

Abstract
Impairment in the regeneration process is a critical determinant for skeletal muscle wasting in chronic diseases and degenerative muscle disorders. Inflammatory cytokines are known to cause significant muscle wasting, however, their role in myofiber regeneration is less clear. In this study we have investigated the role of tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in skeletal muscle regeneration in vivo. Our results show that expression levels of TWEAK and its receptor Fn14 are significantly increased in skeletal muscles of mice after injury. Genetic deletion of TWEAK increased the fiber cross-sectional area and levels of embryonic isoform of myosin heavy chain in regenerating tibial anterior muscle. Conversely, muscle-specific transgenic overexpression of TWEAK reduced the fiber cross-sectional area and levels of the embryonic myosin heavy chain in regenerating muscle. TWEAK induced the expression of several inflammatory molecules and increased interstitial fibrosis in regenerating muscle. Genetic ablation of TWEAK suppressed, whereas overexpression of TWEAK increased, the activation of nuclear factor-kappa B without affecting the activation of Akt or p38 kinase in regenerating myofibers. Primary myoblasts from TWEAK-null mice showed enhanced differentiation in vitro, whereas myoblasts from TWEAK-Tg mice showed reduced differentiation compared with wild-type mice. Collectively, our study suggests that TWEAK negatively regulates muscle regeneration and that TWEAK is a potential therapeutic target to enhance skeletal muscle regeneration in vivo.
Skeletal muscle has remarkable ability to regenerate in response to both genetic and acquired forms of muscle fiber damage.1 Injury to skeletal muscle leads to the activation of a population of undifferentiated muscle precursor cells, commonly referred to as satellite cells, which contribute to the repair process and the formation of new myofibers.1–3 In normal adult skeletal muscle, satellite cells reside between the plasma membrane and basal lamina in a relatively quiescent, nonproliferative state.3 Once activated, these cells undergo proliferation and migration to sites of muscle fiber damage. After several rounds of cellular division, these cells exit the cell cycle and differentiate into post-mitotic myotubes, which then evolve into mature adult fibers.4,5 Although considerable progress has been made towards understanding the process of skeletal muscle regeneration,1–3,5 the role of various intrinsic and extrinsic signals in muscle regeneration remains poorly understood.
Studies in the recent past have provided strong evidence that several cytokines and chemokines produced by both immune and skeletal muscle cells play a critical role in the acquisition and the maintenance of skeletal muscle mass in vivo.6 For example, interleukin (IL)−4 has been found to positively regulate myoblast fusion, regeneration, and growth in vivo.7 Similarly, interferon-γ also promotes myofiber regeneration after injury.8 In contrast, several pro-inflammatory cytokines (such as tumor necrosis factor [TNF]-α, IL-1β and IL-6) while augmenting the proliferation of muscle progenitor cells, inhibit the terminal differentiation/fusion of myoblasts into mature myofibers.9–11 Injection of soluble TNF-α (henceforth TNF) protein at specific time points during regeneration delayed the appearance of regenerating fibers, without exacerbating fiber death following the initial trauma.12 Similarly, overexpression of TNF in skeletal muscle has been found to cause significant inhibition of myofiber regeneration following injury.13 Coincidently, these pro-inflammatory cytokines have also been suggested as the major mediators of skeletal muscle-wasting in chronic diseases.14,15
TNF-like weak inducer of apoptosis (TWEAK) is a pro-inflammatory cytokine belonging to TNF super family ligands.16 TWEAK is initially synthesized as a type II transmembrane protein, cleaved to its soluble form, and signals as a trimerized molecule.16 Generally, TWEAK signaling occurs through its binding to Fn14, a type I transmembrane receptor belonging to the TNF receptor super family.16 However, there are also reports that TWEAK and Fn14 may function independently.17–19 We have previously reported that TWEAK inhibits the differentiation of cultured C2C12 or primary myoblasts into multinucleated myotubes.20 Furthermore, addition of TWEAK to cultured myotubes or its chronic administration in mice causes dramatic loss in skeletal muscle mass.21 More importantly, we have recently demonstrated that TWEAK-Fn14 dyad hasten the loss of skeletal muscle mass and function in response to denervation.22 While insufficient myofiber regeneration contributes significantly to skeletal muscle wasting, the role and the mechanisms of action of TWEAK during muscle regeneration in vivo remain unknown.
Skeletal muscle regeneration is governed by coordinated activation of multiple cell signaling pathways. Activation of extracellular signal-regulated kinase and nuclear factor κB (NF-κB) pathways generally stimulate cell proliferation and coordinately inhibit differentiation, in part by inactivation of MyoD.23–25 In contrast, the activation of p38 mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase/Akt kinase, and NFATc pathways have been found to promote the regeneration and growth of myofibers after injury.26–30 While TWEAK has been found to modulate the activity of many of these signaling pathways in cultured myoblasts and myotubes,20,21,31,32 how TWEAK regulates the activation of various pro-myogenic and anti-myogenic signaling pathways during myofiber regeneration in vivo is not yet known.
In this study using TWEAK-knockout (TWEAK-KO) and skeletal muscle specific TWEAK-transgenic (TWEAK-Tg) mice, we have investigated the role and the mechanisms by which TWEAK regulates skeletal muscle regeneration. Our results demonstrate that skeletal muscle regeneration is significantly improved in TWEAK-KO mice and reduced in TWEAK-Tg mice after cardiotoxin (CTX)-mediated injury. Furthermore, our study suggests that TWEAK inhibits myofiber regeneration by stimulating NF-κB transcription factor and exacerbating inflammatory response without affecting the activation of either p38 MAPK or Akt pathway.
Materials and Methods
Animals
Generation of transgenic mice expressing full-length TWEAK cDNA under the control of muscle creatine kinase promoter has been described previously.21 Because TWEAK-Tg mice were generated in B6D2F1 background, these mice were crossed with C57BL/6 mice for 7 to 8 generations before using for this study. TWEAK-KO mice were kindly provided by Dr. Avi Ashkenazi.33 All of the mice were in the C57BL/6 background and their genotype was determined by PCR from tail DNA. At the age of 8 weeks, 100 μl of 10 μmol/L cardiotoxin (CTX, CalBiochem) dissolved in PBS was injected into the tibial anterior (TA) muscle to induce necrotic injury. At various time points, TA muscle was collected from euthanized mice for biochemical and histology studies. Experimental protocols with mice were approved in advance by the Institutional Animal Care and Use Committee at University of Louisville.
Primary Myoblast Isolation and Fusion Assays
Satellite cells were isolated from hind limb of 8-week-old mice using a method as described.34,35 Briefly, mice were sacrificed and TA, gastrocnemius, and extensor digitorum longus muscles were isolated. Excess connective tissues and fat were cleaned in sterile PBS followed by mincing of skeletal muscle in Dulbecco's Modified Eagle's Medium and enzymatic dissociation with 0.1% pronase. The digested slurry was spun, pelleted, and triturated several times and then passed through 100 μmol/L vacuum filter (Steriflip, Millipore). The filtrate was spun at 1000 × g and resuspended in growth medium containing basic fibroblast growth factor (5 ng/ml). Cells were first re-fed after 3 days of initial plating. During first few passages cells were also enriched by pre-plating. After three passages, the medium of the cells was replaced with differentiation medium (2% horse serum in Dulbecco's Modified Eagle's Medium) and after 72 hours the cultures were stained for myosin heavy chain (using MF20 antibody) and 4,6-diamidino-2-phenylindole, as described.20,31 We used fusion index as a criterion to measure differentiation of cultured myoblasts. The fusion index calculated was a percentage ratio between the total number of nuclei in the myotubes versus the sum of nuclei in the myotubes and myoblasts.
