

Abstract
Alterations of the p53 pathway are among the most frequent aberrations observed in human cancers. We have performed an exhaustive analysis of TP53, p14, p15, and p16 status in a large series of 143 soft tissue sarcomas, rare tumors accounting for around 1% of all adult cancers, with complex genetics. For this purpose, we performed genomic studies, combining sequencing, copy number assessment, and expression analyses. TP53 mutations and deletions are more frequent in leiomyosarcomas than in undifferentiated pleomorphic sarcomas. Moreover, 50% of leiomyosarcomas present TP53 biallelic inactivation, whereas most undifferentiated pleomorphic sarcomas retain one wild-type TP53 allele (87.2%). The spectrum of mutations between these two groups of sarcomas is different, particularly with a higher rate of complex mutations in undifferentiated pleomorphic sarcomas. Most tumors without TP53 alteration exhibit a deletion of p14 and/or lack of mRNA expression, suggesting that p14 loss could be an alternative genotype for direct TP53 inactivation. Nevertheless, the fact that even in tumors altered for TP53, we could not detect p14 protein suggests that other p14 functions, independent of p53, could be implicated in sarcoma oncogenesis. In addition, both p15 and p16 are frequently codeleted or transcriptionally co-inhibited with p14, essentially in tumors with two wild-type TP53 alleles. Conversely, in TP53-altered tumors, p15 and p16 are well expressed, a feature not incompatible with an oncogenic process.
Malignant soft-tissue sarcomas are rare tumors accounting for around 1% of all cancers in adults. They are classified according to their eventual line of differentiation. Molecular approaches have described two main genetics in these tumors. The first one, characterized by simple karyotypes and high-level amplifications of chromosome 12 encompassing MDM2 and CDK4 loci, is observed in well-differentiated or undifferentiated liposarcomas.1–3 The other one, corresponding to complex genomic profiles, is observed in leiomyosarcomas (LMS) and in undifferentiated pleomorphic sarcomas (UPS).2,4 LMS correspond to 10% to 15% of soft-tissue sarcomas and are tumors of poor prognosis with a strong smooth muscle differentiation. They are generally localized to the retroperitoneum, and less frequently to the limbs.5 Undifferentiated sarcomas are less frequent (5% of soft-tissue sarcomas). They are predominantly observed in limbs, and have a slightly better prognosis.5 Similar genomic alterations have been described in these two types of tumors, suggesting that they share common oncogenic pathways. Among these common alterations, deletion of chromosome 13 targeting RB1,6–7 deletion of chromosome 10q encompassing PTEN locus,8 and deletion and/or mutation of TP53 have been described.9–11 TP53, the most frequently altered gene in human cancers,12 encodes a transcription factor which accumulates under stress conditions, leading to cell cycle arrest and apoptosis induction.13 In this article, we report an exhaustive analysis of TP53-CDKN2A-CDKN2B status in a large series of 34 LMS and 109 UPS. Deletions and mutations of TP53 are frequently observed in both groups, particularly in LMS where biallelic inactivations are predominant. Nevertheless, 20% of LMS and 29% of UPS do not present an alteration of this gene. Multiple connections between p53 and p14/p16/p15 pathways have been described.14–16 p15 and p16 proteins are able to induce cell cycle arrest in G1 phase by inhibiting cyclin-dependent kinases CDK4 and CDK6.17 p14 protein is a well-known inhibitor of MDM2, an ubiquitin-ligase targeting p53 to proteasomal degradation.18 This protein is indispensable for oncogenic signaling-mediated activation of p53. Its loss appears as an alternative to TP53 alteration.14 We have thus studied p14 genomic and expression status, and have observed frequent deletion and/or loss of expression of this gene. From our results, it appears that p53/p14 pathway is altered in all analyzed tumors. It has been described in some mouse cellular models that inactivation of either p53 or p14 function is sufficient to bypass senescence, but not to establish permanent cell lines, for which loss of p16 function is required.16 Nevertheless, other mouse cell types could be immortalized by TP53 or p14 alteration only. It has also been described that p15 can act as a p16 substitute for CDK4 inhibition.19 All these observations prompt us to analyze genomic and expression status of these two genes. We have demonstrated that p14, p15 and p16 are very frequently lost all together, and that even in nondeleted tumors their expression seems to be transcriptionally co-regulated. In tumors with two wild-type TP53 alleles, expression of the three genes is lower than in tumors with TP53 alterations. On the contrary, tumors with altered TP53 do not express p14, but overexpress p15 and p16. From these data, it seems that all 143 sarcomas from this series present a p53 pathway inactivation, either through direct TP53 alteration, or by p14 expression loss. Moreover, it has been described that cells deficient for TP53 are less sensitive to p16-induced cell cycle arrest16,20–21: indeed, we can observe that only TP53 altered tumors exhibit a high p15-p16 expression.
Materials and Methods
Tumor Samples, Array-CGH, and Transcriptome Analyses
Tumors were classified as previously described,22 according to histological—clinical features and to a “smooth muscle differentiation scoring” established by immunohistochemistry for all tumors, except 13 (Supplemental Table S1, at http://ajp.amjpathol.org). Nevertheless, according to pathological features and Affymetrix data, these 13 tumors were classified as UPS. Therefore, the series of frozen tumors consists of 34 LMS and 109 UPS. All clinical and pathological data are presented in Supplemental Table S1, at http://ajp.amjpathol.org. All samples were obtained before chemotherapy. None of them is radiation-induced. According to French law, the samples were collected in agreement with the Agence Nationale d'accréditation et d’évaluation en santé (ANAES) recommendations. Experiments were performed in agreement with the Bioethics Law #2004–800, and the Ethics Charter from the National Institute of Cancer (INCa). All these tumors were analyzed by array-CGH as previously described22 and present complex rearranged karyotypes. Tumoral DNA/normal DNA ratios >2 were considered as amplifications, ratios >1.2 and <0.8 were considered as gains and losses, respectively. Computation of genomic alterations observed by array-CGH was provided by the VAMP interface (http://bioinfo.curie.fr/vamp).23 Transcriptomes of 31/34 LMS and 109/109 UPS samples were analyzed using U133Plus 2.0 Affymetrix arrays. For this purpose, data were normalized using the GeneChip Robust Multiarray Averaging (GCRMA) algorithm.24 To compare genes expression profiles, we have performed unpaired t-tests using the Genespring GX software (Agilent Technologies, Santa-Clara, CA). A significant variation corresponds to fold changes >3 with P < 0.01 (Benjamini-Hochberg P value correction).
Cell Line Establishment and Culture Conditions
Cell lines were established and cultured as previously described.22 Each cell line was named as the tumor from which it derives with an additional terminal L (LMS148L, UPS108L, UPS137L, and UPS152L). LMS148L was previously described.22
For proteasome inhibition experiments, cell lines were seeded at a density of 1.5, and 105 cells/well onto 6-well plates and were cultured for 8 hours in medium with 10 μmol/L MG132 (C2211, Sigma, St Louis, MO) or 50 μg/μL ALLN (A6185, Sigma). Total protein extracts were then obtained by crushing cells in Laemmli 1× buffer and a Western blot was performed as described below. The experiment was performed three times.
Real-Time Genomic Quantitative PCR
Genomic DNA was isolated using a standard phenol-chloroform extraction protocol. To determine the copy number status of TP53, p16, p15 and p14, real-time PCR was performed on genomic DNA using TaqMan Universal Master Mix (Applied Biosystems, Foster City, CA). Primers and probes used are presented in Table 1. For the TP53 genomic analysis we used two different couples of primers/probe: one localized in intron 1 (TP53.1) and the other one localized at the junction between intron 3 and exon 4 (TP53.3). LMS and UPS are very rearranged tumors. For normalizing the results, we used three reference genes: GAPDH, ALB and CDK4, to have, for each tumor, at least two of these reference genes in normal copy number (array-CGH). Tumor data were normalized against data obtained for normal DNA. The results were then calculated as previously described.22 For the TP53 study, normal status corresponds to 0.8 ≤ ratio ≤1.2, 0.1 < ratio <0.8 is considered as a hemizygous deletion. When ratio is inferior to 0.1, the deletion is considered as homozygous. For UPS16, the deletion was also considered as homozygous because the corresponding cell line is homozygously deleted. The 0.16 qPCR ratio is thus probably due to normal cells contamination. For CDKN2A/ARF study, same criteria were used.
