

Abstract
Tumor-associated macrophages can potentially kill tumor cells via the high concentrations of nitric oxide (NO) produced by inducible nitric oxide synthase (iNOS); however, tumor-associated macrophages actually support tumor growth, as they are skewed toward M2 activation, which is characterized by low amounts of NO production and is proangiogenic. We show that the mouse renal cell carcinoma cell line, RENCA, which, on stimulation, expresses high levels of iNOS mRNA, loses its ability to express the iNOS protein. This effect is mediated by the microRNA miR-146a, as inhibition of RENCA cells with anti-miR- 146a restores iNOS expression and NO production (4.8 ± 0.4 versus 0.3 ± 0.1 μmol/L in uninhibited cells, P < 0.001). In vivo, RENCA tumor cells do not stain for iNOS, while infiltrating tumor-associated macrophages showed intense staining, and both cell types expressed iNOS mRNA. Restoring iNOS protein expression in RENCA cells using anti-miR-146a increases macrophage-induced death of RENCA cells by 73% (P < 0.01) in vitro and prevents tumor growth in vivo. These results suggest that, in addition to NO production by macrophages, tumor cells must produce NO to induce their own deaths, and some tumor cells may use miR-146a to reduce or abolish endogenous NO production to escape macrophage-mediated cell death. Thus, inhibiting miR-146a may render these tumor cells susceptible to therapeutic strategies, such as adoptive transfer of M1-activated macrophages.
Tumor-associated macrophages (TAMs) typically infiltrate and accumulate in solid tumors,1 and are equipped with efficient killing mechanisms, such as production of high cytotoxic amounts of nitric oxide (NO) by the enzyme inducible nitric oxide synthase (iNOS).2,3 High NO concentrations can either initiate tumor cell apoptosis (cytotoxic effects) or arrest cell cycle (cytostatic effects), as NO or its derivatives cause nitrosative stress that can release cytochrome c, increase nuclear accumulation of wild-type p53, and reduce Bcl-2.4–6 However, the factors that determine whether the mechanism used would lead to cytotoxicity or cytostasis are still unknown.
Expression of iNOS is up-regulated in many tumors, but immunohistochemistry reveals that it is mostly expressed in TAMs and only to a lesser degree in the tumor cells themselves.7 However, TAMs are often ineffective in tumor killing and actually promote tumor growth. The paradox of strong iNOS expression that is correlated8 with aggressiveness and high incidence of metastasis (eg, in breast, gastric, and colorectal carcinomas7), is explained by the levels of NO that are, in fact, generated. While high levels of NO cause tumor-cell death, low amounts of NO actually promote tumor growth, angiogenesis, and metastasis by regulating blood flow, increasing vascular permeability, and up-regulating vascular endothelial growth factor and matrix metalloproteinases.9–11
Control over NO production is subject to the polarization of TAMs toward an alternative or M2 activation by anti-inflammatory mediators that are secreted by tumor cells; eg, interleukin-10, transforming growth factor β, and prostaglandin E2. These mediators can induce arginase-1, which competes with iNOS for the common substrate L-arginine, causing reduced NO generation.9 Hypoxia, characterizing solid tumors, can also reduce NO production by disrupting protein-protein interactions that are required for iNOS activity.12 Thus, although iNOS is highly expressed, the tumor microenvironment reprograms TAMs to generate only low amounts of NO, promoting tumor growth.
Expression of iNOS in tumor cells themselves is still controversial, as many studies found reduced iNOS expression, or even its complete loss, in high-grade carcinomas and in metastatic cells, while other studies observed a positive correlation between high-grade carcinomas and iNOS expression.4,13–18 Thus, production of NO in the tumor is regulated either by controlling macrophage (or other stroma cells) iNOS activity or by regulating iNOS expression in tumor cells, emphasizing the importance of low NO concentrations for tumor cell survival.
Recently, a new family of non-coding small RNA molecules has been identified as regulators of gene expression. These microRNAs (miRNAs) are transcribed as long, hairpin structured primary transcripts, which are processed into short, mature miRNAs that are incorporated into the RNA-induced silencing complex, and recognize sequences of imperfect complementarity in 3′ untranslated regions of target mRNAs. In mammals, RNA-induced silencing complex binding directs the target mRNA predominantly to translational repression or to degradation. Abnormal miRNA expression is often associated with widespread dysregulation of gene expression and is associated with diverse cancer diseases.19
Indeed, miRNAs have emerged as critical components of several canonical signaling pathways, including Myc, p53, and nuclear factor κB (NF-κB), and they often undergo gain- and loss-of-function in cancer.20 Specifically, miR-146a was shown to target IRAK1 and TRAF6, inhibit toll-like receptor signaling and NF-κB activation,21,22 and was implicated in tumorigenesis.23 Thus, loss of miR-146a expression was suggested to contribute to constitutive activation of the NF-κB and progression of tumors.24,25
Although regulation of iNOS is mostly known to be transcriptional and mediated by lipopolysaccharide (LPS)-induced NF-κB and interferon (IFN) γ-induced IRF-1,26,27 the reduction or loss of iNOS expression in tumor cells and the presence of AU-rich elements in the 3′ untranslated regions of iNOS mRNA26 suggest its posttranscriptional regulation by miRNAs. So far, only one study showed that over-expression of miR-146a inhibited the expression of IFNγ and iNOS in mouse splenic lymphocytes,28 but its involvement in regulation of iNOS in tumor cells is still unknown. Here we show that miR-146a specifically mediates the translational inhibition of iNOS in the mouse renal carcinoma RENCA cell line, leading to fast tumor growth rate and possibly protection from macrophage-induced tumor death.
