

Abstract
Treatment options for triple-receptor negative (ER−/PR−/Her2−) and Her2-overexpressing (ER−/PR−/Her2+) breast cancers with acquired or de novo resistance are limited, and metastatic disease remains incurable. Targeting of growth signaling networks is often constrained by pathway redundancy or growth-independent cancer cell cycles. The cell-cycle protein Cdc7 regulates S phase by promoting DNA replication. This essential kinase acts as a convergence point for upstream growth signaling pathways and is therefore an attractive therapeutic target. We show that increased Cdc7 expression during mammary tumorigenesis is linked to Her2-overexpressing and triple-negative subtypes, accelerated cell cycle progression (P < 0.001), arrested tumor differentiation (P < 0.001), genomic instability (P = 0.019), increasing NPI score (P < 0.001), and reduced disease-free survival (HR = 1.98 [95% CI: 1.27–3.10]; P = 0.003), thus implicating its deregulation in the development of aggressive disease. Targeting Cdc7 with RNAi, we demonstrate that p53-mutant Her2-overexpressing and triple-negative breast cancer cell lines undergo an abortive S phase and apoptotic cell death due to loss of a p53-dependent Cdc7-inhibition checkpoint. In contrast, untransformed breast epithelial cells arrest in G1, remain viable, and are able to resume cell proliferation on recovery of Cdc7 kinase activity. Thus, Cdc7 appears to represent a potent and highly specific anticancer target in Her2-overexpressing and triple-negative breast cancers. Emerging Cdc7 kinase inhibitors may therefore significantly broaden the therapeutic armamentarium for treatment of the aggressive p53-mutant breast cancer subtypes identified in this study.
Breast cancer is the most frequently diagnosed malignancy in women in the Western world and accounts for around 16% of all cancer death.1 Despite increasing incidence, these mortality figures are decreasing as a result of widespread screening programs and systemic use of adjuvant hormonal therapy and chemotherapy.2,3 Moreover, targeted therapies for breast cancer are evolving rapidly and are broadening available therapeutic options.4,5 Targeting of Her2/neu with trastuzumab has resulted in remarkable reductions in relapse when combined with chemotherapy in Her2-positive breast cancers.6 However, the majority of patients are Her2-negative, and acquired and de novo resistance further limits this type of therapeutic intervention. This has led to the targeting of additional components of growth and survival signaling pathways including ras, raf, Mek, PI3K, and mTOR.7 It is not yet clear how maximal blockade of vertical signal transduction pathways with a combination of receptor and downstream agents will be tolerated. This approach is further compromised by pathway redundancy and cancer cell cycles becoming independent of upstream growth signaling pathways, so-called autonomous cancer cell cycles.8 In particular, therapeutic options for treatment of basal-like cancers are severely constrained by their estrogen (ER), progesterone (PR), and Her2 triple-receptor negative status. New molecularly targeted therapies are therefore urgently required for aggressive breast cancers if further decline in mortality is to be achieved.
An alternative approach to the vertical targeting of signal transduction pathways is to direct therapeutic interventions downstream at the DNA replication initiation machinery.8 Cdc7 kinase is a core component of this machinery and is therefore a potentially attractive target for cancer therapy.9 Cdc7 kinase phosphorylates and activates the Mcm2-7 replicative helicase, an essential step for the initiation of DNA synthesis at chromosomal replication origins.10–12 Cancer cells have been shown to establish only limited numbers of replication forks under Cdc7 rate-limiting conditions, causing fork stalling/collapse during an abortive S phase that is followed by apoptotic cell death.13,14 Untransformed human fibroblasts, on the contrary, appear to avoid lethal S phase progression in the presence of low Cdc7 levels by eliciting a p53-dependent Cdc7-inhibition checkpoint that arrests cells at the G1/S boundary.13 However, it has not yet been established whether this checkpoint is active in cell types of epithelial lineage, such as mammary epithelial cells. Furthermore, it is currently unclear whether the cell cycle arrest after Cdc7 inhibition is reversible. This is an essential prerequisite in the therapeutic context, as an irreversible cytostatic arrest would cause severe toxicity effects in self-renewing tissues with high turnover (eg, skin, gut mucosa and bone marrow).
The Mcm2-7 replication initiation factors (MCM) have emerged as diagnostic and prognostic biomarkers for cancer.8 More recently, we have reported that combined analysis of MCM expression and biomarkers of S-G2-M cell cycle phase progression (eg, geminin, Plk-1, Aurora A, and histone H3) allows in vivo determination of tumor cell cycle kinetics.8 This has lead to the identification of three discrete tumor cell cycle phenotypes in breast cancer: (I) well-differentiated tumors composed predominantly of MCM-negative cells, indicative of an out-of-cycle state; (II) tumors composed of cells with high MCM but low geminin, Plk-1, Aurora A, and histone H3 phosphorylated on Ser-10 (H3S10ph) levels, indicative of a G1-delayed/arrested state; and (III) tumors showing high MCM and S-G2-M marker expression, indicative of accelerated cell cycle progression (Figure 1).8,15 The accelerated cell cycle phenotype had a higher risk of relapse when compared with out-of-cycle and G1-delayed/arrested phenotypes (hazard ratio [HR] = 3.90) and was tightly associated with the Her2-overexpressing and triple-receptor negative subtypes.15 Because Cdc7 activity is rate-limiting for entry into S phase, the important question arises regarding whether overexpression of this essential kinase might be linked to breast cancers displaying the accelerated cell cycle phenotype (phenotype III) and thereby present an attractive target in Her2-overexpressing and triple-receptor negative cancers. Moreover, a high proportion (40–80%) of these aggressive tumor subtypes harbor p53 mutations,16–19 suggesting that a large proportion of these breast cancers are likely to respond to Cdc7 inhibitors due to an impaired Cdc7-inhibition checkpoint.
Figure 1.
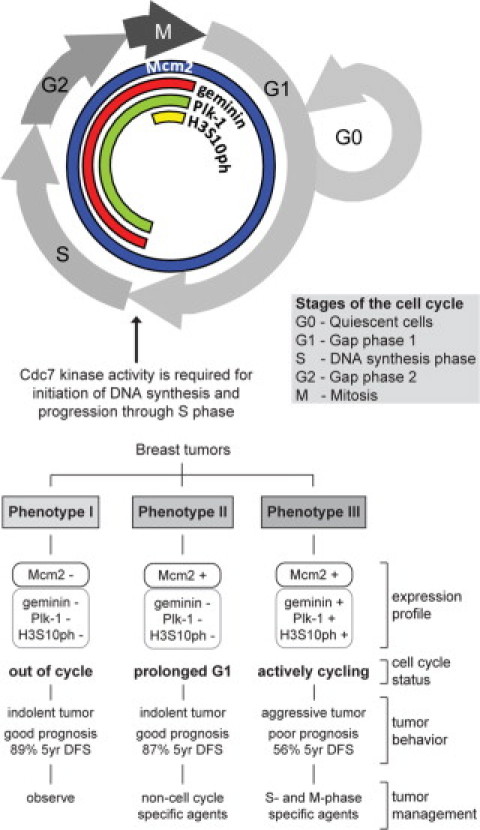
Phase-specific distribution of cell-cycle biomarkers in proliferating cells and out-of-cycle states. Three distinct cell-cycle phenotypes (I, II, and III) are characterized by the differential expression of cell-cycle biomarkers Mcm2, geminin, Plk-1, and histone H3 phosphorylated on Ser-10 (H3S10ph). Prognosis and treatment response can be predicted from the distinct immunoexpression profile displayed by a tumor.15 DFS indicates disease-free survival.
Here we report the first detailed investigation of Cdc7 expression dynamics in breast cancer and investigate Cdc7 as a novel therapeutic target in p53mut Her2-overexpressing and triple-receptor negative tumors. Importantly, we also tested in untransformed breast epithelial cells whether the Cdc7-inhibition checkpoint is reversible, a prerequisite for targeting Cdc7 in cancer therapy.
