

Abstract
The tuberous sclerosis complex 2 (TSC2) gene encodes the protein tuberin, which functions as a key negative regulator of both mammalian target of rapamycin (mTOR) C1-dependent cell growth and proliferation. Loss-of-function mutations of TSC2 result in mTORC1 hyperactivity and predispose individuals to both tuberous sclerosis and lymphangioleiomyomatosis. These overlapping diseases have in common the abnormal proliferation of smooth muscle-like cells. Although the origin of these cells is unknown, accumulating evidence suggests that a metastatic mechanism may be involved, but the means by which the mTOR pathway contributes to this disease process remain poorly understood. In this study, we show that tuberin regulates the localization of E-cadherin via an Akt/mTORC1/CLIP170-dependent, rapamycin-sensitive pathway. Consequently, Tsc2(−/−) epithelial cells display a loss of plasma membrane E-cadherin that leads to reduced cell-cell adhesion. Under confluent conditions, these cells detach, grow in suspension, and undergo epithelial-mesenchymal transition (EMT) that is marked by reduced expression levels of both E-cadherin and occludin and increased expression levels of both Snail and smooth muscle actin. Functionally, the Tsc2(−/−) cells demonstrate anchorage-independent growth, cell scattering, and anoikis resistance. Human renal angiomyolipomas and lymphangioleiomyomatosis also express markers of EMT and exhibit an invasive phenotype that can be interpreted as consistent with EMT. Together, these results suggest a novel relationship between TSC2/mTORC1 and the E-cadherin pathways and implicate EMT in the pathogenesis of tuberous sclerosis complex-related diseases.
Mutation of the TSC2 gene gives rise to the autosomal dominant disorder, tuberous sclerosis complex (TSC), that is characterized by “hamartomas” in the brain, kidney, skin, heart, and lung.1 Genetic and biochemical analyses have highlighted the role of the TSC2 protein, tuberin, in concert with its interacting partner, TSC1 (hamartin), in negatively regulating mammalian target of rapamycin (mTOR) C1 by promoting the hydrolysis of Rheb-GTP.2 Multiple factors including growth factors, energy, and oxygen availability converge on the TSC1/TSC2 complex to modulate mTORC1 activity.3 The best characterized function of mTORC1 is the promotion of protein synthesis through its downstream targets, p70S6K and 4E-BP1. In turn, p70S6K mediates phosphorylation of IRS-1 to inhibit phosphatidylinositol 3-kinase/AKT signaling in a negative feedback mechanism.4 Consequently, the loss of TSC1 or TSC2 leads to an “overgrowth” phenotype with increased cell size and proliferation, characteristic of the hamartomas seen in tuberous sclerosis. However, many of the clinical and pathological features of TSC remain unexplained by our current understanding of the function of these genes.
One such example is the lymphangioleiomyomatosis (LAM) that occurs in ∼40% of females diagnosed with TSC.5 The sporadic form of LAM is also associated with mutation of the TSC2 gene.6 LAM is a unique disease that affects women of childbearing age and is characterized by the infiltration of smooth muscle-like cells in the lung interstitium, which eventually leads to the progressive loss of pulmonary function and cystic destruction of the lung.7 Although LAM is not exclusive to the lungs and can involve the axial lymphatic system and other organs, mortality due to respiratory failure takes place within 8 to 15 years after diagnosis.8 LAM and angiomyolipoma (AML) are classified as perivascular epithelioid cell neoplasms), that is, defined as “mesenchymal tumors composed of histologically and immunohistochemically distinctive perivascular epithelioid cells.”9 These tumors are characterized histologically by their epithelioid appearance and their physical relationship to blood vessels.9 The abnormal cells display a distinct immunophenotype that includes the expression of melanocytic (eg, gp100) and smooth muscle markers (eg, smooth muscle actin) but not epithelial antigens. Because perivascular epithelioid cells have no normal anatomical counterpart, the origin of these tumors remains elusive.
One current theory suggests that pulmonary LAM is the result of a metastatic process in which certain precursor cells migrate to the lung and invade the parenchyma.10 Indeed, primary LAM cells have been shown to be invasive in vitro,11 and these cells have been identified in body fluids, including blood and urine, suggesting that LAM cells are capable of detaching from their primary sites and entering the circulation.12 Further evidence in support of this hypothesis comes from the observations that pulmonary recurrences of LAM after lung transplant contain cells that originated from the organ recipients,13,14 and common patterns of TSC2 mutation have been identified in LAM and lymph node disease from the same individual.15 Finally, the unique immunophenotype in LAM does not reflect an epithelial nor mesenchymal origin but rather a mixture of “epithelioid” and “spindle” cells that is suggestive of a variable differentiation pattern.16 Nonetheless, both populations of cells are believed to be clonally derived. The spindle cells are reported to be more proliferative and express smooth muscle actin, whereas the epithelioid cells express the melanocytic markers (eg, HMB-45) and are less mitotically active. Collectively, the observed behavior of LAM cells with respect to their infiltrative growth pattern, metastatic potential, and altered cell differentiation is reminiscent of cells undergoing epithelial-mesenchymal transition (EMT).17,18 Here, we propose that LAM may be a manifestation of EMT and show that human AML and LAM do indeed express markers of EMT.
One of the critical steps driving EMT is the repression of E-cadherin, resulting in loss of cell-cell adhesion. E-cadherin is expressed in most epithelial cells in which adherens junctions are formed to create the multicellular organization important for the formation and maintenance of bodily compartments. Structural studies highlight the essential role of calcium in the progressive cis-dimerization of the cadherin ectodomain, leading to the formation of a trans-dimer “zipper” between multiple cis-dimers to form cell adhesion.19 In this study, we provide evidence that E-cadherin membrane localization is regulated by the Akt/mTORC1 pathway such that the loss of TSC2 leads to significant reduction in membrane E-cadherin. Consequently, cells deficient in Tsc2 are less adhesive and more prone to detach and to undergo EMT. Our findings highlight a novel functional link between tuberin and E-cadherin activity that may contribute to the pathogenesis of TSC and related disorders.
Materials and Methods
Cell Lines, Plasmids, Reagents and Transfection
LEF2, ERC18M, LEF8, and ERC34 are four independent Tsc2(−/−) (Eker) renal epithelial tumor cell lines20; NRK52E is a wild-type rat kidney epithelial cell line (CRL1571, American Type Culture Collection, Manassas, VA). The Tsc2(−/−) cells were grown in Dulbecco's modified Eagle's medium (DMEM)/F12 with 10% fetal bovine serum (FBS) and NRK52E cells in DMEM with 5% FBS, as recommend. Plasmids for Myc-Tsc1, Flag-Tsc2, Myc-mLST8, HA-Rheb(Q64L), and Myr-Akt and siRNA for Tsc2 (Dharmacon, Lafeyette, CA) and CLIP170 (Ambion, Austin, TX) were transiently transfected into cells as described previously.21 Rapamycin, wortmannin, and LY294002 were purchased from Calbiochem (La Jolla, CA). Antibodies used included E-cadherin, fibronectin, integrin β1, FAK, EEA1, and GM130 (BD Biosciences, San Jose, CA); rpS6, phospho-S6, Akt, phospho-Akt (Ser473), and phospho-Smad2 (Cell Signaling, Beverly, CA); smooth muscle actin (SMA), β-actin, and α-tubulin (Sigma-Aldrich, St. Louis, MO); FAK pY397 (Epitomics, Burlingame, CA); Tsc2 and caveolin-1 (Santa Cruz Biotechnology, Santa Cruz, CA); Snail (Abcam, Cambridge, MA); transforming growth factor (TGF) (Millipore, Billerica, MA); occludin (Invitrogen, Carlsbad, CA); and E-cadherin functional blocking antibody, DECMA-1 (Sigma-Aldrich).
Clinical Samples
Human AML and LAM tissues were obtained from the University of Washington and the National Disease Research Interchange under institutional review board-approved protocols. All samples were de-identified and anonymous.
Immunofluorescence Confocal Analysis
Cells were seeded onto two-well chamber slides (Nalge Nunc International, Rochester, NY) or eight-well chamber slides (Falcon; BD Biosciences Discovery Labware, Bedford, MA) coated with poly l-lysine and allowed to adhere to the slide (16 hours) before they were fixed with 4% paraformaldehyde or cold (−20°C) methanol. After PBS washing and treatment with 0.1% Triton X-100, the cells were blocked with 10% normal goat serum in PBS for 30 minutes at room temperature and then incubated with the indicated antibodies diluted in PBS for 1 hour at room temperature or 4°C overnight. Cells were washed and then incubated with Alexa Fluor 488- or 568-conjugated anti-rabbit and anti-mouse antibodies as appropriate for 1 hour at room temperature. Cells were washed in PBS, mounted, counterstained with 4,6-diamidino-2-phenylindole (DAPI), and visualized with a Leica SP/NT Spectral Confocal microscope.
Immunohistochemical Analysis
Five-micrometer tissue sections on glass slides were deparaffinized, rehydrated, and washed with PBS. After antigen retrieval in 0.1 mmol/L sodium citrate (pH 6.0) and quenching of endogenous peroxidase activity with 3% H2O2, samples were blocked with 5% normal goat serum or 5% normal horse serum before incubation with primary antibodies overnight at 4°C. Negative controls were treated with 5% normal goat serum or normal horse serum without primary antibodies. Signals were processed according to the supplied protocol (Elite ABC Kit). Slides were counterstained with hematoxylin QS and dehydrated and mounted using Permount (Fischer Scientific, Santa Clara, CA).
