

Abstract
The liver is the major site of pigment epithelium-derived factor (PEDF) synthesis. Recent evidence suggests a protective role of PEDF in liver cirrhosis. In the present study, immunohistochemical analyses revealed lower PEDF levels in liver tissues of patients with cirrhosis and in animals with chemically induced liver fibrosis. Delivery of the PEDF gene into liver cells produced local PEDF synthesis and ameliorated liver fibrosis in animals treated with either carbon tetrachloride or thioacetamide. In addition, suppression of peroxisome proliferator-activated receptor gamma expression, as well as nuclear translocation of nuclear factor-kappa B was found in hepatic stellate cells (HSCs) from fibrotic livers, and both changes were reversed by PEDF gene delivery. In culture-activated HSCs, PEDF, through the induction of peroxisome proliferator-activated receptor gamma, reduced the activity of nuclear factor-kappa B and prevented the nuclear localization of JunD. In conclusion, our observations that PEDF levels are reduced during liver cirrhosis and that PEDF gene delivery ameliorates cirrhosis suggest that PEDF is an intrinsic protector against liver cirrhosis. Direct inactivation of HSCs and the induction of apoptosis of activated HSCs may be two of the mechanisms by which PEDF suppresses liver cirrhosis.
Liver cirrhosis is the result of a prolonged repair process in response to repeated hepatocyte damage of various origins. It involves the activation of hepatic stellate cells (HSCs), the production of extracellular matrix and α-smooth muscle actin (α-SMA) by these cells, and eventual fibrosis and hepatic failure.1 In liver cirrhosis, three cell types, activated HSCs, epithelial cells of regenerating bile ducts, and Kupffer cells, secrete monocyte chemoattractant protein-1 (MCP-1), an inflammatory chemokine that facilitates the recruitment of lymphocytes and monocytes.2,3 MCP-1 produced by activated HSCs increases hepatic injury and fibrosis by intensifying inflammation.3,4 Identification of the intrinsic molecules that modulate HSC activation can increase our understanding of the mechanism by which liver cirrhosis develops and can also lead to the development of new therapeutic agents for the treatment of this condition.
Pigment epithelium-derived factor (PEDF) is a 50-kDa secreted glycoprotein that performs several different biological functions. It is the most potent endogenous anti-angiogenic factor involved in the prevention of retinal neovascularization in mice.5 It is also a neuorotropic and neuroprotective factor, protecting neurons against insults such as glutamate toxicity and oxidative damage.6 The binding of different domains of PEDF to different types of receptor is probably the mechanism responsible for these diverse functions.7
Liver is the major organ that synthesizes PEDF.5,8 Recent findings indicate that PEDF may have play a role during the development of liver cirrhosis. Serum PEDF has been found to be decreased in the serum of cirrhosis patients.8 PEDF null mice have been shown by perisinusoidal α-SMA labeling to have activated HSCs.9 These findings suggest a potential role for PEDF as an intrinsic protector against liver cirrhosis. The confirmation of this hypothesis awaits experimental evidence of a reduction of PEDF synthesis in fibrotic liver tissue, an ability of PEDF to counteract the cirrhosis process, and a possible mechanism for its effects on cirrhosis.
Peroxisome proliferator-activated receptor gamma (PPARγ) is a transcription factor that heterodimerizes with retinoid X receptor-α and activates genes involved in lipid homeostasis.10 It can be activated by a variety of ligands, including fatty acids and eicosanoids.10 PPARγ is expressed in quiescent HSCs, and its expression and activity dramatically decrease in myofibroblast-like, activated HSCs in vitro11,12 and in vivo.11,13 In culture-activated HSCs, the expression of PPARγ via an adenoviral vector prevents cell proliferation as well as the production of alpha1(I) collagen, α-SMA, and transforming growth factor-beta1.12 The inhibition of transcription regulator JunD function has been implicated as the mechanism of this effect.12 We have recently shown that PEDF induces PPARγ expression in endothelial cells and that this action is responsible for the anti-angiogenesis action of PEDF.14,15 Whether PEDF induces PPARγ in HSCs and affects the process of liver cirrhosis remains to be determined.
Nuclear factor (NF)-κB is a nuclear transcription activator of genes controlling survival and inflammation.16 MCP-1 is one of the inflammatory factors induced partly by NF-κB.17 Studies using NF-κB inhibitors demonstrate that NF-κB protects HSCs from apoptotic stimuli such as tumor necrosis factor-α (TNF-α) and gliotoxin.18,19 NF-κB induces cellular FLICE-inhibitory protein (c-FLIP) and Bcl-xL and is crucial for inhibition apoptosis induced by TNF-related apoptosis-inducing ligand (TRAIL).20 NF-κB is prevented from the nuclear translocation necessary for its action by its association with cytoplasmic inhibitory proteins, such as IκBα. In response to stimulation, IκB is degraded, leading to the release and nuclear translocation of NF-κB.16
The current study provides two lines of evidence supporting the notion that PEDF is an intrinsic protector against liver cirrhosis. One line of evidence is that production of PEDF by hepatocytes is reduced during cirrhosis and that exogenous PEDF results in partial amelioration of cirrhosis. The other is that the mechanism of this protective effect in an animal model appears to be that PEDF induces HSC inactivation, and that PPARγ participates in the signaling of PEDF through a complex interaction involving the nuclear translocation of NF-κB and JunD.
Materials and Methods
Animal Treatment
To induce liver fibrosis, 6-week-old BALB/c mice (six mice per experimental condition) were injected intraperitoneally twice a week with either CCl4 solution (5 ml/kg body weight as a 1:4 mixture with olive oil) or thioacetamide (TAA) (200 mg/kg) for 3 weeks. Experimental procedures were approved by the Mackay Memorial Hospital Review Board and conducted according to national animal welfare regulations.
Generation of Adeno-Associated Virus PEDF
Adeno-associated virus PEDF (AAV-PEDF) was constructed and purified by the methods of Xiao X et al21 In brief, human PEDF and enhanced green fluorescent protein (EGFP) cDNAs were obtained from plasmid pET-15b-PEDF and p-EGFP-N1 (Clontech, Mountain View, CA), respectively.22 The PEDF cDNA was subcloned into the BamHI/NotI site of the AAV packaging plasmid (pAAV-D+), allowing transcription of PEDF to be driven by the cytomegalovirus promoter. For generation of recombinant AAV, pAAV-D (+)-hPEDF, and AAV helper plasmids (pXR5 and pXX6) were cotransfected into human embryonic kidney 293T cells. This cloning procedure was also used to generate AAV-EGFP. The expression of AAV-PEDF in 293T cells was confirmed by PCR and immunoblotting. The AAV particles were produced by multiple freeze/thaw cycles and cesium chloride density gradient purification. Titers were determined by dot-blot assay in the range of 1.0 to 3.0 × 1013 viral particles/ml. The expression of AAV was examined by immunoblotting of liver proteins from mice injected through the tail vein with AAV-PEDF (2 × 1012 viral particles). Prominent human PEDF expression was found 2 weeks after AAV infection.
Cell Isolation, Culture, and Treatment
Primary rat HSCs were isolated by in situ portal vein perfusion with collagenases from livers of male Sprague-Dawley rats (300 to 450 g). All procedures were performed with the animals under sodium pentobarbital anesthesia. Rat livers were perfused with oxygenated PB-1 solution (4.7 mmol/L KCl, 1.2 mmol/L KH2PO4, 118 mmol/L NaCl, 25 mmol/L NaHCO3, 0.5 mmol/L EGTA, 5.5 mmol/L glucose, pH 7.4) at a flow rate of 35 ml/min for 15 minutes at 37°C, then with oxygenated PB-1 solution containing 2 mmol/L CaCl2, collagenase type I and IV (∼ 90 U/ml; Gibco BRL, Rockville, MD) and 0.001% DNase I (10U/μL; Roche Molecular Biochemicals, Indianapolis, IN) for 20 minutes, followed by oxygenated Ca2+-containing PB-1 solution without collagenases for 5 minutes. This suspension was filtered through nylon gauze (mesh size: 106 μm) and centrifuged 2 times at 40 × g for 5 minutes at room temperature to remove the hepatocytes. The supernatant was centrifuged at 800 × g for 10 minutes at 4°C and then cell pellet was resuspended in 5 ml of PBS, and layered carefully over a 30 ml of 25%/50% Percoll (Sigma, St. Louis, MO), and centrifuged at 800 × g for 15 minutes at 4°C. The cell fraction in the 25% Percoll was gently aspirated, mixed with PBS, and centrifuged at 800 × g for 7 minutes at 4°C by a previously described method.23 After PBS wash for two times, the final cell pellet was resuspended in Dulbecco's modified Eagle's medium (DMEM) supplemented with 20% fetal bovine serum (FBS) and 1% penicillin/streptomycin, and plated on 100-mm plastic dishes. After plating for 24 hours, nonadherent cells and debris were removed by PBS washing and cells were then cultured in 10% FBS-DMEM. Cell purity was verified to be approximately 95% to 98% by vitamin A fluorescence at day 2 after isolation (representative pictures are shown in supplemental Figure S1 at http://ajp.amjpathol.org). HSCs were activated by culture on plastic for 10 days and then passaged by trypsin-EDTA treatment. Subsequently, the activated HSCs were incubated in 10% FBS-DMEM for 2 days and then used for experiments.
HSC-T6 cells, a rat immortalized HSC cell line, retain all features of activated HSCs and were kindly provided by Dr. SL Friedman.24 Cells were grown in Waymouth medium supplemented with 10% FBS at 37°C in a humidified atmosphere of 5% CO2. Recombinant human PEDF derived from stable baby hamster kidney cell transfectants and preserved in 50 mmol/L sodium phosphate buffer (solvent) was obtained from Chemicon (Temecula, CA). Treatments with PEDF (200 ng/ml, unless differently specified), TNF-α, G3335, GW9662, or SN50 (all from Calbiochem, San Diego, CA) were performed after cells were switched to serum-free medium.