Materials and Methods
Study Cohort
Patients diagnosed with invasive breast cancer between 1999 and 2004 were identified retrospectively at University College London Hospital (London, UK). Clinical characteristics of the patient cohort are presented in Supplemental Table 1 (at http://ajp.amjpathol.org). Histological specimens were assessed for histological subtype and tumor grade based on Royal College of Pathologists guidelines and established histological20 and clinical criteria.21 Archival patient breast tissue samples were retrieved from the archives of the UCLH Department of Pathology and included cases spanning histological grades 1–3. The 171 breast cancers were subdivided according to their ER, PR, and Her2 receptor status into three distinct groups: ER+/PR+/Her2± (n = 135), ER−/PR−/Her2+ (n = 8), and ER−/PR−/Her2− (n = 28). These subgroups, commonly termed “luminal,” “Her2,” and “triple-negative” cancers, respectively, approximate to subtypes previously defined by gene expression profiling.16 Breast cancers were characterized according to cell cycle phenotype as described.15 For each patient the Nottingham Prognostic Index (NPI) was calculated as described.21 Randomly selected cases of clinically and histologically normal breast tissue from 21 premenopausal women who had undergone reduction mammoplasty were included in the study. Local research ethics approval for the study was obtained from the Joint UCL/UCLH Committees on the Ethics of Human Research.
Antibodies
Rabbit polyclonal antibody (PAb) against human geminin was generated as described.22 Antibodies were purchased from the following suppliers: Mcm2 (BM28, clone 46), p21 (SX118), and Rb (G3-245) monoclonal antibodies (MAb) from BD Biosciences (Oxford, UK); Mcm2 phosphorylated on Ser-53 (Mcm2S53ph) PAb from Bethyl Laboratories (Montgomery, TX); p53 (Ab-6 DO-1) and poly ADP ribose polymerase (PARP-1) (Ab-2 C-2-10) MAbs from Calbiochem Nottingham, UK); histone γH2A.X and Rb phosphorylated on Ser-807/811 (pRbS807/811ph) PAbs from Cell Signaling (Danvers, MA); ER (1D5), Ki-67 (MIB-1) and PR (PgR 636) MAbs from Dako (Glostrup, Denmark); Cdc7 MAb from MBL International (Woburn, MA); caspase 8 (1-1-37) MAb, histone H3 phosphorylated on Ser-10 (H3S10ph) PAb and Plk-1 (35-206) MAb from Millipore (Watford, UK); caspase 3 (CPP32 4-1-18) MAb from Novus Biologicals (Cambridge, UK); caspase 9 (F7) MAb, cyclin A (C-19) PAb, cyclin B (GNS1) MAb, and cyclin E (C-19) PAb from Santa Cruz Biotechnology (Santa Cruz, CA); and β-actin MAb from Sigma (Dorset, UK).
Immunoexpression Profiling
Paraffin wax-embedded tissue obtained at initial diagnosis was available for all patients. Tissue blocks were chosen that contained representative tumor sample. Immunoexpression profiling was performed as described.15 Primary antibodies were applied at the following dilutions: Cdc7 (1:100), ER (1:200), and PR (1:200). Her2 immunostaining was performed using a HercepTest kit (Dako). Incubation without primary antibody was used as a negative control, and colonic epithelium was used as a positive control. Labeling indices (LI) of the markers in each tumor were determined as described.15,23–25 To evaluate ER and PR expression, the quick (Allred) scoring system was used and positivity was defined as a score >2.26 Her2 expression was assessed using the manufacturer's scoring system.
DNA Image Analysis
For each case, one 40-μm section of paraffin wax-embedded tissue obtained from the same block as that assessed by immunohistochemistry was used to prepare nuclei as described.27 Tumor DNA ploidy status was determined using the Fairfield DNA Ploidy System (Fairfield Imaging, Nottingham, UK) as described.15 For statistical analysis, tetraploid and polyploid tumors were grouped with aneuploid tumors.
Cell Culture, Population Growth Assessment, and Cell Cycle Analysis
BT549 cells (ATCC HTB-122, LGC Standards, Middlesex, UK) and T47D cells (ATCC HTB-133) were cultured in RPMI 1640 medium (Invitrogen, Paisley, UK) supplemented with 10% fetal bovine serum (FBS, Invitrogen) and 10 μg/ml insulin (Sigma). MDAMB157 (ATCC HTB-24) cells were cultured in DMEM (Invitrogen) plus 15% FBS and 10 μg/ml insulin. MDAMB453 and MDAMB231 (ATCC HTB-131 and HTB-26) were cultured in DMEM plus 10% FBS. MCF7 cells (ATCC HTB-22) were grown in MEM (Invitrogen) plus 10% FBS and 10 μg/ml insulin. MCF10A cells (ATCC CRL-10317) were cultured in DMEM/F12 medium (Invitrogen) supplemented with 5% horse serum (Invitrogen), 20 ng/ml human EGF (PrepoTech, Rocky Hill, NJ), 0.5 μg/ml hydrocortisone, 100 ng/ml cholera toxin, and 10 μg/ml insulin (Sigma). Human Mammary Epithelial Cells (HMEpC) were obtained from ECACC (830-05, Health Protection Agency Culture Collections, Salisbury, UK) and cultured at population doublings 10–20 in Mammary Epithelial Cell Growth Medium KIT (PromoCell, Heidelberg, Germany). All cells were cultured at 37°C with 5% CO2. Cell proliferation assessment and flow cytometric cell cycle analysis were performed as described.28,29
CldU and IdU Incorporation Assay
Double-labeling experiments with 5-chloro-2′-deoxyuridine (CldU) and 5-iodo-2′-deoxyuridine (IdU) (Sigma) were performed as described.30 Primary antibodies used were mouse anti–5-bromo-2-deoxyuridine (BrdU) for IdU (B44, BD Biosciences) and rat anti-BrdU for CldU (BU1/75, Abcam, Cambridge, UK). DNA was visualized with DAPI. Confocal fluorescence microscopy of random fields of nuclei was performed as below and images of the DAPI channel (blue), Alexa Fluor 488 channel (green, CldU), and Alexa Fluor 594 channel (red, IdU) were obtained. Three hundred to 500 DAPI-stained nuclei (blue) were routinely counted for each treatment, and the percentage of labeled nuclei was quantified. Images of individual nuclei were acquired at ×1000 magnification.
RNA Interference
Small interfering RNA (siRNA) experiments in transformed cell lines were performed as described29 using a specific RNA duplex targeting Cdc7 mRNA (Thermo Scientific, Chicago, IL): sense 5′-GCT CAG CAG GAA AGG TGT TTT-3′ and antisense 5′-AAC ACC TTT CCT GCT GAG CTT-3′. Nontargeting siRNA (Invitrogen) was used as negative control for all transformed cells. All transfections were performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer's directions, and 75 nmol/L (BT549), 100 nmol/L (MDAMB157 and MDAMB453), or 10 nmol/L (HMEpC) of CDC7 siRNA was used to achieve efficient knockdown. For double-transfection with CDC7 and p53 siRNAs (p53 specific duplex, sense 5′-GGA AGA CUC CAG UGG UAA UUU-3′ and antisense 5′- AUU ACC ACU GGA GUC UUC CUU-3′ and ON-TARGETplus SMARTpool TP53 L-003329–00 [Thermo Scientific]), HMEpC and MCF10A cells were first transfected with 10 nmol/L CDC7 or control (Luciferase siRNA, Ambion, Austin, TX) siRNA. After 24 hours medium was removed and cells were retransfected with control (20 nmol/L), CDC7 (10 nmol/L) plus control (10 nmol/L), or CDC7 (10 nmol/L) plus p53 (9 nmol/L duplex plus 1 nmol/L SMARTpool) siRNA mixtures. Cells were harvested at the indicated time points after the second transfection. Efficient knockdown was assessed by qRT-PCR and/or Western blot.
Real-Time PCR
Total RNA was isolated from cells with the PureLink Microto-Midi Total RNA Purification System (Invitrogen) according to the manufacturer's instructions. Total RNA (40 ng) was reverse transcribed, and real-time PCR was performed using a SuperScript III Platinum SYBR Green OneStep qRTPCR kit (Invitrogen) following the manufacturer's instructions. Reactions were carried out in an Eppendorf Mastercycler ep Realplex Real-Time PCR System (Eppendorf, Cambridge, UK), and results were analyzed with Realplex v1.5 software (Eppendorf). Primer sequences were: CDC7 forward 5′-AACTTGCAGGTGTTAAAAAAG-3′ and reverse 5′-TGAAAGTGCCTTCTCCAAT-3′; GAPDH (invariant control) forward 5′-TCAACTACATGGTTTACATGTTC-3′ and reverse 5′-GATCTCGCTCCTGGAAGAT-3′.