Anchorage Independent Growth Assay
Nonadherent cells were harvested from medium and collected by centrifugation. Adherent cells were harvested by trypsinization and then collected by centrifugation. Collected cells were rinsed with PBS and resuspended in 1 ml of PBS for counting purposes; 105 cells were plated per sample. Plates were prepared as follows: 2.0% agarose solution was mixed with an equal volume of serum-free medium (containing penicillin/streptomycin antibiotics). This 1.0% agarose solution was poured onto 60-mm plates, forming an agarose base. Then a 0.6% agarose solution was mixed with an equal volume of serum-free medium (containing penicillin/streptomycin antibiotics), producing a 0.3% agarose solution. Nine milliliters of this 0.3% agarose solution was mixed with the appropriate volume of cell suspension to generate 3 × 105 cells/10 ml of solution. This cell-agarose solution was then divided equally among three 60-mm plates to produce 1 × 105 cells/plate. After solidifying (∼5 minutes at room temperature), 3 ml of medium (containing serum and penicillin/streptomycin antibiotics) was added to each plate. Every 2 days, plates were refed with fresh medium. Two weeks after plating, plates were supplemented with 500 μl of 0.4% iodonitrotetrazolium solution (Sigma-Aldrich) to visualize colonies. Colonies were counted by light microscopy, and the results represent the average number of colonies per 60-mm plate (three replicates per sample).
Trypan Blue Exclusion Assay
Quantification of viable nonadherent cells was performed using the trypan blue exclusion method. In brief, nonadherent cells were harvested and collected by centrifugation. Cells were rinsed with PBS and then resuspended in 1 ml of PBS. A 10-μl aliquot of cell suspension was incubated with 50 μl of PBS/40 μl of 0.4% trypan blue stain (Invitrogen/Gibco) solution for 5 minutes at room temperature. Viable and nonviable cells were counted by hemacytometer. Results represent the number of viable cells over the total number of cells counted and are expressed as percentages.
Flow Cytometric Analyses
Steady-state cell surface integrin β1 was measured by flow cytometry as described previously.22 In brief, NRK52E and LEF2 cells were detached by 0.05% trypsin-EDTA for 10 minutes at 37°C, washed with PBS, pelleted by centrifugation, and fixed in cold 4% paraformaldehyde-PBS for 20 minutes. Cells were incubated with anti-integrin β1 antibody (BD Biosciences) for 1 hour at room temperature. Surface integrin β1 was quantified by fluorescence-activated cell sorter analysis after staining with an Alexa Fluor 488 goat anti-mouse secondary antibody.
Hanging Drop Assay
HEK293T cells at 50% confluence were transfected with 1.6 μg of empty vector (pcDNA3), Myc-Tsc1, Flag-Tsc2, HA-Myr-Akt (active Akt), HA-Rheb-WT, or Myc-Rheb-Q64L (active Rheb) using Lipofectamine PLUS transfection reagent (Invitrogen). Twenty-four hours after transfection, medium was aspirated, and cells were resuspended in PBS. Half of the cell suspension was reserved for protein analysis. The remaining cell suspension was counted by hemacytometer, and 3 × 104 cells were triturated by passing the cells 10 times through a 200-ml pipette tip to break clumps into individual cells. Cell samples were suspended as hanging drops from the lid of a 24-well cell culture plate and incubated for 2 hours. Cell aggregation was assessed using phase-contrast microscopy. Quantification of the degree of aggregation is presented as the number of cell aggregates (>4 cells) in one field (minimum of 100 cells counted) with the average taken from three to four images per sample. Each cell suspension reserved for protein analysis was lysed in 0.5% Nonidet P-40 buffer and then analyzed by SDS-polyacrylamide gel electrophoresis and Western immunoblot methods to detect exogenous protein expression.
Cell Agitation Assay
A second cell-cell adhesion assay was performed essentially as described previously.23 In brief, cells were trypsinized, washed once, and resuspended in medium. Cells were gently passed three times through a 26-gauge syringe and counted by hemacytometer. Cell suspensions (2 × 104 cells in 200 μl) were placed in 1.5-ml reaction tubes alone or with 2 mmol/L EGTA or a functional blocking anti-E-cadherin antibody (1:100) (Sigma-Aldrich)24 for 2 hours at 37°C and agitated at 300 rpm. After 2 hours, cell suspensions were gently mixed, and five 10-μl drops were placed on a dish analyzed at ×40 magnification using phase-contrast microscopy. Quantification of the degree of aggregation is presented as the average number of cell aggregates (consisting of clusters of >4 cells) per field. Cell viability was assessed using the trypan blue exclusion method. For the rapamycin experiments, cells were treated with either dimethyl sulfoxide (DMSO) or 200 nmol/L rapamycin for 24 hours before the agitation assay was performed.
Colony Scattering Assay
Adherent and nonadherent Tsc2(−/−) cells were plated at a low density (5000 cells/100-mm plate) and the morphology of colonies was analyzed after 6 days of growth. A minimum of 100 colonies from each sample was categorized after scoring of phase-contrast images into two categories: compact (majority of cells in colony have cell-cell contacts) and scattered (cells in colony expressing dendritic structures and with a significant loss of cell-cell contacts). The results were tabulated as the number of compact or scattered colonies divided by the total number of colonies counted.
Western Blot Analysis of Tissues
Samples were homogenized in ice-cold radioimmunoprecipitation buffer (1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, 0.15 mol/L NaCl, 10 mmol/L Tris (pH 7.2), 0.025 mol/L β-glycophosphate (pH 7.2), 2 mmol/L EDTA, and 50 mmol/L sodium fluoride) with protease and kinase inhibitors (0.05 mmol/L 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride, 10 μg/ml aprotinin, 10 μg/ml pepstatin, 1 mmol/L orthovanadate, 10 μg/ml leupeptin, and 1 mmol/L microcystin LR). Protein concentration was measured using the BCA Protein Assay (Pierce, Rockford, IL). Equal amounts of protein were separated by SDS-polyacrylamide gel electrophoresis, transferred to Immobilon-P membranes (Millipore), and blotted with antibodies according to the manufacturer's recommendations.
RT-PCR
Total RNA was isolated from 3 × 107 adherent and nonadherent LEF2 cells according to the protocol for the RNeasy Midi Kit (Qiagen, Valencia, CA), and 2 μg of RNA was subjected to RT according to the manufacturer's protocol (RETROscript, Ambion, Austin, TX). Then 5 μl of RT product was subjected to PCR cycling parameters as follows: one cycle at 95°C, 1 minute; 50 cycles at 94°C, 1 minute; 61°C, 1 minute; 72°C, 2 minutes; with final extension at 72°C, 5 minutes. Final products were analyzed by 2.0% agarose gel. The primer sequences were as follows: rat GAPDH (414 bp) 5′-TGCATCCTGCACCACCAACTGC-3′ (sense) and 5′-AATGCCAGCCCCAGCATCAAAG-3′ (antisense); rat E-cadherin (362 bp) 5′-CCAGTTTTCTCGTCCATGCC-3′ (sense) and 5′-CACTTTCAGCCAGCCTGTCT-3′ (antisense); and rat Snail (487 bp) 5′-TCCGATGAGGACAGTGGCAAAA-3′ (sense) and 5′-GTTTGTGGAGCAAGGACATTC-3′ (antisense).
Anoikis Assay
Twelve-well tissue culture plates were coated with 200 μl (6 mg/ml in 95% ethanol) of poly-(2-hydroxyethyl methacrylate) (Sigma-Aldrich) by incubation overnight at 37°C. To perform the anoikis assay, cells were trypsinized into a single cell suspension, and 104 cells in 2.5 ml of DMEM + 5% FBS (for NRK52E) or DMEM + F12 + 10% FBS (for LEF2), were added to each well. Differences in serum concentration were based on the original/recommended conditions for each cell line from the American Type Culture Collection and our laboratory. Starved samples were incubated with either 0.5% or 1% FBS for NRK52E or LEF2, respectively. Suspended cells were incubated at 37°C for up to 3 days. Cells were harvested, washed, and stained with trypan blue to determine cell viability. For the inhibitor study, cells were treated with 200 nmol/L rapamycin or DMSO (vehicle) every 24 hours for up to 3 days.
Results
Tsc2 Regulates E-Cadherin Localization
We have shown previously that the TSC1/TSC2 complex plays a role in protein transport. Specifically, we demonstrated that cells deficient in Tsc1 or Tsc2 mislocalize plasma membrane proteins such as caveolin-1 and Glut-4 to the cytosol because of mTOR-mediated alterations in microtubule-dependent protein trafficking.21,25,26 We speculated that other caveolae-associated membrane proteins may also be regulated by this pathway. It has been shown that the cell-cell adhesion protein, E-cadherin, is concentrated in caveolae, and down-regulation of caveolin-1 leads to disassembly of the E-cadherin complex.27 Here, we examined the relationship between the TSC2 pathway and E-cadherin localization using indirect immunofluorescence analyses. In wild-type rat renal epithelial cells, NRK52E, E-cadherin localized to the plasma membrane on confluence where it is known to play a critical role in adherens junction formation (Figure 1A). In contrast, the Tsc2(−/−) renal epithelial tumor-derived cells (LEF2) originating from the Eker rat model20 showed a paucity of membrane E-cadherin at confluence (Figure 1B). Instead, most of the E-cadherin signals were found in punctate cytosolic structures. Similar changes in E-cadherin localization were observed in three other independently derived Tsc2(−/−) renal epithelial cell lines: ERC18M, LEF8, and ERC34 (data not shown).
Figure 1.
