

Abstract
Cells within the tumor microenvironment influence tumor growth through multiple mechanisms. Pericytes such as hepatic stellate cells are an important cell within the tumor microenvironment; their transformation into highly motile myofibroblasts leads to angiogenesis, stromal cell recruitment, matrix deposition, and ensuing tumor growth. Thus, a better understanding of mechanisms that regulate motility of pericytes is required. Focal adhesions (FAs) form a physical link between the extracellular environment and the actin cytoskeleton, a requisite step for cell motility. FAs contain a collection of proteins including the Ena/VASP family member, vasodilator-stimulated phosphoprotein (VASP); however, a role for VASP in FA development has been elusive. Using a comprehensive siRNA knockdown approach and a variety of VASP mutants coupled with complementary cell imaging methodologies, we demonstrate a requirement of VASP for optimal development of FAs and cell spreading in LX2 liver myofibroblasts, which express high levels of endogenous VASP. Rac1, a binding partner of VASP, acts in tandem with VASP to regulate FAs. In vivo, perturbation of Ena/VASP function in tumor myofibroblast precursor cells significantly reduces pericyte recruitment to tumor vasculature, myofibroblastic transformation, tumor angiogenesis, and tumor growth, providing in vivo pathobiologic relevance to these findings. Taken together, our results identify Ena/VASP as a significant modifier of tumor growth through regulation of FA dynamics and ensuing pericyte/myofibroblast function within the tumor microenvironment.
Tumor stroma importantly influences tumor growth and progression with pericytes representing an important stromal cell owing to their importance in angiogenesis and their transformation into highly motile tumor myofibroblasts.1 Focal adhesions (FAs) are specialized structures that connect extracellular environment to actin cytoskeleton thus facilitating the signal transduction and actin remodeling requisite for the process of pericyte migration during angiogenesis.2,3 Thus, mechanisms that govern FA assembly and pericyte motility may present potential targets for anti-cancer therapy but are not yet fully defined.
vasodilator-stimulated phosphoprotein (VASP) is a member of the Ena/VASP family of proteins that regulate actin cytoskeleton and cell migration.4 It contains distinct subdomains that facilitate specific protein interactions and actin binding characteristics that culminate in an anticapping/branching function within the cytoskeleton.4,5 Although VASP resides within FAs, its precise role in FA dynamics has not been completely defined, probably due in part to cell-type specific VASP functions.6–8
Because the role of VASP in pericyte function and ensuing effects within the tumor microenvironment remain unexplored, we investigated the role of VASP in FA development in human hepatic stellate cells (HSC), liver pericytes that express a high level of VASP and which on transformation into myofibroblasts, develop a highly motile state that has been postulated to influence tumor growth. Our data reveal a requirement of VASP in FA dynamics and pericyte motility, which requires cooperation of each distinct VASP subdomain. Indeed, overexpression of the EVH2 domain alone results in a dominant negative function that inhibits FA assembly, cell motility, and angiogenesis. We also identify a role of Rac1 as a key binding partner of VASP that promotes its ability to regulate FA and cell motility. Perturbation of Ena/VASP function in tumor myofibroblast precursor cells significantly reduced tumor growth in mice demonstrating an important role for these cell biological mechanisms in microenvironmental regulation of tumor growth. Taken together, our results identify VASP as a regulator of FA development, pericyte motility, and tumor growth and identify several important mechanisms by which this regulation is conferred.
Materials and Methods
Cell Culture
LX2, a well-characterized cell line derived from human liver pericytes that recapitulates many features of an activated HSC phenotype and is more amenable than primary cells to plasmid transfection or viral transduction, was used in this study.9 These cells were cultured in DMEM supplemented with 10% fetal bovine serum, penicillin, and streptomycin.
Generation of VASP Mutant Constructs and Retroviruses
Human VASP cDNA or YFP cDNA were amplified by PCR and inserted into a retroviral vector to generated pMMP-VASPWT-YFP. Similarly, cDNA sequences encoding specific domains of VASP were inserted into the pMMP vector to generate VASP mutant constructs. All constructs were confirmed by sequencing before use. Retroviruses were generated as described.10
Viral Transduction and siRNA Transfection of Cells
Retroviral transduction of cells were performed as described.10,11 The transduction efficiency of both methods was close to 100%. siRNAs (Qiagen) were transfected into cells using Oligofectamine Reagent (Invitrogen, Carlsbad, CA). All biochemical, microscopic, and functional assays were performed at 72 hours post transfection. Protein knockdown was confirmed by Western blot analysis with specific antibodies. siRNAs used include: VASP siRNA(1411–1431) (SI00051359), VASP siRNA (369–389) (SI02664200), VASP siRNA(1824–1834) (SI02664193), VASP siRNA(444–464) (SI03081183), VASP siRNA(616–636) (SI00302127), Rac1 siRNA (318–338) (SI02655051), and negative control siRNA (SI03650318).
Confocal immunofluorescence microscopy (IF) and quantification of IF was performed as described.10 Primary antibodies used include anti-vinculin (Sigma V9131, 1:800), anti-VASP (BD Transduction Laboratory, 610447 1:400), anti-VASP (M4) (Alexis, 1:1000) and anti-Rac1 (BD Transduction Laboratory, 610650, 1:400), anti-smooth muscle α actin (αSMA) (Sigma A2547, 1:1000), anti-desmin (Ab-2) (CalBiochem, 1:100), anti–PDGF-Rβ (28E1) (#3169, 1:100), anti-active Caspase-3 (559565, BD Pharmingen). Confocal microscopy was performed using a LSM 5 Pascal Laser Scanning Microscope (Carl Zeiss Inc., Germany). Fibroblast density and microvessel density (MVD) of tumors were quantitated by using Image Pro-Plus software (version 6.2, MediaCybernetics Inc., Bethesda, MD) as previously described.12
FA Analysis
Cells were immunolabeled with anti-vinculin or anti-paxillin, and confocal images were acquired with a ×10 lens with a zoom factor of 1.5 or 2.5. Cells devoid of mature cortical FA were defined as cells that contained no large mature vinculin- or paxillin-positive FA plaques. Approximate 500 cells per sample were randomly picked from ten different fields, and the number of cells devoid of FA plaques was analyzed and counted. To quantitate FA size and the number of FA per cell, images were acquired with a ×63 lens and analyzed by Image Pro-Plus software.13 To visualize cell-matrix adhesions in myofibroblasts in tumors in situ, 7-μm tumor frozen sections were immunostained with anti-vinculin or anti-paxillin, and confocal images were captured with a ×100 lens.
In Situ Cross-Linking, Immunoprecipitation, and Western Blot Analysis
In situ cross-linking, immunoprecipitation (IP), and Western blot analysis was performed as previously described.14,15 The antibodies used included the following: anti-VASP (BD Transduction Laboratory, 610447 1:1000), anti-VASP (M4) (Alexis, 1:1000), anti-vinculin (Sigma, V9131, 1:1000), anti-Rac1 (BD Transduction Laboratory, 610650, 1:1000), anti-GFP (Sigma, G6795 and G1544, 1:1000), anti–proliferating cell nuclear antigen (PCNA) (BD Transduction Laboratory, 610664, 1:5000), anti-Ki-67 (DAKO, M7249, 1:100), and anti-Ki-67 (ab15580, Abcam, 1:500). Densitometric analysis was done using ImageJ software (National Institutes of Health, Bethesda, MD).
Vascular Tubulogenesis, Cell Spreading Assays, and Boyden Chamber Assays
LX2 cells were seeded onto four-well chamber slides (Lab-Tek, Nalge Nunc International) (15,000 cells per well in serum-free medium) that were previously coated with matrigel (150 μl/well, BD Bioscience, San Jose, CA). Vascular tubes formed on matrigel were photographed and analyzed at different time points post seeding by Image Pro-Plus software.16 Cell spreading assays were performed as previously described.17,18 Chemotaxis was measured by modified Boyden chamber assays (Becton Dickinson, Heidelberg, Germany) with the use of 8-μm pore size polycarbonated filters coated with type I collagen (50 μg/ml) as described previously.15 Briefly, cells (LX2 or primary human HSC [HHSC]) transfected with control or VASP siRNA were suspended in serum free medium and seeded to the upper wells (20,000 cells per well) while lower chambers were filled with 26 μl of medium with or without serum. After four hours incubation at 37°C the polycarbonated filter was removed, and migrated cells on the lower surface were stained with HEMA-3. Cells passed through the filter were quantified from random microscopic fields as we have previously done.15 Each assay was performed in triplicate with six replicates of each group per assay. Results are expressed as the mean number of migrated cells ± SD.