BrdU Cell Proliferation Assay
Cells were seeded on glass coverslips and subjected to RNA interference as described above. Before harvest, cells were pulsed with 10 μmol/L BrdU (Sigma) for 1 hour at 37°C. Cells were fixed with 1% paraformaldehyde and permeabilized with PBS/0.2% Triton X-100. DNA was denatured with 2 N HCl for 1 hour and cells were incubated with anti–BrdU-FITC antibody (Alexis Biochemicals, Exeter, UK), 50 ng/ml propidium iodide (PI), and 50 ng/ml RNase A (both from Sigma). Each coverslip received a final wash with PBS before being mounted in Vectashield (Vector Laboratories, Peterborough, UK) mounting medium. Fluorescence confocal microscopy of random fields of nuclei was performed on a Leica TCS SP confocal microscope (Leica, Buckinghamshire, UK). Images were collected, and pictures of the PI channel (red) and FITC channel (green) were obtained using Leica TCS PowerScan software (Leica). The original magnification was ×200. Three hundred to 500 PI-stained nuclei (red) were routinely counted for each treatment, and the percentage of nuclei incorporating BrdU (green) was quantified.
Western Blot Analysis
Whole cell extracts (WCE) were prepared by cell lysis in modified RIPA buffer (50 mmol/L Tris-Cl pH 7.4, 300 mmol/L NaCl, 0.1% NP40, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 5 mmol/L EDTA, 1 mmol/L EGTA, 100 mmol/L sodium fluoride and 1 mmol/L sodium orthovanadate) followed by sonication for 10 seconds. Fifty micrograms of protein was loaded in each lane, separated by 4–20% SDS-PAGE, and transferred by semidry electroblotting onto Hybond C Extra nitrocellulose membranes (GE Healthcare, Buckinghamshire, UK). Blocking, antibody incubations, and washing steps were performed as described.31
TUNEL Assay
Cells were seeded on glass coverslips and subjected to RNA interference as described above. After 96 hours cells were fixed in 1% paraformaldehyde. The TUNEL assay was performed using an ApopTag Fluorescein Direct In Situ Apoptosis Detection kit (Millipore) according to the manufacturer's instructions. Coverslips were washed in PBS before being mounted in Vectashield with 1.5 μg/ml DAPI. Confocal fluorescence microscopy of random fields of nuclei was performed as described above, and images of the DAPI channel (blue) and FITC channel (green) were obtained. Three hundred to 500 DAPI-stained nuclei (blue) were routinely counted for each treatment, and the percentage of TUNEL-positive nuclei (green) was quantified.
Statistical Analysis
Labeling indices were summarized using median and interquartile range. Relationships between Cdc7 expression and tumor grade, lymph node status, tumor subtype, NPI, DNA ploidy status, and cell cycle phenotype were assessed using a combination of nonparametric Jonckheere-Terpstra, Mann–Whitney, and Kruskal Wallis analysis of variance tests as appropriate (Supplemental Table 2 at http://ajp.amjpathol.org). Analysis of disease-free and overall survival was performed using Kaplan–Meier plots (using high and low categories of Cdc7 as above and below the median value, respectively) and Cox regression (treating Cdc7 expression level as a continuous variable). Hazard ratios (with 95% confidence intervals [CI]) for Cdc7 and cell cycle phenotype were estimated separately in univariate analysis and then combined in multivariate analysis. Patients with incomplete data were excluded from multivariate analysis. All tests were two-sided and used a significance level of 0.05. Analysis was carried out using SPSS 12.0 for Windows (SPSS, Chicago, IL).
Results
Deregulated Cdc7 Expression Is Linked to Mammary Epithelial Tumorigenesis
Cdc7 protein levels are tightly down-regulated in quiescent and differentiated human cells.29 Cdc7 deregulation leading to elevated levels of this essential kinase has been linked to acquisition of the malignant, hyperproliferative phenotype in vitro and in vivo.9 Whether elevated Cdc7 levels are also associated with mammary epithelial tumorigenesis is not known. To address this question, we compared Cdc7 expression at RNA and protein level in asynchronously proliferating normal (HMEpC), hyperproliferating untransformed (MCF10A), and a panel of malignantly transformed (MCF7, BT549, T47D, MDAMB157, and MDAMB231) breast epithelial cell lines. Compared to primary cells, Cdc7 protein levels were between four- and 10-fold higher in hyperproliferating and the malignantly transformed cell lines as determined by Image J densitometry analysis (Figure 2A). Cdc7 mRNA levels were also raised in hyperproliferating and transformed cells, but to a lesser degree (Figure 2C). Next, we sought to investigate whether elevated Cdc7 levels are also associated with malignant transformation in breast epithelial cells in vivo. Epithelial cells of the terminal duct lobular unit appear to reside in a G1 arrested, “licensed” (primed) state, indicated by high MCM protein expression (median: 33.5%) and the absence of S-G2-M markers (<1%) (Figures 1 and 2B). Consistent with the finding that mammary epithelial cells withhold from progression through S-G2-M phase, Cdc7 expression was very low (median: 0.3%) (Figure 2B). In striking contrast, progression to the fully developed malignant phenotype (poorly differentiated, aggressive high-grade [grade 3] tumors) is associated with up-regulation of Cdc7 levels (median values: 0.3% versus 13.9%, P < 0.001) (Figure 2, B and D). This is coupled with cell cycle progression as indicated by a significant increase in the proportion of cells expressing the S-G2-M markers geminin (median values: 0.5% versus 17.2%, P < 0.001), Plk-1 (median values: 0% versus 36%, P < 0.001), Aurora A (median values: 0% versus 11.4%, P < 0.001) and H3S10ph (median values: 0% versus 2.5%, P < 0.001) (Figure 2, B and D and data not shown). Taken together, these findings are in keeping with the rate-limiting effects of Cdc7 on cell proliferation.
Figure 2.
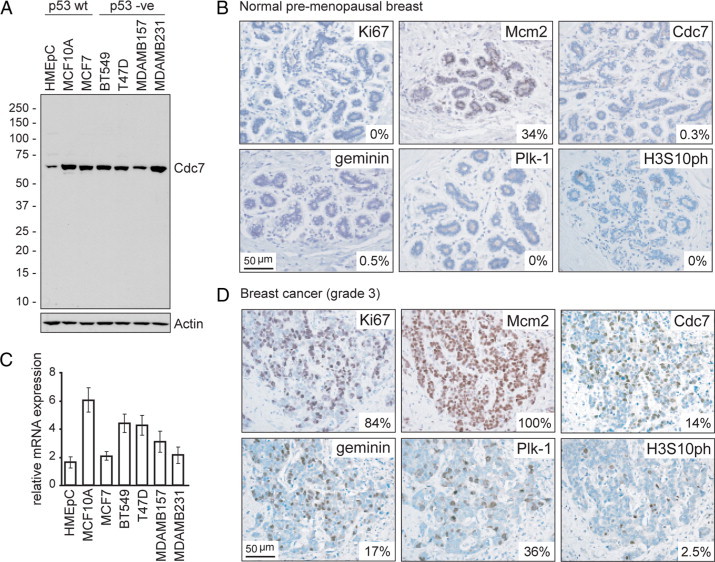
Cdc7 expression in breast epithelial cell lines and tissue. A: Immunoblot analysis of whole cell extracts (WCE) prepared from asynchronously proliferating cultures of normal (HMEpC), hyperproliferating (MCF10A), and malignantly transformed (MCF7, BT549, T47D, MDAMB157, and MDAMB231) breast epithelial cells probed with antibodies against Cdc7 and actin (loading control). C: CDC7 mRNA levels relative to GAPDH (invariant control) for each cell line as determined by qRT-PCR. B and D: Photomicrographs of tissue sections of representative normal premenopausal breast (B) and high-grade (grade 3) breast cancer (D) immunohistochemically stained with antibodies to the indicated proteins (original magnification, ×400). The median labeling index (LI) is shown for each marker. Median values and interquartile ranges for the cohort were as follows: Ki-67 40% (25%−69%); Mcm2 93% (70%−100%); Cdc7 14% (9%−22%); geminin 17% (11%−25%); Plk-1 14% (9%−20%); and H3S10ph 3% (1%−4%).
Increased Cdc7 Expression Is Associated with Arrested Tumor Differentiation and Genomic Instability
Next we examined the relationship between Cdc7 expression and pathological features associated with malignant behavior and clinical outcome in a cohort of 171 breast cancer cases (Supplemental Table 2 at http://ajp.amjpathol.org). Cdc7 expression showed a strong positive correlation with tumor grade (P < 0.001), increasing markedly with the degree of arrested differentiation (Supplemental Fig. S1A at http://ajp.amjpathol.org). Increased Cdc7 expression was also associated with acquisition of genomic instability as defined by DNA ploidy status (P = 0.019; Supplemental Fig. S1B at http://ajp.amjpathol.org). In contrast, no significant association was found between Cdc7 expression and nodal metastases (P = 0.54; Supplemental Fig. S1C at http://ajp.amjpathol.org). Increased Cdc7 expression was also found to positively correlate with increasing NPI score (P < 0.001) (Supplemental Fig. S2 at http://ajp.amjpathol.org).
