

Abstract
Senile plaques formed by β-amyloid peptides (Aβ) and neurofibrillary tangles (NFTs) formed by hyperphosphorylated tau, a microtubule-associated protein, are the hallmark lesions of Alzheimer's disease (AD) in addition to loss of neurons. While several transgenic (Tg) mouse models have recapitulated aspects of AD-like Aβ and tau pathologies, a spatiotemporal mapping paradigm for progressive NFT accumulation is urgently needed to stage disease progression in AD mouse models. Braak and co-workers developed an effective and widely used NFT staging paradigm for human AD brains. The creation of a Braak-like spatiotemporal staging scheme for tau pathology in mouse models would facilitate mechanistic studies of AD-like tau pathology. Such a scheme would also enhance the reproducibility of preclinical AD therapeutic studies. Thus, we developed a novel murine model of Aβ and tau pathologies and devised a spatiotemporal scheme to stage the emergence and accumulation of NFTs with advancing age. Notably, the development of NFTs followed a spatiotemporal Braak-like pattern similar to that observed in authentic AD. More significantly, the presence of Aβ accelerated NFT formation and enhanced tau amyloidosis; however, tau pathology did not have the same effect on Aβ pathology. This novel NFT staging scheme provides new insights into the mechanisms of tau pathobiology, and we speculate that this scheme will prove useful for other basic and translational studies of AD mouse models.
Alzheimer's disease (AD) is characterized by a triad of neuropathological hallmarks including senile plaques, neurofibrillary tangles (NFTs), and neuron loss. Senile plaques are extracellular lesions composed of β-amyloid (Aβ) peptides, whereas NFTs are composed mainly of hyperphosphorylated tau, a microtubule-associated protein. Previous reports using the six-stage NFT progression scheme developed by Braak and co-workers defined a spatiotemporal pattern of tangle accumulation which correlates more closely with the severity of dementia in AD patients than the burden of Aβ plaques.1–3 Despite the fact that the burden of Aβ plaques correlates less well with the degree of dementia, the amyloid cascade hypothesis posits that Aβ influences NFT evolution, although other evidence suggests tangles precede plaque formation.4–6 Thus, the influence of plaques and tangles on each other remains controversial, and methodological limitations of postmortem studies of AD brains precludes unequivocal resolution of this controversy.
Thus, to probe the interplay between plaques and tangles, several transgenic (Tg) mouse models with both plaque and tangle pathology have been described as reviewed elsewhere.7 For example, bigenic mouse models harboring both human mutant tau and APP transgenes display enhanced tau tangle formation when compared to their monogenic counterparts.8–10 These results lend support to the view that plaques influence the development of tangles by augmenting NFT formation. However, the lack of a rigorous strategy to define the spatiotemporal accumulation of NFTs limits clear understanding of the impact plaques exert on progressive tau pathology. Moreover, the availability of a tangle staging scheme would facilitate comparative studies of AD pathology across different Tg mice as well as enable more standardized and rigorous assessment of the effects of potential disease-modifying therapies. The need for standardization of Tg mouse studies was also emphasized in a recent assessment of the failure to reproduce many preclinical studies in Tg mouse models of amyotrophic lateral sclerosis.11 Hence, we addressed this issue by generating a novel bigenic mouse model of Aβ and tau pathology through crossing our P301S mutant tau (PS19) Tg mouse model with the well-known PDAPP model that overexpresses mutant V717F APP.12,13 Using this PS19;PDAPP bigenic model, we staged the spatiotemporal pattern of progressive accumulations of tau pathology and showed an accumulation similar to the Braak NFT staging scheme for AD brains. We then exploited this staging scheme to show that the presence of Aβ alters the progressive accumulation of tau pathology. Thus, by developing a tau staging scheme, we provide insights into the modulation of tau pathology by Aβ. We speculate that our novel staging paradigm will lead to additional insights into mechanisms of AD and prove useful in other basic and translational studies of AD mouse models.
Materials and Methods
Generation of Transgenic Mice
Bigenic F1 hybrids were generated by crossing PS19 and PDAPP Tg mice. The PDAPP mouse model is well described, and it was engineered to encode a human APP minigene carrying the V717F mutation driven by the PDGF promoter.13 The PS19 model harbors the T34 isoform of tau with one N-terminal insert and four microtubule binding repeats (1N4R) encoding the P301S mutation driven by the mouse prion protein promoter.12 Characterization and staging of mice was performed on F1 PS19;PDAPP bigenic hybrids and their monogenic parental strain.
Antibodies Used in this Study
AT8 (Thermo Scientific, Rockford, IL), a monoclonal antibody (mAb) raised to phosphorylated Ser202/Thr205 of tau14 was used as the major immunohistochemistry (IHC) tool to map the spatiotemporal accumulation of tau tangles in our single and bigenic Tg mice. Notably, this mAb does not significantly stain normal tau by most IHC methods but stains only pathologically phosphorylated tau. It has been used as an alternative method to Gallyas staining for diagnostic purposes, yielding reproducible results in interlaboratory studies comparing AT8 to Gallyas silver staining.14–16 Additional anti-tau antibodies used include AT270 mAb reactive to PHF-tau Thr181 (Thermo Scientific), AT180 mAb reactive to PHF-tau Thr231 (Thermo Scientific), polyclonal anti-tau antibody 17025 and mAb T14 that recognizes human tau.12 The distribution of Aβ plaques was monitored using mAb Nab228, which recognizes Aβ amino acids 1–11 but also detects full-length APP, and it is used to stain both mature and immature plaques. Additionally, we used Nab61, a mAb that recognizes an oligomer-specific conformational epitope in Aβ and is used here as an indicator of mature plaque formation.17 Finally, we also used mAb to glial fibrillary acidic protein (GFAP) (Dako, Carpinteria, CA) to monitor reactive astrocytes and mAb to IBA-1 (Wako Chemicals, Richmond, VA) to monitor microglial activation in Tg mouse brains.
Histology, Immunohistochemistry, and Staging
Tissue preparation and IHC was performed on single PS19 and PDAPP as well as bigenic PS19;PDAPP Tg mice at four, eight, and eleven months of age. Mice were anesthetized and transcardially perfused with phosphate buffer saline, pH 7.0 in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania. Brains were surgically removed and fixed in 4% neutral buffered formalin. To maintain consistency of dissection and to facilitate analysis in our cohort of mice, brains were coronally sliced in register with the Allen Mouse Brain Atlas (See http://mouse.brain-map.org) starting with an initial cut parallel to the olfactory tract as it bends toward the median eminence (see Figure 1). Subsequent coronal sections were generated 2 mm anteriorly and posteriorly from the initial cut using a rodent brain matrix (ASI Instruments). Coronal slabs were fixed in neutral buffered formalin and paraffin embedded. Paraffin blocks were sectioned at 6 μm through the entire block. Tissues sections were stained using a Polymer horseradish peroxidase detection system (BioGenex, India) and automatically stained using the i6000 Automated Staining System (BioGenex). Staging was performed by sampling every 20–30 slides in duplicate or triplicate. Bregma levels 0.145, −2.055, and −2.88 were analyzed for spatial distribution of AT8 positive neurons. Mice were scored according to AT8 distribution. Fibrillary tau lesions were detected using Gallyas silver and Thioflavine-S/Lipofuscin autofluorescence quenching protocols as previously described.12
Figure 1.