Rac1 Activity Assay
Rac1 activity was determined by using a Rac1 Activation Assay Kit according to manufacturer recommended protocol (17-283; Millipore, Billerica, MA).
GST Pull-Down Assay
VASP cDNA was inserted into expression vector pGEX 6p-1, GST, or GST-VASP fusion proteins were purified from Escherichia coli (BL21 DE3) using glutathione-Sepharose beads. GST pull-down assay was performed as previously described.19
Subcutaneous Tumor Cell Implantation in Mice
All animal procedures were performed in accordance with the Institutional Animal Care and Use Committee guidelines of Mayo Clinic. Mouse embryonic fibroblasts (MEF) were isolated from 13.5-day-old embryos of C57BL/6 mice. MEF (passage 1) transduced with YFP or EVH2-YFP retroviruses were used for co-implantation. In brief, 1 × 106 LLC and 1 × 106 retroviral transduced MEF were mixed and resuspended in 100 μl PBS and implanted into 8-week-old C57BL/6 mice via subcutaneous injection. Tumor nodules were measured by a caliper, and volumes were determined using a formula (volume = length × width × height × 0.52). Tumor nodules were isolated on the 15th day postimplantation for frozen sections, which were subjected to immunostaining and microscopic analyses. Tumor cell apoptosis was determined by IF for active Caspase-3, and proliferation was determined by Western blot analyses for PCNA and Ki-67, two commonly used cell proliferation markers.
Statistical Analysis
Data were presented as mean ± SEM. Bar graphs, blots, and fluorescent micrographs represent typical experiments reproduced at least three times with similar results. Statistical analyses were performed by two-tailed Student's t-test or two-way analysis of variance if two factors were involved (GraphPad Prism 4; GraphPad Software Inc., La Jolla, CA). Value of P < 0.05 was considered statistically significant.
Results
VASP Is Highly Expressed in HSC and Is Associated with Vinculin
HSC are liver specific pericytes, which are postulated to transdifferentiate into more motile myofibroblasts within the hepatic tumor microenvironment with their enhanced migration capacity contributing to tumor development and progression.20–23 As an initial step to investigate the role of VASP in FA development and cell migration in HSC, we analyzed expression of Ena/VASP proteins by Western blot analysis. VASP is highly expressed in both HHSC and an immortalized human HSC cell line (LX2) as detected by a polyclonal anti-VASP (M4) antibody that recognizes both human and mouse VASP (Figure 1A upper left; MEF, which are a commonly used fibroblastic cell are also shown on the gel). Mena was also detected in HSC and MEF (Figure 1A upper left), while a third member of Ena/VASP proteins, Ena/VASP Like (EVL), was not detected in either cell type (data not shown).
Figure 1.
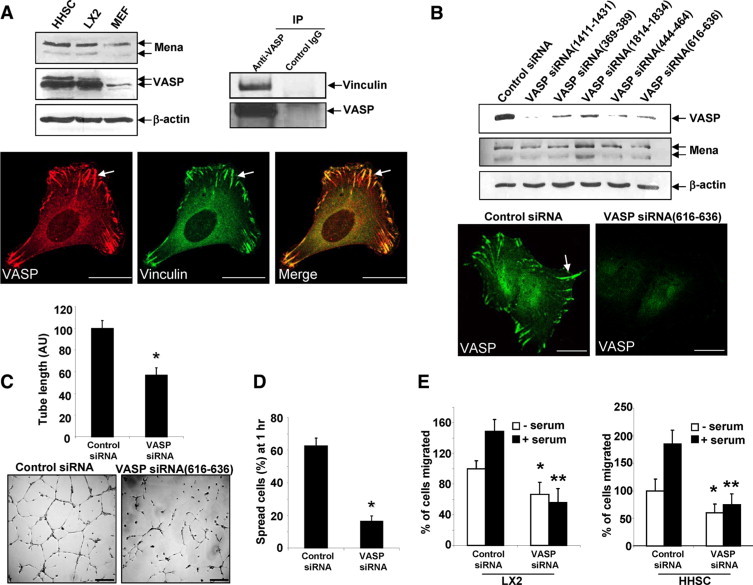
VASP is highly expressed in HSC, and VASP knockdown by specific siRNAs inhibits tubulogenesis, in vitro cell spreading, and migration. A, Upper left: Primary human HSC (HHSC), LX2, and MEF were subjected to Western blot analysis for Ena/VASP proteins. β-actin was used as a loading control. Upper right: Lysates of LX2 cells were subjected to IP using anti-VASP; coprecipitated vinculin was detected by Western blot analysis. Lower: LX2 cells were double-stained with anti-VASP (red) and anti-vinculin (green). Arrows mark colocalization of vinculin and VASP at a large and mature FA plaque. Scale bars = 20 μm. B, Upper: LX2 cells transfected with control siRNA or VASP siRNAs were lysed at 72 hours post transfection for Western blot analysis. The blot was reprobed with anti–β-actin as a protein loading control. Lower: VASP IF shows VASP was eliminated from FA and peripheral plasma membrane by VASP siRNA. Scale bars = 20 μm. C: Seventy-two hours post transfection, LX2 cells transfected with control siRNA or VASP siRNA were subjected to a tubulogenesis assay. Representative photomicrographs of tubes are shown on the bottom, and quantitative data are shown by a bar graph on the top (*P < 0.05, t-test; n = 3 independent experiments). VASP siRNA significantly inhibited vascular tubulogenesis of LX2 cells. Scale bars = 200 μm. D: Seventy-two hours post transfection, LX2 transfected with control siRNA or VASP siRNA were subjected to a cell spreading assay. VASP siRNA significantly inhibited cell spreading on culture dishes (*P < 0.05, t-test; n = 3 independent experiments). E: Cell migration was determined by Boyden chamber assays. VASP knockdown induced a similar inhibition of cell migration in both LX2 cell line and primary HHSC. *P < 0.05, VASP siRNA versus control siRNA in the absence of serum; **P < 0.05, VASP siRNA vs. control siRNA in the presence of serum.
Because vinculin is a commonly used FA marker and interacts with VASP,5,14,24 we focused on this protein in our model system aimed at mechanistically delineating the regulatory role of VASP in FA assembly. VASP colocalizes with vinculin within punctate densities in the cytoplasm as well as cortical FA plaques of cells, as assessed by double IF labeling and confocal microscopy (Figure 1A lower). IP confirmed vinculin and VASP interaction (Figure 1A upper right). The fact that LX2 cells express a high level of VASP and develop abundant mature FA plaques that are positive for VASP and vinculin staining, provided us with a useful cell model in which to study the role of VASP in FA development.
VASP siRNAs Impair FA Assembly and Reduce Tubulogenesis
The precise role of VASP in FA development is unclear and may be cell-type–specific as suggested by differing results in endothelial cells and fibroblast.6–8,25,26 To understand the role of VASP in FA development in HSC, we first used a siRNA approach to knockdown VASP in the LX2 HSC cell line, which recapitulates the phenotype of myofibroblastic, activated HSC. To avoid the possibility of off-target effects of siRNA, we used multiple VASP siRNAs, each targeting against distinct sequence of human VASP transcript (QIAGEN), VASP siRNA (369–389), VASP siRNA, (444–464) VASP siRNA (616–636), VASP siRNA (1411–1431), and VASP siRNA (1814–1834). Among the five VASP siRNAs, VASP siRNA (1814–1834) exclusively targets the 3′-untranslated region (UTR) as further described below. A validated negative control siRNA was used as a control (Allstars, QIAGEN). All VASP siRNAs exhibited comparable effects on reduction of VASP as detected by Western blot analysis at 72 hours post transfection (Figure 1B upper). As anticipated, no significant up-regulation or down-regulation of Mena was detected by VASP siRNAs. Consistent with the Western blot analysis, VASP IF and confocal microscopy revealed that the normal distribution of VASP at mature FA and peripheral plasma membrane was eliminated by VASP siRNA (as an example, VASP siRNA [616–636] is shown in Figure 1B lower). To determine a physiological role of VASP knockdown in cells, a tubulogenesis assay was used to assess pericyte adhesion and migration on matrix with this assay representing a standardized in vitro correlate of in vivo angiogenic capacity of vascular wall cells.27 VASP siRNA significantly inhibited tubulogenesis of LX2 cells (Figure 1C). To further support this observation using a more generalized assay, cell spreading17,18 was measured to test whether this key parameter of tubulogenesis and angiogenesis was affected by VASP siRNAs. As shown in Figure 1D, VASP siRNA significantly inhibited cell spreading as assessed one hour after seeding of cells onto culture dishes. To test whether VASP knockdown affects cell migration, Boyden chamber assays were performed with both LX2 and primary HHSC. In the presence or absence of serum stimulation, inhibition of cell migration was observed in both LX-2 and primary HHSC in response to VASP knockdown (Figure 1E), demonstrating a regulatory role of VASP in cell migration and a relevance of our LX2 model to primary HHSC. Furthermore, multiple VASP siRNAs exhibited a similar effect on inhibition of tubulogenesis, cell spreading, and migration, indicating that adhesion and migration of cells are regulated by VASP (Figure 1B and data not shown).