Indirect Immunofluorescence and Histomorphometric Assays
All experiments involving the evaluation of TWEAK-Tg or TWEAK-KO and control were conducted on at least six mice per group. Cardiotoxin (CTX) or PBS alone injected TA muscle were removed, frozen in isopentane cooled in liquid nitrogen and sectioned in a microtome cryostat. For the assessment of tissue morphology or visualization of fibrosis, 10-μm-thick transverse sections of muscles were stained respectively with the H&E and examined under Nikon Eclipse TE 2000-U microscope (Nikon). Fiber cross-sectional area was analyzed in H&E-stained TA muscle sections using Nikon NIS Elements BR 3.00 software (Nikon). For each muscle, the distribution of fiber cross-sectional area (CSA) was calculated by analyzing 200 to 250 myofibers as described.21,22 Amount of fibrosis in TA muscle sections was determined using Sirius red staining kit following a protocol suggested by manufacturer (American Master Tech).
For immunohistochemistry study, the sections were blocked in 1% bovine serum albumin in PBS for 1 hour, and incubated with anti-embryonic myosin heavy chain (E-MyHC, 1:50 dilution, Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA) in blocking solution at 4°C overnight under humidified conditions. The sections were washed briefly with PBS before incubation with Alexa Fluor 488-conjugated secondary antibody (1:3000, Invitrogen) for 1 hour at room temperature and then washed three times for 30 minutes with PBS. The slides were mounted using fluorescence medium (Vector Laboratories) and visualized at room temperature on Nikon Eclipse TE 2000-U microscope (Nikon), a digital camera (Nikon Digital Sight DS-Fi1), and Nikon NIS Elements BR 3.00 software (Nikon). Image levels were equally adjusted using Abode Photoshop CS2 software (Adobe).
RNA Isolation and Quantitative Real-Time PCR
RNA isolation and quantitative real-time (QRT)-PCR were performed using a method as previously described.20,22 Briefly, RNA was extracted from homogenized TA muscle using TRIzol reagent (Invitrogen) and an RNeasy Mini Kit (Qiagen Inc., Valencia, CA) according to the manufacturer's protocol. The quantification of mRNA expression was carried out using the SYBR Green dye method on 7300 Sequence Detection system (Applied Biosystems, Foster City, CA). Purified RNA (1 μg) was used to synthesize first strand cDNA by reverse transcription system using Ambion's oligo(dT) primer and Qiagen's Omniscript reverse transcriptase kit. The first strand cDNA reaction (0.5 μl) was subjected to real-time PCR amplification using gene-specific primers. The sequences of primers for TWEAK, Fn14, myogenin, TNF-α, IL-β, IL-6, matrix metalloproteinase (MMP)-9, collagen I-a2, chemokine (C-C motif) ligand (CCL)-2, β-actin, and glyceraldehyde-3-phosphate dehydrogenase have been described in our recent publications.20,22,32
Approximately 25 μl of reaction volume was used for the real-time PCR assay that consisted of 2× (12.5 μl) Brilliant SYBR Green QPCR Master mix (Applied Biosystem), 400 nmol/L of primers (0.5 μl each from the stock), 11 μl of water, and 0.5 μl of template. The thermal conditions consisted of an initial denaturation at 95°C for 10 minutes followed by 40 cycles of denaturation at 95°C for 15 s, annealing and extension at 60°C for 1 minute, and, for a final step, a melting curve of 95°C for 15 s, 60°C for 15 s, and 95°C for 15 s. All reactions were carried out in duplicate to reduce variation. Data normalization was accomplished using the endogenous control (glyceraldehyde-3-phosphate dehydrogenase or β-actin), and the normalized values were subjected to a 2−ΔΔCt formula to calculate the fold change between the control and experimental groups.
Western Blot
Quantitative estimation of specific protein was done by Western blot using a method as previously described.21,36,37 TA muscle were washed with PBS and homogenized in lysis buffer A [50 mmol/L Tris-Cl (pH 8.0), 200 mmol/L NaCl, 50 mmol/L NaF, 1 mmol/L dithiothreitol, 1 mmol/L sodium orthovanadate, 0.3% IGEPAL, and protease inhibitors]. Approximately, 100 μg protein was resolved on each lane on 10% SDS-polyacrylamide gel electrophoresis, electrotransferred onto nitrocellulose membrane, and probed using anti-E-MyHC (1:100, Developmental Studies Hybridoma Bank), anti-Fn14 (1:1000, Cell Signaling), anti-phospho-Akt (1:500, Cell Signaling, Inc), anti-total Akt (1:1000, Cell Signaling, Inc), anti-phospho p38 (1:500, Santa Cruz), anti-total p38 (1:1000, Cell Signaling, Inc.), and anti-α-tubulin (1:2000, Cell Signaling, Inc.) and detected by chemiluminescence.
Electrophoretic Mobility Shift Assay
NF-κB activation was analyzed by electrophoretic mobility shift assay (EMSA). Detailed protocol for preparation of nuclear and cytoplasm extracts from skeletal muscle tissues and cultured myotubes and EMSA has been described in detail in our previous publications.20,37,38 In brief, 20 μg of nuclear extracts prepared from control or CTX-injected TA muscle or from cultured primary myotubes were incubated with 16 fmol of 32P end-labeled NF-κB consensus oligonucleotide (Promega) at 37°C for 30 minutes, and the DNA-protein complex was resolved on a 7.5% native polyacrylamide gel. The radioactive bands from the dried gel were visualized and quantified by PhosphorImager (GE Health Care) using ImageQuant TL software.
Statistical Analyses
Results are expressed as mean ± SD. Statistical analyses used Student's t-test to compare quantitative data populations with normal distribution and equal variance. A value of P < 0.05 was considered statistically significant unless otherwise specified.
Results
Expression Levels of TWEAK and Fn14 Are Significantly Increased in Regenerating Muscle Fibers in Vivo
Cardiotoxin (CTX) is a snake venom39 that selectively injures myofibers but leaves nerves, blood vessels, and satellite cells morphologically intact.40 CTX is one of the most widely used agents to induce skeletal muscle injury in mice. Before determining the role of TWEAK in skeletal muscle regeneration in vivo, we first investigated how the expression of TWEAK and its receptor Fn14 is affected in skeletal muscle of mice in response to injury. Left side TA muscle of 8-week-old wild-type C57BL6 mice was injected with CTX in PBS, whereas contralateral right side muscle was injected with PBS alone. After 5 days, the mice were euthanized and TA muscle isolated was used to study the expression of TWEAK and Fn14 by QRT-PCR. As shown in Figure 1A, the mRNA levels of both TWEAK and Fn14 were significantly increased in CTX injected TA muscle compared to contralateral control muscle injected with PBS alone. These data are consistent with a previously published report demonstrating increased expression of TWEAK and Fn14 after CTX-mediated skeletal muscle injury.41
Figure 1.