Table 1.
Primers and Probes Used for Real-Time Genomic Quantitative PCR. Designed with Primer Express Software V2.0 (Applied Biosystems)
| Gene | Primer sequence |
|---|---|
| TP53.1 | |
| Forward | 5′-GAGGCGGGCCGATCAC-3′ |
| Reverse | 5′-CTGACTGCTCTTTTCACCCATCT-3′ |
| Probe | 5′-TGAAGTAAGGAGTTCGAGACCAGCCTGG-3′ |
| TP53.3 | |
| Forward | 5′-CTGACTGCTCTTTTCACCCATCT-3′ |
| Reverse | 5′-ACAGCATCAAATCATCCATTG-3′ |
| Probe | 5′-CAGTCCCCCTTGCCGTCCCA-3′ |
| P16 | |
| Forward | 5′-GGCTGGCTGGTCACCAGA-3′ |
| Reverse | 5′-CGCCCGCACCTCCTCTAC-3′ |
| Probe | 5′-ATGGAGCCTTCGGCTGACTGGCT-3′ |
| P15 | |
| Forward | 5′-GGAAAGAAGGGAAGAGTGTCGTT-3′ |
| Reverse | 5′-CGCGCATTCCGCAGC-3′ |
| Probe | 5′-TTTACGGCCAACGGTGGATTATCCGG-3′ |
| P14 | |
| Forward | 5′-GGTTCTCGCAGTACCATTGAA-3′ |
| Reverse | 5′-TGTTCGCCTCAGTTTCCCA-3′ |
| Probe | 5′-CCTCCCTTCACACAGCCCCTCAATC-3′ |
| GAPDH | |
| Forward | 5′-CCCCACACACATGCACTTACC-3′ |
| Reverse | 5′-CCTAGTCCCAGGGCTTTGATT-3′ |
| Probe | 5′-TAGGAAGGACAGGCAAC-3′ |
| ALB | |
| Forward | 5′-TGAAACATACGTTCCCAAAGAGTT-3′ |
| Reverse | 5′-CTCTCCTTCTCAGAAAGTGTGCATAT-3′ |
| Probe | 5′-TGCTGAAACATTCACCTTCCATGCA-3′ |
| CDK4 | |
| Forward | 5′-TTGCATCGTTCACCGAGATC-3′ |
| Reverse | 5′-CTGGTAGCTGTAGATTCTGGCCA-3′ |
| Probe | 5′-TGACAAGTGGTGGAACAGTCAAGCTGG-3′ |
Real-Time Quantitative Reverse Transcription-PCR
Reverse transcription and real-time PCR were performed as previously described.22 We used TaqMan Gene Expression assays (Applied Biosystems): HS01034249_m1 for TP53; Hs00793225_m1 for p15; Hs00924091_m1 for p14 and Hs99999902_m1 for RPLP0. Specific primers and probe were designed for p16: (F) 5′-CCAACGCACCGAATAGTTACG-3′; (R) 5′-CGGCGCTGCCCATCA-3′ and 5′-TCGGAGGCCGATCCAGGTCATG-3′ (Probe). To normalize the results, we used the RPLP0 gene as a reference gene. Human tumor data were normalized against reference tumor or cell line, for which the genes are expressed to an intermediate level on Affymetrix array: UPS137 for the TP53 expression study and LMS148L for the p14/p15/p16 study. The results were then calculated as previously described.22
Immunohistochemistry
Immunocytochemistry and immunohistochemistry experiments were performed according to the methods previously described,4,22 in particular for α-smooth muscle actin, calponin, transgelin, and caldesmon assays. Expression studies of p53 (1:800, D0–7, Dako, Carpinteria, CA), p16 (CINtec p16 Histology V-Kit, MTM, Heidelberg, Germany) and p14 (1:100, clone 4C6/4, Cell signaling, Danvers, MA) were performed according to manufacturers recommendations.
Fluorescence in Situ Hybridization Assay
Frozen tissues were available for a series of 14 tumors. TP53 fluorescence in situ hybridization experiments were performed on 5 μm-thick tumor sections using Vysis LSI TP53 SpectrumOrange Probe (Abbott laboratories, IL), according to the manufacturer's procedure and using a chromosome 17 centromeric control probe (C17) labeled with fluorescein isothiocyanate-dUTP (Roche, Indianapolis, IN).
Western Blot
Total proteins were extracted by crushing frozen tumor samples in Urea 8M (Interchim Inc, CA). Radioimmunoprecipitation assay buffer was used for cell line proteins extraction. Primary antibodies and dilutions used in this study are as follow: p53 Ab-6 (1:500, clone D0–1, Lab Vision, Thermo Fisher Scientific, CA), p14 (1:500, clone 4C6/4, Cell signaling), p16 (1:2000, sc-468, Santa Cruz Biotechnology, CA), p15 (1:200, sc-612, Santa Cruz Biotechnology), p21 (1:1000, BD Biosciences, CA), and β-actin (1:10000, Sigma-Aldrich). Secondary antibodies were horseradish peroxidase-conjugated: anti-rabbit IgG (1:3000, NA934, Amersham, GE Health care, UK) and anti-mouse IgG (1:3000, NA931V, Amersham).
Mutation Screening
TP53 mutation screening on genomic DNA was assessed as previously described.25 p53 cDNA was also sequenced. Reverse transcription was performed as previously described.22 Protocol is available on request and primers used are as follows: F1 5′-GATTGGCAGCCAGACTGC-3′; R1 5′-CCAGTGTGATGATGGTGAGG-3′; F2 5′-CATGACGGAGGTTGTGAGG-3′; R2 5′-TTCTGACGCACACCTATTGC- 3′. Sequence analysis was performed with SeqScape software v2.5 (Applied Biosystems). To analyze the sequences of p14 and p16, we used for both exons 1 and for common exon 2 primers described by Iwato et al.26 For common exon 3 and for p15 exons 1 and 2, primers were described by Laud et al.27
Functional Analysis of Separated Alleles in Yeast
This test was realized as previously described.25 TP53 gene was considered as mutated when more than 15% of the colonies were red.
Bisulfite Modification and Specific PCR Amplification
Bisulfite modification on 500 ng of gDNA was achieved as previously described.28 Normal DNA and CpGenome Universal Methylated DNA (S7821, Millipore, Upstate Inc, IL) were also treated as negative and positive methylated control DNAs, respectively. Primer pairs used for p14, p16, and p15 are those previously described.28–29
Results
TP53 Alterations Are Very Frequent in Soft Tissue Sarcomas with Complex Genomics
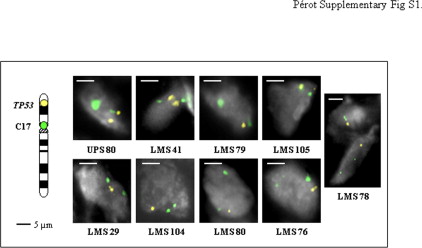
Chromosome 17p13 deletions encompassing the TP53 locus have been previously described in sarcomas, LMS and UPS in particular, suggesting that this gene could be involved in sarcoma oncogenesis.8–9,30 To assess the frequency of this aberration in our series, we analyzed 143 tumors by array-CGH. Among them, 9/34 LMS (26.5%) and 20/109 UPS (18.4%) indeed exhibit a genomic deletion encompassing the TP53 locus (Figure 1).
Figure 1.
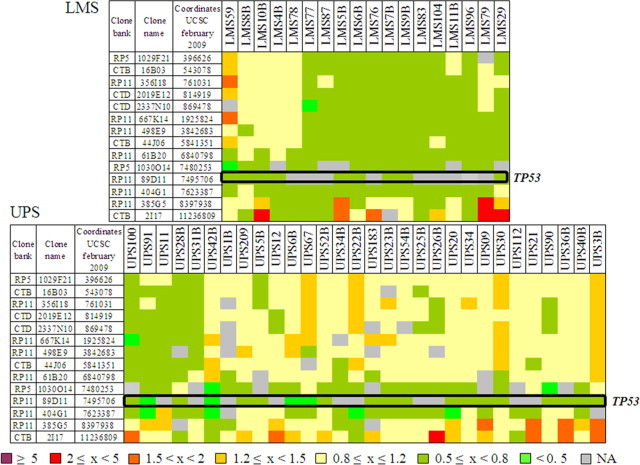
TP53 gene deletions in sarcomas. Array-CGH results on 14 BAC-PAC clones covering the first part of 17p chromosome. Names of these clones and their genomic locations are indicated on the left. Names of studied tumors are indicated at the top. The first graph represents data for LMS and the second one shows those for UPS. Tumoral DNA/normal DNA ratios greater than 2 were considered as amplifications, ratios greater than 1.2 and less than 0.8 were considered gains and losses, respectively. The genomic status (ratio) of each tumor for each locus is indicated by a color code in filled squares, as defined at the bottom. Position of TP53 gene is indicated (open black box).