We hypothesized that VASP knockdown may attenuate cell spreading and tubulogenesis by disturbing FA development. To test this hypothesis, we performed vinculin IF and analyzed FA using fluorescence confocal microscopy. As shown in Figure 2A, signal intensity of FA was markedly reduced in the majority of VASP siRNA transfected cells, indicating that the size and number of mature FA plaques per cell were reduced. To test this further, cells transfected with control or VASP siRNA were analyzed with an alternative FA marker including paxillin and similar results were obtained (Figure 2B). A detailed imaging software approach was used to further dissect the effect of VASP knockdown on FA, including the size of FA and number of FA per cell. In the population of transfected cells that still contain FA, the area of FA and the number of FA per cell were reduced by approximately 50% as compared with control cells (Figure 2B lower panels). In addition to the reduced FA intensity within individual cells as depicted in Figure 2A, we observed that there was a 2–3 fold increase in the population of cells devoid of FA in VASP siRNA-transfected cells (Figure 2C). Representative IF confocal images of cells transfected with VASP siRNA (1411–1434) are shown in Figure 2C lower panels (arrows mark cells devoid of mature FA).
Figure 2.
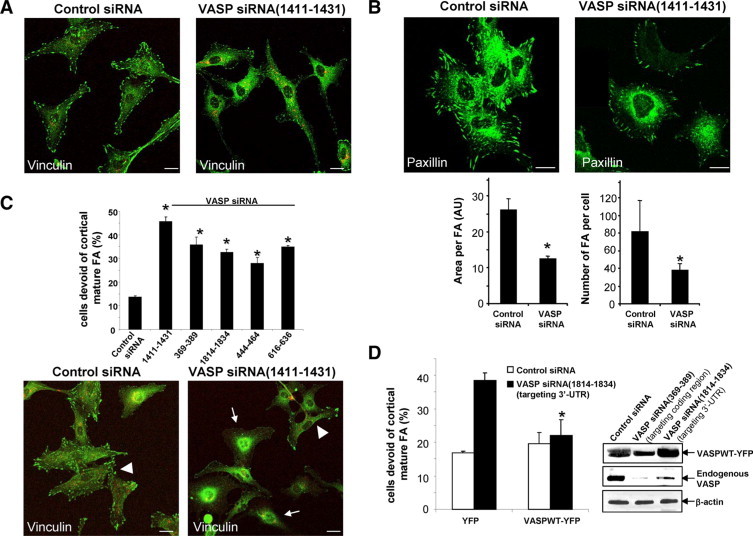
VASP siRNAs impair the development of mature FA. A: At 72 hours post transfection, confocal microscopy of vinculin IF (green) shows that FA signals were reduced by VASP siRNA(1411–1434). Scale bars = 20 μm. Both control and VASP siRNA were prelabeled with Cy3 (red). Scale bars = 20 μm. B, Upper: IF confocal microscopy of an alternative FA marker, paxillin (green), is reduced by VASP siRNA. Scale bars = 20 μm. Lower: High-power confocal images were subjected to FA analysis after background subtraction using Image Pro-Plus software. VASP siRNA significantly reduced the size of FA plaques and number of FA per cell in FA containing cells (*P < 0.05, t-test; n = 15 cells per group). C, Lower: Confocal images of vinculin IF reveal that VASP siRNA (1411–1434) increased the population of cells that are devoid of mature FA plaques (arrows). The arrowhead marks a single cell that still contains FA plaques. Scale bars = 20 μm. Upper: Five distinct VASP siRNAs exhibited a similar effect of increasing the population of cells that are devoid of FA plaques (*P < 0.05, analysis of variance; n = 3 independent experiments). D, Left: LX2 cells transfected with control or VASP siRNA (1814–1834) that targets against 3′-UTR were transduced with retroviruses encoding YFP or VASPWT-YFP. The effect of VASP siRNA (1814–1834) on FA was abolished by VASPWT-YFP fusion protein (*P < 0.05, analysis of variance; n = 3 independent experiments). Right: VASPWT-YFP fusion protein is resistant to targeting and degradation of VASP siRNA (1814–1834). Cells expressing VASPWT-YFP were transfected with various siRNAs, and Western blot analysis demonstrates that endogenous VASP but not VASPWT-YFP was degraded by VASP siRNA (1814–1834). As a control, both endogenous VASP and VASPWT-YFP were targeted by VASP siRNA (369–389), which targets against the coding region of VASP mRNA.
To prove that the impaired FA phenotype that we observed was indeed induced by knockdown of VASP, we performed rescue expression of VASP cDNA from retroviral vector (VASPWT-YFP) in cells transfected with VASP siRNA (1814–1834). Because this specific siRNA targets against 3′-UTR of VASP mRNA and the transcript of VASPWT-YFP does not contain the UTR, VASPWT-YFP is resistant to targeting and degradation by VASP siRNA (1814–1834) (Figure 2D, right panels). In contrast, endogenous VASP was targeted and degraded by VASP siRNA (1814–1834) (Figure 2D, right panels). Based on these data, cells transfected with VASP siRNA (1814–1834) were subjected to YFP (control) or VASPWT-YFP retroviral transduction, and FA plaques were analyzed by vinculin IF. In cells expressing YFP, VASP siRNA (1814–1834) significantly increased the population of cells devoid of FA. However, in cells expressing VASPWT-YFP, the effect of VASP siRNA (1814–1834) on FA was abolished (Figure 2D and see Supplemental Fig. S1 at http://ajp.amjpathol.org). These data indicate that the impaired FA phenotype induced by VASP siRNA was indeed mediated by VASP deficiency. Thus our data, generated with a variety of VASP siRNAs and rigorous controls, highlight a regulatory role of VASP in FA regulation in LX2 cells.
VASP Mutants Recapitulate VASP siRNA Phenotype in FA
Ena/VASP proteins contain three subdomains: an N-terminal domain implicated in protein binding with FPPPP containing proteins (EVH1), a middle domain containing polyproline rich sequences (PRR), which is also a protein binding module, and a C-terminal actin binding domain (EVH2).4,5 Ena/VASP may target to FA by virtue of its EVH1 domain binding with zyxin or vinculin.28,29 The PRR domain is thought to bind SH3 and WW domain–containing proteins, such as c-Abl, Src, as well as the actin monomer binding protein, profilin.5,30 The EVH2 domain contains putative binding blocks for both G- and F-actin as well as a motif that promotes actin oligomerization.31 The mechanisms by which these subdomains cooperate to achieve VASP function are an area of active investigation. Therefore, we next generated a series of YFP-tagged VASP mutant constructs aimed at understanding the mechanistic basis by which the distinct actin binding and protein interaction modules of VASP may interact with vinculin and in turn contribute to FA assembly. These VASP mutants included the VASP N terminus (aa 1–108; EVH1-YFP), a central proline rich region (aa 108–276; PRR-YFP), the VASP C terminus (aa 276–380; EVH2-YFP), and PRRdel-YFP in which the central proline rich region was deleted (Figure 3A). Unlike the focal adhesion targeting of endogenous VASP, none of the mutants localized at FA plaques. Each of the mutants showed a cytoplasmic distribution, with EVH1 and PRR mutants also congregating in a nuclear pattern (Figure 3B), as previously described,32 thus validating our C-terminally YFP-tagged VASP mutant constructs. Interestingly, the actin binding EVH2 domain, unlike the EVH1 domain, also decorated cord-like structures reminiscent of actin bands. Next, cells expressing these VASP mutants were subjected to vinculin IF to study FA formation. We observed prominent colocalization of vinculin with EVH2-YFP or PRRdel-YFP, however colocalization of vinculin with EVH1-YFP and PRR-YFP was not prominent (Figure 3B). Additionally, IP studies confirmed that in addition to the EVH1 domain, vinculin also interacts with the EVH2 of VASP (see Supplemental Figure S2 at http://ajp.amjpathol.org), which corroborates that the C-terminal region of VASP interacts with vinculin.14,33,34 But more importantly, our data show that unlike YFP and VASPWT control constructs, EVH1 and/or EVH2 domain constructs lacking the PRR (EVH1-YFP, EVH2-YFP, and PRRdel-YFP) impair the development of mature FA (Figure 3C). As an example, IF confocal images of FA markers including vinculin and paxillin in cells that express YFP or EVH2-YFP are shown in Figure 4, A and B. To translate the effects of EVH2-YFP–induced FA disassembly to key downstream cellular functions, cells expressing YFP or EVH2-YFP were subjected to a cell spreading assay and a tubulogenesis assay, which reflect coordinated cell adhesion and migration. Similar to VASP siRNA, EVH2 mutant inhibited LX2 cell spreading on cell culture dishes (Figure 4C), and both fluorescent confocal images and quantitative analysis revealed that EVH2-YFP significantly inhibits cell tubulogenesis on matrigel (Figure 4D). In summary, the impaired FA phenotype induced by these VASP mutants (EVH1-YFP, EVH2-YFP, and PRRdel-YFP) was reminiscent of that induced by VASP siRNA, indicating that these VASP mutants, especially exemplified with the EVH2 mutant, function in a “dominant negative” manner by disrupting VASP dependent FA formation, adhesion, and tubulogenesis. Mechanistically, EVH2 binding with vinculin may disrupt FA formation by sequestering vinculin from FA. Based on these data, we later use the EVH2 dominant negative mutant to further interrogate the role of VASP in tumor microenvironment in vivo.