Increased Cdc7 Expression Is Associated with Accelerated Cell Cycle Progression, Breast Cancer Subtype and Reduced Disease-Free Survival
We have previously identified three distinct cell cycle phenotypes in this patient cohort of 171 breast cancers: (I) an “out-of-cycle” state (n = 30); (II) a G1 arrested/delayed state (n = 41); and (III) accelerated S-G2-M phase progression (n = 100) (Figure 1).15 The accelerated cell cycle progression phenotype has a fourfold higher risk of relapse when compared with the out-of-cycle and G1-delayed/arrested phenotypes (HR = 3.90 [95% CI: 1.81–8.4], P < 0.001).15 Extending our reported findings, we noted that Cdc7 expression was significantly increased in phenotype III tumors compared to phenotype I and II tumors (median values: 12.3% versus 1.0% and 4.1% respectively, P < 0.001) (Figure 3A). The link between high Cdc7 expression levels and accelerated cell cycle progression is further reflected in the strong positive correlation between Cdc7 and the S-G2-M phase progression markers geminin (Spearman correlation coefficient = 0.8 [0.74–0.85], P < 0.001), Plk-1 (0.74 [0.66–0.8], P < 0.001), Aurora A (0.59 [0.48–0.68], P < 0.001), and H3S10ph (0.63 [0.53–0.71], P < 0.001).
Figure 3.
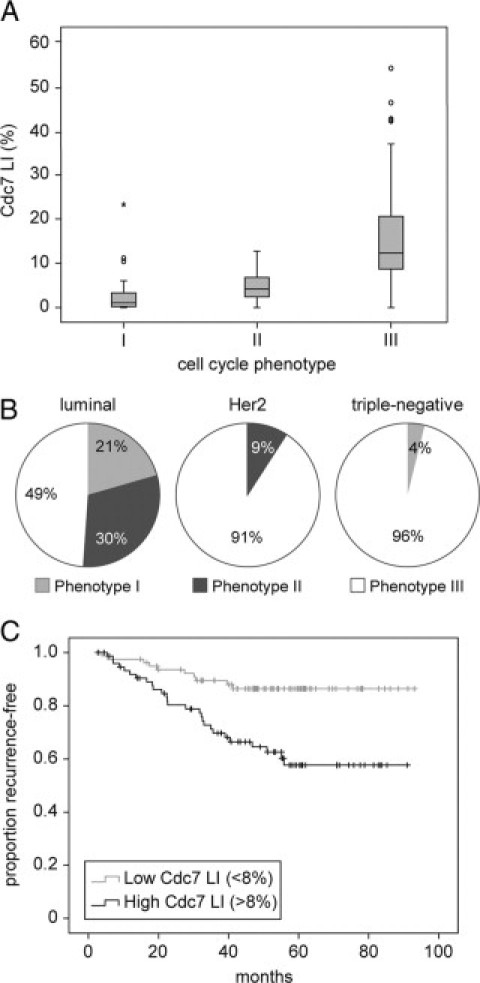
Relationship between Cdc7 expression, cell cycle phenotype, breast cancer subtype, and disease-free survival. A: The median (solid black line), interquartile range (boxed), and range (enclosed by lines) of Cdc7 expression across the breast cancer series are shown according to cell cycle phenotype (see text and Figure 1). Outlying cases are shown by isolated points. B: Relationship between cell-cycle phenotype and breast cancer subtypes. The panels show the proportion of each breast cancer subtype that display cell-cycle phenotypes I, II, and III. C: Kaplan–Meier curves showing an association between Cdc7 and disease-free survival (months from diagnosis to death, recurrence, or last follow-up) across the whole breast cancer series.
Cdc7 expression was also significantly associated with breast cancer subtype. In contrast to “luminal” tumors (ER+/PR+/Her2+/−), Cdc7 expression in “Her2” (ER−/PR−/Her2+) and “triple-negative” (ER−/PR−/Her2−) tumors was significantly elevated (median values: 5.2% versus 19.6% and 20.6% respectively, P < 0.001) (Supplemental Fig. S1D at http://ajp.amjpathol.org). Correspondingly, the majority of Her2 (91%) and triple-negative tumors (96%) were associated with the actively cycling phenotype III (Figure 3B). Taken together, these results establish a strong link between Cdc7 expression and accelerated cell cycle progression in breast cancer and show that increased Cdc7 expression is strongly associated with both Her2-overexpressing and triple-receptor negative breast cancer subtypes.
We have reported that the accelerated cell cycle phenotype (III) has a poor prognosis when compared with the out-of-cycle (I) and G1 delayed/arrested (II) phenotypes (HR = 3.90) and is an independent predictor of survival when compared with NPI in multivariate analysis (HR = 2.71).15 In keeping with this finding, in the same patient cohort Cdc7 is a predictor of disease-free survival in both univariate and multivariate (NPI-adjusted) analyses (HR = 1.98 [1.27–3.10], P = 0.003 and HR = 1.80 [1.43–2.28], P = 0.025 [per 20% increase in Cdc7 levels] respectively) (Figure 3C). These data further implicate Cdc7 deregulation in the development of aggressive disease.
Loss of Cdc7 Function Causes Apoptosis in Her2-Overexpressing and Triple-Receptor Negative Breast Cancer Cells
The majority of Her2-overexpressing and triple-receptor negative breast tumors harbor p53 mutations.16,18 Because loss of functional p53 appears to impair the Cdc7-inhibition checkpoint,13 Cdc7 kinase might be a new therapeutic target in these aggressive tumor subtypes. To test this hypothesis, we have used RNAi against CDC7 in three breast cancer cell lines (BT549, MDAMB157, and MDAMB45) with molecular characteristics approximating to Her2-overexpressing and triple-negative tumors.32,33 BT549 cells (ER−/PR−/Her2 nonamplified, p53mut) were transfected with previously characterized CDC7-siRNA.13 Relative to nontargeting control-siRNA (CO), transfection with CDC7-siRNA (Cdc7KD) reduced CDC7 mRNA levels by >90% at 96 hours posttransfection (Figure 4A). Correspondingly, in WCE Cdc7 protein was undetectable by Western blotting from 48 hours to 96 hours posttransfection, with levels increasing again from 120 hours onwards when the RNAi effect had been washed out (Figure 4A). Consistent with efficient Cdc7 depletion at 72 hours posttransfection, Mcm2 phosphorylation at Ser-53, a known phosphosite for Cdc7,34 was abolished (Figure 4, C and D). In addition, CDC7 down-regulation caused a cessation of cell proliferation. Cdc7-depleted cells showed an increase in cell numbers of just 0.2-fold at 144 hours, compared with increases of 4.0-fold for untreated (UT) cells and 2.2 fold for control-transfected (CO) cells (Figure 4B). BT549 cells depleted of Cdc7 appeared to enter an abortive S phase as demonstrated by a decrease in both the percentage of BrdU-incorporating cells (23% compared to 48% in CO cells) and the intensity of fluorescence, which directly correlates with the amount of BrdU incorporation (Figure 4C). Cdc7-depleted cells exhibited an increase in γH2A.X levels, indicative of DNA double strand breaks (Figure 4C) and, further supporting the notion of an abortive S phase, did not progress to G2/M phase as demonstrated by the lack of cyclin B and histone H3 Ser-10 phosphorylation (Figure 4C).35 Flow cytometry confirmed a progressive decline in the G2/M population in Cdc7-depleted cells and revealed a concomitant increase in the number of cells with less than 2C DNA content (Figure 4C). The detection of a sub-G1 population of cells, as well as morphological changes such as cell shrinkage and nuclear blebbing and abundant TUNEL staining (Figure 4D) compared to controls suggests that failure to elicit the Cdc7-inhibition checkpoint leads to apoptotic cell death in p53mut BT549 cells. As expected, activation of the apoptotic pathway in Cdc7-depleted BT549 cells 96 hours posttransfection was confirmed by a marked decrease in pro-caspase 3, 8, and 9 levels and detection of the apoptotic cleavage product of poly ADP ribose polymerase (PARP-1) (Figure 4D). MDAMB157 (ER−/PR−/Her2 nonamplified, p53mut) and MDAMB453 (ER−/PR−/Her2 amplified, p53mut) cells responded to CDC7 depletion in a similar manner to BT549 cells by entering an abortive S phase followed by induction of apoptosis in a significant proportion of cells (Supplemental Figs. S3 and S4 at http://ajp.amjpathol.org). The in vitro cell line data suggest that loss of p53 function may either contribute to or be sufficient for abrogation of the Cdc7-inhibition checkpoint in mammary epithelium.