Immunohistochemistry
Formalin-fixed, paraffin-embedded, liver tissue arrays (LV805; Biomax, Inc., Rockville, MD) or mice liver specimens were deparaffinized in xylene and rehydrated in a graded series of ethanol concentrations. Slides were blocked with 10% goat serum for 60 minutes and then incubated with primary antibody against human PEDF (1:200 dilution; sc-59641), mouse PEDF (1:200 dilution; sc-74253; Santa Cruz Biotechnology, CA), or CD68 (1:200 dilution; Abcam, Cambridge, MA) overnight at 4°C. The slides were subsequently incubated with the appropriate peroxidase-labeled goat immunoglobulin (1:500 dilution; Chemicon, Temecula, CA) for 20 minutes and then incubated with chromogen substrate (3,3′-diaminobenzidine) for 2 minutes before counterstaining with hematoxylin. Quantification was estimated based on high quality images (1208 × 960 pixels buffer) captured using a Nikon Eclipse 80i light microscope. The total PEDF density score (image intensity/image area) was determined using Image-Pro Plus 4.5.1 software (Media Cybernetics).
Sirius-Red Staining
Deparaffinized liver tissue sections were stained for 1 hour in 0.1% (w/v) Sirius red (Sigma, St. Louis, MO) in a saturated aqueous solution of picric acid, and then rinsed for 30 minutes in 0.01 N acetic acid to remove unbound dye. For semiquantitative analysis of liver fibrosis, 10 fields from each slide were randomly selected under a light microscope, and the red-stained area per total area (mm2/mm2) was measured using the Image-Pro Plus 4.5.1 system.
Immunofluorescence
Deparaffinized liver tissue sections or 4% paraformaldehyde-fixed HSCs were blocked with 10% goat serum and 5% bovine serum albumin for 1 hour. Dual staining was done using primary antibodies against α-SMA (1:100 dilution; ab5694 or ab7817; Abcam), NF-κB p65 (1:200 dilution; #3034; Cell Signaling Technology, Beverly, MA), PPARγ (1:200 dilution; sc-7273; Santa Cruz Biotechnology, CA), or JunD (1:200 dilution; sc-74; Santa Cruz Biotechnology, CA) at 37°C for 2 hours, followed by incubation with the appropriate rhodamine- or fluorescein isothiocyanate-conjugated goat IgG (1:500 dilution; Chemicon, Temecula, CA) for 1 hour at room temperature. Nuclei were located by counterstaining with Hoechst 33258 for 7 minutes. Images were captured using a Zeiss epifluorescence microscope with a CCD camera. To detect nuclear localization of p65 in HSCs of CCl4-treated mice liver, images were captured using a confocal microscope (SP5; Leica) and processed using Advanced Fluorescence software (Leica Application Suite, version 1.6.3).
Semi-Quantitative Reverse Transcriptase PCR
Total RNA was extracted from cells using TRIzol (Invitrogen, Carlsbad, CA). Synthesis of cDNA was performed with 1 μg of total RNA at 50°C for 50 minutes using oligo (dT) primers and reverse transcriptase (RT; Superscript III; Invitrogen) following the manufacturer's instructions. cDNA was equalized in an 18- to 26-cycle amplification reaction (denaturation, 20s, 94°C; annealing, 30s, 57°C; and polymerization, 40s, 72°C) with mouse PEDF primers 5′-CCTCTGTTACTGCCCCTGAG-3′ (forward)/5′-GCCTGCACCCAGTTGTTAAT-3′ (reverse) and rat PPARγ primers 5′-CCCTGGCAAAGCATTTGTAT-3′ (forward)/5′- ACTGGCACCCTTGAAAAATG-3′ (reverse) yielding 177-bp and 221-bp products, respectively. For Bcl-xl and c-FLIP experiments, rat Bcl-xl primers [5′- CCCCAGAAGAAACTGAACCA-3′ (forward)/5′-TGCAATCCGACTCACCAATA-3′ (reverse)] and rat c-FLIP primers [5′-CATTCACCAGGTGGAGGAGT-3′ (forward)/5′- CGGCCTGTGTAATCCTTTGT-3′ (reverse)] were used. The number of cycles for the primer set was selected to be in the linear range of amplification. The PCR products were electrophoresed in a 2% agarose gel containing ethidium bromide and visualized by UV illumination. The intensities of the PCR products were quantified using a FUJI LAS-3000 densitometer and Multi Gauge Ver. 1.01 software (Fujifilm, Tokyo, Japan).
Immunoblot Analysis
Cells were scraped into lysis buffer (150 μl/35-mm well) containing 20 mmol/L HEPES (pH 7.4), 1% SDS, 150 mmol/L NaCl, 1 mmol/L EGTA, 5 mmol/L β-glycerophosphate, 10 mmol/L sodium pyrophosphate, 10 mmol/L sodium fluoride, 100 μmol/L sodium orthovanadate, 10 μg/ml leupeptin, and 10 μg/ml aprotinin. The lysate was incubated on ice for 15 minutes. Total cell lysate was also separated into cytoplasmic and nuclear fractions using the NE-PER nuclear and cytoplasmic extraction kit (Pierce, Rockford, IL) according to the manufacturer's instructions. Each cellular fraction was then resolved by SDS-polyacrylamide gel electrophoresis, electrotransferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA), and processed for immunoblot analysis. Antibodies used in the immunoblot study included type I collagen 1A1, histone HI, JunD, PGC-1, Bax, BclII, Bid, Bad, c-FLIP, Bcl-xL (1:1000 dilution; all from Santa Cruz Biotechnology), p-p65 Ser536 (1:500 dilution; 93H1; Cell Signaling Technology), and MCP-1 (1:1000 dilution; Abcam Ltd). Proteins of interest were detected using the appropriate IgG-HRP secondary antibody and enhanced chemiluminescence reagent. X-ray films were scanned on a Model GS-700 Imaging Densitometer (Bio-Rad Laboratories, Hercules, CA) and analyzed using Labworks 4.0 software. For quantification, blots from at least three independent experiments were used.
Actin Staining
After treatment, primary rat HSCs were fixed for 2 hours with 4% paraformaldehyde, washed by PBS and then permeabilized for 15 minutes with 0.1% Triton X-100. Changes in F-actin structures were detected by 0.33 mmol/L rhodamine-conjugated phalloidin (Sigma) for 20 minutes at room temperature. Stained F-actin was visualized using a Zeiss Axiovert 25 microscope.
PPARγ Small Interfering RNA Treatment
Subconfluent HSC-T6 cells were transfected with a rat PPARγ small interfering (si)RNA (sense sequence: 5′-CACCAUUUGUCAUCUACGAtt-3′) or a mixture of four rat PPARγ siRNAs (SMART-pools; Dharmacon Research, Inc., Lafayette, CO) using INTERFERin siRNA transfection reagent (PolyPlus-Transfection, San Marcos, CA) according to the manufacturer's instructions. The final concentration of siRNA was 10 nmol/L. A species-specific siCONTROL nontargeting siRNA (Dharmacon) was used as a negative control. At 16 hours after siRNA transfection, cells were resuspended in new media for a 24-hour recovery period.
NF-κB Activity Assay
Activated HSCs (1 × 105/well of 6-well plate) were treated with PEDF for 24 hours in serum-free DMEM. After that, NF-κB p50/p65 transcription factor colorimetric assay (SGT510, Chemicon, Temecula, CA) was used to measure the active NF-κB in nuclear extracts following the manufacturer's instructions. Briefly, nuclear extracts from HSCs were prepared using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Pierce, Rockford, IL). Double-stranded biotinylated oligonucleotides containing the consensus sequence for NF-κB binding (5′-GGGACTTTCC-3′) were mixed with nuclear extract and assay buffer. After incubation, the mixture was transferred to a streptavidin-coated enzyme-linked immunosorbent assay kit, processed following manufacturer's instructions, and read at 450 nm using a Spectra MAX GEMINI Reader (Molecular Devices). The specificity of binding was confirmed by competition with unlabeled oligonucleotides.
Evaluation of Apoptosis in Vitro and in Vivo
Activated HSCs (2 × 104/well of 12-well plate) were treated with PEDF for 16 hours in serum-free DMEM and subsequently incubated for another 24 hours in the presence or absence of TNF-α (10 ng/ml). The HSCs were then fixed in 4% (w/v) paraformaldehyde for 16 hours at 4°C and stained using a TdT-mediated dUTP nick-end labeling (TUNEL)–based kit (Roche Molecular Biochemicals, Indianapolis, IN) according to the manufacturer's instructions. The cell number was monitored by counterstaining with Hoechst 33258. The number of nuclei was calculated in ten randomly selected fields of the three different chambers by a Zeiss epifluorescence microscope. To identify apoptosis of activated HSC in vivo, immunofluorescence staining of apoptotic cells was performed using a TUNEL–based kit (Roche). Staining of α-SMA was performed as previously described.25 Sections were observed under a Zeiss epifluorescence microscope (× 200, 10 fields/sample). Images were recorded on Zeiss software.