Materials and Methods
Mice
BALB/c mice (female, 8 weeks old) were kept with a 12-hour light/dark cycle and access to food and water ad libitum under pathogen-free conditions. Mice were cared for in accordance with the procedures approved by the Supervision of Animal Experiments committee at Georg-August University Hospital at Göttingen and outlined in the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Cells
The tumorigenic renal carcinoma RENCA (gift of Dr. Ingo Kausch, Department of Urology, University of Schleswig-Holstein, Campus Lübeck, Germany) and the macrophage-like RAW 264.7 cell lines (ATCC TIB-71), both derived from BALB/c mice, were cultured in Dulbecco's modified Eagle's medium with 10% fetal calf serum, 1% l-glutamine, and antibiotics. Both cell lines were regularly tested for morphological changes and presence of mycoplasma. RAW 264.7 cells were identified as macrophages by their ability to phagocytose zymosan particles, and RENCA cells were tested as cells of epithelial origin by their expression of cytokeratin 18. In some experiments cells were subjected to normoxia (21% O2, 5% CO2, 74% N2) or to hypoxia (in a sealed chamber, Concept 400, Ruskin Technologies, Leeds, UK, with hypoxic environment of O2 < 0.3%, 5% CO2, 95% N2), with or without stimulation with IFNγ (100 U/ml, R&D Systems, Minneapolis, MN) and LPS (1 μg/ml, Escherichia coli 055:B5, Sigma, St. Louis, MO). To avoid possible masking of signals by exogenous stimuli or an immune response, cells were incubated in Dulbecco's modified Eagle's medium without fetal calf serum before their exposure to the experimental conditions or their injection to mice. In all in vitro experiments cell viability was determined using the XTT kit (Biological Industries, Beit-Haemek, Israel).
Determination of Nitrites
Nitrites, the stable product of NO, were determined in RENCA culture supernatants or tumor lysates by mixing equal volumes of the sample and Griess reagent (Sigma) and normalizing to total protein (in lysates). Presence of nitrites produces a chromophoric azo-derivative molecule that absorbs light at 540 nm, and its concentrations were calculated from a nitrite standard curve.
Western Blot Analyses
Lysates from RENCA cells were loaded on a 10% SDS-polyacrylamide gel electrophoresis (20 μg/lane), separated, and transferred onto cellulose nitrate membranes (Schleicher & Schuell, Dassel, Germany). Membranes were blocked with 20% skimmed milk and 1% bovine serum albumin in TBST (0.1% Tween 20, 10 mmol/L Tris pH 8.0, 150 mmol/L NaCl) at room temperature overnight, probed with the diluted (1:1000) mouse monoclonal anti-iNOS (Sigma), washed, and incubated with the 1:5000 diluted horseradish peroxidase-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA). To show equal loading, membranes were stripped and re-probed with anti-α-tubulin (Sigma). To show effects of the proteasome inhibitor MG132 membranes were probed with mouse monoclonal anti-ubiquitin (Biomol, Hamburg, Germany). The enhanced chemiluminescence system (Biological industries) was used for detection and optical density of the bands was quantified using the Bio-Imaging system (Dinco & Renium, Jerusalem, Israel) and TINA software (Raytest, Straubenhardt Germany).
Quantitative Real-Time PCR Analyses
Total RNA was extracted from 106 RENCA cells using RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. RNA integrity and quantity were determined with the Agilent BioAnalyzer 2100 and the Agilent RNA 6000 Nano Kit (Agilent Technologies, Böblingen, Germany). Five hundred nanograms of total RNA were transcribed to cDNA at 37°C for 1 hour using random hexamer primers and Omniscript kit for reverse transcription (Qiagen). Expression of iNOS mRNA was determined by quantitative real-time PCR using the iCycler (BioRad Laboratories, Munich, Germany) and Sybr green (Sybr-Green Supermix, BioRad). Analysis was carried out in duplicates in a volume of 20 μl and a total of 40 cycles, each of 15 seconds at 95°C and 30 seconds at 55°C for iNOS or 56°C for the endogenous reference gene PBGD, which does not change in hypoxia. Product extension was performed at 72° for 30 seconds. The comparative ΔCT method was used for relative quantification, and non-stimulated cells served as a calibrator in each experiment.
In Vivo Mouse Model
Tumors were generated by subcutaneously injecting 2 × 106 RENCA cells into the flanks of BALB/c mice. Tumor size was calculated for each mouse (length × width × 0.5 mm3) at several time points. In different stages of the tumor growth or when tumors were greater than 0.5 cm3, the experiment was stopped and the mice were euthanized for evaluation. Part of the tumor was freshly frozen for evaluation of nitrite concentrations, while other parts were fixed either in HOPE solution or in 4% neutrally buffered formalin and embedded in paraffin for immunohistochemical staining. In some experiments, 14 days after initial injections of RENCA cells and establishment of palpable tumors, 2 × 106 RAW 264.7 cells were injected to the tumor rims every 3 to 4 days, and tumor size was monitored. Alternatively, increasing concentrations of the NO-donor NOC-18 (Alexis Biochemicals, Lausen, Switzerland) were injected into the tumor center in a minimal volume of 25 μl.
Immunohistochemistry
Paraffin sections 3 μm thick were immunostained using mouse monoclonal anti-iNOS antibody (Sigma) in a 1:200 dilution, monoclonal rat anti-mouse pan-macrophage antibody F4/80 (AbD Serotec, Düsseldorf, Germany) in a 1:500 dilution, and rat anti-mouse CD31 endothelial marker in a 1:50 dilution (Acris antibodies, Herford, Germany). A semiquantitative immunoreactive score (IRS) was assigned to negative (0), weak, (1) intermediate (2), or strongly (3) stained cells. Some sections were stained for the nuclear antigen Ki-67, and a Ki-67 proliferation index was calculated (number of strongly stained cells/total number of cells in field).
In Situ Hybridization
Three-micrometer-thick paraffin sections were pre-hybridized in hybridization buffer (50% deionized formamide, 2× standard saline citrate, 10% dextran, and DEPC-treated water) for 20 minutes at 55°C, followed by hybridization with 50 nmol/L of fluorescein-labeled LNA probes for iNOS or a scrambled sequence as negative control (Exiqon, Vedbaek, Denmark). Hybridization was carried out overnight at 52°C followed by successive washes twice with 2× standard saline citrate and 0.1% SDS for 10 minutes, and twice with 0.2× standard saline citrate and 0.1% SDS for 10 minutes, also at 52°C. After hybridization the slides were stained for F4/80 (as described above) using as a secondary antibody Alexa Fluor 555 goat anti-rat (Invitrogen, Darmstadt, Germany), at a dilution of 1:200.