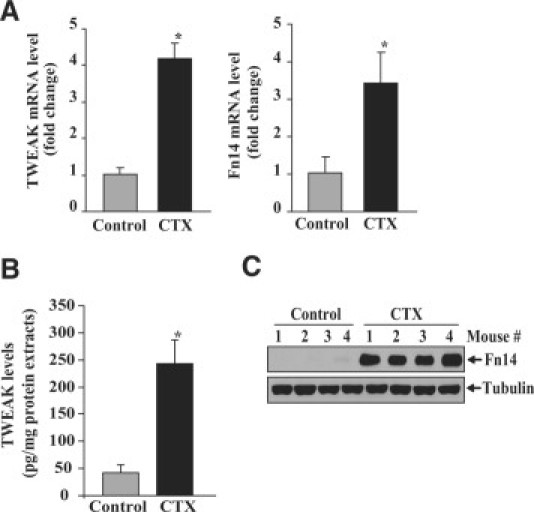
Expression of TWEAK and Fn14 in regenerating skeletal muscle. A: TA muscle of mice was injected with cardiotoxin (CTX); five days later, mRNA levels of TWEAK and Fn14 were measured by QRT-PCR. Data show a significant increase in the transcript levels of TWEAK and Fn14 in CTX-injected TA muscle (n = 4 in each group). B: Quantification using enzyme-linked immunosorbent assay showed a significant increase in protein levels of TWEAK in CTX-injected TA muscle compared with contralateral control TA muscle (n = 4). C: Immunoblotting with anti-Fn14 showed increased protein levels of Fn14 in CTX-injected TA muscle compared with contralateral control TA muscle injected with PBS alone. *P < 0.05, values significantly different from contralateral PBS-injected TA muscle.
We also measured the protein levels of TWEAK in TA muscle extracts using a commercially available enzyme-linked immunosorbent assay kit (R&D Systems). The protein levels of TWEAK were significantly increased in CTX-injected TA muscle compared to those injected with PBS alone (Figure 1B). Furthermore, Western blot analysis of TA muscle extracts showed increased levels of Fn14 protein in CTX-injected TA muscle (Figure 1C). Collectively, these results suggest that both the mRNA and protein levels of TWEAK and Fn14 are significantly increased in regenerating skeletal muscle.
Genetic Ablation of TWEAK Augments Whereas Overexpression of TWEAK Inhibits Myofiber Regeneration
We used TWEAK-KO mice to study the effects of genetic ablation of TWEAK on skeletal muscle regeneration. A previous report has shown that TWEAK-KO mice do not show any overt phenotype; however, a significant increase in the number of natural killer cells in secondary lymphoid organs of TWEAK-KO mice compared with wild-type mice was observed.33 Our recent analysis of skeletal muscle of TWEAK-KO mice further suggested that there is no difference in skeletal muscle structure between TWEAK-KO and wild-type mice at the age of 3 months.22 We also used TWEAK-Tg mice which express 4- to 6-fold higher levels of TWEAK in skeletal muscle compared to littermate wild-type mice.22 We have recently published that TWEAK-Tg mice do not show any muscle phenotype until they were 5 to 6 months old. However, around 6 months of age TWEAK-Tg mice show significant muscle atrophy and fibrosis in skeletal muscle.22 Since 8-week-old TWEAK-KO or TWEAK-Tg mice did not demonstrate any skeletal muscle phenotype and mice at this age are fully developed, we used 8-week-old mice to evaluate the in vivo role of TWEAK in skeletal muscle regeneration. The TA muscle of TWEAK-KO, and TWEAK-Tg mice and wild-type were injected with CTX and 5, 10, and 21 days post-CTX injection, the muscles were isolated, and analyzed after staining with H&E. Although no noticeable difference was observed at fifth day post CTX injection, differences in fiber size became evident at 10 and 21 days after CTX injection in both TWEAK-KO and TWEAK-Tg compared to wild-type mice. Regenerating myofibers in TWEAK-KO mice at 10 and 21 days appeared larger in size compared to wild-type mice whereas size of regenerating myofibers was considerable reduced in TWEAK-Tg mice compared to corresponding littermate wild-type mice (Figure 2).
Figure 2.
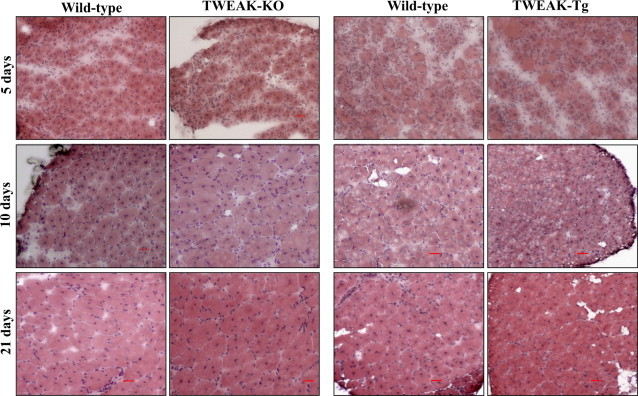
Histological analyses of regenerating TA muscle of TWEAK-KO and TWEAK-Tg mice. TA muscle of TWEAK-KO and TWEAK-Tg mice and the corresponding control mice were given intramuscular injections of CTX followed by isolation of TA muscle at different time points and H&E staining. Representative images of H&E-stained sections demonstrate increased fiber diameter in TA muscle of TWEAK-KO and its reduction in TWEAK-Tg mice compared with the corresponding control mice (n = 6 in each group at indicated time point). Scale bar = 50 μm.
Quantitative estimation of myofiber diameter 10 days post CTX injection showed that fiber CSA was considerably higher in regenerating TA muscle of TWEAK-KO mice compared to age-matched wild-type mice (Figure 3A). In contrast, mean fiber CSA was found to be considerably reduced in TWEAK-Tg mice compared to littermate wild-type mice (Figure 3B). Interestingly, the number of myofibers having more than one nucleus was significantly higher in TWEAK-KO mice compared to wild-type mice (Figure 3C). Conversely, the count of such myofibers was found to be significantly reduced in TWEAK-Tg mice compared to littermate wild-type mice (Figure 3C) suggesting that TWEAK may interfere with the fusion of muscle progenitor cells (ie, satellite cells) with regenerating myofibers.
Figure 3.
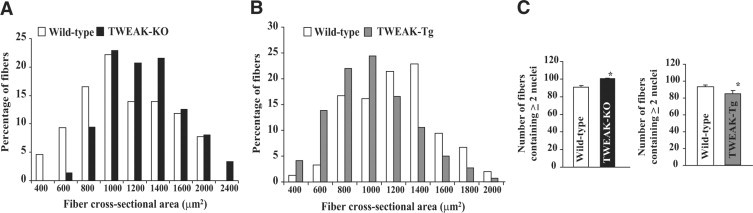
Quantification of fiber cross section area (CSA) in regenerating TA muscle of TWEAK-KO and TWEAK-Tg mice. H&E-stained TA muscle sections from TWEAK-KO and wild-type and TWEAK-Tg and littermate wild-type mice 10 days post-CTX injections were used to calculate the fiber CSA. A: Data show that fiber CSA was increased in TWEAK-KO (n = 6) compared with wild-type mice (n = 6). B: A reduction in fiber CSA was noticed in TWEAK-Tg mice (n = 6) compared with littermate wild-type mice (n = 6). C: Analysis of H&E-stained TA muscle sections 10 days post-CTX injection showed significantly increased number of myofibers containing more than two central nuclei in TWEAK-KO mice compared with wild-type mice. This count was significantly reduced in TWEAK-Tg mice compared with littermate wild-type mice. n = 6 in each group. *P < 0.05, values significantly different from wild-type mice.
TWEAK Inhibits the Levels of E-MyHC in Regenerating TA Muscle
To further investigate the role of TWEAK in skeletal muscle regeneration, we measured the protein levels of E-MyHC in regenerating TA muscle of TWEAK-KO and TWEAK-Tg mice after CTX injection. Staining of TA muscle sections with anti-EMyHC and subsequent quantitative estimation of fiber CSA showed that the size of the E-MyHC-positive myofibers was considerably higher in TWEAK-KO mice compared to wild-type mice. On the other hand, the size of the E-MyHC-positive myofibers was found to be reduced in TWEAK-Tg compared to littermate wild-type mice (Figure 4A) indicating that TWEAK inhibits the growth of regenerating myofibers. We also performed Western blot to quantify the amount of E-MyHC in regenerating TA muscle of TWEAK-KO and TWEAK-Tg mice. Our results showed that the levels of E-MyHC protein were considerably increased in TWEAK-KO and reduced in TWEAK-Tg mice compared to their corresponding control mice (Figures 4, B and C).