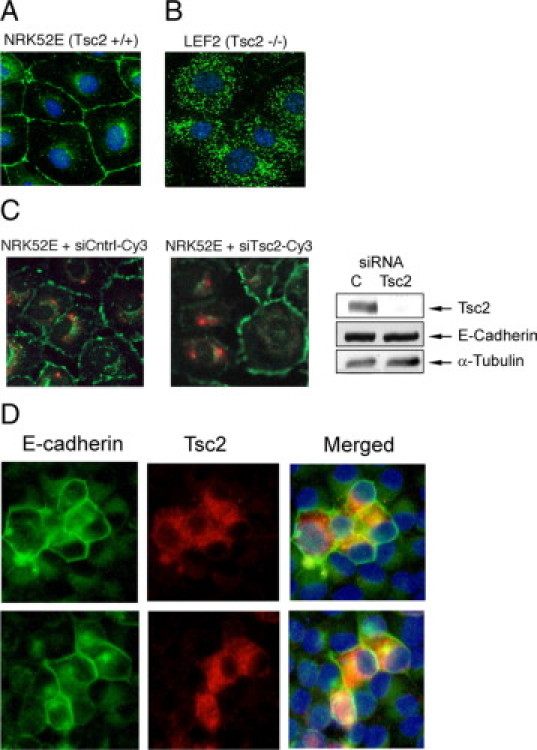
Tsc2 regulates E-cadherin localization. Immunofluorescence analysis of wild-type rat kidney epithelial NRK52E cells (A) and Tsc2(−/−) rat kidney tumor epithelial LEF2 cells (B) using anti-E-cadherin antibodies (green). C: In loss-of-function experiments, NRK52E cells were transiently transfected with Cy3-labeled (red) Tsc2 siRNA or control siRNA (siCntrl) and analyzed for E-cadherin expression (green). Western blot shows effects of siRNA on Tsc2 and E-cadherin expression. α-Tubulin serves as the loading control. C, control siRNA. D: In gain-of-function analyses, LEF2 cells were transiently transfected with Tsc2 expression plasmid (red) and stained for E-cadherin (green). Two examples are shown. Nuclei were stained with DAPI (blue).
To determine whether E-cadherin localization was regulated by TSC2, we performed loss-of-function studies using small interfering RNA (siRNA) in the NRK52E cells. As shown in Figure 1C, cells transfected with Cy3-labeled Tsc2 siRNA, but not with control siRNA, have lost membrane E-cadherin compared with adjacent nontransfected cells. Conversely, replacement of Tsc2 in the LEF2 cells resulted in strong membrane E-cadherin staining compared with that in nontransfected cells including at the interface between the transfected and nontransfected cells (Figure 1D). Although intercellular homophilic interaction stabilizes membrane E-cadherin, its membrane localization is not contingent on such an interaction. For example, EGTA-treated NRK52E cells underwent cell detachment while preserving membrane E-cadherin (Supplemental Figure S1, see http://ajp.amjpathol.org). Taken together, these observations show that modulation of Tsc2 expression regulates E-cadherin localization in renal epithelial cells.
The extent to which the loss of Tsc2 affects other plasma membrane proteins is not known. Because other cell-adhesion molecules such as integrins can be associated with caveolae, we examined the membrane expression of integrin-β1 in the wild-type and Tsc2-mutant cells using flow cytometric analyses as described.22 Unlike E-cadherin, both the level of surface expression and the proportion of cells expressing surface integrin-β1 were similar between the Tsc2(−/−) cells (LEF2 and ERC18M) and the wild-type NRK52E cells (Supplemental Figure S2, A and B, see http://ajp.amjpathol.org). Further, the extent to which FAK is phosphorylated (tyrosine 397) relative to total FAK was equivalent between these cells (Supplemental Figure S2C, see http://ajp.amjpathol.org). Other membrane proteins including ZO-1 and claudin-1 were properly localized to the plasma membrane in the Tsc2(−/−) cells (data not shown). These findings suggest that the effects of TSC2/mTORC1 on protein trafficking are specific to a subset of plasma membrane proteins and do not represent a global defect in post-Golgi transport.
E-Cadherin Resides in the Golgi of Tsc2(−/−) Cells
To further characterize E-cadherin in the Tsc2(−/−) cells, we determined its subcellular localization using dual immunofluorescence analyses. Our earlier study demonstrated a defect in post-Golgi transport of the Tsc2(−/−) cells affecting caveolin-1 and its associated proteins.25 Here, we found that E-cadherin colocalized extensively with GM130, a Golgi marker, in the perinuclear region and partially with caveolin-1 in the periphery of the cells (Supplemental Figure S3, A and B, see http://ajp.amjpathol.org). In contrast, E-cadherin resided in compartments distinct from endosomes (EEA1) and β-catenin (Supplemental Figure S3, C and D, see http://ajp.amjpathol.org), although in cell lysates, E-cadherin was able to immunoprecipitate β- and α-catenin (data not shown), suggesting that it is biochemically intact. In wild-type NRK52E cells, E-cadherin also showed colocalization with GM130 at the perinuclear compartment but not at the membrane, whereas it colocalized with β-catenin at the plasma membrane but not in the cytosol (Supplemental Figure S3, E and F, see http://ajp.amjpathol.org). Together, these data are consistent with our model that Tsc2 regulates the post-Golgi transport of E-cadherin, and consequently, in the absence of tuberin, E-cadherin becomes largely retained in the Golgi.
Membrane Localization of E-Cadherin Is Dependent on mTORC1 and Akt Activities
Next, we explored the mechanism of Tsc2-dependent E-cadherin localization. One of the major signaling events after the loss of Tsc2 is the activation of mTORC1, and we have previously shown that the mislocalization of membrane caveolin-1 in the Tsc2(−/−) cells can be restored by prolonged exposure (ie, >5 hours) to rapamycin.21 The latter restores the disorganized microtubules in the mutant cells toward normality and reestablishes proper protein transport, but this effect requires time (eg, hours) to accomplish.21 To determine whether E-cadherin distribution was sensitive to mTORC1 activity, LEF2 cells were treated with rapamycin (200 nmol/L for 24 hours) followed by immunofluorescence analysis. Figure 2A shows that rapamycin was highly effective in rescuing the membrane localization of E-cadherin in the Tsc2-mutant cells. Conversely, LEF2 cells treated with a phosphatidylinositol 3-kinase inhibitor, wortmannin (1 μmol/L, 24 hours), showed no significant change in the cytosolic distribution of E-cadherin (Figure 2A). In wild-type NRK52E cells, treatment with wortmannin or rapamycin did not affect membrane localization of E-cadherin (Supplemental Figure S4, see http://ajp.amjpathol.org).
Figure 2.
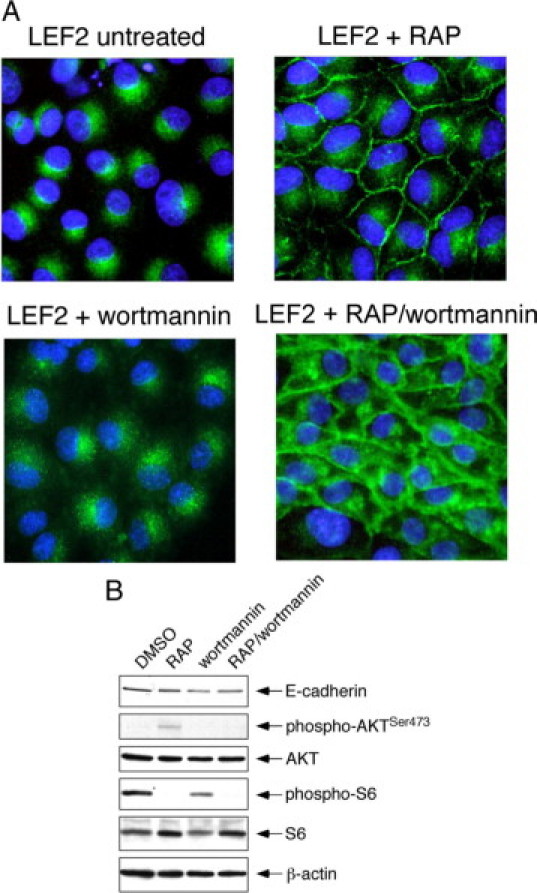
Membrane E-cadherin localization is dependent on mTORC1 activity. A: Effects of rapamycin (RAP, 200 nmol/L, 24 hours), wortmannin (1 μmol/L, 24 hours), or both inhibitors on E-cadherin distribution in LEF2 cells. Green, E-cadherin; blue, nuclei. B: Western blot analysis shows expression of phospho-S6(Ser235/236), phospho-AKT(Ser473), and E-cadherin for the conditions shown in A after a short exposure to inhibitors: rapamycin (RAP) for one hour and wortmannin for ten minutes. β-Actin serves as a loading control.
In the Tsc2-null state, mTORC1 is constitutively active and leads to Akt suppression by S6K1-IRS1 negative feedback.28 Rapamycin inhibits mTORC1 and releases this feedback to activate Akt. To decipher whether the effects of rapamycin on E-cadherin localization were due to changes in mTORC1 and/or Akt activities, we exposed the LEF2 cells to rapamycin and wortmannin concurrently. On treatment with the two inhibitors, the cells showed strong membrane E-cadherin localization but also had increased cytosolic signals compared with cells treated with rapamycin alone (Figure 2A). Immunoblot analysis shows that mTORC1 (as indicated by phospho-S6) and Akt activities were both suppressed in the presence of both inhibitors, whereas rapamycin alone inhibited mTORC1 and augmented Akt activities and wortmannin alone had minimal effects on mTORC1 activity (Figure 2B). These observations suggest that down-regulation of mTORC1 in the absence of Akt activity is sufficient to rescue E-cadherin membrane localization in the Tsc2(−/−) epithelial cells and clearly highlight the role of mTORC1 in regulating E-cadherin trafficking. However, the increased cytosolic E-cadherin expression after both inhibitors suggests that Akt may also influence E-cadherin expression and/or localization.
