 These matricellular proteins have all been shown to support intermediate adhesion in adherent cell types in vitro.25,55,56 It is not known, however, whether these other matricellular proteins signal increased collagen expression or collagen capsule formation through their respective focal adhesion labilizing domains, as does TSP1.
The absence of a correlation between pericapsular cell population and capsular collagen organization indicates that collagen capsule formation in response to local NTD (1-35)-EGFP protein is the result of increased collagen accumulation by fibroblasts, rather than due to stimulation of increased cell migration to the sponge implant. Interestingly, the hep I peptide or NoC1 did not alter the ratio of procollagen to processed collagen in either mouse or human fibroblasts (Figures 8, A, B, and D and 9A).
It is well established that TSP1 indirectly increases collagen transcription and deposition through activation of latent TGF-β via the TSP1 type 1 repeats,57 distinct from the NTD. Therefore, we speculated that TSP1 contains a second site for collagen regulation within amino acids 1-35 of the NTD. We did not detect increased secreted active TGF-β1-3 or phosphorylation of Smad 2 in TSP1 NTD-treated cells, despite a robust increase in collagen I deposition in the ECM. This suggests that the CRT-binding domain of TSP1 does not stimulate collagen by first directly stimulating TGF-β signaling or activation.
Instead, the mechanism by which TSP1 signaling through CRT/LRP1 stimulates expression of fibrillar collagens appears to require Akt signaling. Several studies indicate that phosphorylation of Akt regulates the expression and deposition of collagen I. Blocking Akt with specific inhibitors (including Akt inhibitor II) reduces collagen I mRNA transcript levels in human dermal fibroblasts.58 PI3 kinase, upstream of Akt, is also able to regulate collagen I protein expression. Inhibitors of either PI3 kinase or Akt signaling are reported to reduce collagen production in mesangial cells, normal dermal and scleroderma fibroblasts, lung fibroblasts, and hepatic stellate cells.58–62 In our studies, we were unable to draw conclusions regarding the involvement of PI3 kinase because of the effects of the LY294002 compound on baseline levels of collagen I expression. This observation of the LY294002 effect on collagen production is consistent with other published observations.59–62
As with many other TSP1 receptors, collagen regulation in response to TSP1 might not only be dictated by TSP1 bioavailability but also by the cell surface availability of both CRT and LRP1. CRT is up-regulated in response to stress, and we demonstrated its increased expression in atherosclerotic rabbit arteries.63–65 CRT is best known as a ubiquitous endoplasmic reticulum resident chaperone and intracellular calcium regulator.66 However, work from our lab and others have demonstrated stimulation of intracellular signaling via TSP1 binding to cell surface CRT, which stimulates engagement of LRP1-mediated signaling.19,20,63 LRP1 similarly has dual functions in mediating endocytic clearance of molecules, including TSP1 and matrix metalloproteinases, and it also acts as a signaling receptor.67,68 It is therefore possible that the presence of cell surface CRT dictates whether LRP1 will respond to TSP1 by activating signaling pathways or by internalizing TSP1. Furthermore, the multiplicity of TSP1 actions is potentially regulated by the availability of specific TSP1 domains, either in the context of the entire molecule or as an isolated domain. Recent work from Lee et al demonstrates that TSP1 is cleaved by ADAMTS-1 into N-terminal and C-terminal fragments, both of which are detectable in vivo.24 The NTD of TSP1 is also released by cathepsin G and neutrophil elastase.69,70 Therefore, proteases present in early wound beds might generate locally elevated levels of N-terminal and C-terminal fragments of TSP1.
Fibrous collagen encapsulation is required for stabilization of the atherosclerotic plaque and encapsulation of primary tumors but is detrimental for osseointegration of bone and dental implants and malleability of soft tissue implants. Regardless of the host tissue, collagen encapsulation requires active fibroblasts. The NTD of TSP1 has been shown to participate in adhesion modulation, migration, anoikis survival, and now collagen deposition.15,28,29 It seems unlikely that cells responding to TSP1/CRT signaling are able to accomplish both cell migration and matrix deposition simultaneously. It is more likely that cell density, cell–cell and cell–ECM interactions, and the availability of full-length versus cleaved TSP1 will also play a role in regulating whether the response to the NTD of TSP1 results in cell migration or ECM deposition. The addition of this current work to the previous studies of the TSP1 CRT-binding domain suggest that this interaction coordinates fibroblast responses to injury by inducing a “remodeling phase” with the specific action additionally cued by the tissue environment. This leads us to speculate that the CRT-binding sequence of TSP1 has potential as a protein therapeutic for guided tissue remodeling.