Figure 4.
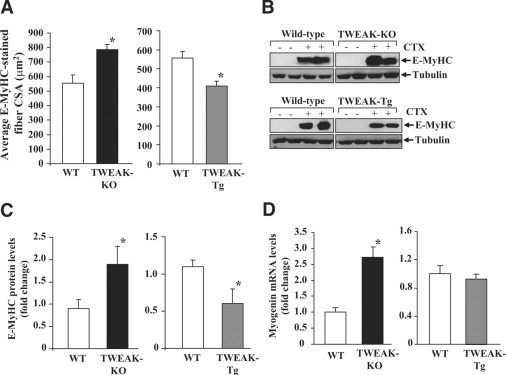
Expression of embryonic myosin heavy chain (E-MyHC) and myogenin in regenerating TA muscle of TWEAK-KO and TWEAK-Tg mice. A: TA muscle of TWEAK-KO, TWEAK-Tg, and the corresponding control mice were given an intramuscular injection of CTX. After 5 days, muscle sections were prepared and immunostained with anti-E-MyHC and average fiber cross-sectional area of E-MyHC-positive fibers was measured. *P < 0.05, values significantly different from wild-type mice. B: Representative immunoblots show protein levels of E-MyHC in CTX-injected TA muscle of TWEAK-KO and its reduced levels in TWEAK-Tg as compared with the corresponding control mice. C: Quantification of Western blot (normalized for tubulin levels) shows a significant increase in protein levels of E-MyHC in CTX-injected TA muscle of TWEAK-KO compared with wild-type mice. The levels of E-MyHC were significantly reduced in CTX-injected TA muscle of TWEAK-Tg compared with littermate wild-type mice. n = 4 in each group. D: Transcript levels of myogenin in CTX-injected TA muscle of TWEAK-KO and TWEAK-Tg mice. n = 3 in each group. *P < 0.01, values significantly different from wild-type mice.
Newly formed myofibers express several myogenic regulatory factors including myogenin. We next sought to determine how the expression of myogenin was affected in regenerating TA muscle of TWEAK-Tg and TWEAK-KO mice. In comparison to control mice, the mRNA level of myogenin was found to be significantly increased in TWEAK-KO mice. The transcript levels of myogenin was somewhat reduced in TWEAK-Tg mice, however, it was not significantly different from littermate wild-type mice (Figure 4D).
TWEAK Stimulates the Expression of Inflammation-Related Molecules in Regenerating Skeletal Muscle
To understand the mechanisms by which TWEAK inhibits skeletal muscle regeneration, we studied the expressions of several inflammatory molecules which are known to inhibit myogenic differentiation. We have recently reported that the transcript levels of inflammation-related cytokines (eg, TNF, IL-1β, and IL-6) are comparable in 3-month-old naive TA muscle of control, TWEAK-Tg, and TWEAK-KO mice.22 Here, we investigated whether the presence or absence of TWEAK in regenerating myofibers affects the expression of inflammatory molecules. TA muscles were isolated from TWEAK-KO, TWEAK-Tg, and their corresponding control mice 5 days post-CTX injection and processed to study the mRNA levels of inflammatory cytokines (eg, TNF-α, IL-1β, IL-6), chemokine (CCL2), and matrix-degrading enzyme (eg, MMP-9) by QRT-PCR assays. Interestingly, the mRNA levels of TNF-α, IL-6, and CCL-2 (but not IL-1β and MMP-9) were significantly reduced in CTX-injected TA muscle of TWEAK-KO mice compared to CTX-injected TA muscle of wild-type mice (Figure 5A). Furthermore, the mRNA levels of TNF-α, IL-1β, IL-6, CCL-2, and MMP-9 were found to be significantly increased in regenerating TA muscle of TWEAK-Tg compared to that of control mice (Figure 5B). These data suggest that TWEAK plays an important role in exacerbating the expression of inflammatory molecules in regenerating myofibers after injury.
Figure 5.
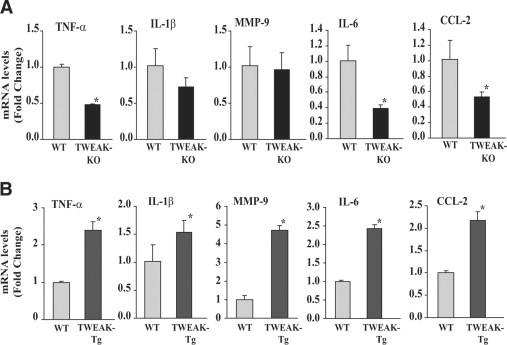
Expression of pro-inflammatory molecules in regenerating TA muscle of TWEAK-Tg and TWEAK-KO mice. A: Fold difference in mRNA levels of TNF-α, IL-1β, MMP-9, IL-6, and CCL-2 in TA muscle of TWEAK-KO and wild-type mice measured five days after CTX injection by QRT-PCR. n = 4 in each group. B: Expression levels of TNF-α, IL-1β, MMP-9, IL-6, and CCL-2 in CTX-injected TA muscle of TWEAK-Tg and littermate wild-type five days post CTX injections. n = 4 in each group. *P < 0.05, values significantly different form wild-type mice. WT, wild-type.
TWEAK Augments Interstitial Fibrosis in Regenerating Myofibers
Fibrosis is an important pathological consequence which can limit the regeneration of myofibers after injury.42 Since TWEAK augmented the expression of a number of inflammatory molecules which are known to induce fibrosis in various organs, we next sought to determine whether the level of fibrosis is affected in regenerating TA muscle of TWEAK-KO, TWEAK-Tg, and corresponding control mice. After 10 days of intramuscular injections of CTX, TA muscle were isolated and the muscle sections prepared were stained with Sirius red dye. As shown in Figure 6A, the level of fibrosis was considerably lower in TWEAK-KO mice compared to that of wild-type mice. In contrast, the extent of fibrosis was found to be increased in CTX-injected TA muscle of TWEAK-Tg mice compared to littermate wild-type mice (Figure 6A). We also quantified the extent of fibrosis by measuring the mRNA levels of collagen I-a2, one of the major collagens in skeletal muscle,22,43 by QRT-PCR technique. Although the levels of collagen I-a2 were comparable in CTX-injected TA muscle of TWEAK-KO and wild-type mice, a significantly increase in mRNA levels of collagen I-a2 was observed in CTX-injected TA muscle of TWEAK-Tg compared to control mice (Figure 6B).
Figure 6.
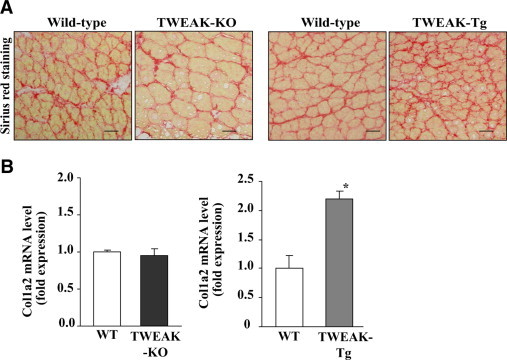
Fibrosis in regenerating TA muscle of TWEAK-KO and TWEAK-Tg mice. A: Muscle sections prepared from TA muscle ten days post CTX injection were stained with Sirius red dye. Representative photomicrographs show that the level of fibrosis is reduced in regenerating TA muscle of TWEAK-KO mice and increased in TWEAK-Tg mice compared with CTX-injected TA muscle of the corresponding wild-type mice. B: Transcript levels of collagen I measured by QRT-PCR in TA muscle ten days post CTX injection were comparable in wild-type and TWEAK-KO mice but were significantly increased in TWEAK-Tg mice compared with littermate wild-type mice. *P < 0.05, values significantly different form wild-type mice. WT, wild-type.