To further address the contribution of Akt and mTORC1 in E-cadherin localization, we manipulated their activities by overexpressing a myristylated (Myr)-Akt mutant or components of the mTOR pathway, mLST8 and Rheb, respectively, as described previously.21 As shown in Figure 3A, LEF2 cells expressing Myr-Akt have patchy membrane E-cadherin, indicating that Akt can partially normalize E-cadherin localization, but the overall effect was significantly less than that of rapamycin treatment. Conversely, overexpression of mLST8 and Rheb(Q64L), an active form of Rheb, in the NRK52E cells led to a reduction in membrane E-cadherin localization (Figure 3B). Western blot analyses were performed to determine whether the Tsc2/mTOR pathway also affects E-cadherin expression. Replacement of Tsc2 in the Tsc2(−/−) cells and expression of mLST8 and Rheb(Q64L) in wild-type cells did not significantly alter E-cadherin steady-state levels, although the results may be affected by transfection efficiency (Supplemental Figure S5, A and B, see http://ajp.amjpathol.org). To further address this possibility, we down-regulated Tsc2 in NRK52E cells by more than 90% and observed no difference in E-cadherin expression (Figure 1C). In addition, when the Tsc2(−/−) cells were treated with rapamycin, E-cadherin expression did not change significantly (Supplemental Figure S5C, see http://ajp.amjpathol.org). Together, these observations suggest that Tsc2 regulates E-cadherin localization without significant effects on its expression and that this phenomenon is mediated predominately through mTORC1 with some contribution from Akt activity, such that rapamycin effectively rescues the defect in the Tsc2(−/−) cells by modulating both mTORC1 and Akt activities.
Figure 3.
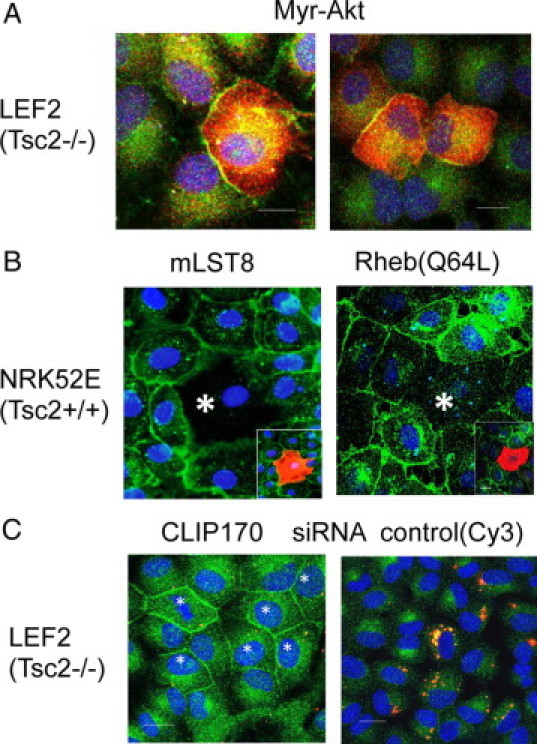
Influence of Akt-mTORC1-CLIP170 on E-cadherin localization. A: Immunofluorescence analysis of E-cadherin (green) in LEF2 cells transfected with Myr-Akt (red). B: NRK52E cells were transfected with mLST8 (left) or Rheb(Q64L) expression plasmids (right). Small insets show transfected cells (asterisk) in red. E-cadherin is shown in green. C: Examples of LEF2 cells that were transiently transfected with CLIP170 or control (Cy3) siRNA, followed by immunofluorescence analysis of E-cadherin (green). Nuclei were stained with DAPI (blue). Asterisks marks the transfected cells.
We previously attributed the defect in microtubule-dependent trafficking in the Tsc2(−/−) cells to the activity of CLIP170, an mTORC1 effector that serves as a microtubule plus-end binding protein.21 To determine the effects of CLIP170 on E-cadherin localization, LEF2 cells were transiently transfected with Cy3-labeled CLIP170 siRNA as described.21 Figure 3C shows examples of cells displaying plasma membrane E-cadherin on down-regulation of CLIP170 but without effects using control siRNA. By counting a minimum of 100 cells each, E-cadherin membrane localization was noted in 70% of LEF2 cells transfected with CLIP170 siRNA compared with 23% of these cells transfected with control siRNA (P < 0.05). This result confirms the role of CLIP170-dependent protein transport on E-cadherin localization functioning downstream of mTORC1.
Tsc2(−/−) Cells Are Less Adhesive
Given that E-cadherin is involved in cell-cell adhesion, we examined the influence of tuberin on this property using two independent assays. First, a hanging drop assay was performed in HEK293T cells that were transiently transfected with Tsc1, Tsc2, Myr-Akt, Rheb (wild-type and Q64L mutant), or control vector. Twenty-four hours after transfection, cell suspensions were placed as hanging drops onto uncoated plates and after 2 hours, the number of cell aggregates of >4 cells were counted. Figure 4A shows the formation of these aggregates found in Tsc1- and Tsc2- but not control vector-transfected HEK293T cells. Figure 4B summarizes the findings expressed as the average number of cell aggregates per 100 cells counted. Only those cells transfected with Tsc1 or Tsc2 formed aggregates in this assay. Although Myr-Akt partially recovered membrane E-cadherin in the Tsc2(−/−) cells (Figure 3A), it was insufficient to promote cell aggregates under these conditions. Next, we used the hanging drop assay to compare the ability of the Tsc2(+/+) and Tsc2(−/−) cells to self-adhere. We found that wild-type NRK52E cells formed aggregates to a much greater extent than the Tsc2-mutant LEF2 and ERC18M cells (Figure 4C) consistent with the role of tuberin in regulating cell-cell adhesion.
Figure 4.
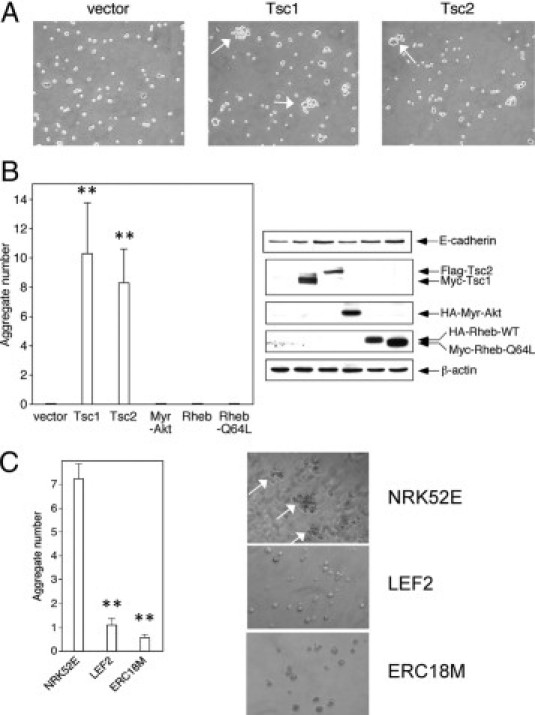
Tsc2 regulates cell-cell adhesion. A: Hanging drop assays were performed in HEK293T cells transfected with indicated vectors. Arrows highlight examples of aggregates of more than four cells 2 hours after plating. B: The number of cell aggregates per 100 cells counted after transfection of the indicated vectors was tabulated. The Western blot shows the expression of E-cadherin and the transgenes used in this experiment. C: Cell aggregation of wild-type (NRK52E) and mutant (LEF2 and ERC18M) cells in hanging drop assays. Graph indicates average number of aggregates per 100 cells counted. Arrows indicate cell aggregates. **P < 0.01.
To confirm this result, we used a cell agitation assay to define the effects of the loss of membrane E-cadherin on cell-cell adhesion. Single cell suspensions were placed in culture medium under continuous agitation at 300 rpm for 2 hours at 37°C, after which the number of cell aggregates per high-power field was counted. The NRK52E cells consistently formed aggregates of four or more cells, and this effect was dependent on intact E-cadherin function because treatment with EGTA or anti-E-cadherin functional blocking antibody24 significantly reduced aggregate formation (Figure 5A). In the Tsc2(−/−) cells, the average number of aggregates was lower than that observed for NRK52E cells, and treatment with rapamycin significantly increased the number of aggregates (Figure 5B). Importantly, the ability of rapamycin to induce aggregates in the Tsc2(−/−) cells was significantly decreased by the treatment with EGTA or anti-E-cadherin functional blocking antibody (Figure 5C). These observations strongly suggest that the effect of Tsc2 on rapamycin-sensitive localization of E-cadherin is functionally relevant to cell-cell adhesion and that the mislocalization of E-cadherin in the Tsc2-mutant cells is, at least in part, responsible for the reduced cell-cell adhesion.
Figure 5.
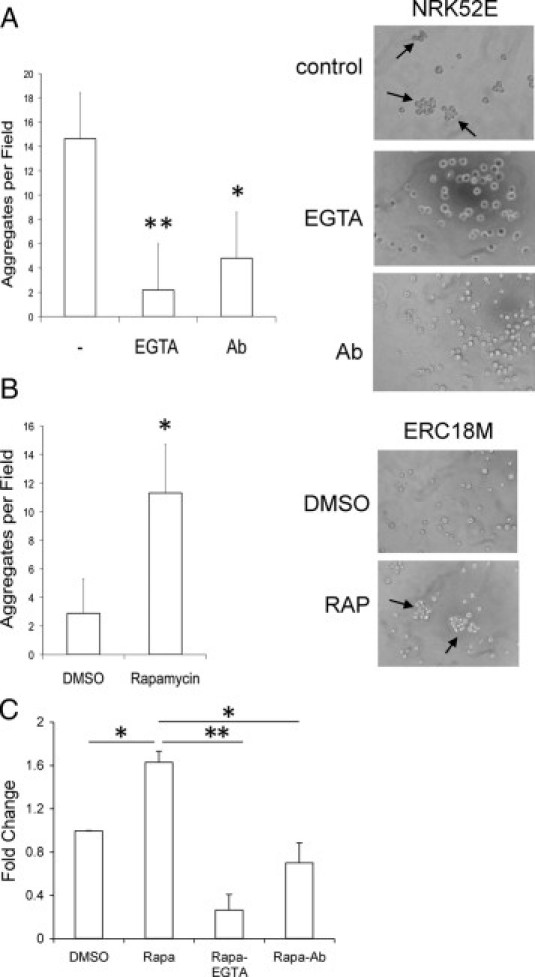
Rapamycin affects E-cadherin-mediated cell-cell adhesion. A: NRK52E cells form aggregates that are dependent on E-cadherin function. Cells were pretreated with EGTA (2 mmol/L) or functional blocking anti-E-cadherin antibody (1:100) (Ab) or untreated followed by cell agitation assay. The number of aggregates per high-power field was tabulated. B: Aggregation of ERC18M [Tsc2(−/−)] cells with or without rapamycin (RAP) in a cell agitation assay. C: Response of the LEF2 [Tsc2(−/−)] cells to rapamycin alone or in combination with EGTA or anti-E-cadherin antibody to form aggregates relative to vehicle (DMSO) control in a cell agitation assay. Each experiment was repeated in triplicate. Arrows highlight cell aggregates. *P < 0.05; **P < 0.01.