Immunoprecipitation
HSC-T6 cells (2 × 105/well in 6-well plates) were incubated for 1 day in 10% FBS- Waymouth medium, and then treated with PEDF for 24 hours in serum-free Waymouth medium. After this treatment, cells were lysed in buffer A (10 mmol/L HEPES pH 7.9, 1.5 mmol/L MgCl2, 10 mmol/L KCl, 0.5% Nonidet P-40 and protease inhibitor cocktail). After 15 minutes on ice, nuclei were cleared by centrifugation and agitated with Protein G Sepharose 4B (Sigma) for 1 hour at 4°C. Pre-cleared lysates were incubated with anti-PPARγ antibody (20 μg; sc-7273; Santa Cruz Biotechnology) or NF-κB p65 (5 μg; #3034; Cell Signaling Technology), and Protein G Sepharose at 4°C for 4 hours. Immunoprecipitates were washed five times with buffer A and processed for SDS-polyacrylamide gel electrophoresis.
Statistics
Results are mean ± SEM, analysis of variance, and linear regression were used for statistical comparisons. P < 0.05 was considered significant.
Results
Reduction of PEDF Protein in Human Cirrhotic Liver
To investigate PEDF expression in cirrhotic liver, tissue array slides from 25 samples of normal liver tissue (NL), 23 samples of cirrhotic tissue (LC), and 7 liver tissue samples from patients infected by hepatitis B virus (HL) were analyzed by immunohistochemistry using anti-human PEDF antibody. Results showed that the PEDF signal was clearly stronger in NL than in LC (Figure 1A, cases 1 and 2, as compared to cases 3 and 4). The intensity of the PEDF signal in NL, LC, and HL was 5.3 ± 1.0, 2.3 ± 1.3, and 4.0 ± 1.8, respectively (Figure 1B). All LC showed prominent collagen deposition in the portal area, as confirmed by Sirius-red staining. Two LC were from patients with hepatitis B virus infection, and their PEDF intensity was 3.3.±0.4 and 3.1 ± 0.5. Of the 25 NL cases, the six showing no Sirius-red staining (Figure 1A, case 1) had a higher average PEDF signal (> 5.3 ± 1.0) than the rest of the NL cases, which showed sporadic Sirius-red staining (Figure 1A, case 2). However, the small case number limits statistical analysis of this trend. PEDF intensity in 5 out of the 7 HL was lower than those of NL, although the degree of Sirius-red staining was similar to NL. There was no difference in PEDF protein levels when tissues were compared according to age and sex.
Figure 1.
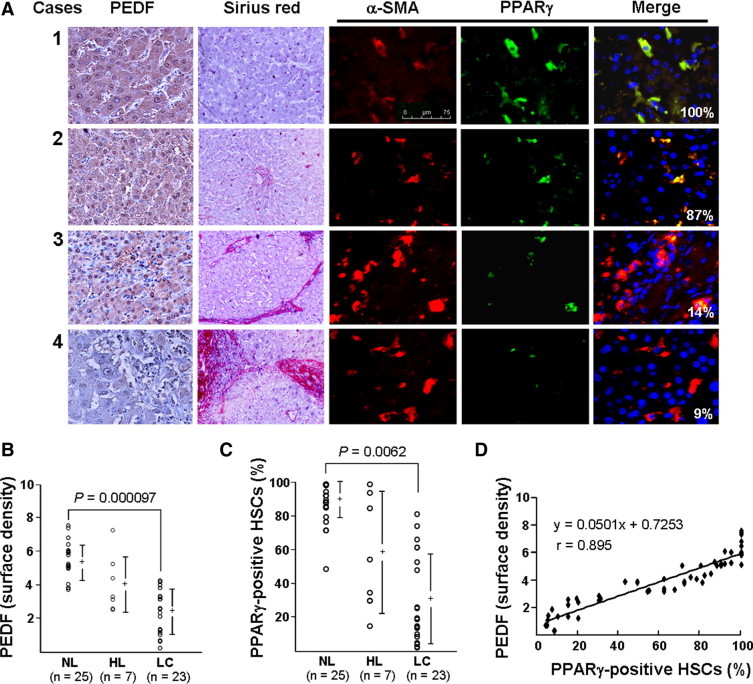
Expression of PEDF and PPARγ in human liver specimens. A: Consecutive sections of liver specimens from normal livers (cases 1 and 2) and cirrhosis livers (cases 3 and 4) are shown in horizontal rows. Immunohistochemical staining of PEDF (brown) and morphometric analyses of liver fibrosis by staining for collagen using Sirius-red are shown (original magnification, ×100). HSCs (α-SMA, red) and PPARγ (green) were detected by immunofluorescence microscopy (original magnification, ×400). PPARγ-positive HSCs (yellow; merge) identified as nuclei stained blue with Hoechst 33258 and cytoplasm stained red with α-SMA were counted using the Image-Pro Plus 4.5.1 computer program. Percentages of PPARγ-positive HSCs of representative pictures are shown. Case 4 shows negative staining for hepatic PEDF and faint staining for HSC PPARγ. B: Relative levels of PEDF in normal liver (NL) were higher than those seen in either HBV-induced inflammatory liver (HL) or cirrhotic liver (LC). Results of anti-PEDF-stained liver sections are expressed as the surface density. “+” represents median and a vertical bar represents SD (SD). C: The percentage of PPARγ-positive HSCs in NL was higher than in HL and LC. Three different researchers, blind to the experimental procedures, determined PPARγ-positive HSCs, which were used as the mean for analysis. D: Correlation between hepatic PEDF protein and PPARγ-positive HSCs in human liver specimens.
PPARγ Levels in HSC Correlate with Levels of Hepatic PEDF Protein
To investigate the expression of PPARγ in HSCs of cirrhotic liver, the same tissue array slides were examined by dual-immunofluorescent staining for PPARγ (green) and α-SMA (red). As shown in Figure 1A, α-SMA-positive HSCs that exhibited typically elongated cytoplasm and the numbers were increased in LC had an obvious reduction of PPARγ content (cases 3 and 4) compared to that seen in NL slices (cases 1 and 2). In addition, PPARγ was expressed mainly in HSCs and only rarely in hepatocytes or other sinusoid cells. The percentage of PPARγ-positive HSCs in NL was 88 ± 12%, whereas in LC and HL it was 32 ± 26% and 59 ± 34%, respectively (Figure 1C). These data indicate that PPARγ expression in HSC is reduced in human cirrhotic liver. Paired correlation analysis showed a positive correlation between hepatic PEDF levels and the fraction of PPARγ-positive HSCs (Figure 1D, r = 0.895).
Carbon Tetrachloride-Induced Liver Damage Reduces PEDF Levels
To verify the change of PEDF expression in liver cirrhosis, a CCl4–induced mouse liver fibrosis model was used. Immunoblotting combined with RT-PCR analyses showed that immediately after the first intraperitoneal injection with CCl4, hepatic PEDF levels decreased markedly compared to control levels. PEDF levels were partially recovered 7 days later (Figure 2A). We also noted that the initiation of the recovery of PEDF mRNA levels (at day 2) occurred earlier than recovery of protein levels (at day 7) (compare Figure 2A to 2B), a finding suggesting that PEDF protein expression was also regulated in some way at the post-transcriptional level. Moreover, immunoblotting and immunohistochemistry showed that PPARγ levels declined along with the reduction in PEDF when mice were exposed to CCl4 for 1 to 5 days and recovered partially at day 7 after CCl4 injection (Figure 2, A and C).
Figure 2.
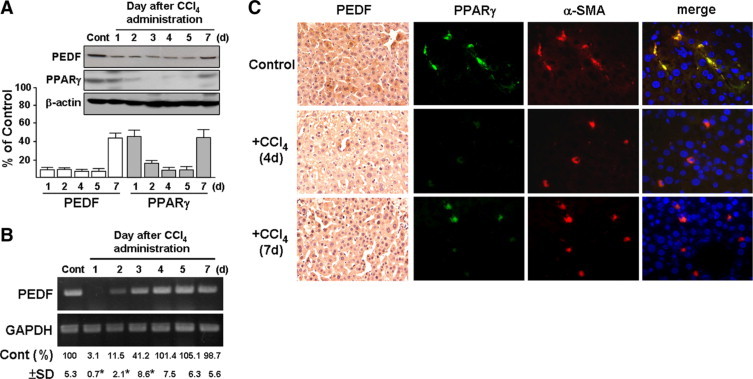
Time-course analysis of liver PEDF and PPARγ protein expression levels in mice treated with CCl4. Mice were injected once with CCl4 and euthanized after one to seven days as indicated. Olive oil treatment was used as a control (Cont). A: Representative immunoblots and densitometric analyses with SD are shown. β-actin was used as a loading control. B: RT-PCR analysis of PEDF. Representative results and quantification of PEDF RNA bands normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) are shown. *P < 0.005 versus control. C: Representative images from consecutive sections of a liver specimen show mouse PEDF (brown) and PPARγ-positive HSCs (yellow) stained by α-SMA (red) and PPARγ (green), respectively. Original magnification, ×400.
PEDF Prevents CCl4- and Thioacetamide-Induced Hepatic Fibrosis
To increase PEDF in the liver, human PEDF gene was delivered into liver cells by an AAV vector. In control experiments, the same vector was used to deliver the EGFP gene. After mice were given intraperitoneal CCl4 injections twice per week for 3 weeks, Sirius red and hematoxylin-stained liver slices from these animals showed marked parenchymal injury and bridging fibrosis (Figure 3A). When mice were injected with AAV-PEDF for 1 week before an additional 3 weeks of CCl4 treatment, immunofluorescence staining using an anti-human PEDF antibody showed PEDF gene delivery by this method to achieve local synthesis of PEDF (green) in hepatocytes(Figure 3B). At the same time, reduced parenchymal injury and smaller areas of fibrosis were observed compared to that seen in the CCl4+AAV-EGFP control group (Figure 3C; 1.1 ± 0.5% versus 6.9 ± 0.9%).
Figure 3.