TWEAK Causes the Activation of NF-κB Transcription Factor in Regenerating Skeletal Muscle of Mice
To further understand the mechanisms by which TWEAK inhibits skeletal muscle regeneration and causes inflammation and fibrosis, we investigated the activation of Akt, p38 MAPK and NF-κB in TA muscle of control, TWEAK-Tg and TWEAK-KO mice after CTX injections. The activation of Akt and p38 MAPK was studied by performing Western blots using antibodies against respective phosphorylated and total protein forms and the DNA-binding activity of NF-κB in nuclear extracts was measured by EMSA. The levels of activation of Akt, p38 MAPK, and NF-κB were significantly increased in CTX-injected TA muscle compared to those injected with PBS alone. However, there was no noticeable difference in the level of phosphorylation of Akt or p38 MAPK in regenerating TA muscle of control, TWEAK-Tg, or TWEAK-KO mice (Figure 7A). Interestingly, the activation of NF-κB was found to be increased in regenerating TA muscle of TWEAK-Tg mice compared to control mice (Figure 7B). In contrast, the activation of NF-κB was dramatically reduced in TWEAK-KO mice compared control mice (Figure 7B). Taken together, these results suggest that TWEAK specifically modulates the activation of NF-κB in regenerating myofibers of mice.
Figure 7.
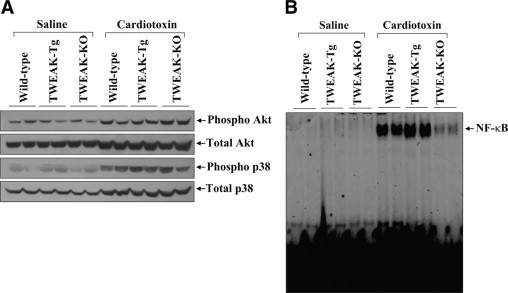
Activation of Akt, p38 MAPK, and NF-κB in regenerating TA muscle of control, TWEAK-Tg, and TWEAK-KO mice. A: Representative immunoblots show no apparent differences in levels of phospho-Akt and phospho p38MAPK in regenerating TA muscle of control, TWEAK-Tg, and TWEAK-KO mice. B: DNA-binding activation of NF-κB measured by EMSA was found to be increased in regenerating TA muscle of TWEAK-Tg mice and reduced in TWEAK-KO mice as compared with control mice.
Genetic Deletion of TWEAK Enhances the Differentiation of Muscle Progenitor Cells into Myotubes in Vitro
We have previous shown that TWEAK receptor Fn14 is essential for the differentiation of myoblasts into myotubes.31 Similarly, defective myotube formation has been reported in satellite cell cultures from Fn14-knockout mice.41 We investigated whether TWEAK plays any role in the differentiation of cultured muscle progenitor cells in vitro. Primary myoblasts from hind-limb muscle of control, TWEAK-Tg, and TWEAK-KO mice were isolated, cultured, and differentiated into myotubes by incubation in differentiation medium for 72 hours. Interestingly, the myotube formation was found to be significantly reduced in cultures prepared from TWEAK-Tg and increased in TWEAK-KO compared to control mice (Figure 8A). Both fusion index and diameter of myotubes were significantly low in TWEAK-Tg mice and high in TWEAK-KO mice (Figure 8, B and C). It is important to note that in TWEAK-Tg mice, TWEAK is expressed using muscle creatine kinase promoter which is not active in undifferentiated myoblasts. Therefore, a reduction in fusion index in TWEAK-Tg cultures suggests that the initial production of TWEAK by differentiated myotubes inhibits the fusion of additional myoblasts to myotubes and hence their growth. These results also suggest that TWEAK by itself is sufficient to inhibit the differentiation of primary myoblasts into myotubes. We also measured the activation of NF-κB in cultured myotubes prepared from control, TWEAK-Tg, and TWEAK-KO mice. As shown in Figure 8D, the DNA-binding activity of NF-κB was noticeable higher in primary cultures from TWEAK-Tg mice compared to control or TWEAK-KO mice after incubation in differentiation medium for 72 hours. These results further indicate that TWEAK inhibits myogenic differentiation and muscle growth potentially through stimulating the activity of NF-κB transcription factor.
Figure 8.
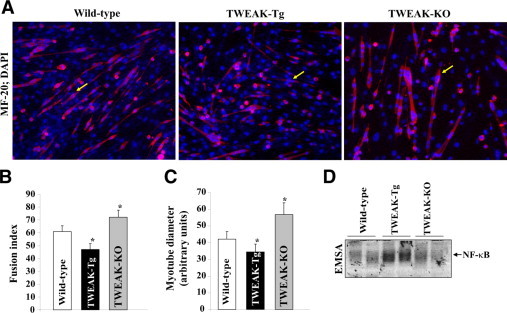
Differentiation of satellite cells prepared from control, TWEAK-Tg, and TWEAK-KO mice. A: Representative MF-20 antibody (for MyHC) stained photomicrographs of primary myoblast cultures after incubation in differentiation medium for 72 hours. Arrow points to a typical and representative myotube in each culture. Analyses of these cultures showed that the (B) fusion index and (C) myotube diameter were significantly reduced in TWEAK-Tg and significantly increased in TWEAK-KO, as compared with wild-type mice. *P < 0.05, values significantly different from that of wild-type mice. D: DNA-binding activity of NF-κB transcription factor was considerably higher in TWEAK-Tg cultures compared with control mice. EMSA, electrophoretic mobility shift assay.
Discussion
Skeletal muscle regeneration is multistep process that involves the participation of a number of autocrine and paracrine factors.1–3 The inflammatory response which starts within hours of injury and lasts up to 7 days involves accumulation of inflammatory cells such as neutrophils and macrophages which help removing tissue debris through phagocytosis in injured skeletal muscle.6 These infiltrating phagocytes in injured skeletal muscle are also known to produce a number of cytokines and chemokines which can affect the proliferation and differentiation of satellite cells and may regulate extracellular matrix remodeling.6 While it is now well-established that the recruitment of macrophages and neutrophils is critical for efficient regeneration of injured muscle, the role of various pro-inflammatory and anti-inflammatory cytokines in skeletal muscle regeneration is less clear. While cell cultures studies have suggested that inflammatory cytokines can modulate both the proliferation and differentiation of myogenic cells, the direct correlation between in vitro and in vivo studies is still lacking. Furthermore, there could also be some redundancy among cytokines regarding their role in skeletal muscle regeneration. This is partly elucidated by studies with TNF. Although TNF strongly affects the survival, proliferation, and differentiation of cultured myoblasts,9,10 skeletal muscle regeneration was normal in TNF-null mice.44
TWEAK is a multifunctional cytokine that is produced by a number of cell types including macrophages and skeletal muscle.16,22,45 In addition to inhibiting myogenic differentiation,20 our recent studies have provided genetic evidence that elevated levels of TWEAK cause muscle atrophy in vivo21 and the TWEAK-Fn14 system is a major mediator of muscle loss in disuse conditions such as denervation.22 Our present study demonstrates that the expression of both TWEAK and Fn14 are increased in skeletal muscle and TWEAK is sufficient to inhibit the regeneration and growth of skeletal muscle in vivo. Deletion of TWEAK in mice not only improves the expression of E-MyHC and myogenin (the markers of muscle regeneration), it also increases the fiber CSA of regenerating myofibers (Figures 2, 3, and 4). This inference is well supported by studies in TWEAK-Tg mice where a 4- to 6-fold increase in TWEAK levels was sufficient to reduce the levels of E-MyHC and fiber CSA in regenerating myofibers (Figures 2, 3, 4). Furthermore, our cell culture experiments demonstrating increased differentiation (fusion index) and myotube diameter in primary muscle cell cultures prepared from TWEAK-KO mice and their reduction in primary muscle cultures from TWEAK-Tg mice provide additional evidence that TWEAK inhibits myogenic differentiation and myotube growth (Figure 8, A, B, and C).