Moreover, 9/34 LMS and 10/109 UPS have non available data for the clone containing TP53, but they present a deletion of at least one TP53 adjacent clone and could thus be also deleted for TP53. As explained below, these putative deletions were confirmed by another alternative approach. In tumors for which frozen samples were available, array-CGH results were confirmed by fluorescence in situ hybridization (Supplemental Figure S1, at http://ajp.amjpathol.org). To definitively validate these data and to search more subtle alterations, we then analyzed by genomic real-time PCR the copy number status of the gene. All deletions detected or suspected by array-CGH were confirmed (see Supplemental Table S2, at http://ajp.amjpathol.org). Moreover, 21 deletions not previously observed were found (4 LMS and 17 UPS). These results indicate that approaches with low resolution such as fluorescence in situ hybridization or Bacterial Artificial Chromosome (BAC)-array are not sufficient to exhaustively assess the frequency of TP53 loss. Finally, 22/34 LMS (64.7%) and 47/109 UPS (43.1%) present a deletion of part or entirety of the TP53 gene. These deletions are more frequent in LMS (P = 0.03, Fisher's exact test), but they are smaller in UPS, targeting the gene more precisely. Among these deletions, 3 are putative homozygous deletions (UPS16, UPS67, and UPS148), and 6 present a breakpoint in the gene (LMS29, LMS76, LMS78, LMS6B, UPS21, and UPS69).
We then analyzed the mutational status of TP53 by sequencing exons 2 to 11 on genomic DNA in all samples, and on the whole cDNA for the mutated tumors. Indeed, TP53 mutations have been previously described in sarcomas but most of the studies only searched for mutations at the hotspot mutation sites (exons 4–5 to 8).10,30–33 Our results are presented in Supplemental Table S3, at http://ajp.amjpathol.org. Twenty mutations were detected in LMS (59%), and 35 in UPS (32%). TP53 mutations are more frequent in LMS (P = 0.008, Fisher's exact test). In 86 tumors, functional analysis of separated alleles in yeast was performed, and was consistent with the sequencing in all tumors but one, UPS32B, for which a mutation was detected only by sequencing (Supplemental Table S2, at http://ajp.amjpathol.org). As usual, most mutations (80% of LMS mutations and 88% of UPS mutations, Figure 2A) are localized in the DNA binding domain. LMS present more intronic mutations than UPS, and mutations which do not affect the DNA binding domain in LMS are preferentially localized in the 5′ side of the coding sequence (Figure 2A). Interestingly, the rate of transition in UPS is dramatically lower than in LMS (P = 0.048, Fisher's exact test) (Figure 2B). Conversely, the percentage of insertion/deletion is higher in UPS. These results suggest that there could be different mutagenesis mechanisms between these two tumor types, even though we could not find any evidence of a particular exposure of the patients to radiations or chemicals. When compared to the distribution of TP53 somatic mutations from the International Agency for Research on Cancer (IARC) database,34 it appears that missense mutations are less frequent in soft tissue sarcomas than in other malignancies (74% in database versus 54 to 55% in our series, Figure 2C). Distribution observed in LMS differs from IARC's one by a higher splice mutation rate, while that of UPS differs by a higher rate of frame shift. Among the 55 mutations detected in our series, 12 are not indexed in International Agency for Research on Cancer or in Universal Mutation Database (UMD) databases. Ten of these twelve mutations are complex mutations (deletions) and the two others are substitutions.
Figure 2.
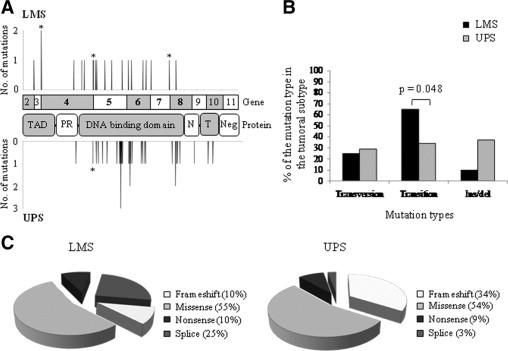
TP53 mutations identified in LMS and UPS. A: Distribution of the identified TP53 mutations along the coding and protein sequences in LMS (top) and UPS (bottom). Asterisks correspond to mutations at splicing sites. TAD, transactivation domain; PR, proline-rich domain; N, NLS sequence; T, tetramerization domain; and Neg, negative regulation domain. Exons 4 to 8 were studied by functional analysis of separated alleles in yeast. B: Comparison of the different types of mutations between LMS and UPS. Significant Fisher's exact test P value is indicated over brackets. C: Pie charts representing the different mutation types in LMS and in UPS.
We classified both tumor groups according to the TP53 alleles status (Figure 3A, and Supplemental Table S2, at http://ajp.amjpathol.org). 2WT corresponds to tumors with two wild-type copies of TP53; 1WT to tumors with one WT copy and a deletion of the other one; 1WT/1MUT to tumors with one WT allele and one mutated allele; 1MUT to tumors with one mutated allele and a deletion of the other one and 2 MUT to tumors with two copies of the mutated allele (isodisomy). The most striking feature is the high frequency of biallelic inactivations of TP53 in LMS (50% − 1MUT and 2MUT), most of them corresponding to a mutation/deletion process. The rate of biallelic inactivation is significantly different between LMS and UPS (P = 1.8 × 10−5, Fisher's exact test). In almost all UPS (87.2%), a wild-type TP53 allele is retained.
Figure 3.
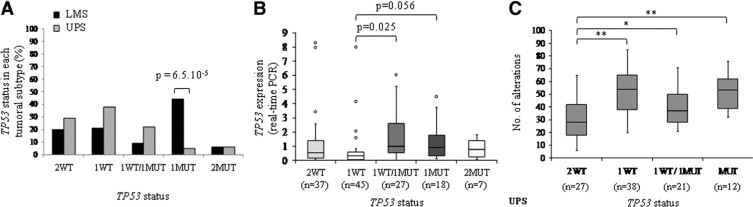
TP53 alleles status in tumors. A: Percentage of tumors with a particular TP53 gene status. 2WT, tumors with two wild-type copies of TP53; 1WT, tumors with one WT copy and a deletion of the other one; 1WT/1MUT, tumors with one WT allele and one mutated allele; 1MUT, tumors with one mutated allele and a deletion of the other one; and 2 MUT, tumors with 2 mutated alleles. Significant Fisher's exact test P value is indicated over brackets. B: Boxplot analysis of TP53 expression in the different groups defined according to TP53 alleles status (real-time PCR). Results for LMS and UPS were similar and thus grouped together. Student's t-test P value is indicated over brackets. C: Boxplot analysis of the alterations number in UPS classified according to their TP53 alleles status. Number of alterations was evaluated according to array-CGH data by the VAMP interface (http://bioinfo.curie.fr/vamp).23 Only UPS data are presented because the LMS series was too small to be really statistically informative. *P < 0.05; **P < 0.01 (Student's t-test) (2WT vs 1WT: P = 1.2.10−6; 2WT vs. 1WT/1MUT: P = 1.4.10−2; 2WT vs. 1or2MUT: P = 2.4.10−3).
We then analyzed by quantitative real-time PCR the expression of TP53 in tumors classified according to their alleles status (Figure 3B). A strong correlation was observed between Affymetrix and PCR data (n = 134, r = 0.8, Pearson's correlation) (Supplemental Table S2, at http://ajp.amjpathol.org). Tumors with 1WT TP53 allele exhibit a half-expression compared to tumors with 2WT TP53 alleles, suggesting a dosage effect. Mutated alleles seem to be well expressed: indeed, expression of the gene is higher in samples with a mutated allele, even when the other one is deleted. As usually described, Western blot and immunohistochemistry were positive only in tumors with TP53 mutations, but not in all mutated tumors (Supplemental Table S2, at http://ajp.amjpathol.org).