Reverse Transfection and Inhibition of miR-146a
The siPORT NeoFX transfection agent (Applied Biosystems/Ambion, Austin, TX) was diluted 1:25 with OPTI-MEM1 medium (Gibco, Invitrogen), combined with 30 nmol/L of the anti-miR-146a inhibitor or its Cy3-labeled negative control (anti-miR-NC), incubated 10 minutes to allow transfection complexes to form and then dispensed into 24-well plates. The amount of 6 × 104 RENCA cells/well were overlaid in suspension over the transfection complexes and gently tilted to evenly distribute the complexes. Cells were incubated at 37°C overnight, following replacement with fresh medium and stimulation with IFNγ and lipopolysaccharide (LPS) for 24 hours. These conditions were calibrated according to the manufacturer's instructions, reaching transfection efficiency of >95%.
Cytotoxicity Assay
The amount of 5 × 104 RENCA cells were uploaded with 5 μmol/L fluorescent CellTracker orange (Invitrogen, Molecular Probes) for 30 minutes, following extensive washes with PBS, before co-culturing them for 24 hours with RAW 264.7 cells at a 2:1 ratio, with or without the combined stimulation and with the NO scavenger PTIO (100 μmol/L, Alexis), or the iNOS inhibitor L-NIL (50 μmol/L, Alexis). Increased fluorescence in the supernatants reflecting increase in RENCA cell death was measured by a fluorimeter (TECAN Safire, Tecan, Crailsheim, Germany), and calculated as fold from non-stimulated RENCA cells.
Directed in Vivo Angiogenesis Assay
The directed in vivo angiogenesis assay kit (Trevigen, Gaithersburg, MD) was used according to the manufacturer's instructions. Silicon tubes (“angioreactors”) were filled with Coultrex basement membrane extract mixed with differing concentrations of NOC-18, PBS as a negative control or a mixture of FGF-2 (30 ng/μl)/vascular endothelial growth factor (0.1 ng/μl) as a positive control. The angioreactors were implanted subcutaneously into a pocket in the dorsal flank of BALB/c mice. After 17 days, the mice were sacrificed and the angioreactors were removed and photographed.
Statistical Analyses
All values are presented as means ± SE. The data were analyzed using repeated measures analysis of variance (analysis of variance), and the Student Newman-Keuls multiple comparisons test was used to evaluate significance between experimental groups. Alternatively, two-group comparisons were analyzed by the two-tailed unpaired t-test. P values exceeding 0.05 were not considered significant.
Results
RENCA Cells Transcribe iNOS mRNA But Do Not Express the Protein in Vitro or in Vivo
The iNOS enzyme, which is strongly induced by the combined stimulation of IFNγ and LPS and is up-regulated by hypoxia,10 could not be detected in vitro by Western blot analysis of RENCA cells (Figure 1A). The minimal amount of nitrites that accumulated on addition of IFNγ and LPS in normoxia (Figure 1B) could suggest low expression of iNOS that was below the level of detection by Western blot. However, this is not likely in view of the large number of cells used (8 × 106 cells/treatment), the high amount of protein loaded (20 μg/lane), and the strong signal observed in RAW 264.7 cells (used as positive control). In hypoxia no nitrites were accumulated despite the stimulation, suggesting that hypoxia inactivates iNOS also in tumor cells, as we showed before for macrophages.10 In comparison, the same number of stimulated RAW 264.7 cells produces 39 ± 3.5 μmol/L nitrites. In contrast to the absence of iNOS protein, real-time PCR analysis revealed that on stimulation with IFNγ and LPS, high amounts of iNOS mRNA were accumulated in RENCA cells (795 ± 300-fold from control cells, P = 0.0195; Figure 1C).
Figure 1.
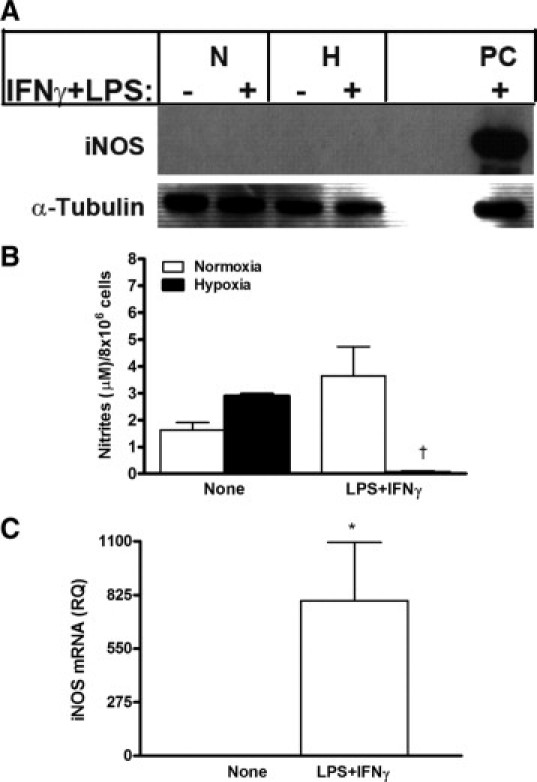
RENCA cells do not express iNOS protein, despite high mRNA expression levels. RENCA cells (8 × 106) were incubated in serum-free medium with or without IFNγ (100 U/ml) and LPS (1 μg/ml) for 24 hours under either normoxic or hypoxic conditions. A: A representative gel showing that Western blot analysis could not detect iNOS protein expression (n = 4). N, normoxia; H, hypoxia; PC, positive control (RAW 264.7 cell lysate). B: Determination of nitrite concentrations in the supernatants (n = 5). C: Accumulation of iNOS mRNA in RENCA cells after a 24-hour incubation in normoxic conditions with or without the addition of IFNγ and LPS as determined by real-time PCR, normalized to the endogenous reference gene PBGD, and calibrated to non-stimulated cells (n = 5). *P < 0.05 relative to no stimulation; †P < 0.05 relative to stimulated RENCA cells in normoxic conditions.