Figure 3.
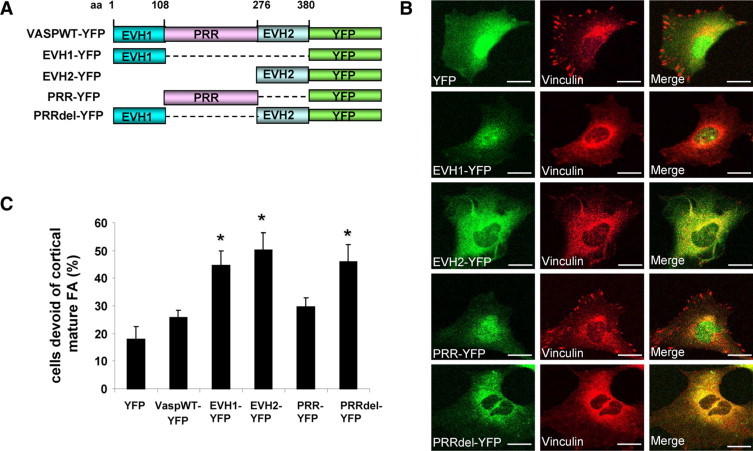
VASP mutants recapitulate VASP siRNA phenotype of FA disruption. A: YFP-tagged retroviral constructs that express deletion or truncation VASP mutants are shown. The structure of full-length wild-type VASP is depicted on the top. B: Cells expressing individual VASP mutant were stained for vinculin (red). There is marked colocalization of vinculin with EVH2-YFP or PRRdel-YFP. Scale bars = 20 μm. C: In contrast to YFP or VASPWT controls, retroviral expression of EVH1, EVH2, or PRRdel mutant significantly increased the population of cells devoid of mature FA (*P < 0.05, analysis of variance; n = 3 independent experiments).
Figure 4.
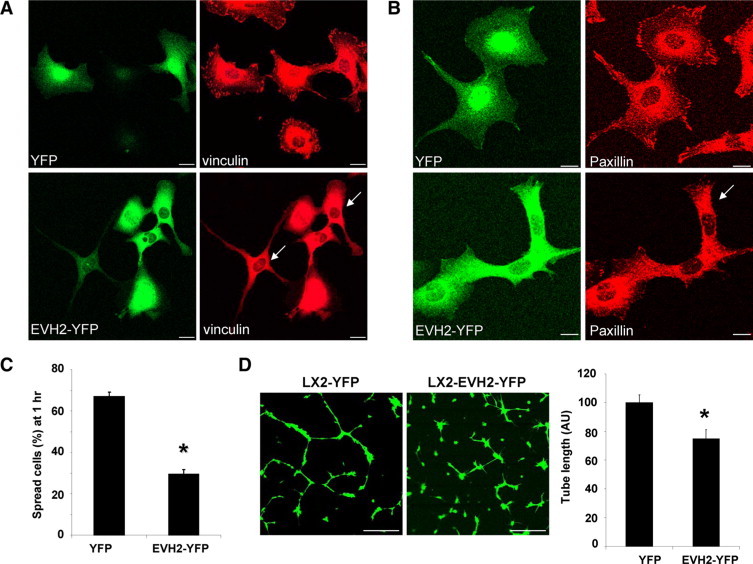
EVH2 mutant impairs FA development, cell spreading, and vascular tubulogenesis of LX2 cells. A: LX2 cells expressing YFP or EVH2-YFP were subjected to vinculin IF. Arrows mark cells that are devoid of mature FA plaques in cells expressing EVH2-YFP. Scale bars = 20 μm. B: FA development was impaired by EVH2-YFP as analyzed with an alternative FA marker, paxillin. Arrow marks a cell that is devoid of mature FA plaques in cells expressing EVH2-YFP. Scale bars = 20 μm. C: Cells expressing YFP or EVH2-YFP were subjected to a cell spreading assay. The EVH2-YFP significantly inhibited cell spreading on cell culture dishes (*P < 0.05, t-test; n = 3 independent experiments). D: LX2 cells that express YFP or EVH2-YFP were subjected to a tubulogenesis assay. Representative confocal images are shown on the left, and quantitative data are shown by a bar graph on the right. The EVH2 mutant significantly reduced tubulogenesis of LX2 cells (*P < 0.05, t-test; n = 3 independent experiments). Scale bars = 200 μm.
Rac1 Acts in Tandem with VASP to Modulate FA Formation
It has been shown that active Rac1 colocalizes with VASP and that Rac1 activity is regulated by VASP in endothelial cells.35,36 Furthermore, Rac1 is a known a regulator of FA assembly.37 These prior observations led us to pursue experiments to delineate a potential role for Rac1 in the process of VASP regulation of FA. We first tested whether VASP interacts with Rac1 using an in vitro binding assay. GST or a GST-VASP fusion protein was incubated with LX2 lysates, the bound Rac1 was detected with anti-Rac1 in a Western blot analysis, suggesting a Rac1-VASP interaction (Figure 5A, left). This was further supported by IF confocal microscopy showing that in LX2 cells that express VASP-YFP, VASP-YFP colocalizes with Rac1 at FA (Figure 5A, right panels). Taken together, these data suggest that VASP interacts with Rac1 in LX2 cells.
Figure 5.