Functions of p53 in apoptosis induction and cell cycle arrest are activated by different stress signals, DNA damages in particular, and its inactivation is associated with an increase of chromosomal instability.35 We assessed in UPS the number of chromosome breakpoints according to TP53 alleles status (Figure 3C). We can observe that breakpoints are significantly less frequent in tumors with 2WT TP53 alleles. This result suggests that TP53 inactivation could be involved in sarcoma genomic instability.
p14 Alteration Could Substitute for TP53 Inactivation or Reinforce It
It is well-known that p14 is implicated in p53 degradation by proteasome, through its interaction with MDM2.18 It has also been recently described that oncogenic signaling-mediated activation of p53 is completely abrogated in absence of p14.14 Moreover, deletion of p19-Arf locus could substitute for mutation of Trp53 in murine model of soft tissue sarcoma in Kras mutated context.36 We thus analyzed the p14 genomic (array-CGH, genomic real-time PCR) and expression status (expression real-time PCR, Affymetrix arrays), to document putative inactivation of this gene which could compensate for a lack of TP53 inactivation. Results are presented in Supplemental Table S4 at http://ajp.amjpathol.org. By array-CGH, 5/34 LMS (14.7%) and 19/109 UPS (17.4%) exhibit a loss of p14 region. This loss was confirmed by genomic PCR, and found in 5 additional LMS and 15 additional UPS. Thus, a p14 deletion is observed in 29.4% LMS and 31.2% UPS. Among them, 3 tumors present very low genomic PCR ratios (<0.1), suggesting that these tumors harbor a homozygous deletion of the gene. The residual signal observed for these samples could be due to contaminant normal cells. For 41 tumors, the genomic PCR ratio is comprised between 0.1 and 0.8, suggesting the loss of at least one allele. Finally, 76 tumors have a normal ratio at p14 locus (0.8≤ratio ≤1.2), and 21 have an increased number of copies (ratio >1.2). We then search for mutations in all tumors, but none was detected. These results were coherent with those previously described.37
Afterward, we analyzed the expression of p14 gene. Affymetrix probe sets (207039_at and 209644_x_at) recognize both p14 and p16 cDNAs, so we designed primers and probe specific for p14 cDNA. As shown in Supplemental Table S4 at http://ajp.amjpathol.org, among tumors for which we could perform the experiment, the 3 tumors with putative homozygous deletions, 24 tumors with 1 copy, 15 tumors with 2 copies and 1 tumor with 3 copies exhibit extremely low or null expression of p14 (ratio tumor/control <0.1). These tumors with no expression of p14 correspond to 3/33 LMS (9.1%) and 40/102 UPS (39.2%). These frequencies are significantly different (n = 135, P = 0.001, exact Fisher's test). Curiously, these tumors with 1, 2 or 3 copies do not express p14. It has been described in human esophageal squamous cell carcinoma that CpG islands hypermethylation of p14 promoter could lead to a reduced expression.29 Nevertheless, we could not find any such methylation in our series (Figure 4A), demonstrating that this mechanism is not involved in p14 down-regulation in human sarcomas. Alternative hypotheses could be a down-regulation by histones methylation/deacetylation as described in different human tumors,38–39 or by a transcriptional inhibition by different regulatory proteins.15 Finally, some tumors with only one copy of p14 express it, but to a lower level than tumors with two copies. All together, these observations suggest that two different mechanisms could be used to decrease p14 expression: the first one, more stringent, leads to an abolition of expression; the other one corresponds to a decrease of expression only by gene dosage effect.
Figure 4.
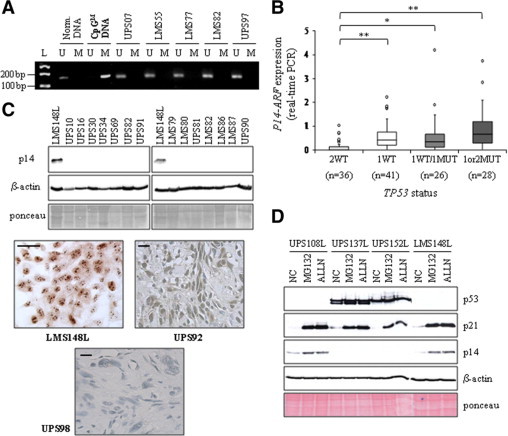
p14 expression in sarcomas. A: Some tumor methylation profiles of the p14 promoter are presented as examples. M: methylated-specific PCR, U: unmethylated-specific PCR. Normal DNA and CpGenome Universal Methylated DNA (CpGM DNA) are used as controls. L, molecular weight ladder. B: Boxplot analysis of p14 expression (real-time PCR). *P < 0.05; **P < 0.01 (Student's t-test) (2WT vs. 1WT: P = 5.7.10−5; 2WT vs. 1WT/1MUT: P = 2.10−2; 2WT vs. 1or2MUT: P = 1.5.10−4). Data for the two tumor types have been grouped because of the similarity of their results. C: Representative p14 Western blot. β-actin and Ponceau staining are shown as loading controls. Examples of p14 immunohisto/cytolabeling are presented. Scale bar = 40 μm. D: p14 Western blot performed after 8 hours of MG132 or ALLN treatment of four sarcoma cell lines. NC: negative control, cell lines cultured in medium with dimethyl sulfoxide. Features of these cell lines are similar to those of the corresponding tumors and are described in the Supplemental Table S4, at http://ajp.amjpathol.org. p21 and p53 are used to see the efficiency of the inhibitors. Ponceau staining and β-actin, loading controls.
We then compared p14 expression according to the TP53 alleles status (Figure 4B and Supplemental Table S4, at http://ajp.amjpathol.org). We could observe that almost all tumors with 2WT TP53 alleles (30/36) present a significantly lower p14 expression than others. These results strongly suggest that loss of p14 expression could substitute for TP53 alterations, but also that this pathway is of paramount importance for soft tissue sarcoma oncogenesis.
Immunohistochemistry and/or Western blot analysis was performed for almost all tumors. We do not observe any expression of p14 protein except in one tumor (UPS92) and two cell lines (UPS108L and LMS148L) (Figure 4C and Supplemental Table S4, at http://ajp.amjpathol.org). This result is coherent in tumors without RNA expression, but unexpected in cases with a good RNA level. Since the p14 protein is degraded by the proteasome,40 we studied in sarcoma cell lines if the lack of protein could be due to a proteasomal degradation. We performed on 3 UPS and 1 LMS cell lines an experiment of proteasome inhibition using MG132 and ALLN (Figure 4D). UPS108L and LMS148L expressed high level of p14 RNA, while UPS137L and 152L do not express the gene, and correspond to negative controls. Proteins p53 and p21, which are described as sensitive to proteasomal degradation,41–42 allow to assess the inhibitors efficiency: indeed, we observed an accumulation of p21 in all cell lines, and an increase of p53 level in the two mutated cell lines (UPS137L and 152L). As shown in Figure 4D, a strong accumulation of p14 is observed in UPS108L and LMS148L. This result demonstrates that, in these cell lines, p14 RNA is translated in protein, which is degraded by proteasome. The absence of detectable protein in tumors could thus be due to the same mechanism.