Acknowledgements
We gratefully acknowledge Dr. Themis Kyriakides (Yale University), Dr. Bin Sun, Dr. Anne Woods, Dr. Namasivayam Ambalavanan, Dr. Janusz Kabarowski, Dr. Brian Parks, and Arlene Bulger (UAB), and Dr. Kurt Hankenson (University of Pennsylvania) for technical advice, assistance, and use of equipment. We thank Dr. Deane Mosher (University of Wisconsin) for NoC1. We acknowledge use of the following UAB Core facilities: the Laboratory for Multi-Modality Imaging Assessment and Molecular Imaging Core, the UAB High Resolution Imaging Facility, and the UAB Comparative Pathology Laboratory. Thank you also to Dr. Yong Zhou (UAB) for helpful discussions.
Footnotes
Supported by National Institutes of Health (NIH) grant HL79644 (J.E.M.-U.), UAB Cardiovascular and Pathophysiology Training grant NIH T32 HL007918 and an American Society of Cell Biology Minority Affairs Council award (M.T.S.), and the University of Alabama at Birmingham MSTPT32 GM008361 (L.V.G.). This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program grant No. C06 RR 15490 from the National Center for Research Resources, NIH.
References
- 1.Midwood KS, Williams LV, Schwarzbauer JE. Tissue repair and the dynamics of the extracellular matrix. Int J Biochem Cell Biol. 2004;36:1031–1037. doi: 10.1016/j.biocel.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Alford AI, Hankenson KD. Matricellular proteins: extracellular modulators of bone development, remodeling, and regeneration. Bone. 2006;38:749–757. doi: 10.1016/j.bone.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 3.Schellings MW, Pinto YM, Heymans S. Matricellular proteins in the heart: possible role during stress and remodeling. Cardiovasc Res. 2004;64:24–31. doi: 10.1016/j.cardiores.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 4.McPherson J, Sage H, Bornstein P. Isolation and characterization of a glycoprotein secreted by aortic endothelial cells in culture. Apparent identity with platelet thrombospondin. J Biol Chem. 1981;256:11330–11336. [PubMed] [Google Scholar]
- 5.Raugi GJ, Mumby SM, Ready CA, Bornstein P. Location and partial characterization of the heparin-binding fragment of platelet thrombospondin. Thromb Res. 1984;36:165–175. doi: 10.1016/0049-3848(84)90338-4. [DOI] [PubMed] [Google Scholar]
- 6.DiPietro LA, Polverini PJ. Angiogenic macrophages produce the angiogenic inhibitor thrombospondin 1. Am J Pathol. 1993;143:678–684. [PMC free article] [PubMed] [Google Scholar]
- 7.Reed MJ, Iruela-Arispe L, O'Brien ER, Truong T, LaBell T, Bornstein P, Sage EH. Expression of thrombospondins by endothelial cells. Injury is correlated with TSP-1. Am J Pathol. 1995;147:1068–1080. [PMC free article] [PubMed] [Google Scholar]
- 8.Majack RA, Cook SC, Bornstein P. Control of smooth muscle cell growth by components of the extracellular matrix: autocrine role for thrombospondin. Proc Natl Acad Sci USA. 1986;83:9050–9054. doi: 10.1073/pnas.83.23.9050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agah A, Kyriakides TR, Lawler J, Bornstein P. The lack of thrombospondin-1 (TSP1) dictates the course of wound healing in double-TSP1/TSP2-null mice. Am J Pathol. 2002;161:831–839. doi: 10.1016/S0002-9440(10)64243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murphy-Ullrich JE, Hook M. Thrombospondin modulates focal adhesions in endothelial cells. J Cell Biol. 1989;109:1309–1319. doi: 10.1083/jcb.109.3.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schultz-Cherry S, Murphy-Ullrich JE. Thrombospondin causes activation of latent transforming growth factor-beta secreted by endothelial cells by a novel mechanism. J Cell Biol. 1993;122:923–932. doi: 10.1083/jcb.122.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Isenberg JS, Ridnour LA, Dimitry J, Frazier WA, Wink DA, Roberts DD. CD47 is necessary for inhibition of nitric oxide-stimulated vascular cell responses by thrombospondin-1. J Biol Chem. 2006;281:26069–26080. doi: 10.1074/jbc.M605040200. [DOI] [PubMed] [Google Scholar]