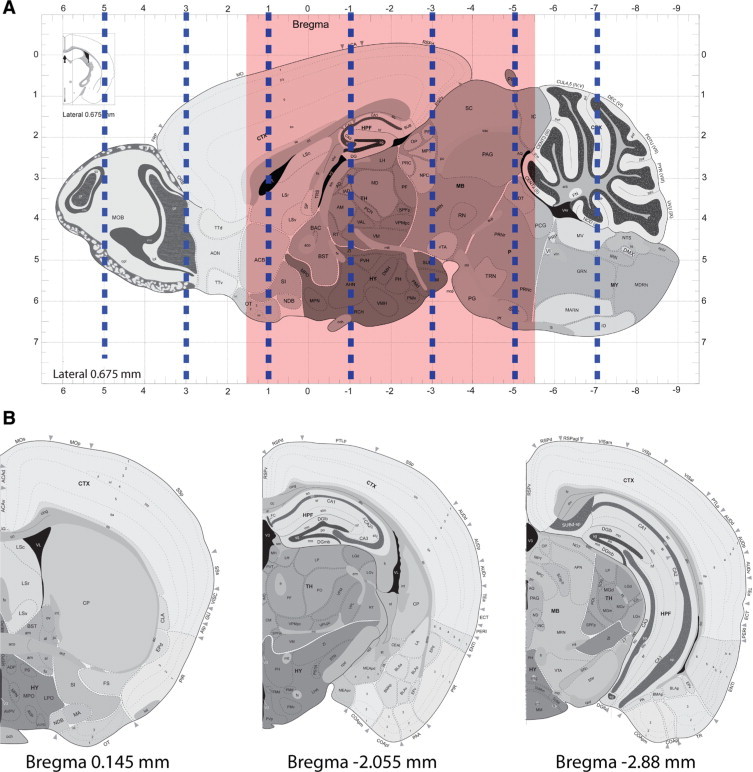
Schematic representation of bregma regions analyzed for staging tau progression. A: Sagittal view of mouse brain indicating the approximate positions of coronal slices generated at 2-mm intervals as indicated by dashed blue lines. Red shaded area indicates structural span analyzed for current study. B: Coronal views of the three bregma regions used for staging tau progression. Mouse illustrations in this and other figures are adapted from the Allen Mouse Brain Atlas (see http://mouse.brain-map.org).
Aβ Plaque Quantification
Tissue sections were stained with Nab228, and Aβ deposits were quantified by the percentage area occupied. Four regions of interest (ROI) were defined representing anterior and posterior cortex (bregmas −2.055 and −2.88, respectively) and anterior and posterior hippocampus (bregmas −2.055 and −2.88, respectively). Bregma region 0.145 was not analyzed due to the rarity of Aβ deposits present within this region. Images of Nab228 staining were grayscaled, and ROI were outlined and threshold against background. ImageJ Particle Analyzer was used to determine area fraction occupied by deposits. All data were analyzed by analysis of variance.
Analysis of Body Weight, Motor Phenotype, and Survival
Cross-sectional body weight and motor phenotype were assessed before perfusion at 4, 8, and 11 months of age on the following mice: i) PS19, ii) PDAPP, iii) PS19;PDAPP, iv) Wild-Type (wt) B6C3H/F1 background. Motor phenotype was scored on a scale of 0 to 1 according to ambulatory ability; Normal (score: 0) or paresis, paralysis, and/or hunchback posture (score: 1). Paresis was defined as those mice displaying partial utility of hindlimbs, with slight impairment to ambulation as indicated by minor dragging of hindlimbs. Paralysis was defined as lack of hindlimb function, with inability to fully stand on hindlimbs. Life span studies were completed on a separate cohort of mice (n = 6–10). Due to the paralysis observed in the PS19 line, we euthanized mice which became moribund and were classified as dead in a Kaplan–Meier Survival Curve, as their lifespan would not exceed more than two weeks when moribund. Statistical analysis on survival curves was performed using the logrank test and cross-sectional body weight was analyzed using a t-test.
Sandwich ELISA Analysis
Aβ 40 and Aβ 42 levels were detected using ELISA protocols previously described.18 Briefly, brain regions were sonicated in RIPA buffer (0.5% sodium deoxycholate, 0.1% SDS, 1% NP-40, 5 mmol/L EDTA in TBS, pH 8.0) containing protease inhibitors (1 μg/ml of pepstatin A, leupeptin, L-1-tosylamido-2-phenylethyl chloromethyl ketone, 1-chloro-3-tosylamido-7-amino-2-heptanone, soybean trypsin inhibitor, and 0.5 mmol/L PMSF) at 2 μl/mg of tissue followed by centrifugation at 100,000g for 20 minutes at 4°C. Resulting pellet was further extracted by sonication with 70% formic acid (FA) at 2 μl/mg of tissue followed by a second centrifugation. Both RIPA and FA lysates were assayed by sandwich ELISA using antibodies Ban50 (anti Aβ1–10), and end specific BC05 and BA27 to distinguish Aβ 40 and Aβ 42, respectively. Statistical analysis was performed using analysis of variance Bonferroni's posttest.
Results
Accelerated Motoric Phenotype in Bigenic PS19;PDAPP Tg Mice
PS19 mice have been shown to exhibit clasping and limb retraction when suspended by the tail, which is followed by limb weakness and progressive paralysis, hunchback posture, and ultimately the inability to feed.12 We investigated whether these motoric phenotypes were accelerated in our bigenic PS19;PDAPP mouse model. We also cross-sectionally measured body weight of our Tg mouse cohorts to determine whether there were differences in body weights between monogenic and bigenic mice. As expected, the body weight of wild-type (WT) and PDAPP mice significantly increased with advancing age as indicated in (Figure 2A). The median weight distribution of PS19 mice showed a slight decrease with a wide range (from 18–41grams) observed at 11 months of age. This disparity in weight is most likely due to variation in the onset of motor phenotype which correlates with pathology observed in PS19 mice. Similarly, a decrease in weight with increasing age was also observed for PS19;PDAPP mice, albeit with a much smaller variation in the range of weight at 11 months of age.
Figure 2.