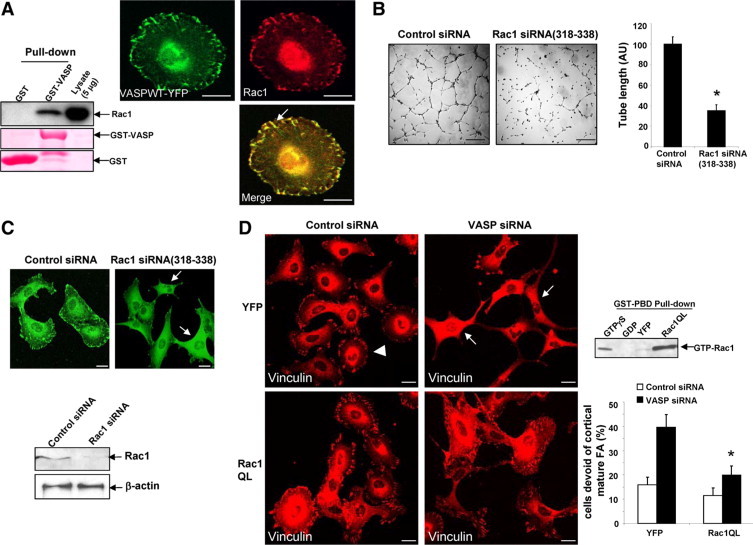
Constitutively active Rac1 reverses the FA phenotype of VASP siRNA. A, Left: GST-VASP interacts with Rac1 in vitro. GST or a GST-VASP fusion protein was incubated with LX2 cell lysates, and the bound Rac1 was detected by Western blot analysis. Ponceau S was used to assess purity of GST and GST-VASP fusion proteins. Right: Cells that express VASPWT-YFP were subjected to Rac1 IF (red). Arrow depicts colocalization of VASP with Rac1. Scale bars = 20 μm. B: At 72 hours post siRNA transfection, cells were harvested for tubulogenesis assays. Representative tubes formed on matrigel are shown on the left, and quantitative data are shown by a bar graph on the right. Scale bars = 200 μm. Rac1 siRNA (318–338) significantly inhibits vascular tubulogenesis of LX2 cells (*P < 0.05; t-test; n = 3 independent experiments). C: Confocal images of vinculin IF show that Rac1 siRNA(318–338) disrupts FA formation (upper, arrows). Scale bars = 20 μm. Lower: Western blot analysis show that Rac1 was efficiently knocked down by Rac1 siRNA (318–338) at 72 hours post siRNA transfection. D, Left: Cells transfected with control or VASP siRNA were subjected to retroviral transduction, respectively to express YFP or Rac1QL (constitutively active Rac1). Rac1QL reversed the FA phenotype of VASP siRNA. Arrowhead marks a single cell that still contains FA plaques. Arrows mark cells that are devoid of mature FA. Scale bars = 20 μm. Lower right: Quantitation of microscopy shows that Rac1QL significantly reduced the population of cells that are devoid of FA in VASP siRNA-transfected cells (*P < 0.05; analysis of variance; n = 3 independent experiments). Upper right: Highly increased Rac1 activity was detected in cells transduced with Rac1QL retroviruses by a Rac1 activity assay. Cell lysates pretreated with GTPγS or GDP were used as positive or negative control, respectively.
To further establish the role of VASP/Rac1 protein interaction in FA assembly in LX2 cells, Rac1 was knocked-down by Rac1 siRNA (318–338). Similar to VASP siRNA, Rac1 siRNA inhibited tubulogenesis and markedly impaired the development of mature FA in LX2 cells (Figure 5, B and C). To test whether Rac1 and VASP are in a common pathway to modulate FA, we knocked down both Rac1 and VASP and examined their effects on FA. No synergistic or additive effect was observed on FA disassembly in response to double siRNA transfection, suggesting that Rac1 and VASP may share a common pathway to regulate FA (see Supplemental Figure S3 at http://ajp.amjpathol.org). These observations also led us to test the prediction that VASP may promote FA formation by binding and activating Rac1 and that therefore a constitutively active Rac1 would act downstream from VASP and would thus reverse the impaired FA phenotype of VASP siRNA. Rac1 cDNA with a point mutation Q61L was inserted into a retroviral vector (Rac1QL) to generate constitutively active Rac1 retroviruses (Figure 5D, upper right). Indeed, as shown in Figure 5D, Rac1QL significantly inhibited the effect of VASP siRNA on FA, supporting a regulatory role of VASP and Rac1 binding and signaling interaction in FA development.
Disruption of Ena/VASP Function in Stromal Pericytes Inhibits Tumor Growth in Vivo
We next extended our molecular model of the regulation of FA assembly by Ena/VASP for functional relevance in an in vivo cancer setting in which pericyte motility, migration, adhesion, and tubulogenesis are essential. To this end, we performed xenotopic tumor studies with Lewis lung carcinoma cells (LLC) and syngeneic MEF (a pericyte/myofibroblast precursor cell). MEF were used in these xenotropic studies because they display more robust viability than murine HSC and syngeneic compatibility not possible with human LX2. Therefore, we first tested whether the EVH2 VASP mutant disrupts FA in MEF similar to what we observed in LX2 pericytes. As shown in Figure 6A, the EVH2 mutant impaired the development of FA of MEF and also significantly inhibited spreading of MEF on cell culture dishes (Figure 6B).
Figure 6.
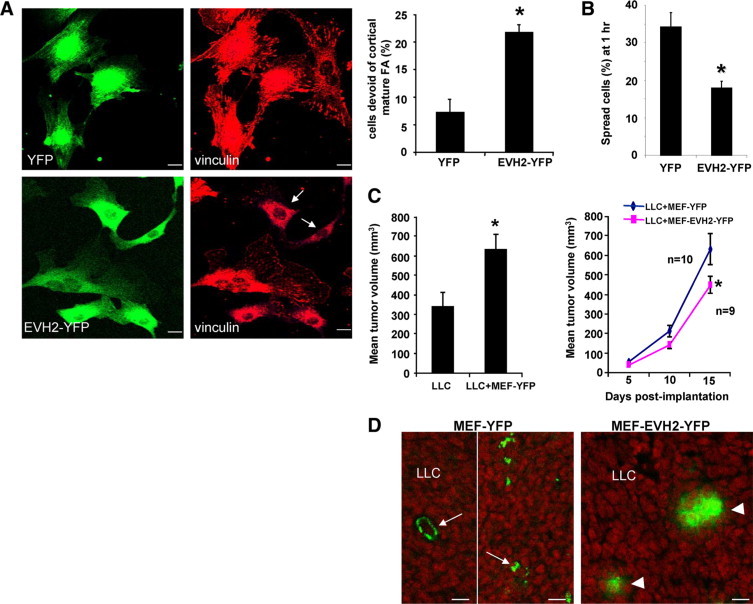
Perturbation of Ena/VASP function in MEF reduces the tumor growth conferred by tumor cell-MEF coimplantation. A: MEF expressing YFP or EVH2 mutant were subjected to vinculin IF. The EVH2 mutant induced impaired FA phenotype in MEF. Representative photomicrographs are shown in the left, and quantitative data are depicted by a bar graph in the right. Arrows mark cells that are devoid of large mature FA. Scale bars = 20 μm (*P < 0.05 t-test; n = 3 independent experiments). B: MEF expressing YFP or EVH2 mutant were subjected to a cell spreading assay. EVH2-YFP significantly inhibits cell spreading of MEF on cell culture dishes (*P < 0.05 t-test; n = 3 independent experiments). C, Left: Coimplantation of MEF with Lewis Lung carcinoma cells (LLC) subcutaneously into syngeneic mice promoted tumor growth compared with LLC alone (*P < 0.05 t-test; n = 9–10 tumors per group). Tumor nodules were measured on the 15th day post implantation. Right: 1 × 106 LLC mixed with 1 × 106 MEF that express either YFP or EVH2-YFP were implanted into syngeneic mice. Tumor nodules were measured and the tumor growth curves are displayed (*P < 0.05 t-test). D: Coimplanted YFP-tagged MEF were detected in tumor nodules on the 15th day post implantation by fluorescence confocal microscopy (green). MEF that express YFP incorporated into both vessels and interdigitation of tumor stroma (arrows, left). Conversely, MEF that express EVH2-YFP failed to distribute throughout the tumor stroma (arrowheads). Scale bars = 20 μm.
Conceptually consistent with prior studies using other tumor and myofibroblast cell types,1,38–40 coimplantation of MEF with LLC into syngeneic mice promotes tumor growth by promoting a prometastatic tumor microenvironment which is rich in highly motile MEF that generate tumor promoting neo-vessels and tumor stimulating growth factors (Figure 6C, left). However, LLC coimplantation with MEF that express EVH2-YFP yielded smaller tumors as compared with LLC coimplantation with MEF that express YFP (Figure 6C; right). Consistent with previous data showing exogenous MEF were efficiently recruited to the tumor vessels,39 analysis of YFP by confocal microscopy of tumor sections revealed that MEF expressing YFP avidly incorporated into tumor stroma including pericyte recruitment to vascular structures (arrows, Figure 6D, left). Conversely, MEF expressing EVH2-YFP and with a deficient FA formation phenotype, incorporated poorly into the tumor stroma and vasculature (arrowheads, Figure 6D right). We next performed more in-depth morphological analysis of the tumor microenvironment focusing on markers that track vascular wall cells including endothelial cells, pericytes, and myofibroblasts. IF and confocal microscopy for mesenchymal cell markers including smooth muscle α actin (α-SMA), desmin, and PDGF-receptor β10 revealed that there was less SMA-positive or desmin-positive fibroblasts in LLC tumors coimplanted with MEF that express EVH2-YFP (Figure 7A). Interestingly, coimplantation of MEF that express EVH2-YFP did not affect recruitment of PDGF-R–positive stromal cells (Figure 7A), consistent with the heterogeneous nature of tumor-associated fibroblasts.10 Because stromal pericytes and myofibroblasts also promote tumor growth by promoting angiogenesis, we also analyzed tumor MVD by performing IF staining with endothelial marker, CD31/PECAM1 in tumor sections from our experimental groups. As shown in Figure 7B, MVD was significantly reduced in LLC tumors coimplanted with MEF that express the defective EVH2-YFP as compared with the YFP control MEF. Next, analyses were performed to analyze for tumor cell apoptosis and proliferation. First, IF staining for active Caspase-3 was performed in tumor sections to test whether coimplanted MEF that express EVH2 affects tumor cell apoptosis thereby inhibiting tumor growth. While these studies did not reveal differences in tumor cell apoptosis between the groups (data not shown), Western blot analyses for two cell proliferation markers PCNA and Ki-67 did reveal reduced tumor cell proliferation in tumors derived from coimplanted MEF that express EVH2 (Figure 7C). Although other mechanisms are certainly at play as well, these studies provide evidence that the regulation of FA assembly by Ena/VASP within tumor stromal cells regulates pericyte/myofibroblast recruitment, tumor angiogenesis, and subsequently tumor growth in vivo, thereby providing a pathobiological context for our earlier mechanistic experimental model.