- 13.Ferrari do Outeiro-Bernstein MA, Nunes SS, Andrade AC, Alves TR, Legrand C, Morandi V. A recombinant NH(2)-terminal heparin-binding domain of the adhesive glycoprotein, thrombospondin-1, promotes endothelial tube formation and cell survival: a possible role for syndecan-4 proteoglycan. Matrix Biol. 2002;21:311–324. doi: 10.1016/s0945-053x(02)00010-0. [DOI] [PubMed] [Google Scholar]
- 14.Iruela-Arispe ML, Bornstein P, Sage H. Thrombospondin exerts an antiangiogenic effect on cord formation by endothelial cells in vitro. Proc Natl Acad Sci USA. 1991;88:5026–5030. doi: 10.1073/pnas.88.11.5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pallero MA, Elzie CA, Chen J, Mosher DF, Murphy-Ullrich JE. Thrombospondin 1 binding to calreticulin-LRP1 signals resistance to anoikis. FASEB J. 2008;22:3968–3979. doi: 10.1096/fj.07-104802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams JC, Kureishy N, Taylor AL. A role for syndecan-1 in coupling fascin spike formation by thrombospondin-1. J Cell Biol. 2001;152:1169–1182. doi: 10.1083/jcb.152.6.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chung J, Wang XQ, Lindberg FP, Frazier WA. Thrombospondin-1 acts via IAP/CD47 to synergize with collagen in alpha2beta1-mediated platelet activation. Blood. 1999;94:642–648. [PubMed] [Google Scholar]
- 18.Dawson DW, Pearce SF, Zhong R, Silverstein RL, Frazier WA, Bouck NP. CD36 mediates the In vitro inhibitory effects of thrombospondin-1 on endothelial cells. J Cell Biol. 1997;138:707–717. doi: 10.1083/jcb.138.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orr AW, Pedraza CE, Pallero MA, Elzie CA, Goicoechea S, Strickland DK, Murphy-Ullrich JE. Low density lipoprotein receptor-related protein is a calreticulin coreceptor that signals focal adhesion disassembly. J Cell Biol. 2003;161:1179–1189. doi: 10.1083/jcb.200302069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goicoechea S, Orr AW, Pallero MA, Eggleton P, Murphy-Ullrich JE. Thrombospondin mediates focal adhesion disassembly through interactions with cell surface calreticulin. J Biol Chem. 2000;275:36358–36368. doi: 10.1074/jbc.M005951200. [DOI] [PubMed] [Google Scholar]
- 21.Streit M, Velasco P, Riccardi L, Spencer L, Brown LF, Janes L, Lange-Asschenfeldt B, Yano K, Hawighorst T, Iruela-Arispe L, Detmar M. Thrombospondin-1 suppresses wound healing and granulation tissue formation in the skin of transgenic mice. EMBO J. 2000;19:3272–3282. doi: 10.1093/emboj/19.13.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Isenberg JS, Calzada MJ, Zhou L, Guo N, Lawler J, Wang XQ, Frazier WA, Roberts DD. Endogenous thrombospondin-1 is not necessary for proliferation but is permissive for vascular smooth muscle cell responses to platelet-derived growth factor. Matrix Biol. 2005;24:110–123. doi: 10.1016/j.matbio.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 23.Iruela-Arispe ML. Regulation of thrombospondin1 by extracellular proteases. Curr Drug Targets. 2008;9:863–868. doi: 10.2174/138945008785909365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee NV, Sato M, Annis DS, Loo JA, Wu L, Mosher DF, Iruela-Arispe ML. ADAMTS1 mediates the release of antiangiogenic polypeptides from TSP1 and 2. EMBO J. 2006;25:5270–5283. doi: 10.1038/sj.emboj.7601400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy-Ullrich JE, Gurusiddappa S, Frazier WA, Hook M. Heparin-binding peptides from thrombospondins 1 and 2 contain focal adhesion-labilizing activity. J Biol Chem. 1993;268:26784–26789. [PubMed] [Google Scholar]
- 26.Orr AW, Pallero MA, Murphy-Ullrich JE. Thrombospondin stimulates focal adhesion disassembly through Gi- and phosphoinositide 3-kinase-dependent ERK activation. J Biol Chem. 2002;277:20453–20460. doi: 10.1074/jbc.M112091200. [DOI] [PubMed] [Google Scholar]