A similar lack of protein expression in RENCA tumor cells was found in vivo, in 14-day-old and 28-day-old tumors, with no necrosis or with a necrotic core, respectively. Expression of iNOS protein was similar in the 14-day-old and 28-day-old tumors (Figure 2, A and D), but was localized primarily in macrophages, which exhibited a strong IRS, whereas tumor cells showed only weak nonspecific staining. In situ hybridization shows that iNOS mRNA is transcribed in the tumor cells in vivo, and is accumulated in the RENCA tumor cells more than in the macrophages (Figure 2F). Despite the fact that almost twofold more macrophages infiltrated the 28-day-old tumors (Figure 2, A, B, E), and these were accumulated in the perinecrotic areas (1.8-fold relative to normoxic areas, P = 0.026), accumulation of nitrites was reduced by 3.4-fold in 28-day-old tumors lysates with established necrosis (Figure 2C). Hypoxia was demonstrated in the 28-day-old, but not in the 14-day-old tumors, both by a 1.6-fold increase (P = 0.0002) in the mean vessel distance that was measured by staining the sections with anti-CD31, and by staining with anti-HIF1α, which localized hypoxia to the perinecrotic areas (data not shown). The low amounts of nitrites accumulated in tumor lysates may reflect TAM activity in normoxic areas of the tumor (eg, in young tumors) that are reduced by hypoxia10 in tumors with large necrotic areas.
Figure 2.
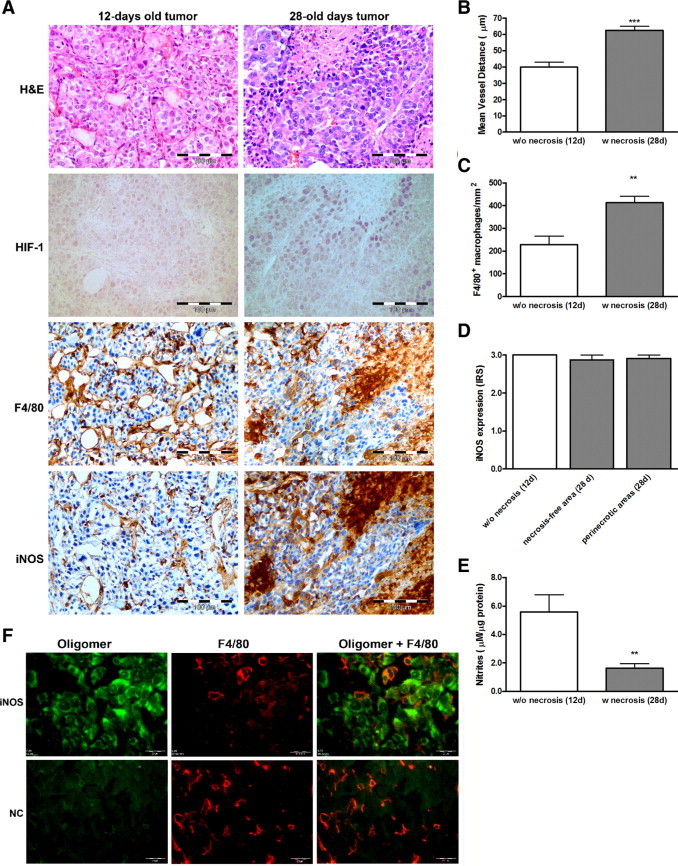
RENCA tumor cells do not express iNOS protein, although infiltrating macrophages do. Tumors were generated by subcutaneously injecting 2 × 106 RENCA cells into the flanks of BALB/c mice (n = 6). Tumors were harvested 14 days (small without necrosis) or 28 days (with a necrotic core) after injection, paraffin-embedded, sectioned, and stained (A) with H&E and for HIF-1, the pan-macrophage marker F4/80, and iNOS. Scale bar = 100 μm. B: The mean vessel distance was estimated by CD31 staining. C: The distribution of macrophages per area was measured. D: The intensity of iNOS expression was evaluated by assigning an immunoreactive score. E: The generation of nitrites was measured. F: Expression of iNOS mRNA (green) was evaluated by in situ hybridization followed by staining for F4/80 (red). NC, scrambled probe as negative control. Scale bar = 20 μm. **P < 0.01, ***P < 0.001 relative to the small, non-necrotic tumors.
Translation of iNOS Is Inhibited in RENCA Cells
The contrast between the high accumulation of iNOS mRNA and the lack of its protein suggested either fast degradation of iNOS protein or inhibition of its translation. We therefore incubated RENCA cells with increasing amounts of the proteasome inhibitor MG132 and tested its efficiency by dose-dependent increase in the accumulation of polyubiquitinated protein conjugates (Figure 3B). However, iNOS protein remained undetected (Figure 3A), ruling out the possibility of fast degradation in the proteasome.
Figure 3.