Tsc2(−/−) Cells Detach and Grow in Suspension
As a consequence of reduced cell adhesion, we anticipated that the Tsc2-mutant cells will display abnormal morphology and behavior in culture. Unlike the wild-type NRK52E cells that grew as a monolayer and exhibited contact inhibition on confluence, the Tsc2(−/−) renal epithelial cells detached from the monolayer and grew in suspension; this was a consistent finding in all four independent cell lines that we examined (LEF2, ERC18M, LEF8, and ERC34) (Figure 6A, Supplemental Figure S6A, see http://ajp.amjpathol.org). The viability of the postconfluent Tsc2(−/−) cells that lifted off the plate (hereby known as nonadherent Tsc2(−/−) cells) was determined using the trypan blue exclusion assay. The majority of these nonadherent cells remained viable under nutrient-rich growth conditions (eg, 10% FBS) and starvation (eg, 0.1% FBS) (Figure 6B). Further, the mutant cells that became nonadherent, but not wild-type cells, were able to grow and form colonies in soft agar (Figure 6C). Thus, the Tsc2(−/−) epithelial cells displayed loss of contact inhibition and acquired anchorage-independent growth.
Figure 6.
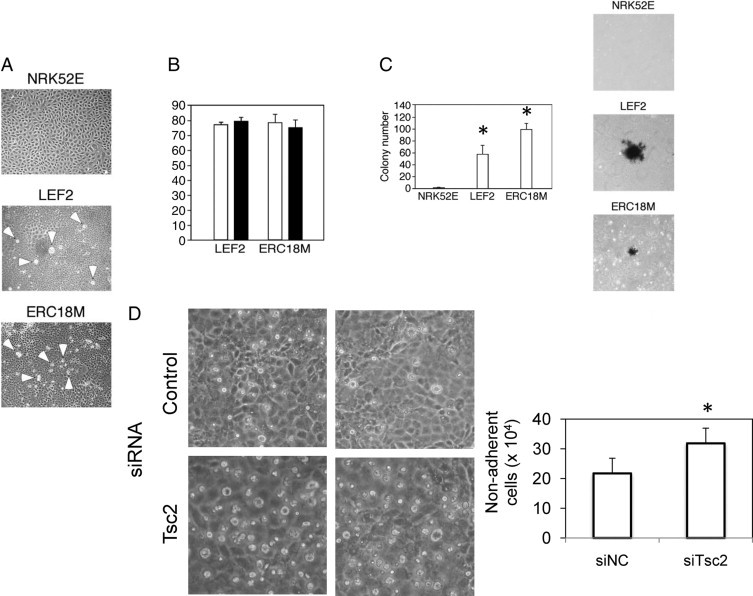
Tsc2(−/−) cells detach and grow independent of anchorage. A: Growth pattern of wild-type NRK52E cells compared with that of the two independently derived Tsc2(−/−) cell lines (LEF2 and ERC18M) at confluence. Arrowheads indicate refractile cells that have detached from the plates. B: Cell viability assessment by trypan blue exclusion of the detached cells under different growth conditions. White bars, 10% FBS; black bars, 0.1% FBS. C: Colony formation in soft agar of wild-type and mutant cells assessed 2 weeks after plating and stained with 0.4% iodonitrotetrazolium solution. Graph indicates number of colonies for each cell type. *P < 0.05, compared with NRK52E. D: Effects of Tsc2 siRNA on postconfluent NRK52E cells. Left: Photomicrographs depicting the relative abundance of detached, refractile cells in control or Tsc2 siRNA-transfected cells. Right: The number of viable (based on trypan blue exclusion), nonadherent cells was counted. *P < 0.05 compared with control (siNC).
When calcium-mediated E-cadherin interaction was disrupted by EGTA treatment, the wild-type NRK52E cells also underwent cell detachment; however, these cells were prone to undergo apoptosis (eg, anoikis) as noted by trypan blue exclusion and caspase-3 activity (Supplemental Figure S7, A–C, see http://ajp.amjpathol.org). Thirty minutes after EGTA treatment, 30% of the cells became detached, but a third of these were not viable, and caspase-3 activity increased by fourfold. When the NRK52E cells were treated with Tsc2 siRNA, significantly more cells became nonadherent and remained viable (Figure 6D), suggesting that the loss of Tsc2 reprograms the cells not only to become less adherent but maintains their viability once detached.
Nonadherent Tsc2(−/−) Epithelial Cells Undergo EMT
EMT often accompanies the loss of E-cadherin function, and we postulate that the Tsc2(−/−) cells are predisposed to undergo EMT on detachment. During EMT, epithelial cells become more motile and resistant to detachment-related cell death (ie, anoikis).17 The molecular signature of EMT includes the down-regulation of epithelial markers and up-regulation of mesenchymal markers. We examined the expression of these markers in nonadherent Tsc2(−/−) cells from four independent cell lines using a combination of immunoblot, RT-PCR, and immunofluorescence analyses (Figure 7, A–C, Supplemental Figure S6B, see http://ajp.amjpathol.org). Compared with the adherent cells, the nonadherent cells showed significant reduction in E-cadherin and occludin expression, accompanied by an induction of smooth muscle actin and Snail expression. Further, expression of SMAD2, but not of SMAD3/4, was significantly higher in the nonadherent mutant cells (Figure 7D). Changes in these molecular markers are consistent with those in the nonadherent cells undergoing EMT. However, the loss of Tsc2 per se is not sufficient for EMT because the adherent Tsc2(−/−) cells do not express these EMT markers. Instead, our observations suggest that the absence of Tsc2 promotes cell detachment and survival whereupon they further undergo EMT.
Figure 7.
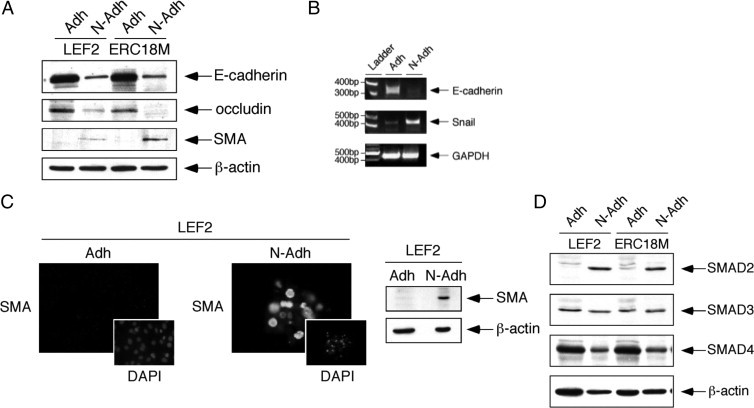
Nonadherent Tsc2(−/−) epithelial cells express markers of EMT. A: Western blot analysis of E-cadherin, occludin, and SMA in adherent (Adh) and nonadherent (N-Adh) cells derived from two Tsc2(−/−) epithelial tumor cell lines, LEF2 and ERC18M. β-Actin served as a loading control. B: RT-PCR analysis of LEF2 cells showing expression of E-cadherin, Snail, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) control in adherent and nonadherent states. C: SMA expression in adherent and nonadherent LEF2 cells analyzed by immunofluorescence analysis (left) and Western blotting analysis (right). DAPI-stained nuclei show corresponding cells in the same fields as the SMA immunofluorescence. D: Expression of SMAD proteins in adherent and nonadherent cells.
On a functional level, we examined the ability of the mutant cells to resist anoikis and to undergo cell scattering. In the anoikis assay, cells were plated onto poly-(2-hydroxyethyl methacrylate) plates to prevent cell attachment, and the number of viable cells was determined by trypan blue exclusion assay over a 72-hour period. When grown under serum-rich conditions, the Tsc2(−/−) LEF2 cells survived significantly better than wild-type NRK52E cells (percent viability at day 3: 56 vs. 28%, P < 0.05) (Figure 8A). However, under relative serum starvation, the majority of the Tsc2(−/−) cells died at 72 hours similar in proportion to the wild-type cells (percent viability: 25 vs. 18%, NS). Thus, cells lacking Tsc2 exhibited anoikis resistance in a growth factor-dependent manner. On the contrary, NRK52E cells were susceptible to anoikis even when placed in serum-rich medium (10% FBS) (data not shown). Next, we assayed the cells in terms of their ability to form tight, compact colonies as one would expect for wild-type epithelial cells. Figure 8B shows the results of the colony-scattering assay in which the proportion of scattered versus compact colonies was assessed based on their colony morphology. The nonadherent LEF2 and ERC18M cells showed significantly fewer compact colonies compared with the adherent parental cells, indicating that the nonadherent cells have altered their morphology consistent with EMT.
Figure 8.