- 27.Orr AW, Pallero MA, Xiong WC, Murphy-Ullrich JE. Thrombospondin induces RhoA inactivation through FAK-dependent signaling to stimulate focal adhesion disassembly. J Biol Chem. 2004;279:48983–48992. doi: 10.1074/jbc.M404881200. [DOI] [PubMed] [Google Scholar]
- 28.Murphy-Ullrich JE. The de-adhesive activity of matricellular proteins: is intermediate cell adhesion an adaptive state? J Clin Invest. 2001;107:785–790. doi: 10.1172/JCI12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orr AW, Elzie CA, Kucik DF, Murphy-Ullrich JE. Thrombospondin signaling through the calreticulin/LDL receptor-related protein co-complex stimulates random and directed cell migration. J Cell Sci. 2003;116:2917–2927. doi: 10.1242/jcs.00600. [DOI] [PubMed] [Google Scholar]
- 30.Yan Q, Murphy-Ullrich JE, Song Y. Structural insight into the role of thrombospondin-1 binding to calreticulin in calreticulin-induced focal adhesion disassembly. Biochemistry. 2010;49:3685–3694. doi: 10.1021/bi902067f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goicoechea S, Pallero MA, Eggleton P, Michalak M, Murphy-Ullrich JE. The anti-adhesive activity of thrombospondin is mediated by the N-terminal domain of cell surface calreticulin. J Biol Chem. 2002;277:37219–37228. doi: 10.1074/jbc.M202200200. [DOI] [PubMed] [Google Scholar]
- 32.Murphy-Ullrich JE, Schultz-Cherry S, Hook M. Transforming growth factor-beta complexes with thrombospondin. Mol Biol Cell. 1992;3:181–188. doi: 10.1091/mbc.3.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonadio J, Smiley E, Patil P, Goldstein S. Localized, direct plasmid gene delivery in vivo: prolonged therapy results in reproducible tissue regeneration. Nat Med. 1999;5:753–759. doi: 10.1038/10473. [DOI] [PubMed] [Google Scholar]
- 34.Midwood KS, Wierzbicka-Patynowski I, Schwarzbauer JE. Preparation and analysis of synthetic multicomponent extracellular matrix. Methods Cell Biol. 2002;69:145–161. doi: 10.1016/s0091-679x(02)69011-9. [DOI] [PubMed] [Google Scholar]
- 35.Van Duyn Graham L, Sweetwyne MT, Pallero MA, Murphy-Ullrich JE. Intracellular calreticulin regulates multiple steps in fibrillar collagen expression, trafficking, and processing into the extracellular matrix. J Biol Chem. 2010;285:7067–7078. doi: 10.1074/jbc.M109.006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hedman K, Kurkinen M, Alitalo K, Vaheri A, Johansson S, Hook M. Isolation of the pericellular matrix of human fibroblast cultures. J Cell Biol. 1979;81:83–91. doi: 10.1083/jcb.81.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taipale J, Miyazono K, Heldin CH, Keski-Oja J. Latent transforming growth factor-beta 1 associates to fibroblast extracellular matrix via latent TGF-beta binding protein. J Cell Biol. 1994;124:171–181. doi: 10.1083/jcb.124.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang L, Dan HC, Sun M, Liu Q, Sun XM, Feldman RI, Hamilton AD, Polokoff M, Nicosia SV, Herlyn M, Sebti SM, Cheng JQ. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res. 2004;64:4394–4399. doi: 10.1158/0008-5472.CAN-04-0343. [DOI] [PubMed] [Google Scholar]
- 39.Zhou Y, Hagood JS, Murphy-Ullrich JE. Thy-1 expression regulates the ability of rat lung fibroblasts to activate transforming growth factor-beta in response to fibrogenic stimuli. Am J Pathol. 2004;165:659–669. doi: 10.1016/s0002-9440(10)63330-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB. An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal Biochem. 1994;216:276–284. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- 41.Carlson CB, Lawler J, Mosher DF. Structure of Thrombospondins. Cell Mol Life Sci. 2008;65:672–686. doi: 10.1007/s00018-007-7484-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schultz-Cherry S, Ribeiro S, Gentry L, Murphy-Ullrich JE. Thrombospondin binds and activates the small and large forms of latent transforming growth factor-beta in a chemically defined system. J Biol Chem. 1994;269:26775–26782. [PubMed] [Google Scholar]