Figure 4.
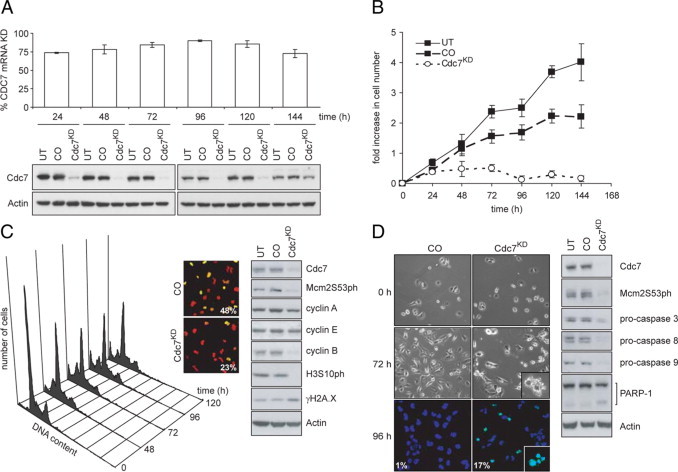
Cdc7 depletion causes apoptosis in BT549 (triple-receptor negative) breast cancer cells. A: Time course of CDC7 mRNA knockdown (KD) in BT549 cells relative to cells transfected with control-siRNA (CO) (upper panel). Immunoblot analysis of untreated (UT), control-siRNA-, and CDC7-siRNA–transfected BT549 whole cell extracts (WCE) probed with the indicated antibodies (lower panel). B: At the indicated time points, cell number was measured in UT, CO, and Cdc7-depleted (Cdc7KD) cell populations. The graph shows fold-increase in cell numbers calculated for each time point relative to the number of cells seeded. C: DNA content of Cdc7KD cells at the indicated times (left panel). At 72 hours posttransfection, BrdU incorporation was assayed. DNA was stained with propidium iodide (PI). Merged images of the BrdU-FITC (green) and PI (red) channels show the percentage of BrdU-positive cells (center panel). WCE from UT, CO, and Cdc7KD cells immunoblotted with the indicated antibodies (right panel). D: Cell death and fragmented apoptotic nuclei (inset) were analyzed by phase-contrast microscopy and positive TUNEL staining in Cdc7KD and CO cells (left panel). DAPI was used to stain DNA. Apoptotic cell death was confirmed in Cdc7KD cells by WCE immunoblotting with the indicated antibodies (right panel).
Loss of Functional p53 Disables the Cdc7-Inhibition Checkpoint in Untransformed Breast Epithelial Cells
To further investigate the link between p53 function and the Cdc7-inhibition checkpoint response, we first asked whether the checkpoint is active in untransformed mammary epithelial cells. To address this question, HMEpC and immortalized nontumorigenic MCF10A cells (p53wt) were first transfected with CDC7-siRNA. Relative to control-siRNA (CO) at 72 hours posttransfection, CDC7-siRNA (Cdc7KD) reduced CDC7 mRNA levels in HMEpC and MCF10A cells by 90% and 95%, respectively (data not shown). At this time point, Cdc7 protein levels were undetectable in WCE prepared from MCF10A cells and were found to be markedly reduced in HMEpC cells (Figure 5A and Supplemental Fig. S5A at http://ajp.amjpathol.org). In both lines, Cdc7 levels eventually recovered to a detectable level after washing out of the RNAi effect (at 96 hours in MCF10A cells and 144 hours in HMEpC, reflecting the shorter population doubling time of the immortalized line). CDC7 down-regulation prevented an increase in cell numbers up to 96 hours posttransfection in both cell lines (Figure 5B and Supplemental Fig. S5B at http://ajp.amjpathol.org). Cell proliferation resumed on recovery of Cdc7 levels at later time points, indicating that untransformed mammary epithelial cells retain the ability to re-enter the cell division cycle after Cdc7 levels are restored. Flow cytometry revealed that the majority of Cdc7-depleted HMEpC and MCF10A cells accumulated with G1 DNA content consistent with either a G1 or early S-phase arrest, while only a small fraction showed G2/M content (Figure 5C and Supplemental Fig. S5C at http://ajp.amjpathol.org). In keeping with the cell cycle profiles, the percentage of cells incorporating BrdU fell from 26% and 34% in control-transfected cells to 6% and 9% in Cdc7-depleted HMEpC and MCF10A cells (Figure 5C; and Supplemental Fig. S5D at http://ajp.amjpathol.org), indicating that the majority of cells had failed to synthesize DNA. Western blotting of WCE prepared from Cdc7-depleted HMEpC and MCF10A cells 72 hours posttransfection showed a marked increase in cyclin E levels, while levels of the S phase cyclin A and the mitotic cyclin B were reduced below the detection limit (Figure 5C and Supplemental Fig. S5E at http://ajp.amjpathol.org). Both Cdc7-depleted cell lines also showed loss of Rb phosphorylation at Ser-807/811, thought to be either Cdk4 or Cdk2 phosphorylation sites,36,37 p53 stabilization and induction of p21 expression (Figure 5C and Supplemental Fig. S5E at http://ajp.amjpathol.org). Only a minute fraction of Cdc7-depleted HMEpC and MCF10A cells with less than 2C (sub G1) DNA content was detected by flow cytometry (Figure 5C and Supplemental Fig. S5C at http://ajp.amjpathol.org). Moreover, compared to control-transfected cells, neither an increase in cells with morphological features of apoptosis and positive TUNEL staining of nuclei (Figure 5D and Supplemental Fig. S5F at http://ajp.amjpathol.org) nor cleavage of pro-caspases 3 and 9 was observed in either cell line (Figure 5D and Supplemental Fig. S5E at http://ajp.amjpathol.org), indicating that arrested cells did not activate the cell death effector machinery. Taken together, the cell cycle phase distribution and cyclin profiles, low CDK activity evident from hypophosphorylated pRb, and the loss of BrdU incorporation support the notion that, in line with other untransformed cell types, breast epithelial cells operate a Cdc7-inhibition checkpoint that arrests the cell cycle at the G1-S boundary in response to low Cdc7 levels.
Figure 5.
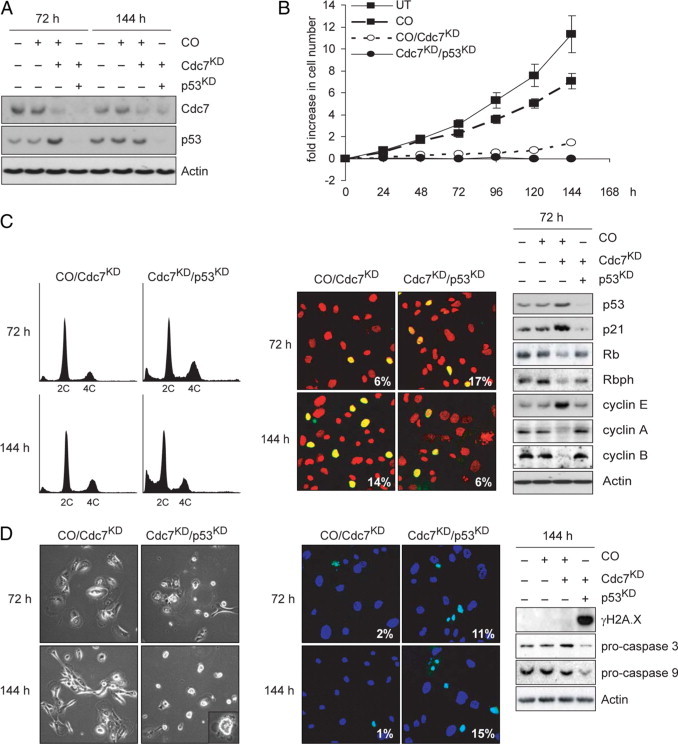
Cell cycle arrest in HMEpC cells following Cdc7 depletion is p53-dependent. A: Immunoblot analysis of WCE prepared from untreated (UT) HMEpC cells and cells transfected with control (CO), CDC7- plus CO- (CO/Cdc7KD), or CDC7- plus p53- (Cdc7KD/p53KD) siRNA probed with antibodies against Cdc7, p53, and actin (loading control). B: At the indicated time points, cell number was measured in UT, CO, CO/Cdc7KD, and Cdc7KD/p53KD cell populations. C: Cell cycle progression analysis of CO/Cdc7KD and Cdc7KD/p53KD HMEpC cells at the indicated time points, determined by DNA content (left panel), percentage of CO/Cdc7KD, and Cdc7KD/p53KD HMEpC cells incorporating BrdU (center panel), and WCE immunoblotting with the indicated antibodies (right panel). D: Apoptotic cell death was detected in Cdc7KD/p53KD HMEpC cells but not in CO or CO/Cdc7KD cells by phase-contrast microscopy (left panel), TUNEL staining (center panel), and immunoblotting for procaspase cleavage (right panel).