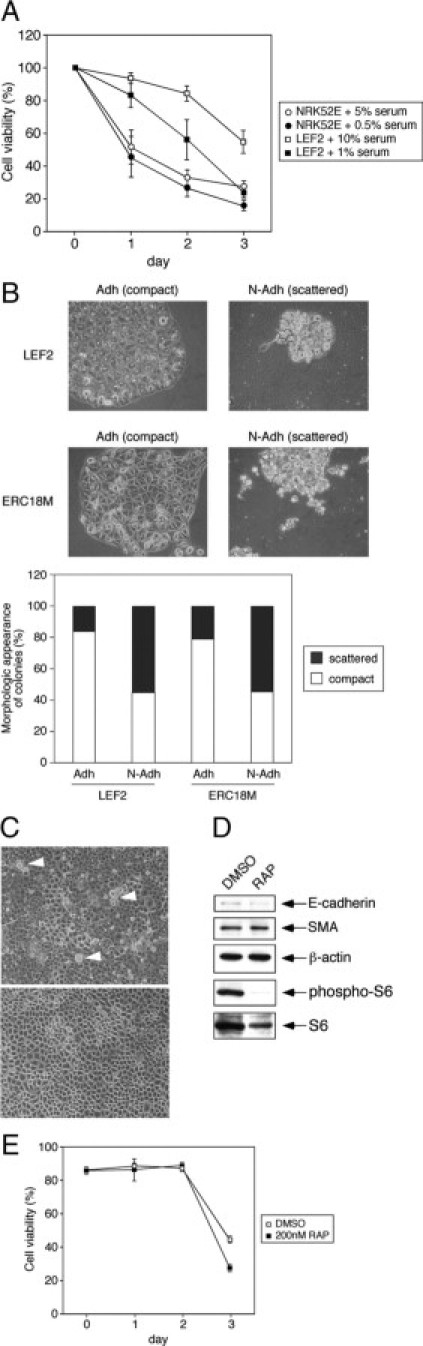
Nonadherent Tsc2(−/−) cells exhibit functional evidence of EMT. A: Anoikis assay of wild-type NRK52E and Tsc2(−/−) LEF2 cells shows the proportion of viable cells when cultured on poly-(2-hydroxyethyl methacrylate) plates over 3 days under high- and low-serum conditions. B: Comparison of adherent (Adh) and nonadherent (N-Adh) Tsc2(−/−) LEF2 and ERC18M cells in the cell scattering assay. Top panels illustrate the appearance of compact and scattered colonies seen in the adherent and nonadherent mutant cells, respectively. The proportion of compact and scattered colonies is tabulated as a percentage of the total number of colonies counted. At least 100 colonies were assessed for each category. C: Effects of rapamycin (RAP, 200 nmol/L) on the morphology of LEF2 cells at confluence. Detaching refractile cells are indicated by arrowheads. DMSO, vehicle control. D: Effects of rapamycin (RAP) on the expression of E-cadherin and SMA in nonadherent LEF2 cells. A change in phospho-S6 indicates the rapamycin effect. β-Actin served as a loading control. E: Effects of rapamycin on anoikis in LEF2 cells.
Given that mTORC1 inhibition led to normalization of E-cadherin localization and cell-cell adhesion, we reasoned that rapamycin treatment might prevent cells from detachment. Indeed, exposure to rapamycin (200 nmol/L, 48 hours) but not to vehicle alone prevented the LEF2 cells from detaching from the confluent monolayer (Figure 8C). The rapamycin-treated LEF2 cells exhibited contact inhibition similar to that seen in wild-type cells. However, rapamycin had no effect on the nonadherent LEF2 cells that have undergone EMT with respect to their expression of EMT markers, E-cadherin and SMA (Figure 8D). Further, mTORC1 inhibition had minimal effects on anoikis resistance with only a slight reduction in the proportion of viable cells after 2 days, which may be attributed to its known effects on cell growth/proliferation (Figure 8E). These findings are consistent with the role of the TSC2 pathway on E-cadherin-dependent function in maintaining cell-cell adhesion and contact inhibition. On loss of TSC2, epithelial cells are prone to detach and undergo EMT, but once initiated, mTORC1 inhibition does not reverse EMT.
Evidence of EMT in AML and LAM
To investigate the in vivo relevance of our findings, we examined tissues from two types of TSC pathological conditions, renal AML and pulmonary LAM, for features of EMT. Figure 9A shows the expression of EMT markers in tissue lysates obtained from frozen samples of AML, LAM, and accompanying normal kidney and lungs. Compared with the normal tissues, these lesions expressed higher levels of smooth muscle actin, fibronectin, Smad 2, and phospho-Smad2, along with phospho-S6. Expression of Snail and TGF-β were also detected in LAM and AML tissues by immunohistochemical analyses. Snail expression was most prominent in the nuclei of the mutant cells in LAM and AML but not in the normal cells (Figure 9B). However, its pattern of expression was found to be heterogeneous. Closer examination of the LAM nodules suggests that the larger epithelioid cells were less immunoreactive to anti-Snail antibody compared with the spindle cells (Figure 9C), suggesting that these two well recognized cell populations in LAM may represent different states of EMT. In addition to Snail, TGF-β was also abundantly expressed in LAM cells, whereas the normal cuboidal epithelium was spared (Figure 9D). We further confirmed by immunofluorescence analysis that E-cadherin is localized diffusely in the cytoplasm of a LAM nodule compared with its membrane localization in normal bronchial epithelium (Figure 9E), consistent with our in vitro studies. Biologically, the growth pattern of LAM is highlighted by the infiltration of LAM cells throughout the adjacent lung parenchyma with a proclivity toward peribronchial and perivascular lymphatics as described previously29 (Figure 10, top panels). Likewise the phospho-S6-expressing AML cells showed an invasive pattern in the kidney with abnormal cells infiltrating and surrounding the normal renal tubules (Figure 10, bottom panels). These features distinguish AML and LAM from other “benign” tumors such as lipoma and hemangioma and are consistent with a display of the in vivo biological phenotype of EMT.
Figure 9.
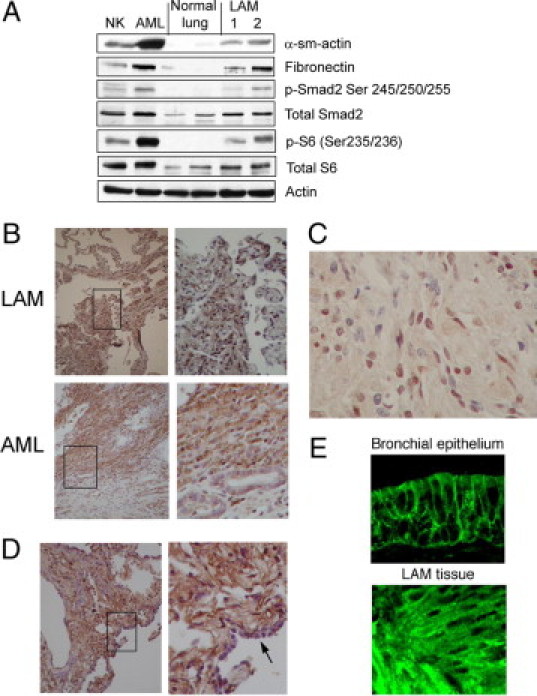
AML and LAM show evidence of EMT. A: Western blot analysis shows the expression of EMT markers (smooth muscle actin [α-sm-actin] and fibronectin), total and phospho (p)-Smad2, total and phospho-S6, and actin (loading control) in tissue lysates from human normal kidney (NK), AML, normal lung, and LAM samples. B: Immunohistochemical analyses of Snail in LAM and AML. Note nuclear staining in the abnormal cells. Right panels: high-power views of the boxed areas. C: High-magnification view of a LAM nodule showing heterogeneous expression of Snail, suggesting distinct populations of LAM cells. D: Immunohistochemical analysis of TGF-β in LAM tissue. Note the absence of TGF-β expression in normal epithelium (arrow in right panel [high-magnification]). E: Immunofluorescence analysis of E-cadherin in LAM compared with normal bronchial epithelium.
Figure 10.
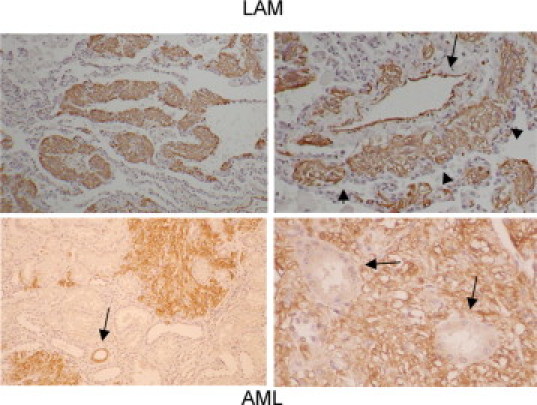
LAM and AML cells exhibit an invasive growth pattern. Immunohistochemical analysis of LAM (top panels) using anti-smooth muscle actin antibody to highlight the LAM cells. Left panel: infiltration of LAM cells (brown) within lung parenchyma. Right panel: tumor cells (arrowheads) surrounding a blood vessel (arrow) in perivascular lymphatic spaces. Bottom panels: phospho-S6 expressing AML cells invading the normal kidney parenchyma. Arrow in left panel indicates a normal collecting duct with phospho-S6 immunoreactivity. Arrows in right panel indicate normal renal tubules surrounded by AML cells.
Discussion
The loss of TSC2 function leads to a spectrum of disease entities that represent underlying abnormalities in cell growth, proliferation, differentiation, and migration.30 Despite extensive characterization of the TSC1/TSC2 pathway in the regulation of mTORC1, the molecular mechanisms linking the biochemical pathway to the biological phenotype remain poorly understood. In this study, we describe a novel role of TSC2 through its effects on E-cadherin localization and function. We show that TSC2 regulates plasma membrane distribution of E-cadherin via an Akt/mTORC1/CLIP170-dependent pathway. Through the activation of mTORC1, the loss of TSC2 in epithelial cells results in the retention of E-cadherin in the Golgi leading to the paucity of membrane E-cadherin. Consequently, cells lacking Tsc2 have reduced cell-cell adhesion and loss of contact inhibition and become nonadherent in vitro. This type of behavior is analogous to the physiological process of cell shedding in the gastrointestinal tract, in which wild-type enterocytes normally migrate to the tips of the villi and detach into the lumen where they undergo anoikis. Early in this process, the loss of membrane E-cadherin has been shown to be necessary for cell detachment and shedding.24 However, rather than undergoing anoikis, the nonadherent Tsc2(−/−) cells acquire the molecular and functional characteristics of EMT that make them resistant to anoikis and display cell scattering and anchorage-independent growth. Human TSC pathological conditions, AML and LAM, show expression of EMT markers including fibronectin, Snail, and TGF-β and display an invasive growth pattern. We propose that some of the unique features found in TSC pathological conditions may be explained by EMT. However, the cell of origin for these lesions is not known currently, and therefore the hypothesis cannot be directly tested.