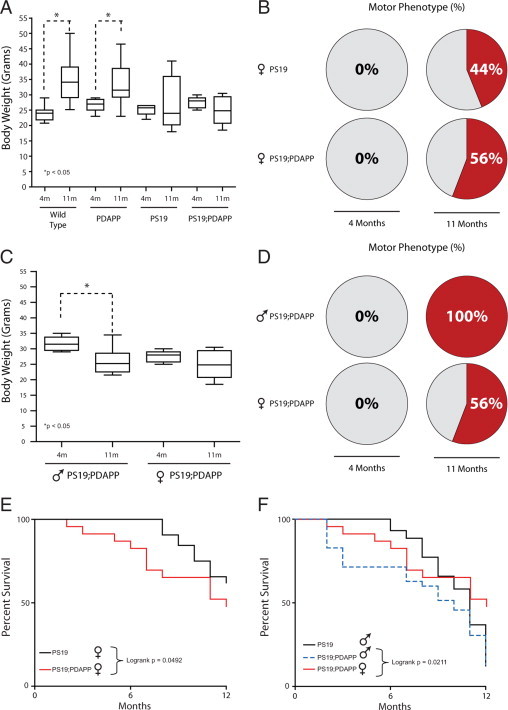
Accelerated motor phenotypes in bigenic PS19;PDAPP mice with reductions in body weight. A: Cross-sectional body weights of WT, PDAPP, PS19, and PS19;PDAPP Tg female mice at 4 and 11 months of age. Bigenic PS19;PDAPP display a more consistent reduction in body weight when compared with monogenic PS19 mice. Additionally, WT and PDAPP mice display significant increases in body weight (*P < 0.05) with age as expected. n = 18 for wild type, n = 5–9 for PDAPP, n = 7–8 for PS19, n = 8–9 for PS19;PDAPP. B: Percentage of female Tg mice displaying motor phenotypes that include paresis, paralysis, and hunchback posture. Mice that display any of these three phenotypes were given a score of 1, whereas mice lacking all three phenotypes were scored as 0. Pie chart represents percentage of mice with a motor score of 1 over two time points (4 and 11 months) and between two Tg mice, that is, PS19, and PS19;PDAPP. n = 9 for PS19, n = 8–9 for PS19;PDAPP. C and D: Comparisons of male and female bigenic PS19;PDAPP mice by cross-sectional body weight (C) and motor phenotype (D) at 4 and 11 months of age. Bigenic male mice display larger decreases in body weight and greater percentages of mice with a motor phenotype. n = 6–8 for PS19;PDAPP males, n = 8–9 for PS19;PDAPP females. E and F: Kaplan–Meier Survival Curves comparing female (E) and male (F) PS19 and bigenic PS19;PDAPP mice. Bigenic mice demonstrate a statistically significant (P = 0.0492) increase in premature death rate with a median survival of 12 months for females (versus 13 months for female PS19 monogenic) and 10 months for males (versus 11 months for male PS19 monogenic). Between male and female bigenic mice, males showed a statistically significant increase in premature death as shown in F (P = 0.0211). n = 32 for PS19 females, n = 44 for PS19 males, n = 24 for PS19;PDAPP females, n = 35 for PS19;PDAPP males.
We next evaluated motoric phenotypes of single PS19 and bigenic PS19;PDAPP mice as indicated by the initial onset of paresis and full paralysis along with the presence of a hunchback posture. Mice, which displayed these phenotypes, were given a score of 1, and mice without any motor phenotypes were scored as 0. The percentage of mice displaying motor phenotype was increased in bigenic PS19;PDAPP (56%) vs. monogenic PS19 mice (44%) at 11 months of age (Figure 2B) with no mice displaying motor phenotypes at 4 months of age. Interestingly, these phenotypic differences seem to be more profound in male bigenic PS19;PDAPP mice with a greater decrease in percentage of body weight and a larger number of those mice displaying some form of motor phenotype at 11 months of age (Figure 2C, 2D).
Finally, we analyzed the life-span of both PS19 and PS19;PDAPP mice and observed a decrease in life-span in the female population when comparing monogenic PS19 with bigenic PS19;PDAPP, with a median survival of 13 and 12 months, respectively. Again, male mice displayed an increase in premature death with a median survival of 10 months for PS19;PDAPP whereas female PS19;PDAPP had a median survival of 12 months (Figure 2, E and F). Taken together, these results suggest that bigenic PS19;PDAPP mice display an accelerated motor phenotype, which affects body weight and survival. Moreover, male bigenic mice displayed an even greater acceleration of phenotype compared to female bigenic mice.
Staging the Progression of Tau Pathology in PS19 Mice
To evaluate the impact of Aβ deposition on tau tangle formation in PS19;PDAPP mice, we compared the progression of tau pathology in monogenic PS19 with the bigenic mice. To accomplish this, we analyzed the progression of abnormally phosphorylated tau detected by the mAb AT8 in PS19 and PS19;PDAPP mouse brains at 4, 8, and 11 months of age. Figure 1A identifies positions of coronal sections generated at 2-mm intervals anteriorly and posteriorly from an initial cut parallel to the olfactory tract as it bends toward the median eminence. Approximately every 20–30 slides of the entire brain were stained for AT8 and sections from bregmas 0.145, −2.055, and −2.88 mm were selected for further analysis (Figure 1B). All images were characterized and rank ordered by the extent of the distribution and intensity of AT8 IHC, and our analysis showed the emergence of six distinct stages among all of the PS19 and PS19;PDAPP mice (Figure 3, A and B). Detailed anatomical distribution and the progression of tau pathology over time were qualitatively scored according to our six stages (see also Supplemental Figures 1–3, http://ajp.amjpathol.org) as summarized below.
Figure 3.
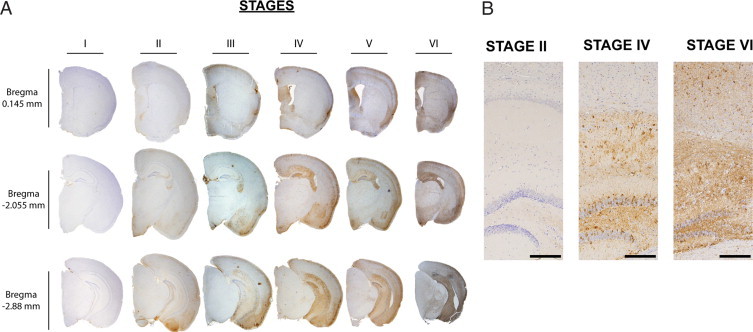
Six stages of AT8 spatial distribution. A: Representative images of six distinct mice, each with a unique stage from I-VI, immunostained with AT8. Each column is an individual mouse while each row is a defined bregma. B: Higher magnification images of Stages II, IV, and VI of the hippocampal column at bregma −2.055 mm, demonstrating progressive tau pathology (calibration bars = 200 mm).
Stage I
Both single PS19 and bigenic PS19;PDAPP mice categorized as Stage I showed rare to sparse AT8 detectable phosphorylated tau immunoreactivity (pTau-ir). pTau-ir is limited in Stage I to the most caudal isocortical regions (eg, bregma −4.28 mm). Affected regions include layers 2/3 of the isocortex and entorhinal cortex. Within the isocortical region, a ventral-dorsal patterning emerged with the most affected regions confined to the ventral cortex, including the perhirhinal, ectorhinal, temporal, and visual cortices. Within the entorhinal cortex, the lateral entorhinal is most affected, while the pre-, post-, and parasubiculum are spared (Supplemental Figure 4, see http://ajp.amjpathol.org). Interestingly, more rostral cortical regions (bregmas rostral to −2.88 mm) lack AT8 immunoreactivity (data not shown).
Stage II
Stage II is similar to Stage I regarding distribution of pTau-ir, but the pTau-ir is more consistently present in Stage II. Additionally, pTau-ir distribution progressed more rostrally throughout the isocortical layers 2/3 and entorhinal layers 2/3 and 4 in Stage II; however, the more severely affected regions are more caudal. Stage II is also marked by the initial presence of AT8 immunostaining in the cortical amygdalar region and piriform area, along with some pTau-ir in the basolateral and medial amygdalar nucleus. Rarely, AT8 pTau-ir is seen in the pyramidal layer of the CA1 field, however this is confined to caudal regions such as bregma −2.88 mm. Stage II is also marked by the occasional involvement of the hypothalamus.