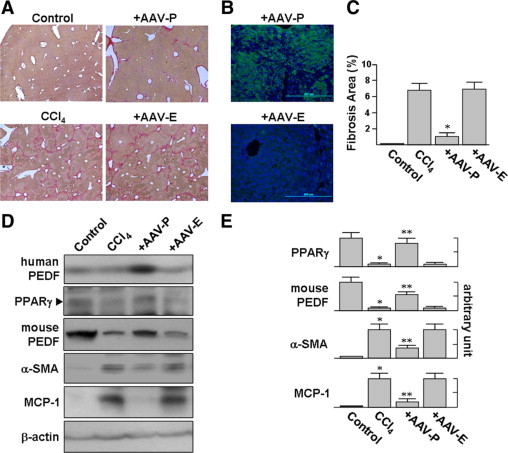
AAV-PEDF protects mice from CCl4-induced hepatic fibrosis. Mice were injected with AAV-PEDF for 1 week and then treated with CCl4 twice per week for 3 weeks. A: There are fewer fibrotic septa, stained by Sirius-red and hematoxylin, in the livers of AAV-PEDF-infected mice (original magnification, ×40). B: Immunofluorescence analysis of liver tissue shows the expression of PEDF in AAV-PEDF-injected animals (original magnification, ×200). C: The average percentages of the areas stained positive with Sirius-red in AAV-PEDF-injected animals are lower than those of the control group. *P < 0.05 versus AAV-EGFP. D and E: AAV-PEDF reduced the accumulation of cirrhosis-related proteins in CCl4-treated mice as evident from the immunoblot analysis of liver lysates with indicated antibodies (D). Representative blots (D) and densitometric analyses (E) from four independent experiments are shown. *P < 0.001 versus control; **P < 0.001 versus AAV-EGFP.
To determine whether the reduced fibrosis seen in mice infected with AAV-PEDF represented suppressed HSC activation, we evaluated the levels of α-SMA and MCP-1 in liver protein extracts (Figure 3, D and E). AAV-PEDF injection diminished α-SMA and MCP-1 levels by a factor of 3.1 and 5, respectively, compared to the levels seen in the CCl4+AAV-EGFP group. AAV-PEDF pretreatment also significantly prevented the decline in PPARγ levels seen in CCl4-treated mice.
The protective effect of AAV-PEDF was also reproducible in TAA-induced liver fibrosis, as indicated by the reduced Sirius-red staining and partial repression of TAA-induced α-SMA and MCP-1 expression seen in liver sections from TAA-treated mice (supplemental Figure S2 at http://ajp.amjpathol.org).
It has been established that mice treated with CCl4 over 4 weeks show a higher grade of inflammation, accompanied by macrophage infiltration.26,27 Immunohistochemistry analysis, using anti-CD68 antibody, of liver sections from mice that had received CCl4 injections for 5 weeks showed an obvious macrophage infiltration in the injured area (supplemental Figure S3 at http://ajp.amjpathol.org). However, liver sections of mice that had received AAV-PEDF displayed a reduction in macrophage infiltration in the injured area compared with the CCl4 or CCl4+AAV-EGFP groups. This observation is consistent with an inhibitory effect of AAV-PEDF on CCl4-induced parenchymal injury and MCP-1 protein expression (Figure 3, A and D). These data suggest that AAV-PEDF may ameliorate inflammation and fibrosis.
PEDF Suppresses the Activation of Cultured HSCs
The protective effect of PEDF against liver fibrosis described above suggests that PEDF prevents the activation of HSCs. This was indeed observed in culture-activated primary rat HSCs, where PEDF treatment for 48 hours significantly reduced α-SMA, Col1a1, and MCP-1 expression compared to solvent treatment alone (Figure 4A).
Figure 4.
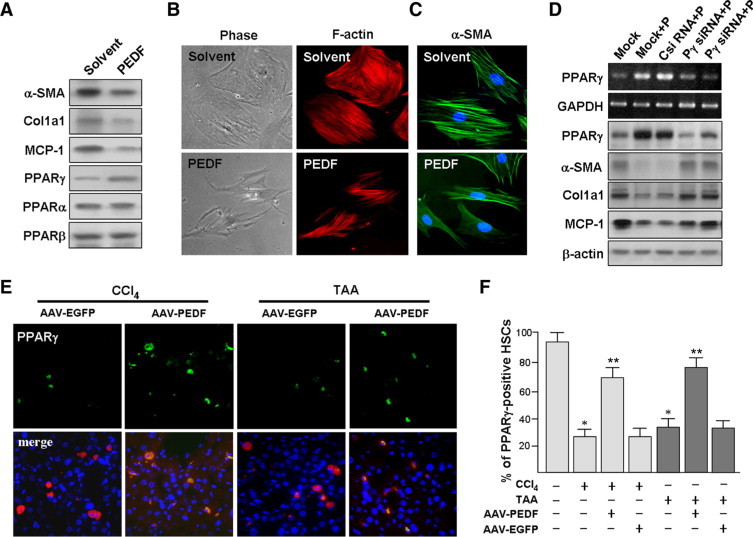
PEDF suppresses primary rat HSC activation. A: Immunoblot analysis of HSC lysates with antibodies as indicated. B: Left panels: Representative phase-contrast micrographs show the modification of culture-activated HSC morphology after PEDF treatment for 48 hours. Right panels: The filamentous actin of cells corresponding to those shown in the left panels was stained with rhodamine-conjugated phalloidin. Original magnification, ×200. C: Immunofluorescence analysis of the expression of α-SMA (green) in HSCs. DNA was visualized with Hoechst 33258 staining. Representative PEDF panels show that PEDF treatment causes a reversal of the activated HSC morphology. D: The inhibitory effect of PEDF on HSC activation is reversed by PPARγ siRNA (Pγ siRNA). HSC-T6 cells were pretreated with siRNAs (C siRNA, control) for 16 hours before exposure to PEDF (P) for an additional 48 hours, and cells were then processed for RT-PCR analysis (blots 1 and 2) and immunoblot analyses (blots 3 to 7). “Mock” indicates transfection reagent-treated cells. E and F: AAV-PEDF prevents CCl4- and TAA-induced HSC PPARγ down-regulation. Mice were injected with AAV-PEDF or AAV-EGFP for 1 week and then treated with CCl4 or TAA twice per week for 3 weeks. E: Representative pictures of four independent experiments show dual immunofluorescence staining of HSCs by α-SMA (red), PPARγ (green), and merged (yellow; PPARγ-positive HSCs). Original magnification, ×400. F: Percentage of PPARγ-positive HSCs. *P < 0.002 versus control mice; **P < 0.05 versus AAV-EGFP.
When the cell morphology of HSCs with and without PEDF exposure was compared, culture-activated HSCs assumed an enlarged, flattened morphology, whereas PEDF exposure resulted in HSCs with dendritic-like morphology and retracted cytoplasm (Figure 4B). We further examined stress fiber architecture, a characteristic feature of HSC activation,28 by rhodamine-phalloidin staining of filamentous (F-) actin and immunofluorescent staining of α-SMA (Figure 4, B and C). Culture-activated HSCs displayed prominent stress fibers and α-SMA staining, whereas PEDF exposure reduced stress fiber and α-SMA formation, an indication of suppression of the activation of HSCs.12
PPARγ Mediates the Suppression of HSCs by PEDF
We investigated whether PEDF could induce PPARγ expression in culture-activated HSCs. Immunoblot analyses revealed that PEDF induced PPARγ expression, but had no such effect on the expression of PPARα or PPARβ (Figure 4A).
In immortalized HSC-T6 cells, PEDF induced mRNA and protein expression of PPARγ in a time- and dose-dependent manner (supplemental Figure S4 at http://ajp.amjpathol.org). PPARγ siRNA pretreatment significantly reduced the suppressive effects of PEDF on HSC activation markers (Figure 4D), indicating that the induction of PPARγ by PEDF is crucial for these effects.
AAV-PEDF Prevents CCl4- and TAA-Induced HSC PPARγ Down-Regulation
To verify that the PEDF signaling described above in cultured HSCs also exists in vivo, we compares changes in PPARγ in cirrhotic liver in the presence and absence of PEDF gene delivery. Immunohistochemistry showed that control animals receiving AAV-EGFP after CCl4 exposure had few PPARγ-positive HSCs, many fewer than animals receiving AAV-PEDF after CCl4 exposure (Figure 4, E and F; 27 ± 5% versus 70 ± 8%) or TAA treatment (34 ± 4% versus 77 ± 5%).
AAV-PEDF Suppresses the Development of Fibrosis in CCl4-Treated Mouse Liver
In a protocol designed to mimic therapy for cirrhosis, mice were injected with CCl4 twice a week for 3 weeks to establish hepatic fibrosis. Then, mice received AAV-PEDF via the tail vein and were continuously injected with CCl4 for further 4 weeks (Figure 5A). The fibrotic area in the liver, quantified by Sirius red staining, was significantly increased (3.3-fold) in mice treated with CCl4 for 7 weeks compared to mice treated for only 3 weeks (Figure 5, B and C). And mice who received AAV-PEDF had a significantly decreased in fibrotic area at week 7 compared to those who received AAV-EGFP (Figure 5C; 16.5 ± 1.6% versus 27.0 ± 3.1%).
Figure 5.