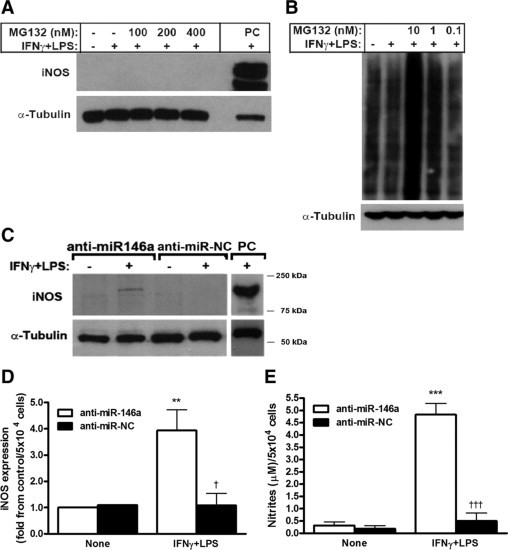
Translation of iNOS mRNA is inhibited by miR-146a. RENCA cells (5 × 106) were incubated under normoxic conditions with IFNγ (100 U/ml) and LPS (1 μg/ml) and with increasing amounts of the proteasomal inhibitor MG132. Two representative Western blot gels showing no expression of iNOS using anti-iNOS (n = 4) (A) and dose-dependent accumulation of polyubiquitinated protein conjugates using anti-polyubiquitin demonstrate the ability of MG132 to block the proteasome (n = 3) (B). C: Representative gel and densitometry analysis (D) of 5 × 104 RENCA cells that were reverse-transfected with either anti-miR-146a or anti-miR-NC (negative control) before their incubation with or without IFNγ and LPS for 24 hours. Transfection of anti-miR-146a restored iNOS expression as well as NO production (E) after stimulation (n = 6). PC, the positive control, was obtained from RAW 264.7 cells stimulated with IFNγ and LPS for 24 hours. **P < 0.01, ***P < 0.001 relative to no stimulation; †P < 0.05, †††P < 0.001 relative to stimulated and anti-miR-146a-transfected RENCA cells.
We next turned to examine if translation of iNOS mRNA is inhibited by the recently discovered machinery of miRNAs. Since among the few specific miRNAs implicated in the regulation of inflammation and cancer, only miR-146a was recently suggested to be involved in the regulation of iNOS in lymphocytes,28 we investigated its involvement in iNOS regulation in RENCA cells. We therefore reverse-transfected RENCA cells with the anti-miR-146a miRNA inhibitor or with its negative control (scrambled sequence). We could show that on stimulation with IFNγ and LPS a protein band corresponding to the iNOS molecular weight now appeared in RENCA cells treated with anti-miR-146a but not with the negative control (Figure 3C), and its expression was fourfold higher (P < 0.05). This iNOS protein was fully active, as 15-fold more nitrites accumulated (Figure 3, D and E, P < 0.001 relative to non-simulated cells and stimulated RENCA cells transfected with negative control).
NO Production by Tumor Cells Is Required to Allow Their Macrophage-Mediated Death
To examine the ability of macrophages to kill tumor cells in vitro, we co-cultured RENCA and RAW 264.7 cells, with and without the combined stimulation, and in the presence of the iNOS inhibitor L-NIL or the NO scavenger PTIO. To monitor only RENCA cell death, these cells were uploaded with the fluorescent dye CellTracker orange. Stimulation of RENCA cells alone or co-culturing with stimulated RAW 264.7 cells at a 2:1 ratio was not sufficient to induce RENCA cell death (Figure 4A) despite nitrite accumulation (12-fold increase, P < 0.001, Figure 4B), whereas addition of L-NIL or PTIO reduced the accumulated nitrites but did not change RENCA cell death. Only when transfected with the anti-miR-146a and in the presence of stimulated RAW 264.7, RENCA cell death increased by 73 ± 19% (P < 0.01), and presence of L-NIL or PTIO reversed this effect (Figure 4C). In contrast, transfection with the negative control and co-incubation with stimulated RAW 264.7 cells did not cause cytotoxicity of RENCA cells, despite the presence of overall similar amounts of nitrites (Figure 4D).
Figure 4.
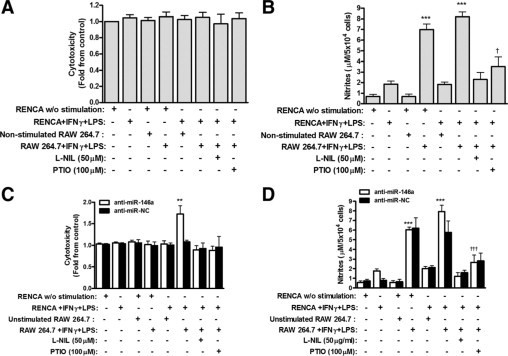
Cytotoxic activity of macrophages depends on tumor cell NO production. RENCA cells (5 × 104) were labeled with CellTracker orange and then co-cultured with unlabeled RAW 264.7 cells at a 2:1 ratio with or without IFNγ (100 U/ml) and LPS (1 μg/ml) and with or without the addition of the iNOS inhibitor, L-NIL, or the NO scavenger, PTIO (n = 11). Alternatively, RENCA cells were first transfected with anti-miR-146a or anti-miR-NC and then co-cultured with RAW 264.7 cells (n = 8) as described above. After 24 hours of incubation, supernatants were collected, and fluorescence was measured for non-transfected (A) and transfected (C) cells, reflecting RENCA cell death and calculated as fold change compared to non-stimulated cells. Nitrite accumulation was measured in non-transfected (B) and transfected (D) cells. **P < 0.01, ***P < 0.001 relative to the control (RENCA cells alone); †P < 0.05 and †††P < 0.001 relative to RENCA and RAW 264.7 with IFNγ and LPS.
We examined the interaction between tumor cells and macrophages in vivo in two sets of experiments. In the first set, the role of macrophage NO production was examined. As hypoxia inhibits macrophage NO production, even in the presence of IFNγ and LPS and induction of high amounts of iNOS protein,10 stimulated RAW 264.7 cells were injected into the normoxic rim of palpable RENCA tumors every 3 to 4 days (Figure 5A). Tumor growth rate, calculated by normalizing the size of the tumor to its size at day 1 (which is actually 14 days after the RENCA cells were injected and tumors became apparent), was impeded only when stimulated RAW 264.7 cells were used (P < 0.05 relative to all other groups), whereas injection of a combination of L-NIL and stimulated RAW 264.7, or of non-stimulated RAW 264.7 cells, did not change tumor growth rate relative to the control (injection of PBS). However, although tumors' growth was slowed down by injection of stimulated RAW 264.7 cells, tumors did not regress. Ki-67 proliferation index was reduced by twofold (P < 0.01) in tumors receiving stimulated RAW 264.7 cell injections in comparison to tumors receiving non-stimulated RAW 264.7 cell injections (data not shown).