Stage III
The development of more consistent and abundant pTau-ir in layers 2/3 of the isocortex is an underlying theme during each stage of development and continues through Stage III. In addition, layer 5 now begins to be affected, although not to the same extent as layers 2/3 because only a few pTau-ir neurons are found scattered throughout layer 5 of the isocortex. Again, a caudal-rostral progression pattern is seen with caudal cortical regions being more affected than rostral regions. Qualitatively, there is an increased abundance of AT8 positive neurons within the cortical amygdala as well as the basolateral and medial amygdalar nucleus. A distinctive feature of stage III is the consistent progression of pTau-ir into the hippocampal formation (HPF), more specifically within the pyramidal layers of CA1 and CA3 as well as in the dentate gyrus. As with the isocortical caudal-rostral patterning, the HPF follows the longitudinal septo-temporal axis and immunoreactivity within the HPF is confined to the more temporal axis.
Stage IV
More abundant appearance of pTau-ir throughout CA1 to CA3 and dispersion into the caudoputamen are hallmarks of Stage IV, wherein tau pathology in the HPF progresses to widely involve the longitudinal septo-temporal axis. Additionally, the hippocampal neuropil in the strata radiatum, oriens, and lucidum are pTau-ir with AT8. The hypothalamus is now more consistently stained and there is a qualitative increase in pTau-ir. Additionally, there is pTau-ir throughout the thalamus and midbrain, along with pTau-ir in the lateral septal nucleus and neurons within layer 5 of the isocortex are more consistently stained.
Stage V
Stage V is similar to Stage IV except AT8 IHC is more consistent and pTau-ir is qualitatively more abundant in all regions previously affected in Stages I–IV. A distinctive feature of Stage V is the penetration of pTau-ir into layers 4 and 6 of the isocortex. There is also a clear increase in AT8 staining throughout most of the isocortical regions in layers 2/3 and 5 along the caudal-rostral axis.
Stage VI
The progressive accumulation of tau pathology culminates in Stage VI wherein the burden of this pathology is the most extensive with evidence of tau mediated neurodegeneration indicated by extensive neuron loss, widespread astrogliosis, microgliosis, and overt volume loss in both the isocortex and HPF. All layers of the isocortex are pTau-ir with AT8 while the HPF is overwhelming AT8-positive. As in Stage IV, the hippocampal neuropil in the strata radiatum, oriens, and lucidum are clearly stained, while the molecular layer of the dentate gyrus is minimally affected. At Stage VI, a dramatic shift to a heavily stained molecular layer is clearly observed. pTau-ir is abundantly present in the perikarya of neurons as well as in their processes in the neuropil throughout most affected regions of the brain. This stage is considered the most advanced because mice become debilitated due to paralysis, and the ability to observe further progression of tau pathology is limited by their truncated lifespan.
Comparative Staging of Tau Neuropathology in Murine Models to Braak in AD Patients
Because our PS19 and PS19;PDAPP models show dramatic progression in the distribution of tau pathology from the entorhinal and neocortex, followed by involvement of the HPF and subcortical structures, to culminate in penetration of all layers of the neocortex, we investigated whether our Tg mouse tau pathology Stages I–VI showed similarity to the trajectory of Braak staging. As described in a recent review by Braak and co-workers,19,20 six distinct stages were outlined (I–VI), and these were subsequently aggregated into three main stages for expedient grading of tau pathology: 1, transentorhinal stage (Stages I/II); 2, limbic stage (Stages III/IV); 3, isocortical stage (Stages V/VI). Briefly, the transentorhinal stage is marked by the initial emergence of NFT in the transentorhinal cortex with more modest involvement of CA1 and rare tau pathology in isocortical regions. The limbic stage of development is highlighted by severe involvement in transentorhinal and entorhinal cortex along with numerous tau tangles and neuropil threads in HPF, with mild tau pathologies seen in the thalamus and amygdala. A more considerable burden of tau lesions is present in the corticomedial complex of amygdala and basolateral nuclei, along with development in putamen and accumbens nucleus.20 The final isocortical stage is highlighted by the invasion of the isocortical association areas. Additionally, there is a continued increasing presence throughout the HPF and development in stratum oriens and radiatum. Hypothalamus, claustrum, and substantia nigra are additionally affected.20
While acknowledging very dramatic differences between the mouse and human brain, we asked whether the staging of tau pathology in our PS19 showed any verisimilitude to the Braak staging of NFTs in human AD brains. Similarly to Braak staging, we clustered mouse tau pathology into three main stages as summarized in Figure 4: i) Entorhinal/Superficial Neocortical Stage, ii) Limbic/Amygdala Stage, and iii) Deep Neocortical/Subcortical Nuclei. Entorhinal/Superficial Neocortical Stage (Stage I/II) is marked by initial pTau-ir in the more caudal cortex involving the temporal association, ectorhinal, and perirhinal corticies. Similarly to Braak there is the appearance of altered pTau-ir in the entorhinal cortical layers 2/3 confined to the most caudal cortical regions. In rare instances, we also observed AT8 detectable pTau-ir in the pyramidal layer of CA1 and rare occurrences in the hypothalamus. Limbic/Amygdala Stage (Stage III/IV) is differentiated from previous stages by the consistent progression into the HPF within the pyramidal layers of CA1, CA2, and CA3, in addition to the granule layers of the dentate gyrus along the septo-temporal axis, with staining throughout the stratum radiatum, oriens, and lucidum. Within this stage we also observed a more consistent progression into cortical amygdala, basolateral, and medial amygdalar nucleus, caudoputamen, thalamus, and hypothalamus. A distinctive feature within Deep Neocortical/Subcortical Nuclei (Stage V/VI) is the progression into deeper layers of the neocortical layers 4, 5, and 6 along the caudal-rostral axis. Additionally, Stage VI is distinguished by an abundance of pTau-ir throughout the molecular layer of the dentate gyrus and increased presence throughout the caudate, putamen, and thalamic regions. Thus, we observed similarities between the different stages of tau pathology in Braak staging of AD brains and our Braak-like staging in monogenic PS19 and bigenic PS19;PDAPP Tg mice (Figure 4A). However, notable dissimilarities between Braak staging and our Braak-like staging in tau Tg mouse models were also observed. For example, the initial appearance of phosphorylated tau pathology within layers 2/3 of the isocortex at the earliest Stages I/II differs from Braak Stages I/II. Additionally, the amygdala and thalamic nuclei in the mice develop at a faster pace, appearing as early as Stage II and III, while Braak staging has their development appearing at Stage III and Stage IV. Overall the Braak-like staging in our murine model is reminiscent of Braak staging, which is highlighted by initial development in entorhinal followed by limbic systems, but the accelerated pathology in the neocortic and subcortical regions distinguishes Braak staging from murine staging.
Figure 4.