To test the supposition that loss of p53 function is sufficient to abrogate the Cdc7-inhibition checkpoint in mammary epithelial cells, we used RNAi to down-regulate p53 in HMEpC cells arrested by Cdc7 depletion. Western blot analysis of WCE prepared from cotransfected cells 72 hours posttransfection shows down-regulation of p53 to below baseline levels found in control cells, which in turn resulted in p21 down-regulation (Figure 5, A and C). Flow cytometry analysis of doubly depleted Cdc7/p53 cells shows an increase in the number of cells with S phase and G2/M DNA content 72 hours posttransfection. The number of cells with less than 2C DNA content progressively increased between 72 and 144 hours, coinciding with a sharp decline in the G2/M population (Figure 5C). Notably, in doubly depleted Cdc7/p53 cells Rb phosphorylation at Ser-807/811 increased, indicating recovery of CDK activity which is in keeping with low p21 levels (Figure 5C). The notion that cells lacking p53 function fail to maintain the Cdc7-inhibition checkpoint arrest, instead accumulating DNA double strand breaks (γH2AX immunostaining; Figure 5D) while progressing through S phase (increase in the percentage of cells incorporating BrdU; Figure 5C) to the G2/M boundary, is further supported by the increase in cyclin A and cyclin B levels (Figure 5C). The decline in the G2/M population in doubly depleted Cdc7/p53 cells and concomitant increase in the number of cells with less than 2C DNA content (Figure 5C) suggests that failure to maintain the Cdc7-inhibition checkpoint leads to apoptotic cell death. As expected, induction of apoptosis in Cdc7/p53 doubly depleted cells was confirmed by the appearance of cells with morphological features of apoptosis, positive TUNEL staining of nuclei and procaspase cleavage (Figure 5D). Thus loss of p53 function in early breast multistep tumorigenesis appears to be sufficient to override the Cdc7-inhibition checkpoint.
The Cdc7-Inhibition Checkpoint–Mediated Cell Cycle Arrest Is Fully Reversible in Breast Epithelial Cells
The RNAi data discussed above show that in untransformed breast epithelial cells, the Cdc7-inhibition checkpoint blocks S phase progression without affecting cell viability. Notably, Cdc7 levels and proliferative capacity were restored between 96 and 144 hours posttransfection (Figure 6, A and B), most likely due to a wash out of the RNAi effect at later time points. An alternative explanation for the recovery of cell proliferation several days posttransfection would be the emergence of a population of proliferating cells that originated from a small fraction of nontransfected cells. To directly test whether arrested cells are able to initiate DNA synthesis after recovery of Cdc7 levels, we performed double-labeling of HMEpC cells with CldU and IdU30 pre- and posttransfection with CDC7-siRNA according to the protocol shown in Figure 6C. Cells were transfected with control- or CDC7-siRNA 45 minutes after the addition of CldU. Directly after the transfection (t0) and 71¼ hours (t72) and 143¼hours (t144) posttransfection, siRNA was washed out, and IdU was added to the medium for the next 45 minutes. Cells were then fixed and studied by confocal fluorescence microscopy with antibodies to CldU (green channel) and IdU (red channel). Representative cells are shown in Figure 6C, revealing different patterns of incorporation of the halogenated nucleotides into DNA. Control-transfected cells that were exponentially proliferating over the course of the experiment continued DNA synthesis at t0 which resulted in replication foci that were labeled green, red, or yellow (green + red) in merged images of the two channels. At t72 and t144, control-transfected cells continued to synthesize DNA as indicated by IdU-incorporation (red pixels), while green pixel intensity per nucleus progressively decreased due to the distribution of incorporated CldU among daughter cells (Figure 6, C and D). In Cdc7-depleted cells only a minute population of 5% were incorporating IdU at t72, whereas the amount of incorporated CldU (green pixel intensity per nucleus) was unaffected, consistent with cell cycle arrest. Importantly, in line with the observed recovery of Cdc7 levels, at t144 cells were able to overcome the cell cycle arrest and initiated DNA synthesis as demonstrated by a 3.5-fold increase in the percentage of cells with double-labeled (yellow) nuclei (Figure 6, C and D). Thus primary breast epithelial cells respond to loss of Cdc7 function with a checkpoint-induced cell cycle arrest that is fully reversible when Cdc7 levels are restored.
Figure 6.
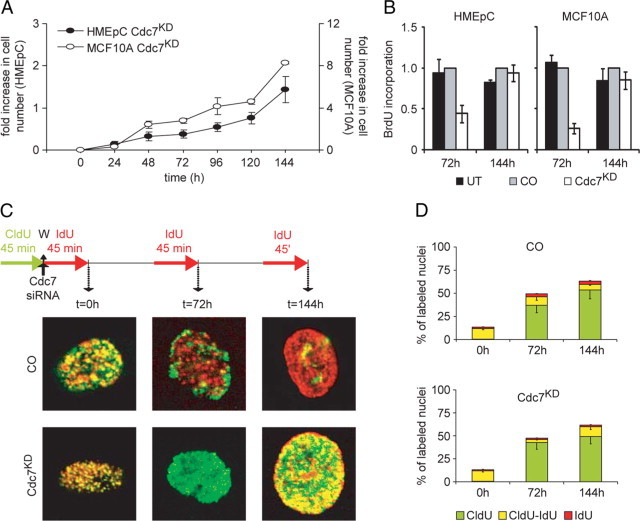
Cell cycle arrest in HMEpC cells after Cdc7 depletion is reversible. A: At the indicated time points, cell number was measured in Cdc7-depleted (Cdc7KD) HMEpC and MCF10A cell populations. B: BrdU incorporation in untreated (UT), control (CO), and CDC7-siRNA transfected HMEpC and MCF10A cells at the indicated time points (relative to CO). C: Cells transfected with CO- or CDC7- (Cdc7KD) siRNA were double-labeled with chlorodeoxyuridine (CldU) and iododeoxyuridine (IdU) as shown (W, washing step). Representative images of individual nuclei for each treatment are shown at high magnification. D: Percentage of nuclei labeled with halogenated nucleotides.
Discussion
The systemic treatment for patients with Her2-negative disease is still limited to endocrine and cytotoxic therapies,2 and increasing use of taxanes and anthracyclines in early stage disease has reduced available therapeutic options after relapse. The vertical targeting of growth signaling pathways, for example with ras and mTOR antagonists, has shown efficacy in advanced breast cancer, but response rates are restricted to around 10% of patients.38,39 Signaling pathway redundancy and establishment of autonomous cancer cell cycles potentially limits the efficacy of this approach. Novel therapeutic interventions are therefore urgently required for the treatment of aggressive Her2-overexpressing and triple-negative breast cancers.