The mislocalization of membrane E-cadherin in the Tsc2-mutant cells is consistent with our earlier observations that protein trafficking is defective because of the loss of TSC2 function. We reported that post-Golgi transport of vesicular stomatitis virus G is impaired in the Tsc2(−/−) cells,25 and consequently, plasma membrane-bound proteins such as caveolin-1 accumulate in the cytosol. This defect is reversible with rapamycin treatment and is dependent on the mTOR effector, CLIP170, and its effects on microtubule organization.21 Recently, we reported that growth factor-stimulated membrane translocation of hexose transporters including Glut4 is also impaired in the Tsc2(−/−) cells, leading to a defect in glucose uptake that is consistent with the clinical observations of reduced fluorodeoxyglucose uptake on positron emission tomography imaging in TSC and LAM.26 In the current study, we add E-cadherin to the list of proteins whose localization is affected by the Akt/mTOR/CLIP170 pathway. The transport of E-cadherin to the plasma membrane has been shown to be dependent on intact microtubules such that pretreatment with nocodazole results in the cytosolic retention of E-cadherin.31 However, the Tsc2-null cells do not exhibit a global defect in membrane transport. For example, other cell adhesion molecules including integrin β1, ZO-1, and claudin-1 were properly localized at the plasma membrane of the Tsc2(−/−) cells. The nature and extent of the proteins whose transport are dependent on the TSC2 pathway remain incompletely defined.
Lu et al27 reported that epidermal growth factor stimulates the internalization of membrane E-cadherin through a caveolae-dependent mechanism, leading to loss of cell-cell adhesion, activation of β-catenin, and the induction of EMT. Our findings are consistent with this model and provide a plausible link between EGF and E-cadherin localization through the activation of Akt/mTORC1. We have previously found that TSC1/TSC2 interacts with the β-catenin degradation complex to modulate its activity.32,33 Here, the loss of membrane E-cadherin may further contribute to the aberrant β-catenin activity in these cells. Indeed, we recently reported that the nonadherent Tsc2-null cells acquire an invasive phenotype that is mediated via β-catenin-dependent, matrix metalloproteinase (MMP) 7 activity.34 As the Tsc2(−/−) cells detach, β-catenin undergoes caspase-dependent cleavage, resulting in transcriptionally active products. For example, an ∼70-kDa truncated β-catenin expressed by the Tsc2-null cells was shown to be a strong coactivator of MMP7 transcription, and the secreted MMP7 promoted their invasive behavior.34 These observations fit nicely with the role of TSC2 in EMT. Because mTORC1 is frequently activated in human cancers, the relevance of our finding deserves further investigation in the context of tumor invasion and metastasis in general.
With respect to the TSC-related disorders, the unique patterns of differentiation and biological behavior observed in LAM and AML have not been adequately explained on the basis of our current knowledge. Findings in this study support an EMT phenotype in human AML and LAM. Functionally, EMT provides a means by which the TSC2-mutant cells can invade and migrate as suggested by the “metastatic” hypothesis of LAM.10 However, a major piece of the puzzle remains unknown: the cellular origin of LAM and AML. Although both disorders are classified as perivascular epithelioid cell neoplasms, there is no normal counterpart for the perivascular epithelioid cell.9 Based on the immunophenotypic profile of these lesions, we speculate that their “epithelial” origin may stem from the neural crest. In vertebrates, the neural crest undergoes EMT to give rise to diverse structures including the peripheral nervous system, melanocytes, facial structures, and some endocrine cells.35 One of the most distinctive characteristics of AML and LAM is their expression of melanocytic proteins such as HMB45/gp100 and Melan-A. AML also expresses other neural crest markers, including NG2 and L1, consistent with this hypothesis.36 The loss of TSC2 in neural crest epithelial cells could potentially promote EMT, resulting in the invasion and migration of the mutant cells to distant sites such as the lung, kidney, and axial lymphatics.
Irrespective of the epithelial origin of AML and LAM, our findings offer insight into some of their common histological features. First, it is well recognized that aberrant cells in these lesions express smooth muscle actin, but they do not otherwise exhibit characteristics typical of smooth muscle cells.16 Indeed, cells undergoing EMT acquire a mesenchymal phenotype without other features of smooth muscle cells. Our data suggest that SMA expression per se may not signify the origin of the LAM or AML cells as mesenchymal, but rather it may reflect a consequence of EMT. Another consistent observation in LAM is the presence of cellular heterogeneity including two distinct populations: spindle-shaped and epithelioid cells. The spindle cells are more proliferative compared with the larger, HMB-45-expressing epithelioid cells.37 We propose that the plasticity of EMT and the reverse process of mesenchymal-epithelioid transition (MET) provide a plausible explanation for the observed heterogeneity. Cells that have undergone EMT acquire a spindle-cell appearance and become more mobile and proliferative, consistent with the preferential Snail expression in these cells. Once the spindle cells have infiltrated the lung parenchyma or other sites, they may undergo MET to become more quiescent with an epithelioid appearance. Both EMT and MET are critical events in the development of organs such as the kidney and ovary as well as cancers.18,38 Whether the phenotypic differences between the two LAM cell populations can be fully explained by the EMT-MET hypothesis remains to be determined. Furthermore, it will be intriguing to investigate whether Snail expression in LAM is associated with clinical outcomes, given that the prognosis of a number of human carcinomas has been shown to correlate with the expression of the Snail family of transcription factors.39–41
Studies have begun to examine the role of mTORC1 inhibition in AML and LAM. Bissler et al42 reported the use of sirolimus in 25 patients with encouraging results. Among those with LAM, there was a significant improvement in pulmonary function while they received sirolimus, but their clinical performance declined toward baseline in the ensuing year of observation (ie, “off” drug). The study provides proof-of-principle that lung function in LAM may be reversible with mTORC1 inhibition. Our data show that the exposure of the adherent Tsc2(−/−) cells to rapamycin significantly reduced the ability of cells to detach, thereby preventing EMT. Thus, our results predict that rapamycin and its analogs would be effective in the prevention of LAM in the at-risk population. However, once the cells undergo EMT, rapamycin does not effectively reverse the process. Therefore, the timing of rapamycin therapy may be an important determinant of success.
In summary, our study suggests a functional link between the TSC2/mTORC1 pathway, E-cadherin localization, and EMT. The clinicopathological observations in LAM as illustrated by the infiltrative growth pattern, metastatic origin, and altered cell differentiation are consistent with this model. Future investigations will focus on defining the role of EMT and MET in LAM, the “neural crest” EMT hypothesis, and the cross-talk between the mTOR and TGFβ pathways in TSC-related pathology. Moreover, given the relevance of Akt/mTOR signaling in human diseases, our findings highlight a potential use of rapamycin and its analogs in preventing EMT that may favorably reduce tumor metastases or attenuate tissue fibrosis. Interestingly, there have been reports suggesting that rapamycin can reduce chronic glomerulosclerosis and liver and cardiac fibrosis in animal models,43–45 but its role in preventing EMT in these settings remains to be defined.
Acknowledgements
We thank Jiangning Li, Ph.D., for valuable assistance with our immunofluorescence analysis. Confocal microscopy was performed at the Keck Microscopy Facility at the University of Washington. LAM tissue was obtained through the University of Washington Medical Center and the National Disease Research Interchange.