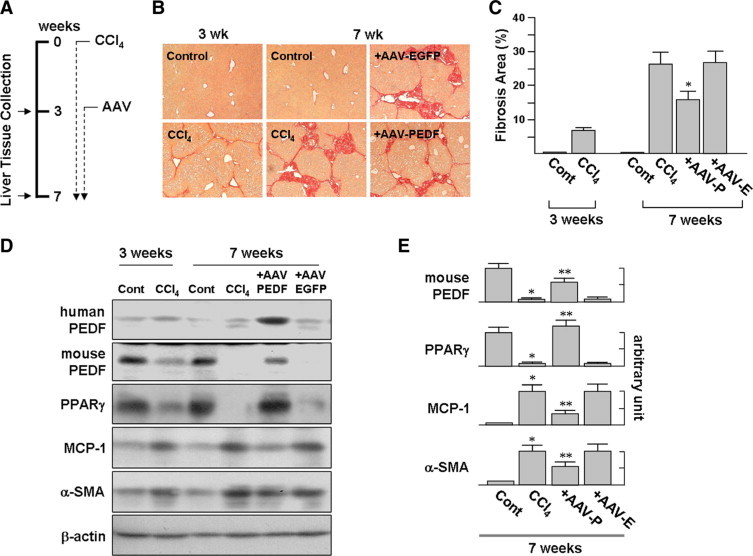
Suppression of CCl4-induced liver fibrosis by PEDF. A: Model of liver fibrosis. Liver fibrosis was induced by intraperitoneal injection of CCl4 twice a week for 3 weeks. Then, mice received a single administration of AAV (2 × 1012 viral particles per mouse) via the tail vein and were continuously injected with CCl4 for an additional 4 weeks. B: Representative Sirius red-stained liver sections. Compared to AAV-EGFP infection, AAV-PEDF infection results in a significant reduction of fibrosis (original magnification, ×100). Representative images of at least three different experiments with six mice in each subgroup are shown. C: Estimation of liver fibrosis by the area of hepatic fibrosis detected by Sirius-red staining shown in B. Data were assessed by analyzing 24 Sirius red-stained liver sections per animal with a computerized image-plus system. D and E: AAV-PEDF prevents the accumulation of cirrhosis-related proteins in CCl4-treated mice. Whole liver protein lysates were extracted for immunoblot analysis with the indicated antibodies. Representative blots (D) and densitometric analyses (E) from three independent experiments are shown. *P < 0.005 versus control; **P < 0.002 versus AAV-EGFP.
Human PEDF protein in mouse liver infected with AAV-PEDF was shown by immunoblotting (Figure 5D). The immunoblots also showed significantly higher expressed PPARγ and mouse PEDF in AAV-PEDF-infected mice compared to AAV-EGFP-infected mice, while at week 7 α-SMA and MCP-1 protein expression in the AAV-PEDF group was decreased by a factor of 1.8-fold and 3.4-fold, respectively, compared to expression of these proteins in the AAV-EGFP group (Figure 5E). The reduced α-SMA protein levels seen in AAV-PEDF-infected mice were correlated to a milder collagen deposition, suggesting that the development of fibrosis is suppressed by AAV-PEDF infection.
PEDF Induces HSC Apoptosis in Vivo
Apoptosis of activated HSC during tissue repair is a known mechanism for termination of HSC proliferation.29,30 We therefore analyzed whether AAV-PEDF could induce HSC apoptosis. Mice were injected with CCl4 for 3 weeks, after which AAV-PEDF was injected for 2 weeks without further CCl4 treatment. We observed prominent HSC apoptosis 2 weeks later (Figure 6A). These HSCs were activated HSC since they could be stained with α-SMA antibody (red). Quantitatively, AAV-PEDF-treated liver had greatly higher numbers of TUNEL-positive HSCs (yellow) compared to the AAV-EGFP-injected controls (Figure 6B; 38.2 ± 7.3% versus 4.9 ± 3.1%). The expression of AAV was confirmed by immunoblot analysis of liver proteins. As shown in Figure 6C, prominent human PEDF expression was found at week 2 after AAV-PEDF infection.
Figure 6.
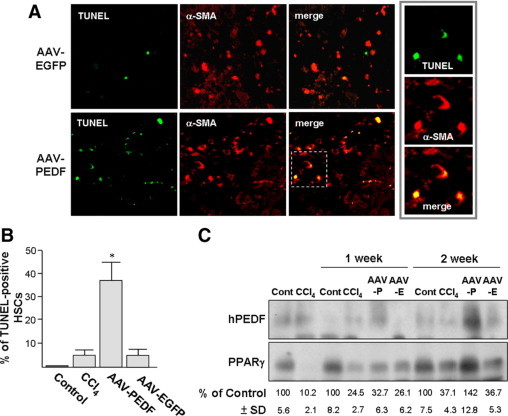
AAV-PEDF induces HSC apoptosis in CCl4-treated mice. Mice were treated with CCl4 twice per week for three weeks and were then injected with AAV (2 × 1012 viral particles per mouse) via the tail vein for two weeks without further CCl4 treatment. A: Liver sections were double-stained with TUNEL to identify apoptotic cells (green) and α-SMA to identify activated HSCs (red). Apoptotic HSCs (yellow) on the superimposed images were quantified under a microscope (×200, 10 fields/liver section) using a digital program. Insets are images before superimposition. B: Percentages of TUNEL-positive HSCs. *P < 0.05 versus AAV-EGFP. C: Immunoblot analyses of liver lysates with antibodies as indicated. Representative blots (top panels) from three independent experiments and six mice per group are shown. Intensities of PPARγ on the immunoblot were determined by densitometry and normalized to β-actin (bottom panels). Cont = olive oil-treated control mice.
There was a significant reduction of PPARγ protein levels induced by CCl4 (36.7 ± 5.3%) in AAV-EGFP-injected controls 2 weeks after termination of CCl4 treatment compared to those treated with olive oil instead of CCl4 (100 ± 7.5%). AAV-PEDF infection reversed the PPARγ reduction to142 ± 12.8%. We also noted that, at 1 and 2 weeks after the termination of CCl4 treatment, endogenous PPARγ levels experienced a spontaneous partial recovery from 10.2 ± 2.1% to 24.5 ± 2.7% at week 1, and 37.1 ± 4.3% at week 2.
PEDF Alters the Intrinsic Anti-Apoptotic Property of HSCs
We also tested whether PEDF could induce apoptosis in HSCs in cell culture. Although PEDF treatment alone did not induce apoptosis of primary culture-activated rat HSCs, PEDF pretreatment for 16 hours increased TNF-α-induced HSC apoptosis from 8 ± 2.1% to 21 ± 3.3% (TUNEL; green, Figure 7A). TNF-α alone (10 ng/ml) did not induce cell apoptosis. This shows the capability of PEDF to induce cultured HSCs into a pro-apoptotic status. The requirement for TNF-α for PEDF-induced apoptosis does not exclude cultured HSC as a valid model system for cirrhosis since TNF-α levels are high in cirrhotic liver.31
Figure 7.
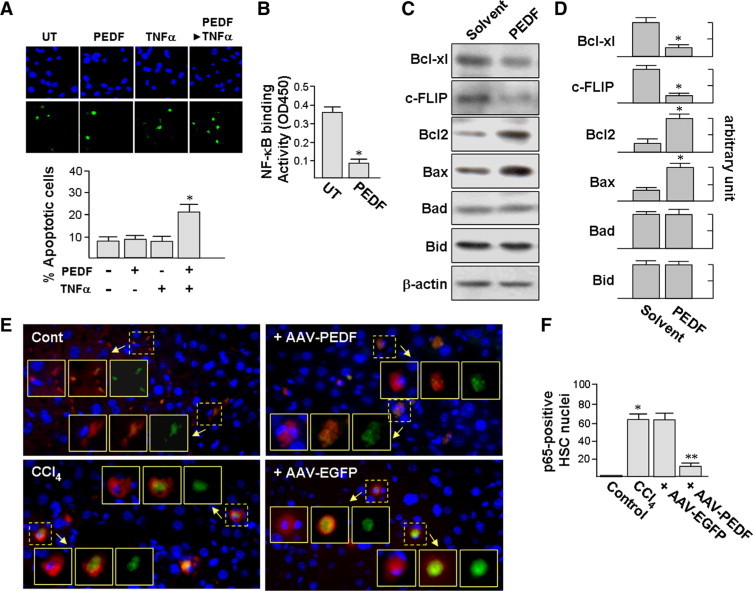
A: PEDF facilitates HSC apoptosis induced by TNF-α. Cells were pretreated with PEDF for 16 hours before exposure to 10 ng/ml TNF-α for an additional 24 hours. TUNEL staining and Hoechst 33258 were used to mark apoptotic cells (green) and nuclei (blue), respectively. A representative image of three independent experiments is shown. The percentages of apoptotic cells were calculated (×200, 10 fields/sample). B: PEDF reduces NF-κB binding to DNA. Nuclear extracts from 1 × 105 primary rat HSCs were measured for NF-κB DNA-binding activity by enzyme-linked immunosorbent assay. *P < 0.05 versus untreated cells. C and D: PEDF affects the expression of apoptosis-related protein. Primary rat HSCs were treated with PEDF or PEDF solvent for 24 hours and then processed for immunoblotting (C). Representative blots (C) and densitometric analyses (D) from three independent results are shown. *P < 0.001 versus control. E and F: PEDF prevents the nuclear translocation of the NF-κB subunit p65/RelA in HSCs in vivo. Mouse treatments were performed as described in the legend of Figure 3. Liver tissues were assayed by dual immunofluorescence staining for α-SMA (red) and p65 (green). Nuclei were stained by Hoechst 33258 (shown in blue). Representative images are from three independent experiments and six mice per group (E). Percentages of p65-positive HSC nuclei were quantified using confocal spectral microscopy (×400, 10 fields/liver section, F). Insets in E from left to right are superimpositions of nucleus and α-SMA, p65 and α-SMA, and p65 alone, respectively. *P < 0.002 versus control mice. **P < 0.001 versus AAV-EGFP.