- 43.Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci USA. 1986;83:4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Greenwood JA, Pallero MA, Theibert AB, Murphy-Ullrich JE. Thrombospondin signaling of focal adhesion disassembly requires activation of phosphoinositide 3-kinase. J Biol Chem. 1998;273:1755–1763. doi: 10.1074/jbc.273.3.1755. [DOI] [PubMed] [Google Scholar]
- 45.Nor JE, DiPiertro L, Murphy-Ullrich JE, Hynes RO, Lawler J, Polverini PJ. Activation of latent TGF-beta by thrombospondin-1 is a major component of wound repair. Oral Biosci Med. 2005;2:153–161. [PMC free article] [PubMed] [Google Scholar]
- 46.Kyriakides TR, Zhu YH, Smith LT, Bain SD, Yang Z, Lin MT, Danielson KG, Iozzo RV, LaMarca M, McKinney CE, Ginns EI, Bornstein P. Mice that lack thrombospondin 2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, an increased vascular density, and a bleeding diathesis. J Cell Biol. 1998;140:419–430. doi: 10.1083/jcb.140.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DiPietro LA, Nissen NN, Gamelli RL, Koch AE, Pyle JM, Polverini PJ. Thrombospondin 1 synthesis and function in wound repair. Am J Pathol. 1996;148:1851–1860. [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou L, Isenberg JS, Cao Z, Roberts DD. Type I collagen is a molecular target for inhibition of angiogenesis by endogenous thrombospondin-1. Oncogene. 2006;25:536–545. doi: 10.1038/sj.onc.1209069. [DOI] [PubMed] [Google Scholar]
- 49.Moura R, Tjwa M, Vandervoort P, Cludts K, Hoylaerts MF. Thrombospondin-1 activates medial smooth muscle cells and triggers neointima formation upon mouse carotid artery ligation. Arterioscler Thromb Vasc Biol. 2007;27:2163–2169. doi: 10.1161/ATVBAHA.107.151282. [DOI] [PubMed] [Google Scholar]
- 50.Nor JE, DiPietro L, Murphy-Ullrich JE, Hynes RO, Lawler J, Polverini PJ. Activation of latent TGF-beta 1 by thrombospondin-1 is a major component of wound repair. Oral Biosci Med. 2005;213:153–161. [PMC free article] [PubMed] [Google Scholar]
- 51.Kyriakides TR, Hartzel T, Huynh G, Bornstein P. Regulation of angiogenesis and matrix remodeling by localized, matrix-mediated antisense gene delivery. Mol Ther. 2001;3:842–849. doi: 10.1006/mthe.2001.0336. [DOI] [PubMed] [Google Scholar]
- 52.Chuang-Tsai S, Sisson TH, Hattori N, Tsai CG, Subbotina NM, Hanson KE, Simon RH. Reduction in fibrotic tissue formation in mice genetically deficient in plasminogen activator inhibitor-1. Am J Pathol. 2003;163:445–452. doi: 10.1016/S0002-9440(10)63674-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bradshaw AD, Reed MJ, Carbon JG, Pinney E, Brekken RA, Sage EH. Increased fibrovascular invasion of subcutaneous polyvinyl alcohol sponges in SPARC-null mice. Wound Repair Regen. 2001;9:522–530. doi: 10.1046/j.1524-475x.2001.00522.x. [DOI] [PubMed] [Google Scholar]
- 54.Konttinen YT, Li TF, Michelsson O, Xu JW, Sorsa T, Santavirta S, Imai S, Virtanen I. Expression of tenascin-C in aseptic loosening of total hip replacement. Ann Rheum Dis. 1998;57:619–623. doi: 10.1136/ard.57.10.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murphy-Ullrich JE, Lane TF, Pallero MA, Sage EH. SPARC mediates focal adhesion disassembly in endothelial cells through a follistatin-like region and the Ca(2+)-binding EF-hand. J Cell Biochem. 1995;57:341–350. doi: 10.1002/jcb.240570218. [DOI] [PubMed] [Google Scholar]
- 56.Murphy-Ullrich JE, Lightner VA, Aukhil I, Yan YZ, Erickson HP, Hook M. Focal adhesion integrity is downregulated by the alternatively spliced domain of human tenascin. J Cell Biol. 1991;115:1127–1136. doi: 10.1083/jcb.115.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schultz-Cherry S, Lawler J, Murphy-Ullrich JE. The type 1 repeats of thrombospondin 1 activate latent transforming growth factor-beta. J Biol Chem. 1994;269:26783–26788. [PubMed] [Google Scholar]