To complete these data, we looked at MDM2 expression on Affymetrix arrays in our series, since p14 acts on p53 pathway through MDM2 inhibition.18 It appears that MDM2 is expressed in all tumors (data not shown), and could thus play its inhibitory role on p53. Moreover, we also verified the expression level of HDMX which is described as a negative regulator of p53, through its transcriptional activity inhibition.43 As shown in Supplemental Figure S2 at http://ajp.amjpathol.org, a very significant overexpression of HDMX was observed in tumors with 2WT TP53 alleles, a feature which could reinforce the effect of p14 loss on p53 inactivation. All together, these data confirm the pivotal role of p53 inactivation in sarcoma oncogenesis.
p16 and p15 Are Inactivated in Tumors with Two Wild-Type TP53 Alleles
p14 and p16 are encoded by two different promoters and alternative reading frames of the CDKN2A locus.15 p15 gene is localized around 8.4 kb centromeric to CDKN2A. This vicinity could explain the frequent loss of these two loci in a wide range of human tumors, sarcoma in particular.37,44–46 Our genomic PCR analysis confirms these data: indeed, we could observe homozygous or hemizygous codeletions of both loci in 38 tumors (Supplemental Table S4, at http://ajp.amjpathol.org). Most codeletions are associated with a decreased expression of p15 and p16, parallel to that observed for p14 and in all cases the 3 mRNAs are expressed in the same manner. Nevertheless, few tumors present only a deletion of p15 (4 tumors) or p16 (6 tumors). We adopt the same analysis strategy than for p14: sequencing, expression analysis and promoter CpG islands methylation study. As previously described in sarcomas,37,47 we found no mutation of p16, and a single mutation of p15 (Supplemental Figure S3, at http://ajp.amjpathol.org). As for p14, some tumors do not exhibit expression of p15 and p16 mRNAs, even if at least one copy of the gene is present. In some instances, we found methylation of p15 and/or p16 promoters CpG islands (Figure 5 and Supplemental Table S4, at http://ajp.amjpathol.org).47 Some of these methylations could participate to the decrease of genes expression in these tumors without deletion of the loci. Nevertheless, in all tumors with methylated promoters, we always observed a high proportion of unmethylated promoter DNA, suggesting that the methylated promoter is present only in a cell subpopulation of the tumor. Thus, these methylated profiles could not explain the null expression observed in some tumors. These results suggest again that a more global inactivation mechanism of the two loci may exist, such as histones methylation/deacetylation or a negative transcriptional regulation.15,38–39
Figure 5.
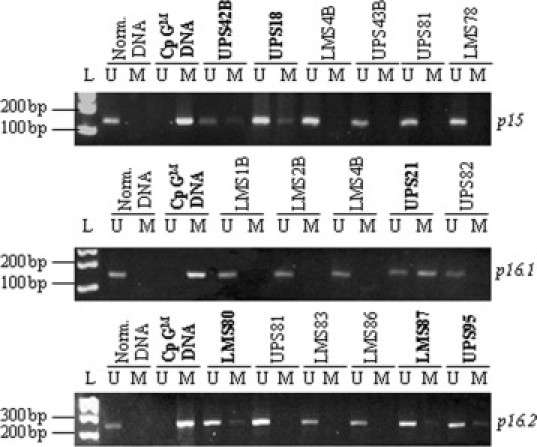
p16 and p15 promoter CpG islands methylation profiles. Some tumor methylation profiles of p16 and p15 promoters are presented. For p16, two regions of methylation sites have been studied (p16.1 and p16.2). Tumors with methylated CpG islands are illustrated in bold characters. L, molecular weight ladder.
It has been previously described that p15 could act as a p16 substitute for CDK4 inhibition, and that it is overexpressed in mouse embryonic fibroblasts with p16 inactivation.19 In our series, both genes are in most instances expressed in the same manner, except in UPS34B, LMS77, UPS37B, UPS113, and LMS5B in which p15 is more expressed, and can fulfill a backup function for p16 (Supplemental Table S4, at http://ajp.amjpathol.org).
As observed for p14 expression, tumors with 2WT TP53 alleles exhibit a significant lower level of p15 and p16 mRNAs than other tumors, a result consistent with the frequent codeletion and/or transcriptional coregulation of the two loci (Figure 6). However, contrary to p14 which is never expressed in tumors at protein level, there is a good correlation between RNA and protein levels for p15 and p16 (Figure 7, A and B, and Supplemental Table S4, at http://ajp.amjpathol.org). We then looked for p16 protein expression in tumors classified according to TP53 allele status (Figure 7C). As observed at RNA level, tumors with 2WT TP53 alleles are those with the lowest p16 expression. Conversely, tumors with altered TP53 exhibit a robust p16 expression. It was previously described in some cellular models that cells with an alteration of TP53 are less sensitive to cell cycle arrest mediated by p16.16,20–21 Thereby, p16 expression in a peculiar cellular context is not necessarily antinomic with an oncogenic process.
Figure 6.
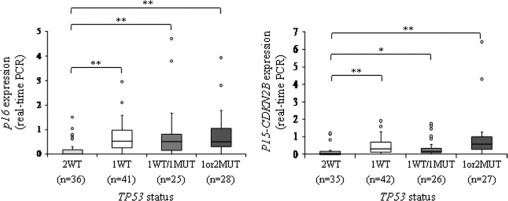
p16 and p15 RNA expression in sarcomas. Boxplot analysis of p16 and p15 expressions (real-time PCR): *P < 0.05; **P < 0.01 (Student's t-test) (for p16: 2WT vs. 1WT: P = 7.6.10−5; 2WT vs. 1WT/1MUT: P = 2.10−2; 2WT vs. 1or2MUT: P = 10−3) (for p15: 2WT vs. 1WT: P = 2.10−3; 2WT vs. 1WT/1MUT: P = 4.7.10−2; 2WT vs. 1or2MUT: P = 10−2). Data for the two tumor types have been grouped because of the similarity of their results.
Figure 7.
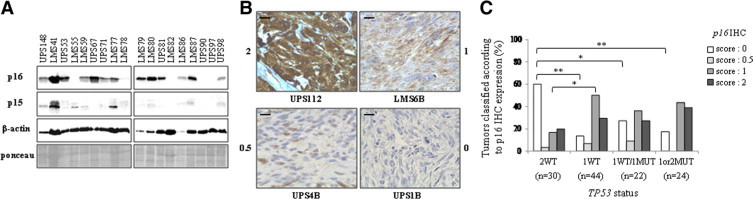
p16 and p15 protein expression in sarcomas. A: Representative p16 and p15 Western blot. β-actin and Ponceau staining, loading controls. B: Examples of p16 immunohistolabeling. Strong positivity, score of 2; positive staining, score of 1; few cells expressing the protein with a weak staining, score of 0.5; and negative staining, score of 0. Scale bar = 40 μm. C: p16 expression scoring repartition in the different TP53 tumor types. p16 immunohistochemistry scores were established as described above. *P < 0.05; **P < 0.01 (Fisher's exact test).
Discussion
To date, publications on TP53 alterations in sarcomas appear heterogeneous, often realized on small series of tumors for which a unique diagnosis, such as malignant fibrous histiocytoma, could correspond to different biological and histological subtypes. Indeed, since these first publications, it has been demonstrated that around 20% of malignant fibrous histiocytoma are actually undifferentiated liposarcomas.1–3 In the same manner, many digestive LMS published more than 10 years ago are in fact GastroIntestinal Stromal Tumor (GIST).48 Moreover, most of these studies have analyzed only hotspot mutation sites, and/or p53 expression by immunohistochemistry, leading to an underestimation of alterations.10–11,30–33 In addition, the other members of the p53 pathway have rarely been studied simultaneously on the same tumors. To obtain an exhaustive overview of the p53 pathway in sarcomas with complex genetics, we have analyzed the different members of the pathway in a series of 109 UPS and 34 LMS.
From this approach, we can conclude that TP53 alterations are highly recurrent in soft tissue sarcomas with complex genetics (80% of LMS and 71% of UPS exhibit at least one alteration: mutation and/or deletion). Nevertheless, we have observed not only that TP53 mutations are less frequent in UPS (59% in LMS versus 32% in UPS), but also that biallelic inactivation is preferentially observed in LMS (50% in LMS versus 12.8% in UPS). Indeed, most UPS retain one wild-type TP53 allele. Moreover, mutation types are slightly different between LMS and UPS, with more frequent frame shifts in UPS and more frequent splice alterations in LMS, suggesting that different mutagenesis mechanisms could occur in the oncogenic processes.
We have established four main groups of tumors according to the TP53 mutation status and its alleles copy number: 2 WT, 1WT, 1WT/1MUT, and 1 or 2 MUT. These situations could lead to different mechanisms of p53 pathway inactivations. For example, 1WT tumors could present an haploinsufficiency, a feature described in Trp53+/− mice as inducing tumors, preferentially sarcomas.49–50 In the same manner, 1WT/1MUT alleles could lead to a dominant negative effect, and some mutations could not only induce a loss of the normal p53 function but could also confer new oncogenic properties.34 Moreover, we can suppose that some premature truncating mutations could generate unstable mRNAs which could be degraded by mRNA decay, a situation equivalent to a loss of one allele.