Here we have identified Cdc7 kinase as a potential therapeutic target in the treatment of Her2-overexpressing and triple-receptor negative breast cancers. Cdc7 lies at the convergence point of upstream growth signaling pathways and may therefore circumvent problems associated with pathway redundancy.8 Moreover, Cdc7 activity is essential for S phase progression and therefore remains a potent target in cancer cell cycles that have become growth independent. Importantly, dysregulation of Cdc7 in breast cancer appears to be associated with the development of an aggressive malignant phenotype. Cdc7 overexpression was observed in immortalized and malignant cell lines when compared with primary mammary epithelial cells. Notably, increased Cdc7 expression was found to be linked to arrested tumor differentiation, genomic instability, reduced disease-free survival, an accelerated cell cycle phenotype, and Her2 (ER−/PR−/Her2+) and triple-receptor negative (ER−/PR−/Her2−) subtypes, implicating Cdc7 deregulation in the development of aggressive disease and thus providing further target validation for Cdc7 in breast cancer. RNAi targeting of Cdc7 resulted in potent cancer-cell-specific killing in p53mut triple-negative and Her2-overexpressing breast cancer cell lines. Primary mammary (HMEpC) and immortalized nontumorigenic (MCF10A) cell lines, on the contrary, avoided lethal S phase entry by activating a p53-dependent Cdc7-inhibition checkpoint resulting in cell cycle arrest at the G1-S boundary. Notably, RNAi targeting of p53 in HMEpC cells arrested by Cdc7 depletion resulted in abrogation of the Cdc7-inhibition checkpoint, which was followed by lethal S-G2-M phase progression, thus recapitulating the response observed in triple-negative and Her2-overexpressing p53mut cancer cell lines. Importantly, loss of p53 function is an early event in mammary multistep tumorigenesis, occurring at the preinvasive stage, with up to 60% of high-grade ductal carcinoma in situ lesions harboring p53 mutations.40–43 Moreover, triple-receptor negative and Her2-overexpressing breast cancer subtypes have a high proportion, 40–80%, of p53 mutations, in keeping with the fact that these aggressive tumors are thought to arise from high-grade ductal carcinoma in situ lesions.16–19,43 These observations indicate that loss of the Cdc7-inhibition checkpoint is likely to occur at an early stage in mammary multistep tumorigenesis and that pharmacological Cdc7 inhibitors currently in development9 may thus have clinical utility not only in the treatment of aggressive Her2-overexpressing and triple-receptor negative tumor types but also in the treatment of ductal carcinoma in situ lesions. In addition, a proportion of luminal type tumors harbor p53 mutations (9 to 13%) display the aggressive cell cycle phenotype (III) and may therefore respond to Cdc7 inhibitors.
The double-labeling synthetic nucleoside incorporation experiments clearly demonstrate that the cell cycle arrest after activation of the Cdc7-inhibition checkpoint in primary somatic cells is reversible on recovery of Cdc7 kinase activity, and secondly, that cells remain fully viable while arrested at the G1-S boundary. This suggests that Cdc7 inhibitors may have limited toxicity in self-renewing tissues with high turnover (eg, gut or the hemopoietic system), thus widening the potential therapeutic window. Interestingly, most systemic chemotherapeutic regimes for breast cancer, whether in the adjuvant setting or for the treatment of metastatic disease, involve combinations of S phase agents that target DNA synthesis (eg, anthracyclines and antimetabolites) and M phase agents with anti-mitotic activity (eg, taxanes, vinca alkaloids and epothilones).2 The Achilles heel of these chemotherapeutic regimens is that S- and M-phase agents also affect normal cycling cells, resulting in marrow suppression (neutropenia), hair loss, and gut toxicity. Activation of the Cdc7-inhibition checkpoint in normal cycling cells using Cdc7 inhibitors before systemic chemotherapy with S- and/or M-phase agents may therefore provide a method of shielding normal cells from chemotherapeutic agents. In support of this concept, we have observed that primary somatic cells arrested by the Cdc7-inhibition checkpoint are completely shielded from anti-mitotic cyclotherapy (S. R. Kingsbury, A. Wollenschlaeger, R. Sainsbury, K. Stoeber, G. H. Williams, manuscript submitted for publication).
In summary, our findings show that Cdc7 kinase is a potent and highly specific anti-cancer target in Her2-overexpressing and triple-receptor negative breast cancers. Pharmacological Cdc7 kinase inhibitors emerging from drug development programs worldwide are therefore likely to significantly broaden the therapeutic armamentarium available for treatment of aggressive p53mut breast cancers, both in the adjuvant and metastatic setting. Analysis of cell cycle phenotype, Cdc7 expression levels, and p53 status may allow prediction of therapeutic response to such inhibitors. Forthcoming clinical trials with Cdc7 inhibitors will provide an opportunity for this novel treatment paradigm to be tested.
Acknowledgements
We thank Joanna Franks and Caroline Richardson for assistance in retrieving archival tissue and recording clinicopathological parameters.
Footnotes
Supported by Cancer Research UK Scientific Program grant C428/A6263 (to K.S. and G.H.W.).
S.R.-A. and I.P. contributed equally to this study.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Web Extra Material
Clinical characteristics of patient cohort
Clinical characteristics of patient cohort
References
- 1.Feuer EJ, Wun LM, Boring CC, Flanders WD, Timmel MJ, Tong T. The lifetime risk of developing breast cancer. J Natl Cancer Inst. 1993;85:892–897. doi: 10.1093/jnci/85.11.892. [DOI] [PubMed] [Google Scholar]
- 2.Miles DW. Recent advances in systemic therapy. When HER2 is not the target: advances in the treatment of HER2-negative metastatic breast cancer. Breast Cancer Res. 2009;11:208. doi: 10.1186/bcr2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.La Vecchia C, Bosetti C, Lucchini F, Bertuccio P, Negri E, Boyle P, Levi F. Cancer mortality in Europe, 2000–2004, and an overview of trends since 1975. Ann Oncol. 2010;21:1323–1360. doi: 10.1093/annonc/mdp530. [DOI] [PubMed] [Google Scholar]
- 4.Petrelli F, Cabiddu M, Cazzaniga ME, Cremonesi M, Barni S. Targeted therapies for the treatment of breast cancer in the post-trastuzumab era. Oncologist. 2008;13:373–381. doi: 10.1634/theoncologist.2007-0173. [DOI] [PubMed] [Google Scholar]
- 5.Di Cosimo S, Baselga J. Targeted therapies in breast cancer: where are we now? Eur J Cancer. 2008;44:2781–2790. doi: 10.1016/j.ejca.2008.09.026. [DOI] [PubMed] [Google Scholar]
- 6.Hortobagyi GN. Trastuzumab in the treatment of breast cancer. N Engl J Med. 2005;353:1734–1736. doi: 10.1056/NEJMe058196. [DOI] [PubMed] [Google Scholar]
- 7.Johnston SR. Targeting downstream effectors of epidermal growth factor receptor/HER2 in breast cancer with either farnesyltransferase inhibitors or mTOR antagonists. Int J Gynecol Cancer. 2006;16(Suppl 2):543–548. doi: 10.1111/j.1525-1438.2006.00692.x. [DOI] [PubMed] [Google Scholar]
- 8.Williams GH, Stoeber K. Cell cycle markers in clinical oncology. Curr Opin Cell Biol. 2007;19:672–679. doi: 10.1016/j.ceb.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 9.Swords R, Mahalingam D, O'Dwyer M, Santocanale C, Kelly K, Carew J, Giles F. Cdc7 kinase - a new target for drug development. Eur J Cancer. 2010;46:33–40. doi: 10.1016/j.ejca.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 10.Machida YJ, Hamlin JL, Dutta A. Right place, right time, and only once: replication initiation in metazoans. Cell. 2005;123:13–24. doi: 10.1016/j.cell.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 11.Remus D, Diffley JF. Eukaryotic DNA replication control: lock and load, then fire. Curr Opin Cell Biol. 2009;21:771–777. doi: 10.1016/j.ceb.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 12.Masai H, You Z, Arai K. Control of DNA replication: regulation and activation of eukaryotic replicative helicase. MCM IUBMB Life. 2005;57:323–335. doi: 10.1080/15216540500092419. [DOI] [PubMed] [Google Scholar]
- 13.Montagnoli A, Tenca P, Sola F, Carpani D, Brotherton D, Albanese C, Santocanale C. Cdc7 inhibition reveals a p53-dependent replication checkpoint that is defective in cancer cells. Cancer Res. 2004;64:7110–7116. doi: 10.1158/0008-5472.CAN-04-1547. [DOI] [PubMed] [Google Scholar]