- 58.Bujor AM, Pannu J, Bu S, Smith EA, Muise-Helmericks RC, Trojanowska M. Akt blockade downregulates collagen and upregulates MMP1 in human dermal fibroblasts. J Invest Dermatol. 2008;128:1906–1914. doi: 10.1038/jid.2008.39. [DOI] [PubMed] [Google Scholar]
- 59.Asano Y, Ihn H, Yamane K, Jinnin M, Mimura Y, Tamaki K. Phosphatidylinositol 3-kinase is involved in alpha2(I) collagen gene expression in normal and scleroderma fibroblasts. J Immunol. 2004;172:7123–7135. doi: 10.4049/jimmunol.172.11.7123. [DOI] [PubMed] [Google Scholar]
- 60.Reif S, Lang A, Lindquist JN, Yata Y, Gabele E, Scanga A, Brenner DA, Rippe RA. The role of focal adhesion kinase-phosphatidylinositol 3-kinase-akt signaling in hepatic stellate cell proliferation and type I collagen expression. J Biol Chem. 2003;278:8083–8090. doi: 10.1074/jbc.M212927200. [DOI] [PubMed] [Google Scholar]
- 61.Ricupero DA, Poliks CF, Rishikof DC, Cuttle KA, Kuang PP, Goldstein RH. Phosphatidylinositol 3-kinase-dependent stabilization of alpha1(I) collagen mRNA in human lung fibroblasts. Am J Physiol Cell Physiol. 2001;281:C99–C105. doi: 10.1152/ajpcell.2001.281.1.C99. [DOI] [PubMed] [Google Scholar]
- 62.Runyan CE, Schnaper HW, Poncelet AC. The phosphatidylinositol 3-kinase/Akt pathway enhances Smad3-stimulated mesangial cell collagen I expression in response to transforming growth factor-beta1. J Biol Chem. 2004;279:2632–2639. doi: 10.1074/jbc.M310412200. [DOI] [PubMed] [Google Scholar]
- 63.Gold LI, Eggleton P, Sweetwyne MT, Van Duyn LB, Greives MR, Naylor SM, Michalak M, Murphy-Ullrich JE. Calreticulin: non-endoplasmic reticulum functions in physiology and disease. FASEB J. 2010;24:665–683. doi: 10.1096/fj.09-145482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu X, Liu X, Zhu X, Tang C. Hypoxic preconditioning induces delayed cardioprotection through p38 MAPK-mediated calreticulin upregulation. Shock. 2007;27:572–577. doi: 10.1097/01.shk.0000246901.58068.a8. [DOI] [PubMed] [Google Scholar]
- 65.Kypreou KP, Kavvadas P, Karamessinis P, Peroulis M, Alberti A, Sideras P, Psarras S, Capetanaki Y, Politis PK, Charonis AS. Altered expression of calreticulin during the development of fibrosis. Proteomics. 2008;8:2407–2419. doi: 10.1002/pmic.200700831. [DOI] [PubMed] [Google Scholar]
- 66.Milner RE, Baksh S, Shemanko C, Carpenter MR, Smillie L, Vance JE, Opas M, Michalak M. Calreticulin, and not calsequestrin, is the major calcium binding protein of smooth muscle sarcoplasmic reticulum and liver endoplasmic reticulum. J Biol Chem. 1991;266:7155–7165. [PubMed] [Google Scholar]
- 67.Lillis AP, Van Duyn LB, Murphy-Ullrich JE, Strickland DK. LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol Rev. 2008;88:887–918. doi: 10.1152/physrev.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen H, Sottile J, Strickland DK, Mosher DF. Binding and degradation of thrombospondin-1 mediated through heparan sulphate proteoglycans and low-density-lipoprotein receptor-related protein: localization of the functional activity to the trimeric N-terminal heparin-binding region of thrombospondin-1. Biochem J. 1996;318:959–963. doi: 10.1042/bj3180959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hogg PJ, Owensby DA, Chesterman CN. Thrombospondin 1 is a tight-binding competitive inhibitor of neutrophil cathepsin G. Determination of the kinetic mechanism of inhibition and localization of cathepsin G binding to the thrombospondin 1 type 3 repeats. J Biol Chem. 1993;268:21811–21818. [PubMed] [Google Scholar]
- 70.Hogg PJ, Jimenez BM, Chesterman CN. Identification of possible inhibitory reactive centers in thrombospondin 1 that may bind cathepsin G and neutrophil elastase. Biochemistry. 1994;33:6531–6537. doi: 10.1021/bi00187a021. [DOI] [PubMed] [Google Scholar]