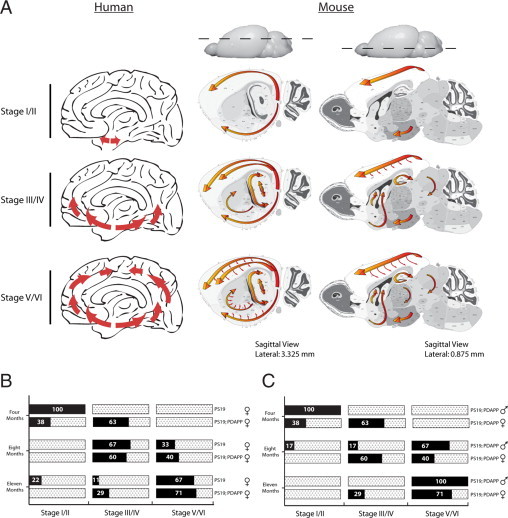
Comparison of Braak staging neurofibrillary changes to murine AT8 pTau-ir. A: Distribution of neurofibrillary development in Alzheimer's pathology and murine staging. Sagittal view of human and mouse brains, with red arrows indicating progression of neurofibrillary changes. Each row indicates defined stages I/II, III/IV, and V/VI. Human pathology initiates in the entorhinal cortex (Stage I/II), then proceeds into the hippocampal formation (HPF) and temporal cortex (Stage III/IV), and finally involves the neocortex (Stage V/VI). Similarly, murine staging begins in the entorhinal cortex with sporadic appearance within layers 2/3 of the neocortex (Stage I/II), which progresses into the HPF and further into the neocortical layers (Stage III/IV), and finally widespread development of pTau-ir in the straitum, thalamus, and deep layers of the neocortex (Stage V/VI). Note: Olfactory bulb, brainstem, and cerebellum were not scored in this study and are therefore not depicted to show pTau-ir. B: Mice at 4, 8, and 11 months of age were scored at bregmas 0.145, −2.055, and −2.88, which resulted in a staging category of I/II, III/IV, and V/VI, respectively. The graph illustrates the percentage of mice that are within a specific staging category (x axis) over a 4-, 8-, and 11-month time point (y axis, left) and within a given genotype (y axis, right). Female mice PS19 versus PS19;PDAPP are shown in B, and males versus female PS19;PDAPP are shown in C.
Expression of Mutant APP in PS19;PDAPP Mice Accelerates Progression of Tau Pathology and Augments Tau Amyloidosis
Our initial characterization of the progression of tau pathology using a Braak-like staging paradigm was conducted using both PS19 monogenic as well as PS19;PDAPP bigenic mice. To investigate whether the progression of pathological pTau-ir is altered in a spatiotemporal manner in our bigenic mouse model of AD pathology by the human mutant APP transgene, we mapped the progression of AT8 detectable pTau-ir and found similar spatial distribution of pTau-ir in our monogenic and bigenic Tg mouse models. However, we noted that the PS19;PDAPP mice entered each stage at an earlier age than their monogenic counterparts (Figure 4B). Comparing female bigenic PS19;PDAPP to female PS19 monogenic Tg mice, we noted the development and progression of staging to occur more consistently and rapidly within the bigenic model. Figure 4B shows that the majority of bigenic mice enter Stage III/IV at four months of age, whereas the monogenic Tg mice are predominately at Stage I/II at this age. Moreover, by 11 months of age, 70% of the PS19;PDAPP mice are at Stage V/VI with the rest (∼30%) at Stage III/IV, but 22% of the monogenic PS19 mice are still at Stage I/II with 11% at Stage III/IV and 67% at Stage V/VI. Thus, this greater variation in the progressive development of tau pathology that we observed in our PS19 monogenic tau mice was reduced in the bigenic PS19;PAPP mice. The accelerated and consistent manner of tau progression within the bigenic Tg mice does not seem to be gender-specific (Figure 4C).
To determine whether tau pathology in our mouse models showed the histochemical properties of amyloids similar to authentic AD NFTs, we compared the number of tangle bearing neurons detected with Thioflavin S (ThS) using brain sections at each age group from monogenic PS19 and PS19;PDAPP bigenic mice. Bigenic mice develop ThS-positive tangles at a much earlier age than monogenic PS19. Additionally, a greater proportion of bigenic mice develop ThS-positive NFTs when compared to monogenic mice at both 8 and 11 months of age (Figure 5A). Once again we observe no profound differences between male and female bigenic PS19; PDAPP mice (Figure 5A). Although the distribution of ThS stained NFTs and AT8 immunopositive hyperphosphorylated tau IHC are similar, ThS stained NFTs display a much more limited regional involvement, with more consistent accumulation in the ventral and more posterior cortical regions which include the cortical amygdala regions, lateral entorhinal cortex along with pyramidal layer of the ventral subiculum (Figure 5, B and C). Within the HPF the CA1, CA2, and CA3 along with the granular layer of the dentate gyrus appeared more vulnerable to accumulate ThS positive NFTs, but the reasons for this are not clear. There is limited ThS-positive NFTs in the more rostral cortex, and when present, these NFTs are predominantly found in layers 2/3 of piriform and cortical amygdalar regions. Argyrophilic neurofibrillary changes were also observed when sections were stained with Gallyas silver (Figure 5C). Taken together, we observed an effect of the overexpression of mutant APP and the presence of Aβ in our PS19;PDAPP bigenic mice. Specifically, Aβ accelerated tau tangle formation and augmented tau amyloidosis compared with monogenic PS19 mice.
Figure 5.
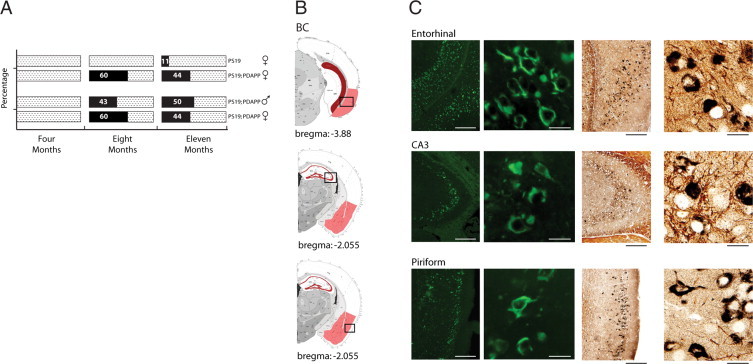
Accelerated neurofibrillary tangle formation in bigenic mice. A: PS19 and PS19; PDAPP Tg mice were stained with ThS and analyzed for NFT formation. The percentage of mice displaying positive ThS staining was plotted over a 4-, 8-, and 11-month time course. Comparison of ThS-positive tangles between male and female PS19;PDAPP Tg mice over a 4-, 8-, and 11-month time course demonstrates a consistent increase in tangle formation regardless of sex. B: Graphical illustration of ThS-positive deposits throughout the mouse brain. Red colorization indicates the presence of ThS-positive tangles. Black boxes correspond to photographic images immediately adjacent to illustration. C: Representative photographic images of ThS and Gallyas Silver staining in entorhinal, CA3, and piriform regions. Calibration bars (ThS and Gallyas Silver): low magnification 200 mm, high magnification 20 mm).
Tau Pathology Does Not Alter Aβ Plaques
Aβ deposition in PDAPP models has been characterized extensively, and there is a well-described increase in Aβ plaque load with advancing age in entorhinal and cingulate cortex as well as in HPF.13,21–24 Similar to these previous studies, we observed limited Aβ deposits at 8 months of age within the corpus callosum, stratum oriens, and radiatum of CA1, visual cortex, molecular layer of the dentate gyrus, and retrosplenial area of both our PS19;PDAPP bigenic and PDAPP monogenic mice. At 11 months of age we observed a substantial increase of Aβ deposition in similar regions in both lines of mice. More notably, our PS19;PDAPP bigenic model reconfirms previous observations of the vulnerability of the perforant pathway as our model recapitulates the deposition of Aβ in the lateral entorhinal cortex and the molecular layer of the DG.