- 14.Blow JJ, Gillespie PJ. Replication licensing and cancer–a fatal entanglement? Nat Rev Cancer. 2008;8:799–806. doi: 10.1038/nrc2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loddo M, Kingsbury SR, Rashid M, Proctor I, Holt C, Young J, El-Sheikh S, Falzon M, Eward KL, Prevost T, Sainsbury R, Stoeber K, Williams GH. Cell-cycle-phase progression analysis identifies unique phenotypes of major prognostic and predictive significance in breast cancer. Br J Cancer. 2009;100:959–970. doi: 10.1038/sj.bjc.6604924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Eystein LP, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calza S, Hall P, Auer G, Bjohle J, Klaar S, Kronenwett U, Liu ET, Miller L, Ploner A, Smeds J, Bergh J, Pawitan Y. Intrinsic molecular signature of breast cancer in a population-based cohort of 412 patients. Breast Cancer Res. 2006;8:R34. doi: 10.1186/bcr1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan DS, Marchio C, Jones RL, Savage K, Smith IE, Dowsett M, Reis-Filho JS. Triple negative breast cancer: molecular profiling and prognostic impact in adjuvant anthracycline-treated patients. Breast Cancer Res Treat. 2008;111:27–44. doi: 10.1007/s10549-007-9756-8. [DOI] [PubMed] [Google Scholar]
- 19.Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S, Deming SL, Geradts J, Cheang MC, Nielsen TO, Moorman PG, Earp HS, Millikan RC. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295:2492–2502. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 20.Bloom HJ, Richardson WW. Histological grading and prognosis in breast cancer; a study of 1409 cases of which 359 have been followed for 15 years. Br J Cancer. 1957;11:359–377. doi: 10.1038/bjc.1957.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rampaul RS, Pinder SE, Elston CW, Ellis IO. Prognostic and predictive factors in primary breast cancer and their role in patient management: the Nottingham Breast Team. Eur J Surg Oncol. 2001;27:229–238. doi: 10.1053/ejso.2001.1114. [DOI] [PubMed] [Google Scholar]
- 22.Wharton SB, Hibberd S, Eward KL, Crimmins D, Jellinek DA, Levy D, Stoeber K, Williams GH. DNA replication licensing and cell cycle kinetics of oligodendroglial tumours. Br J Cancer. 2004;91:262–269. doi: 10.1038/sj.bjc.6601949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dudderidge TJ, Stoeber K, Loddo M, Atkinson G, Fanshawe T, Griffiths DF, Williams GH. Mcm2. Geminin, and KI67 define proliferative state and are prognostic markers in renal cell carcinoma. Clin Cancer Res. 2005;11:2510–2517. doi: 10.1158/1078-0432.CCR-04-1776. [DOI] [PubMed] [Google Scholar]
- 24.Kulkarni AA, Loddo M, Leo E, Rashid M, Eward KL, Fanshawe TR, Butcher J, Frost A, Ledermann JA, Williams GH, Stoeber K. DNA replication licensing factors and aurora kinases are linked to aneuploidy and clinical outcome in epithelial ovarian carcinoma. Clin Cancer Res. 2007;13:6153–6161. doi: 10.1158/1078-0432.CCR-07-0671. [DOI] [PubMed] [Google Scholar]
- 25.Shetty A, Loddo M, Fanshawe T, Prevost AT, Sainsbury R, Williams GH, Stoeber K. DNA replication licensing and cell cycle kinetics of normal and neoplastic breast. Br J Cancer. 2005;93:1295–1300. doi: 10.1038/sj.bjc.6602829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harvey JM, Clark GM, Osborne CK, Allred DC. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J Clin Oncol. 1999;17:1474–1481. doi: 10.1200/JCO.1999.17.5.1474. [DOI] [PubMed] [Google Scholar]
- 27.Haroske G, Giroud F, Reith A, Bocking A. 1997 ESACP consensus report on diagnostic DNA image cytometry. Part I: basic considerations and recommendations for preparation, measurement and interpretation European Society for Analytical Cellular Pathology. Anal Cell Pathol. 1998;17:189–200. doi: 10.1155/1998/390837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eward KL, Obermann EC, Shreeram S, Loddo M, Fanshawe T, Williams C, Jung HI, Prevost AT, Blow JJ, Stoeber K, Williams GH. DNA replication licensing in somatic and germ cells. J Cell Sci. 2004;117:5875–5886. doi: 10.1242/jcs.01503. [DOI] [PubMed] [Google Scholar]
- 29.Kulkarni AA, Kingsbury SR, Tudzarova S, Hong HK, Loddo M, Rashid M, Rodriguez-Acebes S, Prevost AT, Ledermann JA, Stoeber K, Williams GH. Cdc7 kinase is a predictor of survival and a novel therapeutic target in epithelial ovarian carcinoma. Clin Cancer Res. 2009;15:2417–2425. doi: 10.1158/1078-0432.CCR-08-1276. [DOI] [PubMed] [Google Scholar]
- 30.Seiler JA, Conti C, Syed A, Aladjem MI, Pommier Y. The intra-S-phase checkpoint affects both DNA replication initiation and elongation: single-cell and -DNA fiber analyses. Mol Cell Biol. 2007;27:5806–5818. doi: 10.1128/MCB.02278-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kingsbury SR, Loddo M, Fanshawe T, Obermann EC, Prevost AT, Stoeber K, Williams GH. Repression of DNA replication licensing in quiescence is independent of geminin and may define the cell cycle state of progenitor cells. Exp Cell Res. 2005;309:56–67. doi: 10.1016/j.yexcr.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 32.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo WL, Stilwell JL, Pinkel D, Albertson DG, Waldman FM, McCormick F, Dickson RB, Johnson MD, Lippman M, Ethier S, Gazdar A, Gray JW. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mackay A, Tamber N, Fenwick K, Iravani M, Grigoriadis A, Dexter T, Lord CJ, Reis-Filho JS, Ashworth A. A high-resolution integrated analysis of genetic and expression profiles of breast cancer cell lines. Breast Cancer Res Treat. 2009;118:481–498. doi: 10.1007/s10549-008-0296-7. [DOI] [PubMed] [Google Scholar]
- 34.Montagnoli A, Valsasina B, Brotherton D, Troiani S, Rainoldi S, Tenca P, Molinari A, Santocanale C. Identification of Mcm2 phosphorylation sites by S-phase-regulating kinases. J Biol Chem. 2006;281:10281–10290. doi: 10.1074/jbc.M512921200. [DOI] [PubMed] [Google Scholar]
- 35.Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H, Sen S, Allis CD, Sassone-Corsi P. Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol. 2002;22:874–885. doi: 10.1128/MCB.22.3.874-885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Connell-Crowley L, Harper JW, Goodrich DW. Cyclin D1/Cdk4 regulates retinoblastoma protein-mediated cell cycle arrest by site-specific phosphorylation. Mol Biol Cell. 1997;8:287–301. doi: 10.1091/mbc.8.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chi Y, Welcker M, Hizli AA, Posakony JJ, Aebersold R, Clurman BE. Identification of CDK2 substrates in human cell lysates. Genome Biol. 2008;9:R149. doi: 10.1186/gb-2008-9-10-r149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnston SR, Hickish T, Ellis P, Houston S, Kelland L, Dowsett M, Salter J, Michiels B, Perez-Ruixo JJ, Palmer P, Howes A. Phase II study of the efficacy and tolerability of two dosing regimens of the farnesyl transferase inhibitor. R115777, in advanced breast cancer. J Clin Oncol. 2003;21:2492–2499. doi: 10.1200/JCO.2003.10.064. [DOI] [PubMed] [Google Scholar]
- 39.Chan S, Scheulen ME, Johnston S, Mross K, Cardoso F, Dittrich C, Eiermann W, Hess D, Morant R, Semiglazov V, Borner M, Salzberg M, Ostapenko V, Illiger HJ, Behringer D, Bardy-Bouxin N, Boni J, Kong S, Cincotta M, Moore L. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol. 2005;23:5314–5322. doi: 10.1200/JCO.2005.66.130. [DOI] [PubMed] [Google Scholar]
- 40.Arpino G, Laucirica R, Elledge RM. Premalignant and in situ breast disease: biology and clinical implications. Ann Intern Med. 2005;143:446–457. doi: 10.7326/0003-4819-143-6-200509200-00009. [DOI] [PubMed] [Google Scholar]
- 41.Nofech-Mozes S, Spayne J, Rakovitch E, Hanna W. Prognostic and predictive molecular markers in DCIS: a review. Adv Anat Pathol. 2005;12:256–264. doi: 10.1097/01.pap.0000184177.65919.5e. [DOI] [PubMed] [Google Scholar]
- 42.Meijnen P, Peterse JL, Antonini N, Rutgers EJ, van de Vijver MJ. Immunohistochemical categorisation of ductal carcinoma in situ of the breast. Br J Cancer. 2008;98:137–142. doi: 10.1038/sj.bjc.6604112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiechmann L, Kuerer HM. The molecular journey from ductal carcinoma in situ to invasive breast cancer. Cancer. 2008;112:2130–2142. doi: 10.1002/cncr.23430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clinical characteristics of patient cohort
Clinical characteristics of patient cohort