Footnotes
Supported in part by National Institutes of Health (grants R01-CA077882 and R21-HL094934) and the LAM Foundation.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Web Extra Material
References
- 1.Gomez MR. In: Definition and criteria for diagnosis. Tuberous Sclerosis Complex. ed 3. Gomez MR, Sampson JR, Whittemore VH, editors. Oxford University Press; New York: 1999. pp. 10–23. [Google Scholar]
- 2.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005;37:19–24. doi: 10.1038/ng1494. [DOI] [PubMed] [Google Scholar]
- 4.Tremblay F, Brule S, Hee Um S, Li Y, Masuda K, Roden M, Sun XJ, Krebs M, Polakiewicz RD, Thomas G, Marette A. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc Natl Acad Sci USA. 2007;104:14056–14061. doi: 10.1073/pnas.0706517104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheadle JP, Reeve MP, Sampson JR, Kwiatkowski DJ. Molecular genetic advances in tuberous sclerosis. Hum Genet. 2000;107:97–114. doi: 10.1007/s004390000348. [DOI] [PubMed] [Google Scholar]
- 6.Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci USA. 2000;97:6085–6090. doi: 10.1073/pnas.97.11.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson SR, Tattersfield AE. Lymphangioleiomyomatosis. Semin Respir Crit Care Med. 2002;23:85–92. doi: 10.1055/s-2002-25298. [DOI] [PubMed] [Google Scholar]
- 8.Finlay G. The LAM cell: what is it, where does it come from, and why does it grow? Am J Physiol Lung Cell Mol Physiol. 2004;286:L690–L693. doi: 10.1152/ajplung.00311.2003. [DOI] [PubMed] [Google Scholar]
- 9.Folpe AL. Neoplasms with perivascular epitheloid cell differentiation (PEComas) In: Fletcher CDM, Unni KK, Mertens F, editors. World Health Organization Classification of Tumors. Pathology & Genetics: Tumors of Soft Tissue and Bone. IARC Press; Lyon, France: 2002. pp. 221–222. [Google Scholar]
- 10.Henske EP. Metastasis of benign tumor cells in tuberous sclerosis complex. Genes Chromosomes Cancer. 2003;38:376–381. doi: 10.1002/gcc.10252. [DOI] [PubMed] [Google Scholar]
- 11.Goncharova EA, Goncharov DA, Lim PN, Noonan D, Krymskaya VP. Modulation of cell migration and invasiveness by tumor suppressor TSC2 in lymphangioleiomyomatosis. Am J Respir Cell Mol Biol. 2006;34:473–480. doi: 10.1165/rcmb.2005-0374OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crooks DM, Pacheco-Rodriguez G, DeCastro RM, McCoy JP, Jr, Wang JA, Kumaki F, Darling T, Moss J. Molecular and genetic analysis of disseminated neoplastic cells in lymphangioleiomyomatosis. Proc Natl Acad Sci USA. 2004;101:17462–17467. doi: 10.1073/pnas.0407971101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bittmann I, Dose TB, Muller C, Dienemann H, Vogelmeier C, Lohrs U. Lymphangioleiomyomatosis: recurrence after single lung transplantation. Hum Pathol. 1997;28:1420–1423. doi: 10.1016/s0046-8177(97)90233-1. [DOI] [PubMed] [Google Scholar]
- 14.Bittmann I, Rolf B, Amann G, Lohrs U. Recurrence of lymphangioleiomyomatosis after single lung transplantation: new insights into pathogenesis. Hum Pathol. 2003;34:95–98. doi: 10.1053/hupa.2003.50. [DOI] [PubMed] [Google Scholar]
- 15.Karbowniczek M, Astrinidis A, Balsara BR, Testa JR, Lium JH, Colby TV, McCormack FX, Henske EP. Recurrent lymphangiomyomatosis after transplantation: genetic analyses reveal a metastatic mechanism. Am J Respir Crit Care Med. 2003;167:976–982. doi: 10.1164/rccm.200208-969OC. [DOI] [PubMed] [Google Scholar]
- 16.Krymskaya VP. Smooth muscle like cells in pulmonary lymphangioleiomyomatosis. Proc Am Thorac Soc. 2008;5:119–126. doi: 10.1513/pats.200705-061VS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, Thompson EW. Epithelial—mesenchymal and mesenchymal—epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213:374–383. doi: 10.1002/jcp.21223. [DOI] [PubMed] [Google Scholar]
- 19.Pertz O, Bozic D, Koch AW, Fauser C, Brancaccio A, Engel J. A new crystal structure. Ca2+ dependence and mutational analysis reveal molecular details of E-cadherin homoassociation. EMBO J. 1999;18:1738–1747. doi: 10.1093/emboj/18.7.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin F, Wienecke R, Xiao GH, Maize JC, Jr, DeClue JE, Yeung RS. Suppression of tumorigenicity by the wild-type tuberous sclerosis 2 (Tsc2) gene and its C-terminal region. Proc Natl Acad Sci USA. 1996;93:9154–9159. doi: 10.1073/pnas.93.17.9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang X, Yeung RS. Regulation of microtubule-dependent protein transport by the TSC2/mammalian target of rapamycin pathway. Cancer Res. 2006;66:5258–5269. doi: 10.1158/0008-5472.CAN-05-4510. [DOI] [PubMed] [Google Scholar]
- 22.Teckchandani A, Toida N, Goodchild J, Henderson C, Watts J, Wollscheid B, Cooper JA. Quantitative proteomics identifies a Dab2/integrin module regulating cell migration. J Cell Biol. 2009;186:99–111. doi: 10.1083/jcb.200812160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wendeler MW, Praus M, Jung R, Hecking M, Metzig C, Gessner R. Ksp-cadherin is a functional cell-cell adhesion molecule related to LI-cadherin. Exp Cell Res. 2004;294:345–355. doi: 10.1016/j.yexcr.2003.11.022. [DOI] [PubMed] [Google Scholar]
- 24.Fouquet S, Lugo-Martinez VH, Faussat AM, Renaud F, Cardot P, Chambaz J, Pincon-Raymond M, Thenet S. Early loss of E-cadherin from cell-cell contacts is involved in the onset of Anoikis in enterocytes. J Biol Chem. 2004;279:43061–43069. doi: 10.1074/jbc.M405095200. [DOI] [PubMed] [Google Scholar]
- 25.Jones KA, Jiang X, Yamamoto Y, Yeung RS. Tuberin is a component of lipid rafts and mediates caveolin-1 localization: role of TSC2 in post-Golgi transport. Exp Cell Res. 2004;295:512–524. doi: 10.1016/j.yexcr.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 26.Jiang X, Kenerson H, Aicher L, Miyaoka R, Eary J, Bissler J, Yeung RS. The tuberous sclerosis complex regulates trafficking of glucose transporters and glucose uptake. Am J Pathol. 2008;172:1748–1756. doi: 10.2353/ajpath.2008.070958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu Z, Ghosh S, Wang Z, Hunter T. Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of β-catenin, and enhanced tumor cell invasion. Cancer Cell. 2003;4:499–515. doi: 10.1016/s1535-6108(03)00304-0. [DOI] [PubMed] [Google Scholar]
- 28.Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932–1940. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
- 29.Kumasaka T, Seyama K, Mitani K, Sato T, Souma S, Kondo T, Hayashi S, Minami M, Uekusa T, Fukuchi Y, Suda K. Lymphangiogenesis in lymphangioleiomyomatosis: its implication in the progression of lymphangioleiomyomatosis. Am J Surg Pathol. 2004;28:1007–1016. doi: 10.1097/01.pas.0000126859.70814.6d. [DOI] [PubMed] [Google Scholar]
- 30.Mak BC, Yeung RS. The tuberous sclerosis complex genes in tumor development. Cancer Invest. 2004;22:588–603. doi: 10.1081/cnv-200027144. [DOI] [PubMed] [Google Scholar]
- 31.Stehbens SJ, Paterson AD, Crampton MS, Shewan AM, Ferguson C, Akhmanova A, Parton RG, Yap AS. Dynamic microtubules regulate the local concentration of E-cadherin at cell-cell contacts. J Cell Sci. 2006;119(Pt 9):1801–1811. doi: 10.1242/jcs.02903. [DOI] [PubMed] [Google Scholar]
- 32.Mak BC, Takemaru K, Kenerson HL, Moon RT, Yeung RS. The tuberin-hamartin complex negatively regulates β-catenin signaling activity. J Biol Chem. 2003;278:5947–5951. doi: 10.1074/jbc.C200473200. [DOI] [PubMed] [Google Scholar]
- 33.Mak BC, Kenerson HL, Aicher LD, Barnes EA, Yeung RS. Aberrant β-catenin signaling in tuberous sclerosis. Am J Pathol. 2005;167:107–116. doi: 10.1016/s0002-9440(10)62958-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barnes EA, Kenerson HL, Mak BC, Yeung RS. The loss of tuberin promotes cell invasion through the β-catenin pathway. Am J Respir Cell Mol Biol. 2009 doi: 10.1165/rcmb.2008-0335OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pla P, Moore R, Morali OG, Grille S, Martinozzi S, Delmas V, Larue L. Cadherins in neural crest cell development and transformation. J Cell Physiol. 2001;189:121–132. doi: 10.1002/jcp.10008. [DOI] [PubMed] [Google Scholar]
- 36.Lim SD, Stallcup W, Lefkove B, Govindarajan B, Au KS, Northrup H, Lang D, Fisher DE, Patel A, Amin MB, Arbiser JL. Expression of the neural stem cell markers NG2 and L1 in human angiomyolipoma: are angiomyolipomas neoplasms of stem cells? Mol Med. 2007;13:160–165. doi: 10.2119/2006-00070.Lim. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonetti F, Pea M, Martignoni G, Zamboni G, Iuzzolino P. Cellular heterogeneity in lymphangiomyomatosis of the lung. Hum Pathol. 1991;22:727–728. doi: 10.1016/0046-8177(91)90298-4. [DOI] [PubMed] [Google Scholar]
- 38.Baum B, Settleman J, Quinlan MP. Transitions between epithelial and mesenchymal states in development and disease. Semin Cell Dev Biol. 2008;19:294–308. doi: 10.1016/j.semcdb.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 39.Elloul S, Elstrand MB, Nesland JM, Trope CG, Kvalheim G, Goldberg I, Reich R, Davidson B. Snail, Slug, and Smad-interacting protein 1 as novel parameters of disease aggressiveness in metastatic ovarian and breast carcinoma. Cancer. 2005;103:1631–1643. doi: 10.1002/cncr.20946. [DOI] [PubMed] [Google Scholar]
- 40.Shioiri M, Shida T, Koda K, Oda K, Seike K, Nishimura M, Takano S, Miyazaki M. Slug expression is an independent prognostic parameter for poor survival in colorectal carcinoma patients. Br J Cancer. 2006;94:1816–1822. doi: 10.1038/sj.bjc.6603193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waldmann J, Feldmann G, Slater EP, Langer P, Buchholz M, Ramaswamy A, Saeger W, Rothmund M, Fendrich V. Expression of the zinc-finger transcription factor Snail in adrenocortical carcinoma is associated with decreased survival. Br J Cancer. 2008;99:1900–1907. doi: 10.1038/sj.bjc.6604755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, Schmithorst VJ, Laor T, Brody AS, Bean J, Salisbury S, Franz DN. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krämer S, Wang-Rosenke Y, Scholl V, Binder E, Loof T, Khadzhynov D, Kawachi H, Shimizu F, Diekmann F, Budde K, Neumayer HH, Peters H. Low-dose mTOR inhibition by rapamycin attenuates progression in anti-thy1-induced chronic glomerulosclerosis of the rat. Am J Physiol Renal Physiol. 2008;294:F440–F449. doi: 10.1152/ajprenal.00379.2007. [DOI] [PubMed] [Google Scholar]
- 44.Neef M, Ledermann M, Saegesser H, Schneider V, Reichen J. Low-dose oral rapamycin treatment reduces fibrogenesis, improves liver function, and prolongs survival in rats with established liver cirrhosis. J Hepatol. 2006;45:786–796. doi: 10.1016/j.jhep.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 45.Gao XM, Wong G, Wang B, Kiriazis H, Moore XL, Su YD, Dart A, Du XJ. Inhibition of mTOR reduces chronic pressure-overload cardiac hypertrophy and fibrosis. J Hypertens. 2006;24:1663–1670. doi: 10.1097/01.hjh.0000239304.01496.83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.