To assess potential changes in Aβ plaque load due to concomitant tau pathology, we quantified Aβ-ir plaque deposition in bigenic PS19;PDAPP and monogenic PDAPP mice (Figure 6A). Due to the septo-temporal axis distribution of pTau-ir, we analyzed plaque load along a similar caudal-rostral axis within the HPF as well as in a limited cortical region and derived four main ROI that overlap with bregma regions analyzed for pTau-ir staging (Figure 6): ROI I – Anterior Cortex (within bregma −2.055): defined as the retrosplenial area, parietal association, somatosensory, auditory, temporal association, ectorhinal and perirhinal cortices; ROI II – Posterior Cortex (within bregma: −2.88): defined as the retrosplenial area, parietal association, auditory, temporal association, ectorhinal and perirhinal cortices; ROI III – Anterior Hippocampus (within bregma −2.055); and ROI IV – Posterior Hippocampus (within bregma −2.88). Sections were stained with Nab228 and quantified using ImageJ software. Bigenic PS19;PDAPP and monogenic PDAPP mice at 8 and 11 months of age were analyzed for plaque load, and we observed a statistically significant increase in plaque load with increasing age from 8 to 11 months for ROI III (Anterior Hippocampus) and ROI IV (Posterior Hippocampus) (Figure 6J). Regarding ROI I and II, we noted that fewer plaques were present as compared to hippocampal regions, which is most likely age dependent. Both ROI I and II were delineated to analyze cortical deposition; however, plaque deposits in these regions increase with age >12 months and is more limited at younger ages. Our analysis was restricted to a maximal age of 11 months, and therefore lower plaque burden in ROI I and II is not unexpected. We observed no statistically signification changes in plaque load due to PS19 in both ROI I and II. Interestingly, monogenic PDAPP mice showed a trend toward increased Aβ deposits compared to their bigenic counterpart at 11 months of age, which may be due to the overwhelming loss of neurons that secrete Aβ in the bigenic PS19;PDAPP mice thereby leading to an overall reduction in Aβ secretion (Figure 6, B, C, F, G, and J). In contrast, the monogenic PDAPP mice show no overt neuronal loss and therefore levels of secreted Aβ constantly rise with age and this may contribute to an increasing plaque load. Due to the decrease in survival of the bigenic mice, more extended analysis of the effect of Aβ and plaque load on tau pathology beyond 11 months is not possible. Additionally, ELISA quantification of hippocampal soluble (RIPA) and insoluble (FA) fractions of Aβ1–40 and Aβ1–42 showed no statistical significance between age matched groups, but demonstrated a trend of increased Aβ levels in monogenic compared to bigenic counterparts which is similar to our observations of Aβ-ir plaque deposition (Supplemental Figure 5, see http://ajp.amjpathol.org).
Figure 6.
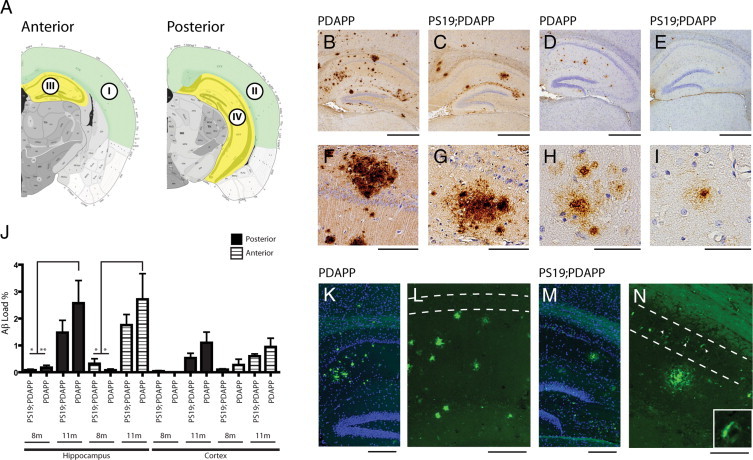
Quantitative analyses of hippocampal and cortical plaque deposition. A: Illustrative depiction of areas analyzed for Aβ load. Anterior and posterior cortical regions are highlighted in green, while anterior and posterior hippocampus regions are highlighted in yellow. Roman numerals designate ROI I–IV. B–I: Brain sections of 11-month-old monogenic PDAPP and bigenic PS19;PDAPP female mice stained with Nab228 (B, C, F, and G), Nab61 (D, E, H, and I), and Thioflavine-S (ThS) (K–N). B and C compares Nab228 staining between PDAPP and PS19;PDAPP, which highlights a tendency for increased Aβ deposits in monogenic PDAPP Tg mice. F and G are higher magnifications of B and C, respectively (Calibration bars: low magnification 0.5 mm, high magnification 100 mm). D and E compare Nab61 staining between PDAPP and PS19;PDAPP mice, demonstrating the low occurrence of mature senile plaques. H and I are higher magnifications of D and E, respectively (Calibration bars: low magnification 0.5 mm, high magnification 50 mm). J: Sections stained with Nab228 and ROI threshold was determined using ImageJ software and graphically represented as a percentage of Aβ load over a given area of ROI. In both PDAPP and PS19;PDAPP Tg mice, an increasing plaque burden is evident with increasing age, which is more predominant in monogenic PDAPP Tg mice within the hippocampal regions. Data were analyzed by analysis of variance and error bars as SEM (*P < 0.01, **P < 0.05). n = 5–9 for PS19 at 8 and 11 months, n = 4–7 for PS19;PDAPP at 8 and 11 months. K and M are ThS staining in PDAPP and PS19;PDAPP, respectively. ThS and DAPI images were overlaid to demonstrate the spatial distribution of plaque deposition within the hippocampus. L and N are ThS images of panels K and M, respectively. White dashed lines denotes CA1 regions of the hippocampus; in L, there is a lack of ThS-positive NFT, while in N, several ThS-positive NFTs are observed (arrowheads) within the CA1 region. Inset in N is higher magnification of ThS-positive NFTs found in the CA1 region. Calibration bars: 200 mm (K and M); 100 mm (L and N).
In addition to Nab228 analysis, we used an oligomeric specific antibody, Nab61, which stains mature plaques.17 We observed at 11 months of age a minimum amount of staining with Nab61 in both PDAPP and PS19;PDAPP, indicating that most Aβ deposits are not highly enriched in oligomeric Aβ species (Figure 6, D, E, H, and I). We also stained sections from these mice with ThS and observed a low abundance of ThS positive Aβ plaques relative to those that are Nab228 positive (Figure 6, K–N). These observations suggest that most Aβ deposits are not composed of Aβ species that are assembled into ThS-positive amyloid fibrils. Hence, the presence of NFTs does not accelerate formation of Aβ plaques or augment Aβ amyloidosis.
Discussion
In this study, we generated a novel bigenic mouse model of AD-like Aβ and tau pathology to: i) study the molecular pathogenesis of disease; ii) use as a model of AD-like Aβ and tau pathology; iii) determine whether tau progresses in a similar fashion as AD related changes outlined by Braak; and iv) assess whether Aβ influences the progression of tau pathology. After devising a staging scheme for tau pathology with verisimilitude to Braak staging for authentic tau pathology in AD brains, we exploited this paradigm to show that Aβ accelerated NFT formation and enhanced tau amyloidosis, but tau pathology did not have the same effect on Aβ pathology. This prompts us to speculate that this tau pathology staging scheme will prove useful in other basic and translational studies of AD mouse models.