It has been reported that the down-regulation of NF-κB sensitizes culture-activated rat HSCs to apoptotic death via TNF-α.18,19,30 We investigated the influence of PEDF on NF-κB activity in cultured HSCs. The DNA-binding activity of NF-κB was significantly decreased by ∼75% after PEDF treatment for 24 hours (Figure 7B). Culture-activated HSC treatment with PEDF or an inhibitor of NF-κB nuclear translocation (SN50) for 24 hours reduced the mRNA and protein levels of the NF-κB-responsive genes Bcl-xL and c-FLIP (supplemental Figure S5 at http://ajp.amjpathol.org and Figure 7C and 7D). Culture-activated HSCs also showed a significant increase in Bcl-2 protein and Bax protein by 4.1-fold and 2.7-fold, respectively, comparing to solvent-stimulated cells. In contrast, PEDF did not affect protein levels of Bad and Bid. These observations suggest that the suppression of NF-κB activity by PEDF may render HSCs sensitive to TNF-α-mediated apoptosis.
To seek in vivo evidence of PEDF on NF-κB, we investigated, by double immunostaining for p65 (a NF-κB subunit) and HSC marker α-SMA, whether AAV-PEDF treatment is able to block CCl4–induced NF-κB nuclear translocation in hepatic HSCs. As shown in Figure 7E, nuclear localization of p65 was detected in HSCs of CCl4-treated mice, while AAV-PEDF-infected mice showed weak nuclear but stronger cytoplasmic staining of p65 in HSCs, an indication that translocation of p65 into HSC nuclei was blocked. Infection with AAV-PEDF significantly reduced the CCl4-induced p65 nuclear translocation from 62 ± 8% to 12 ± 5% compared to the AAV-EGFP-treated group (Figure 7F). Our in vivo observation supports the existence of a potential role of NF-κB in PEDF-induced apoptosis.
PEDF Suppresses JunD and NF-κB Nuclear Translocation in HSCs via a PPARγ-Dependent Mechanism
To elucidate the molecular mechanism involved in PEDF signaling in inactivation of HSCs, we investigated the influence of PEDF on nuclear translocation of JunD. Immunofluorescence analysis of JunD showed a prominent nuclear accumulation in solvent-treated cells and barely detectable PPARγ (Figure 8A). In contrast, PEDF treatment for 48 hours prevented the nuclear localization of JunD and increased both nuclear and cytoplasmic PPARγ levels. GW9662, a PPARγ antagonist, blocked this action of PEDF as was evident from the increase seen in nuclear JunD. Immunoblotting of nuclear extracts of HSC-T6 cells showed that JunD was dramatically decreased in PEDF-treated cells (Figure 8B). siRNA targeting of PPARγ abolished the inhibitory effect of PEDF on nuclear localization of JunD. These findings indicate that PEDF, through PPARγ, prevents the nuclear localization of JunD. We also found that PEDF caused a decrease in nuclear localization of NF-κB, evidence supporting an inhibitory effect of PEDF on DNA-binding activity of NF-κB (Figure 7B).
Figure 8.
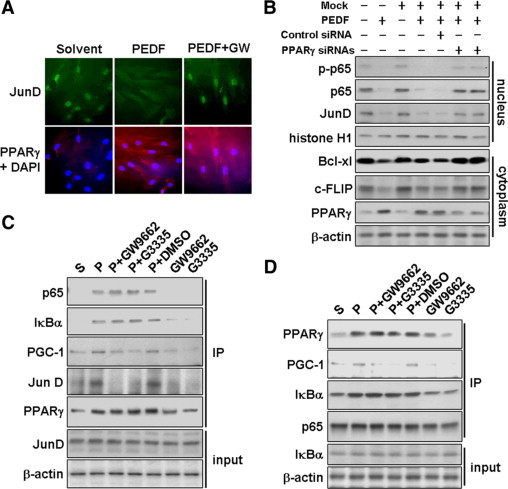
A and B: PEDF prevents JunD and NF-κB nuclear translocation in a PPARγ-dependent manner. A: Primary rat HSCs were assayed by dual immunofluorescence staining for PPARγ (red) and JunD (green). Cells were pretreated with 10 μmol/L PPARγ antagonist (GW9662) for 1 hour and then with or without PEDF for an additional 48 hours. Nuclei were stained with 4′,6-diamidino-2-phenylindinole. B: The inhibitory effect of PEDF on the nuclear translocation of JunD and NF-κB in HSC-T6 cells is blocked by PPARγ siRNA pretreatment. Nuclear and cytoplasmic fractions of HSC-T6 cells were prepared and subjected to immunoblot analysis with antibodies as indicated. Antibodies against histone H1 were used to validate the fractionation procedure. C and D: PPARγ associates with JunD and p65/IκBα. HSC-T6 cells were pretreated with 20 μmol/L PPARγ antagonist (GW9662 or G3335) or DMSO solvent as indicated for 1 hour and then with or without PEDF for an additional 24 hours before the preparation of cell lysates. Immunoblotting with various antibodies as indicated was performed on respective anti-PPARγ (C) or anti-p65 (D) immunoprecipitates. Lysates were also analyzed for total JunD, IκBα, and β-actin before immunoprecipitation (input).
PPARγ Sequestrates p65-IκB Complex and JunD in Cytoplasm
One possible explanation of the reduced JunD and p65 observed in the nuclei is that cytoplasmic PPARγ may directly associate with JunD and p65 and in this way prevent their nuclear translocation. Indeed, JunD and p65 could be precipitated using an anti-PPARγ antibody from PEDF-treated HSC-T6 cells (Figure 8C). Exposure to PPARγ antagonists (GW9662 or G3335, 20 μmol/L) for 24 hours abolished PPARγ binding to JunD and PGC-1 (a well-documented PPARγ cofactor) but did not affect the interaction between PPARγ and the p65-IκB complex. The PEDF-induced increase in the p65-IκB complex in cytoplasm was further confirmed by an immunoprecipitation assay using anti-p65 antibody (Figure 8D). These observations support a model in which PPARγ directly promotes the sequestration of JunD and NF-κB in the cytoplasm.
Discussion
In this study, we found a significant reduction in PEDF levels in all human cirrhotic tissues. This observation, obtained from clinical specimens, was also seen in a mouse model of liver fibrosis. When exogenous PEDF was supplied by gene delivery, the degree of pathology of the liver cirrhosis was reduced, as evident from the reduced area of fibrosis and decreased levels of cirrhosis marker proteins. It has been proposed that the suppression of HSC activation is important for the prevention and resolution of liver fibrosis11–13; however, few endogenous factors have the ability to suppress HSC activation. Our study has demonstrated, both in vitro and in vivo, that PEDF can prevent HSC activation via induction of PPARγ.
In addition to directly suppressing HSC activation, PEDF may also suppress HSC indirectly through suppression of inflammation. Recent animal studies have shown PEDF to have an anti-inflammatory action. PEDF can decrease inflammatory injury from occurring in retina and kidney in a rat model of diabetes via a decrease in the levels of inflammatory factors such as MCP-1.32,33 A suppression of inflammation after CCl4 exposure is evident from the reduced expression of MCP-1 and the reduction of macrophage infiltration caused by PEDF treatment. Interestingly, suppression of inflammation by PEDF may be an effect rather than a cause of HSC inactivation since HSC is one of the major sources of inflammatory cytokines in the liver.1–3
Regarding the mechanism responsible for PEDF depletion after CCl4 treatment, our finding of a reduction in PEDF mRNA levels suggests that an inhibition of PEDF transcription is responsible. However, a recent experimental report in rat hepatic steatosis provides persuasive evidence that matrix metalloproteinase (MMP)-2/9 can degrade PEDF, and this action represents another possible mechanism for PEDF reduction in CCl4-induced liver fibrosis.9 The question then, however, is how the exogenous PEDF in our experiments was able to escape degradation by MMPs. As shown via immunofluorescence assay, exogenous human PEDF was detected throughout liver tissue. Moreover, immunoblotting indicates the presence of PEDF of the proper size in liver extracts. This suggests that the MMPs are likely to be overwhelmed by the efficient expression of PEDF driven by CMV promoter. Additionally, since tissue inhibitor of metalloproteinase is reportedly also synthesized in the liver along with MMPs after CCl4 treatment, there may be a fine balance between MMPs and tissue inhibitor of metalloproteinase.34,35
Our data have also shown that the majority of PEDF-induced PPARγ in HSCs remains in the cytoplasm, rather than the nucleus. Similar subcellular retention of PPARγ has been reported by others in macrophage-like RAW 264 cells, differentiating 3T3-L1 preadipocytes, and normal human urothelial cells, and this cytoplasmic retention is related to the nitration, lipid droplet interaction, and tyrosine phosphorylation of PPARγ.36–38 However, our previous studies demonstrated PEDF-induced nuclear accumulation of PPARγ in endothelial cells.14 These observations suggest that although PEDF can induce PPARγ expression in different types of cells, the subcellular partition of PPARγ is cell type-dependent.
The expression of α-SMA, Col1a1, and MCP-1 is upregulated in part by JunD during the development of cirrhosis or glomerulonephritis.12,17,39 The JunD null mouse is protected against CCl4-induced HSC activation and fibrosis.40 Our in vitro studies have demonstrated that cytoplasmic PPARγ regulates JunD function by causing the retention of JunD in the cytoplasm. This might be the mechanism by which PPARγ arrests HSC activation. Interestingly, the association between JunD and PPARγ is abolished by PPARγ antagonists, suggesting that endogenous PPARγ ligands are required for this physical interaction.