Figure 7.
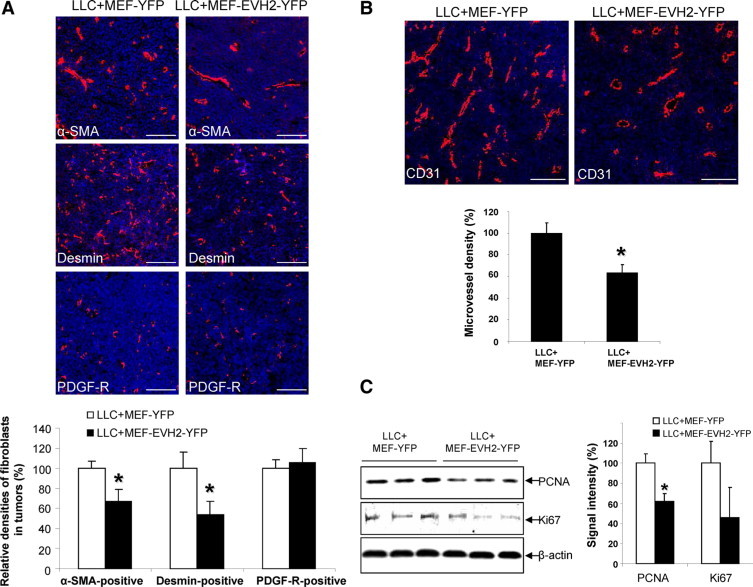
Perturbation of Ena/VASP function in MEF inhibits pericyte incorporation and vascular density of tumors derived from tumor cell-MEF coimplantation. A: On the 15th day post implantation, tumor sections were cryo-sectioned for IF analysis for stromal cell markers, including α-SMA, desmin, and PDGFRβ. Representative IF confocal images are shown on the upper panels, and quantitative data are shown by a bar graph in the bottom. Fewer α-SMA-positive and desmin-positive tumor associated fibroblasts were detected in tumors coimplanted with MEF that express EVH2-YFP as compared with YFP control (*P < 0.05 t-test, n = 6). Scale bars = 100 μm. B: Tumor cryo-sections were subjected to CD31 IF staining, and MVD was quantitated. Reduced MVD was observed in tumors coimplanted with MEF that express EVH2-YFP as compared with YFP control (*P < 0.05 t-test, n = 6). Scale bars = 100 μm. C, Left: Tumors derived from coimplantation were subjected to Western blot analyses for cell proliferation markers, PCNA and Ki-67. The expression levels of PCNA and Ki-67 are reduced in tumors coimplanted with MEF that express EVH2-YFP as compared with YFP control. β-actin was used as a loading control. Right: Quantitative data showing the expression levels of PCNA and Ki-67 in tumors derived from the coimplantation experiments.
Discussion
In this study we make a number of novel observations highly pertinent to pericyte cells that reside within the tumor microenvironment; we demonstrate that: i) Ena/VASP is required for optimal FA development, ii) the effect of Ena/VASP on FAs is mediated by Rac1 activity, and iii) targeted perturbation of Ena/VASP (by virtue of EVH2 mutant overexpression) in tumor myofibroblast precursor cells inhibits tumor growth. These observations lead us to a model in which Ena/VASP modulates FAs in pericytes, thereby influencing the tumor microenvironment, and in turn tumor growth (Figure 8). In this model, VASP binds vinculin by virtue of its EVH1 and EVH2 domains. These domains, though mediating VASP vinculin interaction, are insufficient to recruit the protein complex to FA. Instead, optimal FA development requires functional full-length VASP and its interaction with, and activation of, Rac1. Thus, perturbation of Ena/VASP function of tumor pericytes/myofibroblasts impairs FA assembly, thereby resulting in altered pericyte recruitment to tumor vasculature, reduced tumor angiogenesis, and attenuated tumor growth.
Figure 8.
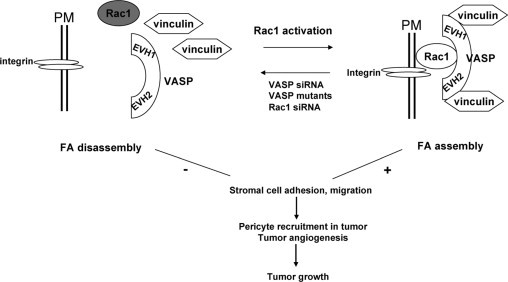
Schematic representation of Ena/VASP proteins in FA of pericytes and tumor growth. VASP binds vinculin by virtue of its EVH1 and EVH2 domains. FA assembly also requires VASP-Rac1 interaction and Rac1 activation as revealed by defective FA phenotypes induced by siRNA targeting of VASP or Rac1 and rescue of the VASP siRNA defective FA phenotype by constitutively active Rac1. Perturbation of this signaling paradigm in vivo using the dominant negative EVH2 domain of VASP impairs FA and cell spreading of tumor associated myofibroblasts, thereby culminating in altered pericyte recruitment to tumor vasculature, reduced tumor angiogenesis, and impaired tumor growth. Myofibroblasts also likely influence tumor growth through angiogenesis-independent mechanisms as well (not shown).
Enhanced expression of Ena/VASP proteins has been recently described in diverse tumor cells with the proposition that this may promote tumor progression and metastasis.41–44 However, indirect tumor growth effects of Ena/VASP expressed within stromal cells such as pericytes have not been previously explored. Indeed, tumor stroma promotes angiogenesis and tumor growth by releasing growth factors, chemokines, and extracellular matrix and is thus now an increasingly recognized anti-cancer target.1 In the liver, quiescent HSC activation into myofibroblasts has been identified as one of key events occurring in the hepatic tumor microenvironment that can promote tumor growth by multiple mechanisms including matrix deposition, growth factor production, and angiogenesis.22,45–49 Our tumoral coimplantation studies with pericyte precursor cells expressing the EVH2 mutant, which we show acts as a dominant negative mutant of Ena/VASP function, provide a mechanistic basis for a role of pericyte motility in the regulation of tumor growth because pericyte motility regulates pericyte recruitment to vessels, a key step in the angiogenic process.16,38,39 However, the inhibitory effects of the EVH2 mutant are likely due to both antiangiogenesis-dependent and antiangiogenesis-independent effects of EVH2 overexpressing MEF because myofibroblasts in the tumor microenvironment regulate tumor cell proliferation though multiple redundant and distinct mechanisms.1,10 Presently, the ability to image and evaluate the function of FA in vivo is limited.50 Indeed, our initial attempts to image FA in our tumor sections in vivo using IF for FA markers revealed mostly a cell–cell junction pattern of staining reminiscent of 3-D matrix adhesions described by others (data not shown).50–52 Future imaging advances in this area will be necessary to allow for better assessment of FA function in vivo. Thus, these studies not only uncover new biological functions of Ena/VASP in the regulation of FA and pericyte motility but they also demonstrate that Ena/VASP proteins within tumor stroma may present potential therapeutic targets for antitumor therapy by virtue of their effects within the tumor microenvironment.
Prior studies suggest a cell-type specific role of VASP in the regulation of FA. For example, VASP plays a regulatory role on FA in endothelial cells.6,8 However, in mouse cardiac fibroblasts, the effects of VASP on FA were not as evident.7,25 Ena/VASP proteins are required for neuritogenesis,53 and, interestingly, the HSC used in this study may share a common origin with neuronal cells as suggested by expression of the neuronal makers, glial fibrillary acid protein (GFAP) and NG2 chondroitin sulfate proteoglycan.15 The possible neuronal lineage of these cells in addition to their high level of VASP expression may account for the robust effect of VASP on FA development observed in our studies. The identification of Rac1 within a VASP/Rac1 protein complex is opportune, owing to its putative role as a VASP signaling partner and its requisite role in the process of FA assembly.35–37 Recently, a Mena-Rac1 association was identified in glioblastoma cell lines,54 further conceptually supporting our data demonstrating a VASP-Rac1 interaction.