Although tau pathology is found in the olfactory bulb, brainstem, and spinal cord of the PS19 mice, we focused our study primarily on the neocortex and hippocampus for the purpose of developing a staging paradigm and assessing the effects of Aβ on tau pathology. Also, unlike authentic AD, tau pathology in Tg mice is highly dependent on the tau transgene, levels of tau expression, and other less well-understood factors. Thus, dissimilarities between the human Braak tangle staging scheme and our Braak-like staging method are to be expected. Indeed, while our staging scheme closely approximated the Braak staging of tau pathology in AD brains, we observed an initial appearance of pTau-ir in layers 2/3 of the isocortex at Stages I/II, and amygdala and thalamic nuclei development proceeding at a faster pace, appearing at Stages II/III compared to Braak Stages III/IV. Although NFT formation in our model is found in regions not observed in AD brain, there still remains a parallel orderly sequence of pathological development between our models and the pathological progression of AD tau pathology, in that entorhinal pathology precedes limbic alterations, and widespread neocortical pathology is indicative of late stage progression.
The clearest parallel between our murine models and AD progression is the development of entorhinal to hippocampal NFTs. Anatomical consideration of NFT progression in AD brains suggests a vulnerability of the perforant pathway. The perforant pathway is a well-characterized neuronal pathway, with a collection of fibers emanating from the entorhinal cortex and “perforating” the subiculum en route to the hippocampus formation, where these fibers cross and run parallel to the hippocampal fissure and into the molecular layer of the dentate gyrus and region CA3.25–28 Disruption of the perforant pathway is presumed to be an underlying pathophysiological event in the course of AD.29–31 The similarity of NFT pathological progression in our mouse model emanating from the entorhinal cortex and proceeding into the HPF suggests that the perforant pathway is also an area of vulnerability. Whether the development of pTau-ir through this circuit is dependent on pre- and postsynaptic connection or a consequence of overexpressing a tau transgene in a murine model, or perhaps a combination of both, remains unresolved.
We observed similar staging between PS19 and PS19;PDAPP Tg mice, although bigenic mice entered stages at earlier time points and in a more consistent manner than their monogenic PS19 counterparts. More interestingly, bigenic mice displayed an enhancement of argyrophilic and ThS-positive NFT changes in the HPF and cortical amygdalar regions. PDAPP mice have been shown to develop Aβ plaques throughout the hippocampus and cortex. Previous reports have demonstrated that the PDAPP Tg model develops Aβ amyloid sheets sweeping through the hippocampus and cerebral cortex.21 Our bigenic PS19;PDAPP mouse model displays similar characteristics of Aβ deposition throughout the hippocampus, however there is limited distribution in the cortex. This limitation is age-dependent, as cortical deposition increases with age >12 months. Our bigenic models have a truncated lifespan; therefore they may not fully recapitulate all aspects of pathology found in the monogenic PDAPP model. Despite this limitation, we observed robust pathology at 11 months of age in the hippocampus. The coincidence of plaque development and ThS NFT changes at 8 months of age, which were not observed in monogenic PS19 at comparable ages, suggest that Aβ may influence or accelerate argyrophilic and ThS changes of NFT. The amyloid cascade hypothesis postulates that over time, elevated levels of Aβ aggregate and cause an inflammatory response and neuritic injury leading to disruption of neuronal metabolism and ionic homeostasis, ultimately leading to altered kinase activity affecting the hyperphosphorylation of tau and NFT formation.6,32,33
The hypothesized molecular pathogenesis of the amyloid cascade has never been fully recapitulated in Tg mouse models, and questions regarding the linearity of the amyloid cascade hypothesis have arisen due to the abundant neurodegenerative diseases whose major pathological findings are filamentous tau lesions in the absence of senile plaques. Such tauopathies argue for tau as a central mediator of neurodegeneration; however, the influences of Aβ are still prevalent, as several studies have shown that Aβ accelerates tau pathology.8–10,34 Lewis8 and Gotz34 showed that Aβ increased the frequency of NFT in specific cortical regions as indicated by Gallyas-positive NFT. Although Gallyas staining is a common technique to study NFT pathology, it is not a specific technique to ascertain whether β-pleated structures exist in NFT which is the characteristic feature of amyloids. Instead ThS is commonly used to stain amyloid structures as it binds to β-pleated sheets. The application of ThS or similar techniques was absent in Lewis and Gotz study, and therefore it is uncertain whether there was an enhancement of amyloid tau formation. We used two techniques, Gallyas and ThS, to examine NFT development in our model. Through the use of our staging paradigm, we were able to demonstrate an augmentation of amyloid tau in our bigenic PS19;PDAPP model; however, our study was limited to three time points, 4, 8, and 11 months, which limited our analysis to define whether amyloid tau forms before or after plaque deposition. At 8 and 11 months of age, all PS19;PDAPP mice displayed plaque deposits and most displayed NFT. At 4 months of age, we did not observe any Gallyas- or ThS-positive neurons, nor did we observe any plaque deposits. Finer temporal and biochemical resolution at ages 5–7 months may allow us to ascertain whether soluble monomeric or oligomeric Aβ contributes to NFT changes, or whether plaque deposition is a requirement for the observed enhancement of amyloid tau. Our PS19 model demonstrates that Aβ is not a requirement for amyloid tau formation, however there exists an augmented amyloid tau formation that is observed in our bigenic PS19;PDAPP model, the mechanisms of which remain largely unknown. We believe our model will allow us to begin to address these questions, specifically the interrelationship of APP and tau.
We have emphasized alterations in tau due to mutant APP, however previous studies have shown an acceleration of Aβ deposits in a bigenic APP/tau Tg mouse35 which disagrees with our findings. By crossing two Tg models, Tg2576 and VLW lines which express human mutant APP and triple tau mutations (G272V, P301L and R406W), respectively, they achieve an enhancement of tau pathology and an acceleration of amyloid deposition.35 What remains unclear is whether the increase in amyloid deposition in specific vulnerable brain regions correlates with increased tau pathology. Although they do not propose a specific mechanistic relationship of tau influencing Aβ deposits, they suggest a rational argument that tau and Aβ influence each other and postulate that overexpression of mutant tau may interfere with lysosomal functionality and therefore influence the production and clearance of Aβ and hence possibly leading to their observed increased Aβ pathology.35 Our model lacks any enhanced amyloid deposition, but instead shows an opposite trend, with bigenic PS19;PDAPP displaying a reduction of Aβ deposits, which may be a consequence of the overt neuronal loss observed in our models. To address whether tau influences Aβ in our PS19;PDAPP model, future biochemical experiments will analyze Aβ40/42 production before NFT formation and neuronal loss to ascertain whether there are any alterations in APP metabolism when compared to monogenic PDAPP.
In conclusion, we have recapitulated a plaque and tangle model in a distribution similarly displayed in AD brains and suggest that Aβ influences the progression of NFT, particularly in the perforant pathway. Many Tg models currently used in neurodegenerative research lack a comprehensive staging analysis. Although not a prerequisite for the study of pathogenesis of these diseases, staging mice allows investigators to pose questions regarding pathological progression in neuronal networks, to better design behavior paradigms that would test regional cortical disturbances, and allow one to focus therapeutic interventions.
Acknowledgements
We thank Theresa Schuck and Susan Leight for excellent technical support. We also thank Leigh Selesner for her assistance with experiments.