In our series, 20% of LMS and 29% of UPS do not exhibit a TP53 alteration. Among these tumors, 17/37 present a deletion of p14 locus, with a subsequent complete loss of RNA expression, and 15/37 present only a complete loss of RNA expression without deletion of the gene. The 5 remaining tumors do not exhibit p14 protein except a cell line, UPS108L, which has a slight protein expression. All studied sarcomas seem to have a defective p53 pathway. To see if tumors exhibiting different ways of p53 pathway inactivation present differentially expressed genes, we have performed unpaired t-tests on Affymetrix data, in LMS and in UPS. We have thus compared gene expression profiles of tumors with at least one TP53 mutation versus tumors with 2 wild-type alleles (2WT) (and thus with p14 alteration) and we have compared tumors with a biallelic TP53 inactivation versus 2WT tumors. We did not find any significantly differentially expressed genes (data not shown), whatever the analysis and the histotype. These results have to be confirmed in a larger set of tumors, because for some tumor groups the number of samples may be not enough consistent to have significant results. However, these negative results suggest that both genetics are equivalent for p53 pathway inactivation and that p14 loss could be an alternative to TP53 alteration. Moreover, it has been previously described in different tumoral models, including sarcomas, that alterations of genes implicated in the p53 pathway, such as MDM2/MDMX amplification/overexpression or p14 inactivation, could lead to an efficient pathway defect.51–53 All these observations tend to show that all studied sarcomas present an inactivation of the p53 pathway, either by direct alteration of the gene, or by inactivation of p14.
Nevertheless, loss of p14 do not necessarily compensate for all p53 functions, since p14-independent activation of TP53 could be induced through other signaling pathways such as ATM/ATR.35 Thus, in tumors with one (1WT) or two wild-type (2WT) TP53 alleles, a functional p53 pathway could be stimulated in some instances, whereas in tumors with direct alterations of TP53, the pathway is completely abrogated. These two situations could notably lead to different chromosomal instability levels. Indeed, tumors with 2 WT TP53 alleles exhibit more simple genomic profiles as documented by the lower number of alterations. These results fit well with some data suggesting that a DNA damage pathway could be stimulated independently of p14 function.35 In this hypothesis, loss of p14 would not participate to chromosomal instability.
Furthermore, we have observed that all tumors do not exhibit detectable level of p14 protein, even if the mRNA is expressed, and whatever the status of TP53. These data suggest that p14 could play other important roles in sarcoma oncogenesis, independently of p53. Few data documenting this possibility are currently available in the literature.15–16 Moreover, most tumors (n = 105) exhibit both events, a feature which has been described in mouse models as shortening the delay of tumor development.16 This p53 pathway inactivation could also be reinforced by the higher expression observed for HDMX, a potent inhibitor of p53 transcriptional activity.43
Finally, according to our results it seems that there is no difference of TP53 target genes expression at the RNA level, between tumors with a direct TP53 alteration and tumors with p14 inactivation. However, it could be of great interest to study the micro(mi)RNAs expression in these tumors. Indeed, it has been described that many miRNAs are under direct TP53 transcriptional control, the mir-34 family, mir-192, 195 and 215 for example.54–55 The study of their expression profile could show if there is a difference between tumors according to their TP53 alleles status, if there is a dosage effect between tumors with one or two wild-type alleles and if some mutated forms of p53 could modify the miRNAs expression. Moreover, we could study in sarcoma cell lines, if miRNAs expression could vary under different stress conditions and functions of the cell line TP53 and/or p14 status.
Frequent codeletions of the CDKN2A/CDKN2B loci are described in a wide range of human cancers.15,19 In our series, we have observed 38 occurrences of such codeletions (26.6%) with subsequent loss of mRNA expression. Moreover, 15 tumors with at least one copy of both loci do not express p14/p15/p16 mRNAs (qPCR ratio <0.1) and 12 tumors express the three mRNAs at very low level (qPCR ratio <0.3). Most of these tumors (19/27) have 2WT TP53 alleles. These results suggest that in these tumors the two loci are either codeleted or transcriptionally coregulated. In this last instance, we could hypothesize that a global mechanism of regulation is involved, such as histone methylation/deacetylation or inhibition by a common regulator.15,38–39 Thus, the combined deficiency for p14/p15/p16 could lead to a synergistic effect in sarcomas development, as described in mouse models.15,19
Conversely, in most tumors with TP53 alterations, p15 and p16 are well expressed at mRNA and protein levels. This induction of expression could be due to oncogenic stress, and may not be antinomic with tumor progression in a TP53 inactivated context.16,20–21
To conclude, the p53 pathway abrogation appears as a pivotal event in soft tissue sarcoma oncogenesis. The precise delineation of the mechanisms of this pathway inactivation could be of extreme interest for the management of tumors. For example, tumors with 2WT TP53 alleles and p14 loss could benefit from drugs preventing p53 degradation, such as Nutlin or RITA.56 On the other hand, drugs such as PRIMA-1 or Ellepticine could restore the transcriptional transactivation function to mutant p53.56
Acknowledgements
We warmly thank Xavier Sastre-Garau, Yves-Marie Robin, Agnès Leroux and Isabelle Valo for providing tumor samples.
Footnotes
Supported by grants from INSERM, Institut Curie, INCa, Conticanet network, Ligue Nationale Contre le Cancer and Fondation de France. The construction of the human BAC array was supported by grants from the Carte d'Identité des Tumeurs (CIT) program of the Ligue Nationale Contre le Cancer. G.P. was supported by grants from Conticanet network and Association Olivier Chappe.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Web Extra Material
{kind=link}
{kind=link}
{kind=link}
Clinical and pathologic data.
TP53 alleles and expression status in tumors.
Description of TP53 mutations observed in tumors. For LMS 103 *, the donor site alteration leads to a maintain of all intron 7 in the cDNA. Abbreviations: del, deletion; ins, insertion; LMS, leiomyosarcoma; UPS, undifferentiated pleomorphic sarcoma; MFH, malignant fibrous histiocytoma; S, sarcoma; OS, osteosarcoma; LPS, liposarcoma; SS, synovialosarcoma. UMD database: a mutation is considered as infrequent, below 10 records; frequent, between 11 and 50 records; hotspot, higher than 50 records.
p14, p16, and p15 genomic and expression status in tumors. Results for real-time genomic PCR are presented as the mean of ratios obtained with two or three reference genes. Hemizygous deletion corresponds to 0.1 less than ratio less than 0.8, homozygous deletion corresponds to 0.1 less than ratio and normal status corresponds to 0.8 less than or equal to ratio less than or equal to 1.2. UPS18B is indicated with a *, which means that a mutation of p15 was found in this sample. Abbreviations: WT, wild-type; MUT, mutated; LMS, leiomyosarcoma; UPS, undifferentiated pleomorphic sarcoma; NA, not available; IHC, immunohistochemistry; WB, western blot; M, promoter methylation detected by bisulfite assay (for at least one of the two studied regions in case of p16 study).