In summary, these studies identify VASP as a key regulator of tumor growth through its effects on FA dynamics, migration, and angiogenic capacity of pericytes. Substantial mechanistic studies reveal a key role for Rac1 as a key effector molecule by which these effects are achieved. It is anticipated that further insights pertaining to the cell biology of tumor microenvironment will eventually lead to new therapies and approaches to treat cancer by targeting this tumoral compartment.
Acknowledgements
We thank Dr. Raul Urrutia for helpful suggestions and critical review of the data.
Footnotes
Supported through R01 DK (V.S.), P30 NIDDK Center grant, K01 CA, and an ALF/AASLD 2006 Sheila Sherlock grant (N.K.).
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Contributor Information
Ningling Kang, Email: kang.ningling@mayo.edu.
Vijay Shah, Email: shah.vijay@mayo.edu.
Web Extra Material
References
- 1.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 2.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 3.Wehrle-Haller B, Imhof B. The inner lives of focal adhesions. Trends Cell Biol. 2002;12:382–389. doi: 10.1016/s0962-8924(02)02321-8. [DOI] [PubMed] [Google Scholar]
- 4.Kwiatkowski AV, Gertler FB, Loureiro JJ. Function and regulation of Ena/VASP proteins. Trends Cell Biol. 2003;13:386–392. doi: 10.1016/s0962-8924(03)00130-2. [DOI] [PubMed] [Google Scholar]
- 5.Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol. 2003;19:541–564. doi: 10.1146/annurev.cellbio.19.050103.103356. [DOI] [PubMed] [Google Scholar]
- 6.Smolenski A, Poller W, Walter U, Lohmann SM. Regulation of human endothelial cell focal adhesion sites and migration by cGMP-dependent protein kinase I. J Biol Chem. 2000;275:25723–25732. doi: 10.1074/jbc.M909632199. [DOI] [PubMed] [Google Scholar]
- 7.Galler AB, Garcia Arguinzonis MI, Baumgartner W, Kuhn M, Smolenski A, Simm A, Reinhard M. VASP-dependent regulation of actin cytoskeleton rigidity, cell adhesion, and detachment. Histochem Cell Biol. 2006;125:457–474. doi: 10.1007/s00418-005-0091-z. [DOI] [PubMed] [Google Scholar]
- 8.Schlegel N, Waschke J. Impaired integrin-mediated adhesion contributes to reduced barrier properties in VASP-deficient microvascular endothelium. J Cell Physiol. 2009;220:357–366. doi: 10.1002/jcp.21772. [DOI] [PubMed] [Google Scholar]
- 9.Xu L, Hui A, Albanis E, Arther M, O'Byrne S, Blaner W, Mukherjee P, Friedman S, Eng F. Human hepatic stellate cell lines. LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54:142–151. doi: 10.1136/gut.2004.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Decker NK, Abdelmoneim SS, Yaqoob U, Hendrickson H, Hormes J, Bentley M, Pitot H, Urrutia R, Gores GJ, Shah VH. Nitric oxide regulates tumor cell cross-talk with stromal cells in the tumor microenvironment of the liver. Am J Pathol. 2008;173:1002–1012. doi: 10.2353/ajpath.2008.080158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang-Decker NCS, Chatterjee S, Yao J, Egan J, Semela D, Mukhopadhyay D, Shah V. Nitric oxide promotes endothelial cell survival signaling through S-nitrosylation and activation of dynamin-2. J Cell Sci. 2007;120:492–501. doi: 10.1242/jcs.03361. [DOI] [PubMed] [Google Scholar]
- 12.de la Taille A, Katz AE, Bagiella E, Buttyan R, Sharir S, Olsson CA, Burchardt T, Ennis RD, Rubin MA. Microvessel density as a predictor of PSA recurrence after radical prostatectomy. A comparison of CD34 and CD31. Am J Clin Pathol. 2000;113:555–562. doi: 10.1309/02W2-KE50-PKEF-G2G4. [DOI] [PubMed] [Google Scholar]
- 13.Das A, Yaqoob U, Mehta D, Shah VH. FXR Promotes Endothelial Cell Motility Through Coordinated Regulation of FAK and MMP-9. Arterioscler Thromb Vasc Biol. 2009;29:562–570. doi: 10.1161/ATVBAHA.108.182725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huttelmaier S, Mayboroda O, Harbeck B, Jarchau T, Jockusch BM, Rudiger M. The interaction of the cell-contact proteins VASP and vinculin is regulated by phosphatidylinositol-4,5-bisphosphate. Curr Biol. 1998;8:479–488. doi: 10.1016/s0960-9822(98)70199-x. [DOI] [PubMed] [Google Scholar]
- 15.Lee J, Decker N, Chatterjee S, Yao J, Friedman S, Shah V. Mechanisms of nitric oxide interplay with Rho GTPase family members in modulation of actin membrane dynamics in pericytes and fibroblasts. Am J Pathol. 2005;166:1861–1870. doi: 10.1016/S0002-9440(10)62495-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Semela D, Das A, Langer DA, Kang N, Leof E, Shah VH. Platelet-derived growth factor signaling through ephrin-B2 regulates hepatic vascular structure and function. Gastroenterology. 2008;135:671–679. doi: 10.1053/j.gastro.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knox P. Kinetics of cell spreading in the presence of different concentrations of serum or fibronectin-depleted serum. J Cell Sci. 1984;71:51–59. doi: 10.1242/jcs.71.1.51. [DOI] [PubMed] [Google Scholar]
- 18.Runyan RB, Versalovic J, Shur BD. Functionally distinct laminin receptors mediate cell adhesion and spreading: the requirement for surface galactosyltransferase in cell spreading. J Cell Biol. 1988;107:1863–1871. doi: 10.1083/jcb.107.5.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao S, Yao Y, McCabe T, Yao Q, Katusic Z, Sessa W, Shah V. Direct interaction between eNOS and dynamin-2: Implications for NOS function. J Biol Chem. 2001;276:14249–14256. doi: 10.1074/jbc.M006258200. [DOI] [PubMed] [Google Scholar]
- 20.Hautekeete ML, Geerts A. The hepatic stellate (Ito) cell: its role in human liver disease. Virchows Arch. 1997;430:195–207. doi: 10.1007/BF01324802. [DOI] [PubMed] [Google Scholar]
- 21.Friedman SL. Mechanisms of disease: mechanisms of hepatic fibrosis and therapeutic implications. Nat Clin Pract Gastroenterol Hepatol. 2004;1:98–105. doi: 10.1038/ncpgasthep0055. [DOI] [PubMed] [Google Scholar]
- 22.Olaso E, Salado C, Egilegor E, Gutierrez V, Santisteban A, Sancho-Bru P, Friedman S, Vidal-Vanaclocha F. Proangiogenic role of tumor-activated hepatic stellate cells in experimental melanoma metastasis. Hepatology. 2003;37:674–685. doi: 10.1053/jhep.2003.50068. [DOI] [PubMed] [Google Scholar]
- 23.Kaimori A, Potter J, Kaimori JY, Wang C, Mezey E, Koteish A. Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J Biol Chem. 2007;282:22089–22101. doi: 10.1074/jbc.M700998200. [DOI] [PubMed] [Google Scholar]
- 24.Reinhard M, Rudiger M, Jockusch BM, Walter U. VASP interaction with vinculin: a recurring theme of interactions with proline-rich motifs. FEBS Lett. 1996;399:103–107. doi: 10.1016/s0014-5793(96)01295-1. [DOI] [PubMed] [Google Scholar]
- 25.Bear JE, Loureiro JJ, Libova I, Fassler R, Wehland J, Gertler FB. Negative regulation of fibroblast motility by Ena/VASP proteins. Cell. 2000;101:717–728. doi: 10.1016/s0092-8674(00)80884-3. [DOI] [PubMed] [Google Scholar]
- 26.Hoffman LM, Jensen CC, Kloeker S, Wang CL, Yoshigi M, Beckerle MC. Genetic ablation of zyxin causes Mena/VASP mislocalization, increased motility, and deficits in actin remodeling. J Cell Biol. 2006;172:771–782. doi: 10.1083/jcb.200512115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lynch PM, Schmid-Schonbein GW, Parker LH, Schmidt M, Jin S-W, Gray AM, Beis D, Pham T, Frantz G, Paliert S, Hillan K, Stainier DYR, de Sauvage FJ, Ye W. The endothelial-cell-derived secreted factor Egf17 regulates vascular tube formation. Nature 2004; 428:754–758. Lymphat Res Biol. 2004;2:96–100. doi: 10.1089/lrb.2004.2.96. Literature watch. [DOI] [PubMed] [Google Scholar]