Footnotes
Supported by National Institutes of Health grant AG11542, CART fund, T32-GM07229 and T32-AG000255, and the Marian S. Ware Program for Drug Discovery. V.M.-Y.L. is the John H. Ware III Professor for Alzheimer's disease research. L.M.P. is supported by a Beca de Postgrado from Fundacion Caja Madrid.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Web Extra Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1.Braak H, Braak E. Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271–278. doi: 10.1016/0197-4580(95)00021-6. [DOI] [PubMed] [Google Scholar]
- 2.Bondareff W, Mountjoy CQ, Roth M, Hauser DL. Neurofibrillary degeneration and neuronal loss in Alzheimer's disease. Neurobiol Aging. 1989;10:709–715. doi: 10.1016/0197-4580(89)90007-9. [DOI] [PubMed] [Google Scholar]
- 3.Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18:351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- 5.Schonheit B, Zarski R, Ohm TG. Spatial and temporal relationships between plaques and tangles in Alzheimer-pathology. Neurobiol Aging. 2004;25:697–711. doi: 10.1016/j.neurobiolaging.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 6.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 7.McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer's disease in transgenic mice. Trends Genet. 2006;22:281–289. doi: 10.1016/j.tig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 9.Perez M, Ribe E, Rubio A, Lim F, Moran MA, Ramos PG, Ferrer I, Isla MT, Avila J. Characterization of a double (amyloid precursor protein-tau) transgenic: tau phosphorylation and aggregation. Neuroscience. 2005;130:339–347. doi: 10.1016/j.neuroscience.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 10.Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. 2003;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 11.Scott S, Kranz JE, Cole J, Lincecum JM, Thompson K, Kelly N, Bostrom A, Theodoss J, Al-Nakhala BM, Vieira FG, Ramasubbu J, Heywood JA. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler. 2008;9:4–15. doi: 10.1080/17482960701856300. [DOI] [PubMed] [Google Scholar]
- 12.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 13.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 14.Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189:167–169. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- 15.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del TK. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alafuzoff I, Pikkarainen M, Al-Sarraj S, Arzberger T, Bell J, Bodi I, Bogdanovic N, Budka H, Bugiani O, Ferrer I, Gelpi E, Giaccone G, Graeber MB, Hauw JJ, Kamphorst W, King A, Kopp N, Korkolopoulou P, Kovacs GG, Meyronet D, Parchi P, Patsouris E, Preusser M, Ravid R, Roggendorf W, Seilhean D, Streichenberger N, Thal DR, Kretzschmar H. Interlaboratory comparison of assessments of Alzheimer disease-related lesions: a study of the BrainNet Europe Consortium. J Neuropathol Exp Neurol. 2006;65:740–757. doi: 10.1097/01.jnen.0000229986.17548.27. [DOI] [PubMed] [Google Scholar]
- 17.Lee EB, Leng LZ, Zhang B, Kwong L, Trojanowski JQ, Abel T, Lee VM. Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem. 2006;281:4292–4299. doi: 10.1074/jbc.M511018200. [DOI] [PubMed] [Google Scholar]
- 18.Lee EB, Skovronsky DM, Abtahian F, Doms RW, Lee VM. Secretion and intracellular generation of truncated Abeta in beta-site amyloid-beta precursor protein-cleaving enzyme expressing human neurons. J Biol Chem. 2003;278:4458–4466. doi: 10.1074/jbc.M210105200. [DOI] [PubMed] [Google Scholar]
- 19.Braak H, Rub U, Schultz C, Del TK. Vulnerability of cortical neurons to Alzheimer's and Parkinson's diseases. J Alzheimers Dis. 2006;9:35–44. doi: 10.3233/jad-2006-9s305. [DOI] [PubMed] [Google Scholar]
- 20.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 21.Reilly JF, Games D, Rydel RE, Freedman S, Schenk D, Young WG, Morrison JH, Bloom FE. Amyloid deposition in the hippocampus and entorhinal cortex: quantitative analysis of a transgenic mouse model. Proc Natl Acad Sci USA. 2003;100:4837–4842. doi: 10.1073/pnas.0330745100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Su Y, Ni B. Selective deposition of amyloid-beta protein in the entorhinal-dentate projection of a transgenic mouse model of Alzheimer's disease. J Neurosci Res. 1998;53:177–186. doi: 10.1002/(SICI)1097-4547(19980715)53:2<177::AID-JNR6>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 23.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT. Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci. 1997;17:7053–7059. doi: 10.1523/JNEUROSCI.17-18-07053.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Witter MP, Wouterlood FG, Naber PA, Van HT. Anatomical organization of the parahippocampal-hippocampal network. Ann NY Acad Sci. 2000;911:1–24. doi: 10.1111/j.1749-6632.2000.tb06716.x. [DOI] [PubMed] [Google Scholar]
- 26.Steward O. Topographic organization of the projections from the entorhinal area to the hippocampal formation of the rat. J Comp Neurol. 1976;167:285–314. doi: 10.1002/cne.901670303. [DOI] [PubMed] [Google Scholar]
- 27.Steward O, Scoville SA. Cells of origin of entorhinal cortical afferents to the hippocampus and fascia dentata of the rat. J Comp Neurol. 1976;169:347–370. doi: 10.1002/cne.901690306. [DOI] [PubMed] [Google Scholar]
- 28.Hjorth-Simonsen A, Jeune B. Origin and termination of the hippocampal perforant path in the rat studied by silver impregnation. J Comp Neurol. 1972;144:215–232. doi: 10.1002/cne.901440206. [DOI] [PubMed] [Google Scholar]
- 29.Thal DR, Holzer M, Rub U, Waldmann G, Gunzel S, Zedlick D, Schober R. Alzheimer-related tau-pathology in the perforant path target zone and in the hippocampal stratum oriens and radiatum correlates with onset and degree of dementia. Exp Neurol. 2000;163:98–110. doi: 10.1006/exnr.2000.7380. [DOI] [PubMed] [Google Scholar]
- 30.Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL. Alzheimer's disease: cell-specific pathology isolates the hippocampal formation. Science. 1984;225:1168–1170. doi: 10.1126/science.6474172. [DOI] [PubMed] [Google Scholar]
- 31.Hyman BT, Van Hoesen GW, Kromer LJ, Damasio AR. Perforant pathway changes and the memory impairment of Alzheimer's disease. Ann Neurol. 1986;20:472–481. doi: 10.1002/ana.410200406. [DOI] [PubMed] [Google Scholar]
- 32.Suh YH, Checler F. Amyloid precursor protein, presenilins, and alpha-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer's disease. Pharmacol Rev. 2002;54:469–525. doi: 10.1124/pr.54.3.469. [DOI] [PubMed] [Google Scholar]
- 33.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 34.Gotz J, Chen F, van DJ, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 35.Ribe EM, Perez M, Puig B, Gich I, Lim F, Cuadrado M, Sesma T, Catena S, Sanchez B, Nieto M, Gomez-Ramos P, Moran MA, Cabodevilla F, Samaranch L, Ortiz L, Perez A, Ferrer I, Avila J, Gomez-Isla T. Accelerated amyloid deposition, neurofibrillary degeneration and neuronal loss in double mutant APP/tau transgenic mice. Neurobiol Dis. 2005;20:814–822. doi: 10.1016/j.nbd.2005.05.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.