It was interesting to note that genetic deletion or overexpression of TWEAK did not affect the cellular infiltrates in the injured skeletal muscle (Figure 2). Our QRT-PCR assays did not show any significant difference in the expression levels of markers for either satellite cells (ie, Pax-7) or macrophages (ie, Mac1 and CD68) at different time points after injection of CTX in skeletal muscle (not depicted) suggesting that TWEAK may not be directly involved in the proliferation of muscle progenitor or inflammatory cells in regenerating tissues. Rather, elevated levels of TWEAK in regenerating muscle exaggerate the expression of inflammatory molecules as supported by our results that the mRNA levels of several pro-inflammatory cytokines such as TNF-α, IL-1β, IL-6, and CCL2 were significantly reduced in TWEAK-KO mice (Figure 5A) and the expression of all these molecules and a matrix-degrading protease MMP-9 was increased in TWEAK-Tg mice compared to wild-type mice (Figure 5B). Furthermore, increased fibrosis in regenerating skeletal muscle of TWEAK-Tg mice and its reduction in TWEAK-KO mice further support our inference that TWEAK causes sustained inflammation leading to fibrosis (Figure 6). Although it is not yet clear whether TWEAK independently augments the fibrosis in regenerating muscle tissues or it does in association with other inflammatory molecules, two independent studies have recently demonstrated that the elevated levels of TWEAK augment fibrosis in skeletal and cardiac muscle of mice.22,46
Our results also demonstrate that fiber CSA of regenerating myofibers was higher in TWEAK-KO mice and was reduced in TWEAK-Tg mice (Figure 3, A and B) indicating that TWEAK affects the growth of regenerating myofibers in vivo. Although the exact mechanisms by which TWEAK regulates the size of regenerating myofibers is not clear, two possible scenarios can be discussed. First, it is possible that TWEAK by itself or in association with other inflammatory cytokines, blocks the fusion of satellite cells with regenerating myofibers resulting in reduced differentiation and growth of myofibers.9,20 On the other hand, TWEAK may also directly act on regenerating myofibers leading to their reduced mass. Although TWEAK is constitutively expressed in skeletal muscle, its receptor is not expressed in adult muscle.22 We have recently reported that catabolic conditions such as denervation and immobilization up-regulates the expression of Fn14 in skeletal muscle and subsequent interaction of TWEAK with Fn14 leads to the activation of ubiquitin-proteasome system resulting in atrophy.22 Since CTX-mediated injury increases the expression of both TWEAK and Fn14 in injured skeletal muscle (Figure 1), the interaction of TWEAK to its receptor may lead to the activation of proteolytic pathways resulting in reduced fiber size in regenerating myofibers. Furthermore, increased fibrosis could also potentially block the migration of satellite cells and hence reduced regeneration. A similar study has previously shown that genetic deletion of myostatin in mice improves skeletal muscle regeneration by reducing the extent of fibrosis in injured muscles.42
Several recent reports have suggested that NF-κB transcription factor plays a critical role in regulation of skeletal muscle mass and regeneration.47 Activation of NF-κB causes severe muscle wasting, in part, through the increased expression of muscle-specific E3 ubiquitin ligase MuRF1.48,49 Furthermore, activated NF-κB inhibits the differentiation of cultured myoblasts into myotubes11,50,51 and blocking NF-κB rescues myogenic differentiation in response to pro-inflammatory cytokines including TWEAK.9,10,21 More recently, Mourkioti et al52 have shown that depletion of IkappaB kinase β, an upstream activator of NF-κB, improves the regeneration and reduces fibrosis in skeletal muscle after CTX-mediated injury. Our results demonstrate that the level of activation of NF-κB is increased in TWEAK-Tg mice and it is inhibited in TWEAK-KO mice (Figure 7B). Similar increased activation of NF-κB was found in primary muscle cultures from TWEAK-Tg mice (Figure 8D). Inhibition of NF-κB in regenerating muscle of TWEAK-KO mice is consistent with recently published reports from our group and others that TWEAK is a potent activator of NF-κB in skeletal muscle and several other cell types including immune cells which infiltrate skeletal muscle tissue after injury.16,20–22,33 Since NF-κB is a prominent inflammation-related transcription factor which induces the activation of a number of inflammatory cytokines, chemokines, cell adhesion molecules, and matrix-degrading enzymes, inhibition of NF-κB might help limiting the level of inflammation in regenerating skeletal muscle.52,53 Indeed, our results show that the expression of several inflammatory molecules which contain consensus NF-κB binding sequence in their promoter/enhancer regions54 is reduced in skeletal muscle of TWEAK-KO mice after CTX-mediated injury (Figure 5A). Coincidently, these results are also in agreement with our recently published report demonstrating that TWEAK causes the activation of NF-κB in denervated skeletal muscle leading to atrophy.22
It is also noteworthy that TWEAK did not affect the activation of Akt or p38 MAPK signaling pathways in regenerating skeletal muscle after CTX-mediated injury in vivo (Figure 7A). Both Akt and p38 MAPK are known to stimulate myogenesis and we have previously shown that TWEAK affects the activation of both these kinases in cultured myoblasts and/or myotubes.21,31,32 However, it appears that the role of TWEAK in the activation of Akt and p38 MAPK is compensated by other factors and that TWEAK specifically modulates the activation of NF-κB in regenerating skeletal muscle.
While our study suggests that TWEAK inhibits skeletal muscle regeneration, previously Girgenrath et al41 have reported that Fn14-knockout mice exhibit delayed muscle regeneration compared to wild-type mice in response to CTX-mediated injury. Furthermore, primary myoblasts from Fn14-knockout mice demonstrated reduced proliferation and defective myotube formation.41 In contrast, our results from multiple experiments showed that primary myoblasts from TWEAK-KO mice differentiate better than control mice and the diameter of myotubes was larger than control mice (Figure 8, A and B). The reasons for differential effects of deletion of TWEAK and Fn14 in skeletal muscle regeneration and cultured myotubes are not yet clearly understood. However, similar to our results, the expression of some of the inflammatory molecules was found to be reduced in regenerating myofibers of Fn14-knockout mice as well.41 It is possible that Fn14 functions independent of TWEAK during muscle regeneration. This possibility is also supported by a previous report from us where we showed that the knockdown of Fn14 receptor using RNA interference technique inhibits the expression of myogenic regulatory factors and differentiation of C2C12 and primary myoblasts into myotubes independent of TWEAK.31 Indeed, there are also other published reports suggesting that in some cell types TWEAK may function independent of Fn14 receptor.17–19 Although more investigations are required to understand the differential effects of TWEAK and Fn14 in skeletal muscle regeneration, the results of the present study provide first genetic evidence that TWEAK is a negative regulator of skeletal muscle regeneration and growth in vivo. Blocking TWEAK activity may help improving skeletal muscle regeneration in response to normal life injuries, chronic diseases, and in various muscular disorders such as muscular dystrophy where insufficient muscle regeneration contributes significantly to skeletal muscle loss.