References
- 1.Chibon F, Mariani O, Derré J, Malinge S, Coindre JM, Guillou L, Lagacé R, Aurias A. A subgroup of malignant fibrous histiocytomas is associated with genetic changes similar to those of well-differentiated liposarcomas. Cancer Genet Cytogenet. 2002;139:24–29. doi: 10.1016/s0165-4608(02)00614-3. [DOI] [PubMed] [Google Scholar]
- 2.Chibon F, Mariani O, Mairal A, Derré J, Coindre JM, Terrier P, Lagacé R, Sastre X, Aurias A. The use of clustering software for the classification of comparative genomic hybridization data. An analysis of 109 malignant fibrous histiocytomas. Cancer Genet Cytogenet. 2003;141:75–78. doi: 10.1016/s0165-4608(02)00664-7. [DOI] [PubMed] [Google Scholar]
- 3.Coindre JM, Mariani O, Chibon F, Mairal A, De Saint Aubain Somerhausen N, Favre-Guillevin E, Bui NB, Stoeckle E, Hostein I, Aurias A. Most malignant fibrous histiocytomas developed in the retroperitoneum are dedifferentiated liposarcomas: a review of 25 cases initially diagnosed as malignant fibrous histiocytoma. Mod Pathol. 2003;16:256–262. doi: 10.1097/01.MP.0000056983.78547.77. [DOI] [PubMed] [Google Scholar]
- 4.Derré J, Lagacé R, Nicolas A, Mairal A, Chibon F, Coindre JM, Terrier P, Sastre X, Aurias A. Leiomyosarcomas and most malignant fibrous histiocytomas share very similar comparative genomic hybridization imbalances: an analysis of a series of 27 leiomyosarcomas. Lab Invest. 2001;81:211–215. doi: 10.1038/labinvest.3780229. [DOI] [PubMed] [Google Scholar]
- 5.Fletcher CDM, Unni KK, Mertens F. Pathology and genetics of tumours of soft tissue and bone. IARC Press; Lyon: 2002. World Health Organisation classification of tumours. [Google Scholar]
- 6.Mairal A, Terrier P, Chibon F, Sastre X, Lecesne A, Aurias A. Loss of chromosome 13 is the most frequent genomic imbalance in malignant fibrous histiocytomas. A comparative genomic hybridization analysis of a series of 30 cases. Cancer Genet Cytogenet. 1999;111:134–138. doi: 10.1016/s0165-4608(98)00227-1. [DOI] [PubMed] [Google Scholar]
- 7.Chibon F, Mairal A, Fréneaux P, Terrier P, Coindre JM, Sastre X, Aurias A. The RB1 gene is the target of chromosome 13 deletions in malignant fibrous histiocytoma. Cancer Res. 2000;60:6339–6345. [PubMed] [Google Scholar]
- 8.Otaño-Joos M, Mechtersheimer G, Ohl S, Wilgenbus KK, Scheurlen W, Lehnert T, Willeke F, Otto HF, Lichter P, Joos S. Detection of chromosomal imbalances in leiomyosarcoma by comparative genomic hybridization and interphase cytogenetics. Cytogenet Cell Genet. 2000;90:86–92. doi: 10.1159/000015640. [DOI] [PubMed] [Google Scholar]
- 9.Sandberg AA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: leiomyosarcoma. Cancer Genet Cytogenet. 2005;161:1–19. [PubMed] [Google Scholar]
- 10.Yoo J, Lee HK, Kang CS, Park WS, Lee JY, Shim SI. p53 gene mutations and p53 protein expression in human soft tissue sarcomas. Arch Pathol Lab Med. 1997;121:395–399. [PubMed] [Google Scholar]
- 11.Das P, Kotilingam D, Korchin B, Liu J, Yu D, Lazar AJ, Pollock RE, Lev D. High prevalence of p53 exon 4 mutations in soft tissue sarcoma. Cancer. 2007;109:2323–2333. doi: 10.1002/cncr.22680. [DOI] [PubMed] [Google Scholar]
- 12.Soussi T, Ishioka C, Claustres M, Beroud C. Locus specific mutation databases: pitfalls and good practice based on the p53 experience. Nat Rev Cancer. 2006;6:83–90. doi: 10.1038/nrc1783. [DOI] [PubMed] [Google Scholar]
- 13.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 14.Efeyan A, Serrano M. p53: guardian of the genome and policeman of the oncogenes. Cell Cycle. 2007;6:1006–1010. doi: 10.4161/cc.6.9.4211. [DOI] [PubMed] [Google Scholar]
- 15.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 16.Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 17.Ortega S, Malumbres M, Barbacid M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta. 2002;1602:73–87. doi: 10.1016/s0304-419x(02)00037-9. [DOI] [PubMed] [Google Scholar]
- 18.Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nature Cell Biol. 1999;1:20–26. doi: 10.1038/8991. [DOI] [PubMed] [Google Scholar]
- 19.Krimpenfort P, Ijpenberg A, Song JY, van der Valk M, Nawijn M, Zevenhoven J, Berns A. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature. 2007;448:943–946. doi: 10.1038/nature06084. [DOI] [PubMed] [Google Scholar]
- 20.Bardeesy N, Bastian BC, Hezel A, Pinkel D, DePinho RA, Chin L. Dual inactivation of RB and p53 pathways in RAS-induced melanomas. Mol Cell Biol. 2001;21:2144–2153. doi: 10.1128/MCB.21.6.2144-2153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, Lowe SW. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109:335–346. doi: 10.1016/s0092-8674(02)00734-1. [DOI] [PubMed] [Google Scholar]
- 22.Pérot G, Derré J, Coindre JM, Tirode F, Lucchesi C, Mariani O, Gibault L, Guillou L, Terrier P, Aurias A. Strong smooth muscle differentiation is dependent on myocardin gene amplification in most human retroperitoneal leiomyosarcomas. Cancer Res. 2009;69:2269–2278. doi: 10.1158/0008-5472.CAN-08-1443. [DOI] [PubMed] [Google Scholar]
- 23.La Rosa P, Viara E, Hupé P, Pierron G, Liva S, Neuvial P, Brito I, Lair S, Servant N, Robine N, Manié E, Brennetot C, Janoueix-Lerosey I, Raynal V, Gruel N, Rouveirol C, Stransky N, Stern MH, Delattre O, Aurias A, Radvanyi F, Barillot E. VAMP: visualization and analysis of array-CGH, transcriptome and other molecular profiles. Bioinformatics. 2006;22:2066–2073. doi: 10.1093/bioinformatics/btl359. [DOI] [PubMed] [Google Scholar]
- 24.Shedden K, Chen W, Kuick R, Ghosh D, Macdonald J, Cho KR, Giordano TJ, Gruber SB, Fearon ER, Taylor JM, Hanash S. Comparison of seven methods for producing Affymetrix expression scores based on False Discovery Rates in disease profiling data. BMC Bioinformatics. 2005;6:26. doi: 10.1186/1471-2105-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manié E, Vincent-Salomon A, Lehmann-Che J, Pierron G, Turpin E, Warcoin M, Gruel N, Lebigot I, Sastre-Garau X, Lidereau R, Remenieras A, Feunteun J, Delattre O, de Thé H, Stoppa-Lyonnet D, Stern MH. High frequency of TP53 mutation in BRCA1 and sporadic basal-like carcinomas but not in BRCA1 luminal breast tumors. Cancer Res. 2009;69:663–671. doi: 10.1158/0008-5472.CAN-08-1560. [DOI] [PubMed] [Google Scholar]
- 26.Iwato M, Tachibana O, Tohma Y, Arakawa Y, Nitta H, Hasegawa M, Yamashita J, Hayashi Y. Alterations of the INK4a/ARF locus in human intracranial germ cell tumors. Cancer Res. 2000;60:2113–2115. [PubMed] [Google Scholar]
- 27.Laud K, Marian C, Avril MF, Barrois M, Chompret A, Goldstein AM, Tucker MA, Clark PA, Peters G, Chaudru V, Demenais F, Spatz A, Smith MW, Lenoir GM, Bressac-de Paillerets B, French Hereditary Melanoma Study Group comprehensive analysis of CDKN2A (p16INK4A/p14ARF) and CDKN2B genes in 53 melanoma index cases considered to be at heightened risk of melanoma. J Med Genet. 2006;43:39–47. doi: 10.1136/jmg.2005.033498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xing EP, Nie Y, Song Y, Yang GY, Cai YC, Wang LD, Yang CS. Mechanisms of inactivation of p14ARF, p15INK4b, and p16INK4a genes in human esophageal squamous cell carcinoma. Clin Cancer Res. 1999;5:2704–2713. [PubMed] [Google Scholar]
- 30.Latres E, Drobnjak M, Pollack D, Oliva MR, Ramos M, Karpeh M, Woodruff JM, Cordon-Cardo C. Chromosome 17 abnormalities and TP53 mutations in adult soft tissue sarcomas. Am J Pathol. 1994;145:345–355. [PMC free article] [PubMed] [Google Scholar]
- 31.Castresana JS, Rubio MP, Gómez L, Kreicbergs A, Zetterberg A, Barrios C. Detection of TP53 gene mutations in human sarcomas. Eur J Cancer. 1995;31A:735–738. doi: 10.1016/0959-8049(95)00121-x. [DOI] [PubMed] [Google Scholar]