Figure 5.
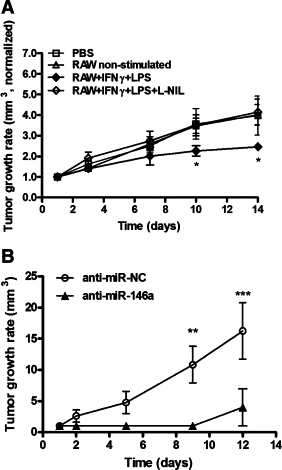
Tumor cell production of NO may be required to prevent tumor growth. A: RENCA cells (2 × 106) were injected into the flanks of BALB/c mice. After 14 days (day 1 in graph), tumors became visible and/or palpable. PBS or 2 × 106 RAW 264.7 cells that were not stimulated, stimulated with IFNγ (100 U/ml) and LPS (1 μg/ml), or stimulated with the iNOS inhibitor L-NIL were injected into the normoxic rim of the tumors every 3 or 4 days. Tumor growth rate was calculated by the tumor volume (length × width × 0.5 mm3) and normalized to the size of each tumor on day 1 (n = 6 in each group). B: RENCA cells (2 × 106) were first reverse-transfected with anti-miR-146a or anti-miR-NC and then similarly injected. Tumor growth rate was calculated as described previously (n = 6 in each group). *P < 0.05, **P < 0.01, ***P < 0.001 relative to the other groups at that time.
In the second set of experiments, the role of tumor cell NO production was examined. RENCA cells were transfected with either anti-miR-146a or the negative control and then injected into BALB/c mice, where endogenous macrophages could infiltrate the growing tumor (Figure 5B). The tumor growth rate in mice injected with RENCA cells that were transfected with the negative control steadily progressed (P < 0.001 relative to start of measurements at 14 days after injection, and to the anti-miR-146a injected group), showing accelerated growth relative to untreated RENCA cells (Figure 5A). In contrast, five of six mice injected with RENCA cells that were transfected with anti-miR-146a did not show any signs of tumor growth, and only one mouse developed a palpable tumor 24 days after injection.
Low Amounts of NO Are Proangiogenic
To demonstrate the role of NO production in the tumoral context, we injected increasing amounts of the NO-donor NOC-18 into palpable tumors every 3 to 4 days (day 1 marks the beginning of injections, 14 days after RENCA cells were subcutaneously injected). Figure 6A shows accelerated tumor growth rate in the groups receiving low-dose injections of NOC-18 (0.5 and 5 μm/ml), whereas growth was impeded in the group injected with the high NOC-18 concentration (50 μg/ml). Fourteen days after injections of NOC-18 started, the group receiving 0.5 μg/ml NOC-18 had significantly larger tumors than the PBS control group (P < 0.01) and the groups receiving 25 μg/ml (P < 0.01) and 50 μg/ml (P < 0.001) groups. In addition, we used the directed in vivo angiogenesis assay and implanted silicone cylinders filled with increasing concentrations of NOC-18 mixed with basement membrane extract. Figure 6B clearly demonstrates the proangiogenic effects that low NOC-18 concentrations had on blood vessel growth into the cylinder, that were reduced in a dose-dependent manner as concentrations increased.
Figure 6.
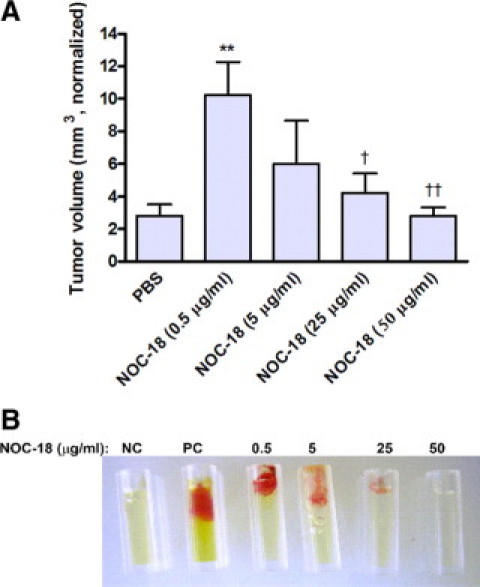
Low concentrations of NO are proangiogenic. A: RENCA cells (2 × 106) were injected into the flanks of BALB/c mice. After 14 days (day 1 in graph), tumors became visible and/or palpable. Increasing concentrations of the NO donor NOC-18 were then injected every 3–4 days into the center of the tumor in a minimal volume. Tumor growth rate was calculated as described previously (n = 4 in each group). **P < 0.01 relative to PBS; †P < 0.05, ††P < 0.01 relative to NOC-18 at 0.5 μg/ml. B: Silicon tubes were filled with PBS (negative control, NC), a mixture of FGF-2 (30 ng/μl) and vascular endothelial growth factor (0.1 ng/μl) (positive control, PC), or with increasing amounts of NOC-18, all mixed with basement membrane extract. The tubes were implanted subcutaneously into the flanks of BALB/c mice, removed after 17 days, and photographed (n = 4).
Discussion
The role of NO in tumor biology is complex and not fully understood. As NO can either support tumor growth or lead to eradication of tumor cells,29 regulating its amounts in the tumor microenvironment becomes critical for tumor cells. In agreement with previous studies,30 we show that low amounts of the NO donor NOC-18, either injected into tumors or present in cylinders, were proangiogenic, and high NO concentrations, whether injected into the tumor or contributed by activated macrophages, reduced tumor growth rate or even arrested it (cytostasis). The impeded growth rate combined with the reduced Ki-67 index suggests an antiproliferative/cytostatic effect, but as the tumors did not regress (cytotoxic effect), it is still unclear whether tumor cells die or simply cease to proliferate.