Acknowledgements
We are highly grateful to Dr. Avi Ashkenazi (Genentech South San Francisco, CA) for providing TWEAK-knockout mice.
Footnotes
Supported by a National Institute of Health grant (RO1 AG029623) and a Clinical and Translational Science Pilot Grant Basic Award from the University of Louisville to A.K.
None of the authors disclosed any relevant financial relationships.
References
- 1.Charge SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- 2.Dhawan J, Rando TA. Stem cells in postnatal myogenesis: molecular mechanisms of satellite cell quiescence, activation and replenishment. Trends Cell Biol. 2005;15:666–673. doi: 10.1016/j.tcb.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 3.Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Relaix F, Marcelle C. Muscle stem cells. Curr Opin Cell Biol. 2009;21:748–753. doi: 10.1016/j.ceb.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 5.Tedesco FS, Dellavalle A, Diaz-Manera J, Messina G, Cossu G. Repairing skeletal muscle: regenerative potential of skeletal muscle stem cells. J Clin Invest. 2010;120:11–19. doi: 10.1172/JCI40373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tidball JG, Villalta SA. Regulatory interactions between muscle and the immune system during muscle regeneration. Am J Physiol Regul Integr Comp Physiol. 2010 doi: 10.1152/ajpregu.00735.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horsley V, Jansen KM, Mills ST, Pavlath GK. IL-4 acts as a myoblast recruitment factor during mammalian muscle growth. Cell. 2003;113:483–494. doi: 10.1016/s0092-8674(03)00319-2. [DOI] [PubMed] [Google Scholar]
- 8.Cheng M, Nguyen MH, Fantuzzi G, Koh TJ. Endogenous interferon-gamma is required for efficient skeletal muscle regeneration. Am J Physiol Cell Physiol. 2008;294:C1183–C1191. doi: 10.1152/ajpcell.00568.2007. [DOI] [PubMed] [Google Scholar]
- 9.Langen RC, Schols AM, Kelders MC, Wouters EF, Janssen-Heininger YM. Inflammatory cytokines inhibit myogenic differentiation through activation of nuclear factor-kappaB. FASEB J. 2001;15:1169–1180. doi: 10.1096/fj.00-0463. [DOI] [PubMed] [Google Scholar]
- 10.Langen RC, Van Der Velden JL, Schols AM, Kelders MC, Wouters EF, Janssen-Heininger YM. Tumor necrosis factor-alpha inhibits myogenic differentiation through MyoD protein destabilization. FASEB J. 2004;18:227–237. doi: 10.1096/fj.03-0251com. [DOI] [PubMed] [Google Scholar]
- 11.Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS., Jr NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science. 2000;289:2363–2366. doi: 10.1126/science.289.5488.2363. [DOI] [PubMed] [Google Scholar]
- 12.Moresi V, Pristera A, Scicchitano BM, Molinaro M, Teodori L, Sassoon D, Adamo S, Coletti D. Tumor necrosis factor-alpha inhibition of skeletal muscle regeneration is mediated by a caspase-dependent stem cell response. Stem Cells. 2008;26:997–1008. doi: 10.1634/stemcells.2007-0493. [DOI] [PubMed] [Google Scholar]
- 13.Coletti D, Moresi V, Adamo S, Molinaro M, Sassoon D. Tumor necrosis factor-alpha gene transfer induces cachexia and inhibits muscle regeneration. Genesis. 2005;43:120–128. doi: 10.1002/gene.20160. [DOI] [PubMed] [Google Scholar]
- 14.Argiles JM, Busquets S, Toledo M, Lopez-Soriano FJ. The role of cytokines in cancer cachexia. Curr Opin Support Palliat Care. 2009;3:263–268. doi: 10.1097/SPC.0b013e3283311d09. [DOI] [PubMed] [Google Scholar]
- 15.Spate U, Schulze PC. Proinflammatory cytokines and skeletal muscle. Curr Opin Clin Nutr Metab Care. 2004;7:265–269. doi: 10.1097/00075197-200405000-00005. [DOI] [PubMed] [Google Scholar]
- 16.Winkles JA. The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat Rev Drug Discov. 2008;7:411–425. doi: 10.1038/nrd2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanabe K, Bonilla I, Winkles JA, Strittmatter SM. Fibroblast growth factor-inducible-14 is induced in axotomized neurons and promotes neurite outgrowth. J Neurosci. 2003;23:9675–9686. doi: 10.1523/JNEUROSCI.23-29-09675.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Polek TC, Talpaz M, Darnay BG, Spivak-Kroizman T. TWEAK mediates signal transduction and differentiation of RAW264.7 cells in the absence of Fn14/TweakR. Evidence for a second TWEAK receptor. J Biol Chem. 2003;278:32317–32323. doi: 10.1074/jbc.M302518200. [DOI] [PubMed] [Google Scholar]
- 19.Bover LC, Cardo-Vila M, Kuniyasu A, Sun J, Rangel R, Takeya M, Aggarwal BB, Arap W, Pasqualini R. A previously unrecognized protein-protein interaction between TWEAK and CD163: potential biological implications. J Immunol. 2007;178:8183–8194. doi: 10.4049/jimmunol.178.12.8183. [DOI] [PubMed] [Google Scholar]
- 20.Dogra C, Changotra H, Mohan S, Kumar A. Tumor necrosis factor-like weak inducer of apoptosis inhibits skeletal myogenesis through sustained activation of nuclear factor-kappaB and degradation of MyoD protein. J Biol Chem. 2006;281:10327–10336. doi: 10.1074/jbc.M511131200. [DOI] [PubMed] [Google Scholar]
- 21.Dogra C, Changotra H, Wedhas N, Qin X, Wergedal JE, Kumar A. TNF-related weak inducer of apoptosis (TWEAK) is a potent skeletal muscle-wasting cytokine. FASEB J. 2007;21:1857–1869. doi: 10.1096/fj.06-7537com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mittal A, Bhatnagar S, Kumar A, Lach-Trifilieff E, Wauters S, Li H, Makonchuk DY, Glass DJ, Kumar A. The TWEAK–Fn14 system is a critical regulator of denervation-induced skeletal muscle atrophy in mice. J Cell Biol. 2010;188:833–849. doi: 10.1083/jcb.200909117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS., Jr NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. 1999;19:5785–5799. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perry RL, Parker MH, Rudnicki MA. Activated MEK1 binds the nuclear MyoD transcriptional complex to repress transactivation. Mol Cell. 2001;8:291–301. doi: 10.1016/s1097-2765(01)00302-1. [DOI] [PubMed] [Google Scholar]
- 25.Perry RL, Rudnick MA. Molecular mechanisms regulating myogenic determination and differentiation. Front Biosci. 2000;5:D750–D767. doi: 10.2741/perry. [DOI] [PubMed] [Google Scholar]
- 26.Lluis F, Perdiguero E, Nebreda AR, Munoz-Canoves P. Regulation of skeletal muscle gene expression by p38 MAP kinases. Trends Cell Biol. 2006;16:36–44. doi: 10.1016/j.tcb.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Pavlath GK, Horsley V. Cell fusion in skeletal muscle—central role of NFATC2 in regulating muscle cell size. Cell Cycle. 2003;2:420–423. [PubMed] [Google Scholar]
- 28.Schertzer JD, Lynch GS. Comparative evaluation of IGF-I gene transfer and IGF-I protein administration for enhancing skeletal muscle regeneration after injury. Gene Ther. 2006;13:1657–1664. doi: 10.1038/sj.gt.3302817. [DOI] [PubMed] [Google Scholar]