PPARγ is considered to be a therapeutic target for the treatment of liver fibrosis.11–13 However, PPARγ levels are depleted in activated HSCs.11,12 A recent study using an adenoviral vector delivering PPARγ to rat livers found that PPARγ overexpression attenuates both HSC activation and the fibrosis induced by bile duct ligation.13 This suggests that a critical level of PPARγ is required to prevent the activation of HSCs and supports our model that PEDF can protect against liver cirrhosis by maintaining PPARγ levels.
Induction of apoptosis of activated HSC during tissue repair is as an important mechanism for termination of HSC proliferation.29,30 Our result is the first to show that PEDF can induce apoptosis of activated HSCs in vivo. Our studies also show that PEDF suppresses the expression of the NF-κB-responsive genes, Bcl-xl and c-FLIP, in cultured HSCs, and the nuclear translocation of NF-κB in activated HSCs in vivo. These actions may be the reason that PEDF gene delivery leads activated HSCs to become susceptible to toxic cytokines such as TNF-α.31
Our immunoprecipitation assay showed that PPARγ binds NF-κB and prevents its translocation into the nucleus. Interestingly, this binding is not abolished by PPARγ antagonists, suggesting that the biologically active site of PPARγ is not required for the physical interaction between of PPARγ and NF-κB. A novel finding by the immunoprecipitation assay is that the level of IκBα is markedly increased in the complex containing PPARγ and NF-κB. It seems PPARγ, when included into IκB-NF-κB complex, stabilizes this complex. Our finding provides an alternative PPARγ-mediated transcription repression mechanism occurring before NF-κB associates with its promoter.
In summary, we have made the novel observation that exogenous PEDF can prevent chemically-induced liver fibrosis in mice. Culture-activated HSCs are PEDF responsive cells, as shown by a PEDF-induced increase in PPARγ expression and reduction in activation markers and NF-κB-responsive anti-apoptosis proteins. We suggest that PEDF represents a potential antifibrotic factor in the liver.
Acknowledgements
We thank Dr. Sung-Liang Yu for helpful discussions, and Jin-Man Chen and Chu-Ping Ho for excellent technical support.
Footnotes
Supported by grants from the National Science Council, Taiwan (NSC 97-2314-B-195-014-MY3) and Mackay Memorial Hospital (MMH-E- 98006).
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Web Extra Material
The purity of primary rat HSCs was confirmed by vitamin A autofluorescence. A: Representative phase-contrast micrograph shows the morphology of primary rat HSCs on plastic dishes isolated on day 2. B: Vitamin A autofluorescence of cells shown in A were visualized by fluorescence microscopy.
AAV-PEDF prevents liver fibrosis and suppresses the accumulation of cirrhosis-related proteins in TAA-treated mice. A: AAV-PEDF prevents liver fibrosis. Morphometric analysis of hepatic fibrosis was assessed by detection of collagen in liver using Sirius-red staining (original magnification, ×40). B: The distribution of AAV-PEDF-expressed human PEDF in adjacent liver sections was detected by immunofluorescence staining using a human PEDF-specific antibody (original magnification, ×200). C: Estimation of collagen deposition area. *P < 0.05 compared with AAV-EGFP. D and E: Liver extracts were subjected to immunoblot analysis with the antibodies indicated. Representative blots (D) and densitometric analyses (E) from three independent experiments are shown. *P < 0.05 versus control; #P < 0.001 versus AAV-EGFP.
AAV-PEDF reduces CCl4-induced inflammatory infiltration. Mice were injected with AAV-PEDF for 1 week and then treated with CCl4 twice per week for 5 weeks. AAV-EGFP-infected mice were used as a control. Liver sections were immunostained with anti-CD68 antibody and counterstained with hematoxylin. Mice receiving either CCl4 or CCl4 plus AAV-EGFP showed prominent macrophage infiltration. Mice receiving AAV-PEDF had mild macrophage infiltration. Original magnification, ×100.
PEDF (5 nM) induces PPARγ mRNA (A) and protein (B) expression in a time-dependent manner, as assayed by RT-PCR and immunoblot analysis. *p< 0.05 versus solvent. C: PEDF induces PPARγ protein in a dose-dependent manner (*P< 005).
PEDF suppresses NF-kB-responsive gene expression. Primary rat HSCs were treated with 5 nM PEDF or solvent, or 10 μM SN50 for 24 h and then analyzed by RT-PCR (blots 1–3) as well as immunoblot analysis (blots 4–6). Representative blots of three independent results are shown.
References
- 1.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 2.Simpson KJ, Henderson NC, Bone-Larson CL, Lukacs NW, Hogaboam CM, Kunkel SL. Chemokines in the pathogenesis of liver disease: so many players with poorly defined roles. Clin Sci (Lond) 2003;104:47–63. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 3.Marra F, DeFranco R, Grappone C, Milani S, Pastacaldi S, Pinzani M, Romanelli RG, Laffi G, Gentilini P. Increased expression of monocyte chemotactic protein-1 during active hepatic fibrogenesis: correlation with monocyte infiltration. Am J Pathol. 1998;152:423–430. [PMC free article] [PubMed] [Google Scholar]
- 4.Mitchell C, Couton D, Couty JP, Anson M, Crain AM, Bizet V, Rénia L, Pol S, Mallet V, Gilgenkrantz H. Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice. Am J Pathol. 2009;174:1766–1775. doi: 10.2353/ajpath.2009.080632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dawson DW, Volpert OV, Gillis P, Crawford SE, Xu H, Benedict W, Bouck NP. Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science. 1999;285:245–248. doi: 10.1126/science.285.5425.245. [DOI] [PubMed] [Google Scholar]
- 6.Yabe T, Wilson D, Schwartz JP. NFkappaB activation is required for the neuroprotective effects of pigment epithelium-derived factor (PEDF) on cerebellar granule neurons. J Biol Chem. 2001;276:43313–43319. doi: 10.1074/jbc.M107831200. [DOI] [PubMed] [Google Scholar]
- 7.Filleur S, Volz K, Nelius T, Mirochnik Y, Huang H, Zaichuk TA, Aymerich MS, Becerra SP, Yap R, Veliceasa D, Shroff EH, Volpert OV. Two functional epitopes of pigment epithelial-derived factor block angiogenesis and induce differentiation in prostate cancer. Cancer Res. 2005;65:5144–5152. doi: 10.1158/0008-5472.CAN-04-3744. [DOI] [PubMed] [Google Scholar]
- 8.Matsumoto K, Ishikawa H, Nishimura D, Hamasaki K, Nakao K, Eguchi K. Antiangiogenic property of pigment epithelium-derived factor in hepatocellular carcinoma. Hepatology. 2004;40:252–259. doi: 10.1002/hep.20259. [DOI] [PubMed] [Google Scholar]
- 9.Chung C, Shugrue C, Nagar A, Doll JA, Cornwell M, Gattu A, Kolodecik T, Pandol SJ, Gorelick F. Ethanol exposure depletes hepatic pigment epithelium-derived factor, a novel lipid regulator. Gastroenterology. 2009;136:331–340. doi: 10.1053/j.gastro.2008.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hihi AK, Michalik L, Wahli W. PPARs: transcriptional effectors of fatty acids and their derivatives. Cell Mol Life Sci. 2002;59:790–798. doi: 10.1007/s00018-002-8467-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyahara T, Schrum L, Rippe R, Xiong S, Yee HF, Jr, Motomura K, Anania FA, Willson TM, Tsukamoto H. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J Biol Chem. 2000;275:35715–35722. doi: 10.1074/jbc.M006577200. [DOI] [PubMed] [Google Scholar]
- 12.Hazra S, Xiong S, Wang J, Rippe RA, Krishna V, Chatterjee K, Tsukamoto H. Peroxisome proliferator-activated receptor gamma induces a phenotypic switch from activated to quiescent hepatic stellate cells. J Biol Chem. 2004;279:11392–11401. doi: 10.1074/jbc.M310284200. [DOI] [PubMed] [Google Scholar]
- 13.Yang L, Chan CC, Kwon OS, Liu S, McGhee J, Stimpson SA, Chen LZ, Harrington WW, Symonds WT, Rockey DC. Regulation of peroxisome proliferator-activated receptor-gamma in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2006;291:G902–G911. doi: 10.1152/ajpgi.00124.2006. [DOI] [PubMed] [Google Scholar]
- 14.Ho TC, Chen SL, Yang YC, Liao CL, Cheng HC, Tsao YP. PEDF induces p53-mediated apoptosis through PPAR gamma signaling in human umbilical vein endothelial cells. Cardiovasc Res. 2007;76:213–223. doi: 10.1016/j.cardiores.2007.06.032. [DOI] [PubMed] [Google Scholar]
- 15.Ho TC, Chen SL, Yang YC, Lo TH, Hsieh JW, Cheng HC, Tsao YP. Cytosolic phospholipase A2-{alpha} is an early apoptotic activator in PEDF-induced endothelial cell apoptosis. Am J Physiol Cell Physiol. 2009;296:C273–C284. doi: 10.1152/ajpcell.00432.2008. [DOI] [PubMed] [Google Scholar]
- 16.Neumann M, Naumann M. Beyond IkappaBs: alternative regulation of NF-kappaB activity. FASEB J. 2007;21:2642–2654. doi: 10.1096/fj.06-7615rev. [DOI] [PubMed] [Google Scholar]