- 32.Taubert H, Würl P, Meye A, Berger D, Thamm B, Neumann K, Hinze R, Schmidt H, Rath FW. Molecular and immunohistochemical p53 status in liposarcoma and malignant fibrous histiocytoma: identification of seven new mutations for soft tissue sarcomas. Cancer. 1995;76:1187–1196. doi: 10.1002/1097-0142(19951001)76:7<1187::aid-cncr2820760714>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 33.Leach FS, Tokino T, Meltzer P, Burrell M, Oliner JD, Smith S, Hill DE, Sidransky D, Kinzler KW, Vogelstein B. p53 Mutation and MDM2 amplification in human soft tissue sarcomas. Cancer Res. 1993;53:2231–2234. [PubMed] [Google Scholar]
- 34.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9:701–713. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 35.Meek DW. Tumour suppression by p53: a role for the DNA damage response? Nat Rev Cancer. 2009;9:714–723. doi: 10.1038/nrc2716. [DOI] [PubMed] [Google Scholar]
- 36.Kirsch DG, Dinulescu DM, Miller JB, Grimm J, Santiago PM, Young NP, Nielsen GP, Quade BJ, Chaber CJ, Schultz CP, Takeuchi O, Bronson RT, Crowley D, Korsmeyer SJ, Yoon SS, Hornicek FJ, Weissleder R, Jacks T. A spatially and temporally restricted mouse model of soft tissue sarcoma. Nat Med. 2007;13:992–997. doi: 10.1038/nm1602. [DOI] [PubMed] [Google Scholar]
- 37.Orlow I, Drobnjak M, Zhang ZF, Lewis J, Woodruff JM, Brennan MF, Cordon-Cardo C. Alterations of INK4A and INK4B genes in adult soft tissue sarcomas: effect on survival. J Natl Cancer Inst. 1999;91:73–79. doi: 10.1093/jnci/91.1.73. [DOI] [PubMed] [Google Scholar]
- 38.Magdinier F, Wolffe AP. Selective association of the methyl-CpG binding protein MBD2 with the silent p14/p16 locus in human neoplasia. Proc Natl Acad Sci USA. 2001;98:4990–4995. doi: 10.1073/pnas.101617298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen CT, Weisenberger DJ, Velicescu M, Gonzales FA, Lin JC, Liang G, Jones PA. Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2′-deoxycytidine. Cancer Res. 2002;62:6456–6461. [PubMed] [Google Scholar]
- 40.Pollice A, Vivo M, La Mantia G. The promiscuity of ARF interactions with the proteasome. FEBS Lett. 2008;582:3257–3262. doi: 10.1016/j.febslet.2008.09.026. [DOI] [PubMed] [Google Scholar]
- 41.Chen X, Barton LF, Chi Y, Clurman BE, Roberts JM. Ubiquitin-independent degradation of cell-cycle inhibitors by the REGgamma proteasome. Mol Cell. 2007;26:843–852. doi: 10.1016/j.molcel.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972–1975. doi: 10.1126/science.1091362. [DOI] [PubMed] [Google Scholar]
- 43.Finch RA, Donoviel DB, Potter D, Shi M, Fan A, Freed DD, Wang CY, Zambrowicz BP, Ramirez-Solis R, Sands AT, Zhang N. mdmx is a negative regulator of p53 activity in vivo. Cancer Res. 2002;62:3221–3225. [PubMed] [Google Scholar]
- 44.Krug U, Ganser A, Koeffler HP. Tumor suppressor genes in normal and malignant hematopoiesis. Oncogene. 2002;21:3475–3495. doi: 10.1038/sj.onc.1205322. [DOI] [PubMed] [Google Scholar]
- 45.Murao K, Kubo Y, Ohtani N, Hara E, Arase S. Epigenetic abnormalities in cutaneous squamous cell carcinomas: frequent inactivation of the RB1/p16 and p53 pathways. Br J Dermatol. 2006;155:999–1005. doi: 10.1111/j.1365-2133.2006.07487.x. [DOI] [PubMed] [Google Scholar]
- 46.Mertens F, Fletcher CD, Dal Cin P, De Wever I, Mandahl N, Mitelman F, Rosai J, Rydholm A, Sciot R, Tallini G, Van den Berghe H, Vanni R, Willén H. Cytogenetic analysis of 46 pleomorphic soft tissue sarcomas and correlation with morphologic and clinical features: a report of the CHAMP Study Group. Chromosomes and MorPhology. Genes Chromosomes Cancer. 1998;22:16–25. doi: 10.1002/(sici)1098-2264(199805)22:1<16::aid-gcc3>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 47.Kawaguchi K, Oda Y, Saito T, Yamamoto H, Tamiya S, Takahira T, Miyajima K, Iwamoto Y, Tsuneyoshi M. Mechanisms of inactivation of the p16INK4a gene in leiomyosarcoma of soft tissue: decreased p16 expression correlates with promoter methylation and poor prognosis. J Pathol. 2003;201:487–495. doi: 10.1002/path.1419. [DOI] [PubMed] [Google Scholar]
- 48.el-Rifai W, Sarlomo-Rikala M, Andersson LC, Miettinen M, Knuutila S. DNA copy number changes in gastrointestinal stromal tumors: a distinct genetic entity. Ann Chir Gynaecol. 1998;87:287–290. [PubMed] [Google Scholar]
- 49.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 50.Venkatachalam S, Shi YP, Jones SN, Vogel H, Bradley A, Pinkel D, Donehower LA. Retention of wild-type p53 in tumors from p53 heterozygous mice: reduction of p53 dosage can promote cancer formation. EMBO J. 1998;17:4657–4667. doi: 10.1093/emboj/17.16.4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, Teunisse A, Lam S, Ramos Y, Mohan A, Johnson D, Wilson M, Rodriguez-Galindo C, Quarto M, Francoz S, Mendrysa SM, Guy RK, Marine JC, Jochemsen AG, Dyer MA. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–66. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- 52.Müller CR, Paulsen EB, Noordhuis P, Pedeutour F, Saeter G, Myklebost O. Potential for treatment of liposarcomas with the MDM2 antagonist Nutlin-3A. Int J Cancer. 2007;121:199–205. doi: 10.1002/ijc.22643. [DOI] [PubMed] [Google Scholar]
- 53.Park YB, Park MJ, Kimura K, Shimizu K, Lee SH, Yokota J. Alterations in the INK4a/ARF locus and their effects on the growth of human osteosarcoma cell lines. Cancer Genet Cytogenet. 2002;133:105–111. doi: 10.1016/s0165-4608(01)00575-1. [DOI] [PubMed] [Google Scholar]
- 54.Hermeking H. p53 enters the microRNA world. Cancer Cell. 2007;12:414–418. doi: 10.1016/j.ccr.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 55.Bailey SG, Sanchez-Elsner T, Stephanou A, Cragg MS, Townsend PA. Regulating the genome surveillance system: miRNAs and the p53 super family. Apoptosis. 2010;15:541–552. doi: 10.1007/s10495-010-0456-1. [DOI] [PubMed] [Google Scholar]
- 56.Bouchet BP, de Fromentel CC, Puisieux A, Galmarini CM. p53 as a target for anti-cancer drug development. Crit Rev Oncol Hematol. 2006;58:190–207. doi: 10.1016/j.critrevonc.2005.10.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clinical and pathologic data.
TP53 alleles and expression status in tumors.
Description of TP53 mutations observed in tumors. For LMS 103 *, the donor site alteration leads to a maintain of all intron 7 in the cDNA. Abbreviations: del, deletion; ins, insertion; LMS, leiomyosarcoma; UPS, undifferentiated pleomorphic sarcoma; MFH, malignant fibrous histiocytoma; S, sarcoma; OS, osteosarcoma; LPS, liposarcoma; SS, synovialosarcoma. UMD database: a mutation is considered as infrequent, below 10 records; frequent, between 11 and 50 records; hotspot, higher than 50 records.
p14, p16, and p15 genomic and expression status in tumors. Results for real-time genomic PCR are presented as the mean of ratios obtained with two or three reference genes. Hemizygous deletion corresponds to 0.1 less than ratio less than 0.8, homozygous deletion corresponds to 0.1 less than ratio and normal status corresponds to 0.8 less than or equal to ratio less than or equal to 1.2. UPS18B is indicated with a *, which means that a mutation of p15 was found in this sample. Abbreviations: WT, wild-type; MUT, mutated; LMS, leiomyosarcoma; UPS, undifferentiated pleomorphic sarcoma; NA, not available; IHC, immunohistochemistry; WB, western blot; M, promoter methylation detected by bisulfite assay (for at least one of the two studied regions in case of p16 study).