- 29.Tureckova J, Wilson EM, Cappalonga JL, Rotwein P. Insulin-like growth factor-mediated muscle differentiation: collaboration between phosphatidylinositol 3-kinase-Akt-signaling pathways and myogenin. J Biol Chem. 2001;276:39264–39270. doi: 10.1074/jbc.M104991200. [DOI] [PubMed] [Google Scholar]
- 30.Chen SE, Jin B, Li YP. TNF-alpha regulates myogenesis and muscle regeneration by activating p38 MAPK. Am J Physiol Cell Physiol. 2007;292:C1660–C1671. doi: 10.1152/ajpcell.00486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dogra C, Hall SL, Wedhas N, Linkhart TA, Kumar A. Fibroblast growth factor inducible 14 (Fn14) is required for the expression of myogenic regulatory factors and differentiation of myoblasts into myotubes. Evidence for TWEAK-independent functions of Fn14 during myogenesis. J Biol Chem. 2007;282:15000–15010. doi: 10.1074/jbc.M608668200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H, Mittal A, Paul PK, Kumar M, Srivastava DS, Tyagi SC, Kumar A. Tumor necrosis factor-related weak inducer of apoptosis augments matrix metalloproteinase-9 production (MMP-9) in skeletal muscle through the activation of nuclear factor-kappa B-inducing kinase and p38 mitogen-activated protein kinase: a potential role of MMP-9 in myopathy. J Biol Chem. 2008;284:4439–4450. doi: 10.1074/jbc.M805546200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maecker H, Varfolomeev E, Kischkel F, Lawrence D, LeBlanc H, Lee W, Hurst S, Danilenko D, Li J, Filvaroff E, Yang B, Daniel D, Ashkenazi A. TWEAK attenuates the transition from innate to adaptive immunity. Cell. 2005;123:931–944. doi: 10.1016/j.cell.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 34.Apponi LH, Leung SW, Williams KR, Valentini SR, Corbett AH, Pavlath GK: Loss of nuclear poly(A)-binding protein 1 causes defects in myogenesis and mRNA biogenesis. Hum Mol Genet 19:1058–1065 [DOI] [PMC free article] [PubMed]
- 35.Rando TA, Blau HM. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J Cell Biol. 1994;125:1275–1287. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dogra C, Changotra H, Wergedal JE, Kumar A. Regulation of phosphatidylinositol 3-kinase (PI3K)/Akt and nuclear factor-kappa B signaling pathways in dystrophin-deficient skeletal muscle in response to mechanical stretch. J Cell Physiol. 2006;208:575–585. doi: 10.1002/jcp.20696. [DOI] [PubMed] [Google Scholar]
- 37.Li H, Mittal A, Makonchuk DY, Bhatnagar S, Kumar A. Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum Mol Genet. 2009;18:2584–2598. doi: 10.1093/hmg/ddp191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar A, Boriek AM. Mechanical stress activates the nuclear factor-kappaB pathway in skeletal muscle fibers: a possible role in Duchenne muscular dystrophy. FASEB J. 2003;17:386–396. doi: 10.1096/fj.02-0542com. [DOI] [PubMed] [Google Scholar]
- 39.Fletcher JE, Jiang MS. Possible mechanisms of action of cobra snake venom cardiotoxins and bee venom melittin. Toxicon. 1993;31:669–695. doi: 10.1016/0041-0101(93)90375-s. [DOI] [PubMed] [Google Scholar]
- 40.Couteaux R, Mira JC, d'Albis A. Regeneration of muscles after cardiotoxin injury. I. Cytological aspects. Biol Cell. 1988;62:171–182. [PubMed] [Google Scholar]
- 41.Girgenrath M, Weng S, Kostek CA, Browning B, Wang M, Brown SA, Winkles JA, Michaelson JS, Allaire N, Schneider P, Scott ML, Hsu YM, Yagita H, Flavell RA, Miller JB, Burkly LC, Zheng TS. TWEAK, via its receptor Fn14, is a novel regulator of mesenchymal progenitor cells and skeletal muscle regeneration. EMBO J. 2006;25:5826–5839. doi: 10.1038/sj.emboj.7601441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCroskery S, Thomas M, Platt L, Hennebry A, Nishimura T, McLeay L, Sharma M, Kambadur R. Improved muscle healing through enhanced regeneration and reduced fibrosis in myostatin-null mice. J Cell Sci. 2005;118:3531–3541. doi: 10.1242/jcs.02482. [DOI] [PubMed] [Google Scholar]
- 43.Kjaer M. Role of extracellular matrix in adaptation of tendon and skeletal muscle to mechanical loading. Physiol Rev. 2004;84:649–698. doi: 10.1152/physrev.00031.2003. [DOI] [PubMed] [Google Scholar]
- 44.Collins RA, Grounds MD. The role of tumor necrosis factor-alpha (TNF-alpha) in skeletal muscle regeneration. Studies in TNF-alpha(−/−) and TNF-alpha(−/−)/LT-alpha(−/−) mice. J Histochem Cytochem. 2001;49:989–1001. doi: 10.1177/002215540104900807. [DOI] [PubMed] [Google Scholar]
- 45.Chicheportiche Y, Bourdon PR, Xu H, Hsu YM, Scott H, Hession C, Garcia I, Browning JL. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem. 1997;272:32401–32410. doi: 10.1074/jbc.272.51.32401. [DOI] [PubMed] [Google Scholar]
- 46.Jain M, Jakubowski A, Cui L, Shi J, Su L, Bauer M, Guan J, Lim CC, Naito Y, Thompson JS, Sam F, Ambrose C, Parr M, Crowell T, Lincecum JM, Wang MZ, Hsu YM, Zheng TS, Michaelson JS, Liao R, Burkly LC. A novel role for tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in the development of cardiac dysfunction and failure. Circulation. 2009;119:2058–2068. doi: 10.1161/CIRCULATIONAHA.108.837286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, Malhotra S, Kumar A. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J Mol Med. 2008;86:1113–1126. doi: 10.1007/s00109-008-0373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 49.Cai D, Frantz JD, Tawa NE, Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell. 2004;119:285–298. doi: 10.1016/j.cell.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 50.Acharyya S, Villalta SA, Bakkar N, Bupha-Intr T, Janssen PM, Carathers M, Li ZW, Beg AA, Ghosh S, Sahenk Z, Weinstein M, Gardner KL, Rafael-Fortney JA, Karin M, Tidball JG, Baldwin AS, Guttridge DC. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest. 2007;117:889–901. doi: 10.1172/JCI30556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bakkar N, Wang J, Ladner KJ, Wang H, Dahlman JM, Carathers M, Acharyya S, Rudnicki MA, Hollenbach AD, Guttridge DC. IKK/NF-kappaB regulates skeletal myogenesis via a signaling switch to inhibit differentiation and promote mitochondrial biogenesis. J Cell Biol. 2008;180:787–802. doi: 10.1083/jcb.200707179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mourkioti F, Kratsios P, Luedde T, Song YH, Delafontaine P, Adami R, Parente V, Bottinelli R, Pasparakis M, Rosenthal N. Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J Clin Invest. 2006;116:2945–2954. doi: 10.1172/JCI28721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oakley F, Mann J, Nailard S, Smart DE, Mungalsingh N, Constandinou C, Ali S, Wilson SJ, Millward-Sadler H, Iredale JP, Mann DA. Nuclear factor-kappaB1 (p50) limits the inflammatory and fibrogenic responses to chronic injury. Am J Pathol. 2005;166:695–708. doi: 10.1016/s0002-9440(10)62291-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kumar A, Takada Y, Boriek AM, Aggarwal BB. Nuclear factor-kappaB: its role in health and disease. J Mol Med. 2004;82:434–448. doi: 10.1007/s00109-004-0555-y. [DOI] [PubMed] [Google Scholar]