Only endogenous NO production by the tumor cells themselves, achieved by transfection of the miR-146a inhibitor, resulted in their increased death on addition of activated RAW 264.7 cells in vitro or in vivo. Despite the differences in the methodology used to achieve tumor cell iNOS expression, this result is similar to previous studies.17 We conclude that in vivo, transfected RENCA cells were killed by macrophages at early stages, reflecting the efficiency of the innate immune system in eliminating tumor cells before skewing of macrophage activation occurs,31 as almost all mice showed no visible tumors after prolonged time. The accelerated tumor growth rate observed in control mice injected with anti-miR-NC could be due to targeting of a specific transcript responsible for cell cycle control by the random sequence, or to its association with the RNA-induced silencing complex causing displacement of other miRNAs. In general, reduced iNOS expression in tumor cells, or even its complete loss, may protect tumor cells from macrophage-induced death. Therefore, the role of NO production in tumor cells as a regulator or initiator of cytostatic versus cytotoxic events merits further investigation.
Despite the presence of the strong inducers IFNγ and LPS, RENCA cells did not express iNOS protein both in vitro and in vivo, although high induction of iNOS mRNA was shown in vitro. The novel finding is that loss of iNOS protein expression is exerted by translational inhibition specifically mediated by miR-146a. This miRNA molecule has already been implicated in tumorigenesis, as a regulator of NF-κB activation,21 which is needed for iNOS induction, and its involvement in iNOS regulation in lymphocytes was suggested.28 However, analysis by different algorithms (miRBase, MIRANDA, TARGETSCAN, PicTar) could not predict iNOS as a target for direct binding by miR-146a, and no homology between miR-146a and iNOS mRNA 3′ untranslated regions was found. It is possible that direct binding cannot be predicted due to complex secondary structure of iNOS mRNA, or that the effect is indirect, as anti-miR-146a may target additional transcripts, which we did not check. For example, NF-κB may be a central target as it controls many genes, including COX-2 that is involved in cell death,30 and tumor necrosis factor α, which could work together with NO to induce cell death.32 In any case, the mechanism allowing miR-146a to regulate iNOS translation should be studied further.
Macrophage therapy for the treatment of cancer, which includes their ex vivo activation and adoptive transfer back to the host, has always been attractive because of their potential to home in on the tumor and kill its cells. However, previous attempts have failed, and could show no beneficial effect in humans, or only inhibition of tumor growth and metastasis without regression of the primary tumors in animal models.33 In retrospect, this approach failed to take into account the effects of the tumoral microenvironment, which includes transforming growth factor β, interleukin-10, prostaglandin E2 and hypoxia, on the skewing of macrophages from M1 to M2 activation. This skewing helps the tumor to escape macrophage killing by reducing NO and tumor necrosis factor α production, suppressing Th1 responses through the recruitment of myeloid-derived suppressor cells and T regulatory cells, and producing growth factors and cytokines needed for angiogenesis and tumor promotion.31 Specifically, reduction of NO production leading to reduced killing is achieved by the cytokine-derived induction of arginase-1,29 which competes with iNOS for the mutual substrate L-arginine, and by the hypoxia-induced inactivation of iNOS.10 Our strategy to inject activated macrophages to the normoxic rim of tumors every 3 to 4 days may have provided enough M1-activated macrophages and delayed their exposure to the tumoral microenvironment. However, we too, could only show impeded tumor growth rate, although we demonstrated that it was NO-dependent, as addition of L-NIL reversed it. Our finding that at least in some tumor cells, expression of iNOS is needed to allow their macrophage-induced death can explain this result, and emphasizes the importance of tumor cell–macrophage interactions.
Finally, our results may suggest a new therapeutic approach, where adoptive transfer of M1-activated macrophages will be combined with the ability to up-regulate tumor cell iNOS expression in well established tumors by inhibiting miRNA function, specifically the activity of miR-146a. The establishment of protocols to transfect tumor cells in existing tumors with appropriate miRNA inhibitors, and the ability of macrophages to regress such tumors merit further investigation.
Footnotes
Supported by the State of Lower Saxony and the Volkswagen Foundation, Hannover, Germany (ZN 2024).
References
- 1.Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104:2224–2234. doi: 10.1182/blood-2004-03-1109. [DOI] [PubMed] [Google Scholar]
- 2.Xie Q, Nathan C. The high-output nitric oxide pathway: role and regulation. J Leukoc Biol. 1994;56:576–582. doi: 10.1002/jlb.56.5.576. [DOI] [PubMed] [Google Scholar]
- 3.Lee VY, McClintock DS, Santore MT, Budinger GR, Chandel NS. Hypoxia sensitizes cells to nitric oxide-induced apoptosis. J Biol Chem. 2002;277:16067–16074. doi: 10.1074/jbc.M111177200. [DOI] [PubMed] [Google Scholar]
- 4.Xie K, Huang S. Contribution of nitric oxide-mediated apoptosis to cancer metastasis inefficiency. Free Radic Biol Med. 2003;34:969–986. doi: 10.1016/s0891-5849(02)01364-3. [DOI] [PubMed] [Google Scholar]
- 5.Brune B. Nitric oxide: NO apoptosis or turning it ON? Cell Death Differ. 2003;10:864–869. doi: 10.1038/sj.cdd.4401261. [DOI] [PubMed] [Google Scholar]