- 28.Niebuhr K, Ebel F, Frank R, Reinhard M, Domann E, Carl UD, Walter U, Gertler FB, Wehland J, Chakraborty T. A novel proline-rich motif present in ActA of Listeria monocytogenes and cytoskeletal proteins is the ligand for the EVH1 domain, a protein module present in the Ena/VASP family. EMBO J. 1997;16:5433–5444. doi: 10.1093/emboj/16.17.5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gertler FB, Niebuhr K, Reinhard M, Wehland J, Soriano P. Mena, a relative of VASP and Drosophila Enabled, is implicated in the control of microfilament dynamics. Cell. 1996;87:227–239. doi: 10.1016/s0092-8674(00)81341-0. [DOI] [PubMed] [Google Scholar]
- 30.Reinhard M, Giehl K, Abel K, Haffner C, Jarchau T, Hoppe V, Jockusch BM, Walter U. The proline-rich focal adhesion and microfilament protein VASP is a ligand for profilins. EMBO J. 1995;14:1583–1589. doi: 10.1002/j.1460-2075.1995.tb07146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bachmann C, Fischer L, Walter U, Reinhard M. The EVH2 domain of the vasodilator-stimulated phosphoprotein mediates tetramerization. F-actin binding, and actin bundle formation. J Biol Chem. 1999;274:23549–23557. doi: 10.1074/jbc.274.33.23549. [DOI] [PubMed] [Google Scholar]
- 32.Grosse R, Copeland JW, Newsome TP, Way M, Treisman R. A role for VASP in RhoA-Diaphanous signalling to actin dynamics and SRF activity. EMBO J. 2003;22:3050–3061. doi: 10.1093/emboj/cdg287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haffner C, Jarchau T, Reinhard M, Hoppe J, Lohmann SM, Walter U. Molecular cloning, structural analysis and functional expression of the proline-rich focal adhesion and microfilament-associated protein VASP. EMBO J. 1995;14:19–27. doi: 10.1002/j.1460-2075.1995.tb06971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brindle NP, Holt MR, Davies JE, Price CJ, Critchley DR. The focal-adhesion vasodilator-stimulated phosphoprotein (VASP) binds to the proline-rich domain in vinculin. Biochem J. 1996;318:753–757. doi: 10.1042/bj3180753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schlegel N, Burger S, Golenhofen N, Walter U, Drenckhahn D, Waschke J. The role of VASP in regulation of cAMP- and Rac 1-mediated endothelial barrier stabilization. Am J Physiol Cell Physiol. 2008;294:C178–C188. doi: 10.1152/ajpcell.00273.2007. [DOI] [PubMed] [Google Scholar]
- 36.Schlegel N, Waschke J. VASP is involved in cAMP-mediated Rac 1 activation in microvascular endothelial cells. Am J Physiol Cell Physiol. 2009;296:C453–C462. doi: 10.1152/ajpcell.00360.2008. [DOI] [PubMed] [Google Scholar]
- 37.Guo F, Debidda M, Yang L, Williams DA, Zheng Y. Genetic deletion of Rac1 GTPase reveals its critical role in actin stress fiber formation and focal adhesion complex assembly. J Biol Chem. 2006;281:18652–18659. doi: 10.1074/jbc.M603508200. [DOI] [PubMed] [Google Scholar]
- 38.Abramsson A, Lindblom P, Betsholtz C. Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest. 2003;112:1142–1151. doi: 10.1172/JCI18549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abramsson A, Berlin O, Papayan H, Paulin D, Shani M, Betsholtz C. Analysis of mural cell recruitment to tumor vessels. Circulation. 2002;105:112–117. doi: 10.1161/hc0102.101437. [DOI] [PubMed] [Google Scholar]
- 40.Vonlaufen A, Joshi S, Qu C, Phillips PA, Xu Z, Parker NR, Toi CS, Pirola RC, Wilson JS, Goldstein D, Apte MV. Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 2008;68:2085–2093. doi: 10.1158/0008-5472.CAN-07-2477. [DOI] [PubMed] [Google Scholar]
- 41.Philippar U, Roussos ET, Oser M, Yamaguchi H, Kim HD, Giampieri S, Wang Y, Goswami S, Wyckoff JB, Lauffenburger DA, Sahai E, Condeelis JS, Gertler FB. A Mena invasion isoform potentiates EGF-induced carcinoma cell invasion and metastasis. Dev Cell. 2008;15:813–828. doi: 10.1016/j.devcel.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han G, Fan B, Zhang Y, Zhou X, Wang Y, Dong H, Wei Y, Sun S, Hu M, Zhang J, Wei L. Positive regulation of migration and invasion by vasodilator-stimulated phosphoprotein via Rac1 pathway in human breast cancer cells. Oncol Rep. 2008;20:929–939. [PubMed] [Google Scholar]
- 43.Hu LD, Zou HF, Zhan SX, Cao KM. EVL (Ena/VASP-like) expression is up-regulated in human breast cancer and its relative expression level is correlated with clinical stages. Oncol Rep. 2008;19:1015–1020. [PubMed] [Google Scholar]
- 44.Toyoda A, Kawana H, Azuhata K, Yu J, Omata A, Kishi H, Higashi M, Harigaya K. Aberrant expression of human ortholog of mammalian enabled (hMena) in human colorectal carcinomas: implications for its role in tumor progression. Int J Oncol. 2009;34:53–60. [PubMed] [Google Scholar]
- 45.Theret N, Musso O, Turlin B, Lotrian D, Bioulac-Sage P, Campion JP, Boudjema K, Clement B. Increased extracellular matrix remodeling is associated with tumor progression in human hepatocellular carcinomas. Hepatology. 2001;34:82–88. doi: 10.1053/jhep.2001.25758. [DOI] [PubMed] [Google Scholar]
- 46.Vidal-Vanaclocha F. The prometastatic microenvironment of the liver. Cancer Microenviron. 2008;1:113–129. doi: 10.1007/s12307-008-0011-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Antoine M, Tag CG, Gressner AM, Hellerbrand C, Kiefer P. Expression of E-selectin ligand-1 (CFR/ESL-1) on hepatic stellate cells: implications for leukocyte extravasation and liver metastasis. Oncol Rep. 2009;21:357–362. [PubMed] [Google Scholar]
- 48.Amann T, Bataille F, Spruss T, Muhlbauer M, Gabele E, Scholmerich J, Kiefer P, Bosserhoff AK, Hellerbrand C. Activated hepatic stellate cells promote tumorigenicity of hepatocellular carcinoma. Cancer Sci. 2009;100:646–653. doi: 10.1111/j.1349-7006.2009.01087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsusue R, Kubo H, Hisamori S, Okoshi K, Takagi H, Hida K, Nakano K, Itami A, Kawada K, Nagayama S, Sakai Y. Hepatic stellate cells promote liver metastasis of colon cancer cells by the action of SDF-1/CXCR4 axis. Ann Surg Oncol. 2009;16:2645–2653. doi: 10.1245/s10434-009-0599-x. [DOI] [PubMed] [Google Scholar]
- 50.Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708–1712. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- 51.Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J Cell Biol. 2003;163:583–595. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. 2009;28:4326–4343. doi: 10.1038/onc.2009.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kwiatkowski AV, Rubinson DA, Dent EW, Edward van Veen J, Leslie JD, Zhang J, Mebane LM, Philippar U, Pinheiro EM, Burds AA, Bronson RT, Mori S, Fassler R, Gertler FB. Ena/VASP Is Required for neuritogenesis in the developing cortex. Neuron. 2007;56:441–455. doi: 10.1016/j.neuron.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 54.Higashi M, Ishikawa C, Yu J, Toyoda A, Kawana H, Kurokawa K, Matsuda M, Kitagawa M, Harigaya K. Human Mena associates with Rac1 small GTPase in glioblastoma cell lines. PLoS One. 2009;4:e4765. doi: 10.1371/journal.pone.0004765. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.