- 17.Roebuck KA, Carpenter LR, Lakshminarayanan V, Page SM, Moy JN, Thomas LL. Stimulus-specific regulation of chemokine expression involves differential activation of the redox-responsive transcription factors AP-1 and NF-kappaB. J Leukoc Biol. 1999;65:291–298. doi: 10.1002/jlb.65.3.291. [DOI] [PubMed] [Google Scholar]
- 18.Son G, Iimuro Y, Seki E, Hirano T, Kaneda Y, Fujimoto J. Selective inactivation of NF-kappaB in the liver using NF-kappaB decoy suppresses CCl4-induced liver injury and fibrosis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G631–G639. doi: 10.1152/ajpgi.00185.2007. [DOI] [PubMed] [Google Scholar]
- 19.Elsharkawy AM, Wright MC, Hay RT, Arthur MJ, Hughes T, Bahr MJ, Degitz K, Mann DA. Persistent activation of nuclear factor-kappaB in cultured rat hepatic stellate cells involves the induction of potentially novel Rel-like factors and prolonged changes in the expression of IkappaB family proteins. Hepatology. 1999;30:761–769. doi: 10.1002/hep.510300327. [DOI] [PubMed] [Google Scholar]
- 20.Travert M, Ame-Thomas P, Pangault C, Morizot A, Micheau O, Semana G, Lamy T, Fest T, Tarte K, Guillaudeux T. CD40 ligand protects from TRAIL-induced apoptosis in follicular lymphomas through NF-kappaB activation and up-regulation of c-FLIP and Bcl-xL. J Immunol. 2008;181:1001–1011. doi: 10.4049/jimmunol.181.2.1001. [DOI] [PubMed] [Google Scholar]
- 21.Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsao YP, Ho TC, Chen SL, Cheng HC. Pigment epithelium-derived factor inhibits oxidative stress-induced cell death by activation of extracellular signal-regulated kinases in cultured retinal pigment epithelial cells. Life Sci. 2006;79:545–550. doi: 10.1016/j.lfs.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 23.Titos E, Clària J, Planagumà A, López-Parra M, Villamor N, Párrizas M, Carrió A, Miquel R, Jiménez W, Arroyo V, Rivera F, Rodés J. Inhibition of 5-lipoxygenase induces cell growth arrest and apoptosis in rat Kupffer cells: implications for liver fibrosis. FASEB J. 2003;17:1745–1747. doi: 10.1096/fj.02-1157fje. [DOI] [PubMed] [Google Scholar]
- 24.Kim Y, Ratziu V, Choi SG, Lalazar A, Theiss G, Dang Q, Kim SJ, Friedman SL. Transcriptional activation of transforming growth factor beta1 and its receptors by the Kruppel-like factor Zf9/core promoter-binding protein and Sp1. Potential mechanisms for autocrine fibrogenesis in response to injury. J Biol Chem. 1998;273:33750–33758. doi: 10.1074/jbc.273.50.33750. [DOI] [PubMed] [Google Scholar]
- 25.Anan A, Baskin-Bey ES, Bronk SF, Werneburg NW, Shah VH, Gores GJ. Proteasome inhibition induces hepatic stellate cell apoptosis. Hepatology. 2006;43:335–344. doi: 10.1002/hep.21036. [DOI] [PubMed] [Google Scholar]
- 26.Kanno K, Tazuma S, Nishioka T, Hyogo H, Chayama K. Angiotensin II participates in hepatic inflammation and fibrosis through MCP-1 expression. Dig Dis Sci. 2005;50:942–948. doi: 10.1007/s10620-005-2669-7. [DOI] [PubMed] [Google Scholar]
- 27.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Enzan H, Himeno H, Iwamura S, Saibara T, Onishi S, Yamamoto Y, Hara H. Immunohistochemical identification of Ito cells and their myofibroblastic transformation in adult human liver. Virchows Arch. 1994;424:249–256. doi: 10.1007/BF00194608. [DOI] [PubMed] [Google Scholar]
- 29.Saile B, Knittel T, Matthes N, Schott P, Ramadori G. CD95/CD95L-mediated apoptosis of the hepatic stellate cell. A mechanism terminating uncontrolled hepatic stellate cell proliferation during hepatic tissue repair Am J Pathol. 1997;151:1265–1272. [PMC free article] [PubMed] [Google Scholar]
- 30.Oakley F, Trim N, Constandinou CM, Ye W, Gray AM, Frantz G, Hillan K, Kendall T, Benyon RC, Mann DA, Iredale JP. Hepatocytes express nerve growth factor during liver injury: evidence for paracrine regulation of hepatic stellate cell apoptosis. Am J Pathol. 2003;163:1849–1858. doi: 10.1016/S0002-9440(10)63544-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weber LW, Boll M, Stampfl A. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol. 2003;33:105–136. doi: 10.1080/713611034. [DOI] [PubMed] [Google Scholar]
- 32.Zhang SX, Wang JJ, Gao G, Shao C, Mott R, Ma JX. Pigment epithelium-derived factor (PEDF) is an endogenous anti-inflammatory factor. FASEB J. 2006;20:323–325. doi: 10.1096/fj.05-4313fje. [DOI] [PubMed] [Google Scholar]
- 33.Wang JJ, Zhang SX, Mott R, Chen Y, Knapp RR, Cao W, Ma JX. Anti-inflammatory effects of pigment epithelium-derived factor in diabetic nephropathy. Am J Physiol Renal Physiol. 2008;294:F1166–F1173. doi: 10.1152/ajprenal.00375.2007. [DOI] [PubMed] [Google Scholar]
- 34.Aram G, Potter JJ, Liu X, Torbenson MS, Mezey E. Lack of inducible nitric oxide synthase leads to increased hepatic apoptosis and decreased fibrosis in mice after chronic carbon tetrachloride administration. Hepatology. 2008;47:2051–2058. doi: 10.1002/hep.22278. [DOI] [PubMed] [Google Scholar]
- 35.Peng Z, Fernandez P, Wilder T, Yee H, Chiriboga L, Chan ES, Cronstein BN. Ecto-5′-nucleotidase (CD73)-mediated extracellular adenosine production plays a critical role in hepatic fibrosis. FASEB J. 2008;22:2263–2272. doi: 10.1096/fj.07-100685. [DOI] [PubMed] [Google Scholar]
- 36.Shibuya A, Wada K, Nakajima A, Saeki M, Katayama K, Mayumi T, Kadowaki T, Niwa H, Kamisaki Y. Nitration of PPARgamma inhibits ligand-dependent translocation into the nucleus in a macrophage-like cell line. RAW 264 FEBS Lett. 2002;525:43–47. doi: 10.1016/s0014-5793(02)03059-4. [DOI] [PubMed] [Google Scholar]
- 37.Thuillier P, Baillie R, Sha X, Clarke SD. Cytosolic and nuclear distribution of PPARgamma2 in differentiating 3T3–L1 preadipocytes. J Lipid Res. 1998;39:2329–2338. [PubMed] [Google Scholar]
- 38.Varley CL, Stahlschmidt J, Lee WC, Holder J, Diggle C, Selby PJ, Trejdosiewicz LK, Southgate J. Role of PPARgamma and EGFR signalling in the urothelial terminal differentiation programme. J Cell Sci. 2004;117:2029–2036. doi: 10.1242/jcs.01042. [DOI] [PubMed] [Google Scholar]
- 39.Behmoaras J, Bhangal G, Smith J, McDonald K, Mutch B, Lai PC, Domin J, Game L, Salama A, Foxwell BM, Pusey CD, Cook HT, Aitman TJ. Jund is a determinant of macrophage activation and is associated with glomerulonephritis susceptibility. Nat Genet. 2008;40:553–559. doi: 10.1038/ng.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smart DE, Green K, Oakley F, Weitzman JB, Yaniv M, Reynolds G, Mann J, Millward-Sadler H, Mann DA. JunD is a profibrogenic transcription factor regulated by Jun N-terminal kinase-independent phosphorylation. Hepatology. 2006;44:1432–1440. doi: 10.1002/hep.21436. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The purity of primary rat HSCs was confirmed by vitamin A autofluorescence. A: Representative phase-contrast micrograph shows the morphology of primary rat HSCs on plastic dishes isolated on day 2. B: Vitamin A autofluorescence of cells shown in A were visualized by fluorescence microscopy.
AAV-PEDF prevents liver fibrosis and suppresses the accumulation of cirrhosis-related proteins in TAA-treated mice. A: AAV-PEDF prevents liver fibrosis. Morphometric analysis of hepatic fibrosis was assessed by detection of collagen in liver using Sirius-red staining (original magnification, ×40). B: The distribution of AAV-PEDF-expressed human PEDF in adjacent liver sections was detected by immunofluorescence staining using a human PEDF-specific antibody (original magnification, ×200). C: Estimation of collagen deposition area. *P < 0.05 compared with AAV-EGFP. D and E: Liver extracts were subjected to immunoblot analysis with the antibodies indicated. Representative blots (D) and densitometric analyses (E) from three independent experiments are shown. *P < 0.05 versus control; #P < 0.001 versus AAV-EGFP.
AAV-PEDF reduces CCl4-induced inflammatory infiltration. Mice were injected with AAV-PEDF for 1 week and then treated with CCl4 twice per week for 5 weeks. AAV-EGFP-infected mice were used as a control. Liver sections were immunostained with anti-CD68 antibody and counterstained with hematoxylin. Mice receiving either CCl4 or CCl4 plus AAV-EGFP showed prominent macrophage infiltration. Mice receiving AAV-PEDF had mild macrophage infiltration. Original magnification, ×100.
PEDF (5 nM) induces PPARγ mRNA (A) and protein (B) expression in a time-dependent manner, as assayed by RT-PCR and immunoblot analysis. *p< 0.05 versus solvent. C: PEDF induces PPARγ protein in a dose-dependent manner (*P< 005).
PEDF suppresses NF-kB-responsive gene expression. Primary rat HSCs were treated with 5 nM PEDF or solvent, or 10 μM SN50 for 24 h and then analyzed by RT-PCR (blots 1–3) as well as immunoblot analysis (blots 4–6). Representative blots of three independent results are shown.