- 6.Leon L, Jeannin JF, Bettaieb A. Post-translational modifications induced by nitric oxide (NO): implication in cancer cells apoptosis. Nitric Oxide. 2008;19:77–83. doi: 10.1016/j.niox.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 7.Fitzpatrick B, Mehibel M, Cowen RL, Stratford IJ. iNOS as a therapeutic target for treatment of human tumors. Nitric Oxide. 2008;19:217–224. doi: 10.1016/j.niox.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Donnini S, Ziche M. Constitutive and inducible nitric oxide synthase: role in angiogenesis. Antioxid Redox Signal. 2002;4:817–823. doi: 10.1089/152308602760598972. [DOI] [PubMed] [Google Scholar]
- 9.Weigert A, Brune B. Nitric oxide, apoptosis and macrophage polarization during tumor progression. Nitric Oxide. 2008;19:95–102. doi: 10.1016/j.niox.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 10.Xu W, Liu LZ, Loizidou M, Ahmed M, Charles IG. The role of nitric oxide in cancer. Cell Res. 2002;12:311–320. doi: 10.1038/sj.cr.7290133. [DOI] [PubMed] [Google Scholar]
- 11.Morbidelli L, Donnini S, Ziche M. Role of nitric oxide in tumor angiogenesis. Cancer Treat Res. 2004;117:155–167. doi: 10.1007/978-1-4419-8871-3_11. [DOI] [PubMed] [Google Scholar]
- 12.Daniliuc S, Bitterman H, Rahat MA, Kinarty A, Rosenzweig D, Lahat N. Hypoxia inactivates inducible nitric oxide synthase in mouse macrophages by disrupting its interaction with alpha-actinin 4. J Immunol. 2003;171:3225–3232. doi: 10.4049/jimmunol.171.6.3225. [DOI] [PubMed] [Google Scholar]
- 13.Ozel E, Pestereli HE, Simsek T, Erdogan G, Karaveli FS. Expression of cyclooxygenase-2 and inducible nitric oxide synthase in ovarian surface epithelial carcinomas: is there any correlation with angiogenesis or clinicopathologic parameters? Int J Gynecol Cancer. 2006;16:549–555. doi: 10.1111/j.1525-1438.2006.00567.x. [DOI] [PubMed] [Google Scholar]
- 14.Crowell JA, Steele VE, Sigman CC, Fay JR. Is inducible nitric oxide synthase a target for chemoprevention? Mol Cancer Ther. 2003;2:815–823. [PubMed] [Google Scholar]
- 15.Moochhala S, Chhatwal VJ, Chan ST, Ngoi SS, Chia YW, Rauff A. Nitric oxide synthase activity and expression in human colorectal cancer. Carcinogenesis. 1996;17:1171–1174. doi: 10.1093/carcin/17.5.1171. [DOI] [PubMed] [Google Scholar]
- 16.Hao XP, Pretlow TG, Rao JS, Pretlow TP. Inducible nitric oxide synthase (iNOS) is expressed similarly in multiple aberrant crypt foci and colorectal tumors from the same patients. Cancer Res. 2001;61:419–422. [PubMed] [Google Scholar]
- 17.Xie K, Fidler IJ. Therapy of cancer metastasis by activation of the inducible nitric oxide synthase. Cancer Metastasis Rev. 1998;17:55–75. doi: 10.1023/a:1005956721457. [DOI] [PubMed] [Google Scholar]
- 18.Juang SH, Xie K, Xu L, Shi Q, Wang Y, Yoneda J, Fidler IJ. Suppression of tumorigenicity and metastasis of human renal carcinoma cells by infection with retroviral vectors harboring the murine inducible nitric oxide synthase gene. Hum Gene Ther. 1998;9:845–854. doi: 10.1089/hum.1998.9.6-845. [DOI] [PubMed] [Google Scholar]
- 19.Kent OA, Mendell JT. A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene. 2006;25:6188–6196. doi: 10.1038/sj.onc.1209913. [DOI] [PubMed] [Google Scholar]
- 20.Lotterman CD, Kent OA, Mendell JT. Functional integration of microRNAs into oncogenic and tumor suppressor pathways. Cell Cycle. 2008;7:2493–2499. doi: 10.4161/cc.7.16.6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baltimore D, Boldin MP, O'Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008;9:839–845. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- 23.Williams AE, Perry MM, Moschos SA, Larner-Svensson HM, Lindsay MA. Role of miRNA-146a in the regulation of the innate immune response and cancer. Biochem Soc Trans. 2008;36:1211–1215. doi: 10.1042/BST0361211. [DOI] [PubMed] [Google Scholar]
- 24.Bhaumik D, Scott GK, Schokrpur S, Patil CK, Campisi J, Benz CC. Expression of microRNA-146 suppresses NF-kappaB activity with reduction of metastatic potential in breast cancer cells. Oncogene. 2008;27:5643–5647. doi: 10.1038/onc.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin SL, Chiang A, Chang D, Ying SY. Loss of mir-146a function in hormone-refractory prostate cancer. RNA. 2008;14:417–424. doi: 10.1261/rna.874808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aktan F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004;75:639–653. doi: 10.1016/j.lfs.2003.10.042. [DOI] [PubMed] [Google Scholar]
- 27.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dai R, Phillips RA, Zhang Y, Khan D, Crasta O, Ahmed SA. Suppression of LPS-induced Interferon-gamma and nitric oxide in splenic lymphocytes by select estrogen-regulated microRNAs: a novel mechanism of immune modulation. Blood. 2008;112:4591–4597. doi: 10.1182/blood-2008-04-152488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lechner M, Lirk P, Rieder J. Inducible nitric oxide synthase (iNOS) in tumor biology: the two sides of the same coin. Semin Cancer Biol. 2005;15:277–289. doi: 10.1016/j.semcancer.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 30.Lala PK, Chakraborty C. Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncol. 2001;2:149–156. doi: 10.1016/S1470-2045(00)00256-4. [DOI] [PubMed] [Google Scholar]
- 31.Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P, Mantovani A. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18:349–355. doi: 10.1016/j.semcancer.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 32.Saio M, Radoja S, Marino M, Frey AB. Tumor-infiltrating macrophages induce apoptosis in activated CD8(+) T cells by a mechanism requiring cell contact and mediated by both the cell-associated form of TNF and nitric oxide. J Immunol. 2001;167:5583–5593. doi: 10.4049/jimmunol.167.10.5583. [DOI] [PubMed] [Google Scholar]
- 33.Andreesen R, Hennemann B, Krause SW. Adoptive immunotherapy of cancer using monocyte-derived macrophages: rationale, current status, and perspectives. J Leukoc Biol. 1998;64:419–426. doi: 10.1002/jlb.64.4.419. [DOI] [PubMed] [Google Scholar]