

Abstract
Rac1, a subunit of NADPH oxidase, plays an important role in directed endothelial cell motility. We reported previously that Rac1 activation was necessary for choroidal endothelial cell migration across the retinal pigment epithelium, a critical step in the development of vision-threatening neovascular age-related macular degeneration. Here we explored the roles of Rac1 and NADPH oxidase activation in response to vascular endothelial growth factor treatment in vitro and in a model of laser-induced choroidal neovascularization. We found that vascular endothelial growth factor induced the activation of Rac1 and of NADPH oxidase in cultured human choroidal endothelial cells. Further, vascular endothelial growth factor led to heightened generation of reactive oxygen species from cultured human choroidal endothelial cells, which was prevented by the NADPH oxidase inhibitors, apocynin and diphenyleneiodonium, or the antioxidant, N-acetyl-l-cysteine. In a model of laser-induced injury, inhibition of NADPH oxidase with apocynin significantly reduced reactive oxygen species levels as measured by dihydroethidium fluorescence and the volume of laser-induced choroidal neovascularization. Mice lacking functional p47phox, a subunit of NADPH oxidase, had reduced dihydroethidium fluorescence and choroidal neovascularization compared with wild-type controls. Taken together, these results indicate that vascular endothelial growth factor activates Rac1 upstream from NADPH oxidase in human choroidal endothelial cells and increases generation of reactive oxygen species, contributing to choroidal neovascularization. These steps may contributed to the pathology of neovascular age-related macular degeneration.
Age-related macular degeneration (AMD) is a leading cause of visual impairment in elderly individuals1–4 and is estimated to affect approximately 14 million people worldwide.5 Ninety percent of legal blindness from AMD is due to neovascularization that originates from endothelial cells in the choroid and grows into neurosensory retina as choroidal neovascularization (CNV).5 Although it is recognized that genetic polymorphisms, such as those involved in the alternative pathway of the complement system, have been strongly associated with increased risk of advanced AMD,6–11 AMD does not manifest at birth but rather in the seventh or eighth decades of life. The current thinking is that a genetic predisposition accompanied by exogenous, chronic, repeated stresses could lead to tissue damage and dysfunction that becomes apparent only later in life.10,12,13 In support of this theory is clinical evidence of independent and sometimes additive risk of advanced AMD from environmental factors.14
Several environmental factors associated with increased risk of AMD also increase oxidative stress. Examples include blue light-induced photochemically released oxidants15 and cigarette smoke-related compounds.16 Other clinical evidence supports oxidative stress in advanced AMD. Proteomic analysis of Bruch's membrane from human eyes with AMD revealed the presence of oxidized compounds.17 The Eye Disease Case Control Study found an association between reduced prevalence of AMD and high dietary intake of antioxidants.18 The placebo-controlled, double-masked multicenter clinical Age-Related Eye Disease Study provided strong support for the relationship between AMD and oxidants by finding reduced progression to advanced AMD and vision loss in subjects who took certain antioxidants and zinc supplements.19
Laboratory evidence provides mechanistic support for oxidative stress in the pathophysiology of AMD. Conditioned media from hydrogen peroxide-treated retinal pigment epithelium (RPE) induced an angiogenic phenotype in cocultured choroidal endothelial cells.20 Glutathione-depleted RPE, thus susceptible to oxidative damage, had increased expression of vascular endothelial growth factor (VEGF) and its receptors compared with the control.20 A mouse model lacking the antioxidant enzyme, CuZn superoxide dismutase, developed features of AMD, namely drusen, CNV, and dysfunction in RPE cell junctions and barrier properties.21 Photo-oxidation of bis-retinoid lipofuscin in cultured RPE cells resulted in released complement activation fragments, providing evidence that oxidation activated complement and also linked photo-oxidative stress and the complement system.22 Although evidence supports oxidative damage in AMD, gaps remain in our understanding of the steps involved in the pathophysiology of advanced vision-threatening AMD.
We reported previously that activation of the small Rho GTPase, Rac1, was necessary for choroidal endothelial cell migration across the RPE, a critical step in the development of neovascular AMD.23 Further, the activation of Rac1 was found downstream of VEGF-VEGF receptor 2 signaling.24 Rac1 is important in directed endothelial cell motility and is also an important subunit of the enzyme complex, NADPH oxidase. In its inactive state, NADPH oxidase consists of membrane-bound subunits, ie, gp91phox and p22phox, and cytosolic subunits, p67phox and p47phox, in addition to Rac1 in endothelial cells.25 When activated, the subunits aggregate and the active enzyme generates reactive oxygen species (ROS) and triggers angiogenic signaling in endothelial cells (ECs).25 In this study, we investigated the role of NADPH oxidase in the pathogenesis of CNV.
Materials and Methods
Reagents
VEGF165 was purchased from R&D Systems, (Minneapolis, MN). Apocynin, diphenyleneiodonium (DPI), and N-acetyl-l-cysteine (NAC) were obtained from Sigma-Aldrich (St. Louis, MO).
Cell Culture
Isolation of Human Choroidal Endothelial Cells
Choroidal endothelial cells (ECs) were obtained from donor eyes of adults younger than 30 years of age from the North Carolina Eye Bank, Inc. (Winston-Salem, NC). Within 24 hours of death, the vitreous and retina were removed from the eyecup. The RPE layer was removed by repeated titration with Hanks' balanced salt solution (Invitrogen, San Diego, CA). The choroidal layer was peeled from the sclera, minced into small pieces, and washed with Hanks' balanced salt solution. The pieces were incubated with 500 μg/ml collagenase 1A (Roche, Nutley, NJ) and 1.2 U/ml Dispase (Invitrogen) for 45 minutes at 37°C with constant agitation. After digestion, the segments were filtered through a 33-μm mesh filter (BD Biosciences, San Jose, CA). The enzymes were neutralized with an equal volume of endothelial growth media (EGM-2, Lonza, Walkersville, MD) with 10% fetal bovine serum (FBS). The filtered cells were centrifuged at 400g at 4°C for 5 minutes and resuspended in 0.5 ml of Hanks' balanced salt solution with 5% FBS. The cells were then incubated with CD31-coated Dynabeads (Invitrogen) for 30 minutes at room temperature with gentle rotation. The CD31-positive cells were separated with the Dynal Magnetic Particle Concentrator (Invitrogen). The cells-bead complex was washed with Hanks' balanced salt solution/5% FBS and reapplied to the magnet. The wash step was repeated five times. The cells were then plated onto a T-75 flask in EGM-2 with 10% FBS at 37°C with 5% CO2. EC identification was based on positive staining for CD31, VE-cadherin, and von Willebrand factor and by uptake of acetylated low-density lipoprotein. Choroidal ECs were maintained in EGM-2 with 10% FBS and used through passage 4.
For in vitro studies, choroidal ECs were grown until 90% confluence and were pretreated for 2 hours in serum-free EBM-2 with 500 μmol/L apocynin, 1 mmol/L NAC, 1 μmol/L DPI, or PBS. Fifteen minutes before collection, choroidal ECs were incubated in 10 ng/ml VEGF165 (R&D Systems).
RPE Studies
Human ARPE-19 cells (American Type Culture Collection, Rockville, MD), up to passage 18, were grown to 80% confluence in Dulbecco's modified Eagle's medium-Nutrient Mixture F-12 (DMEM/F12) (Invitrogen, Carlsbad, CA) and supplemented with 10% FBS. These cells constitutively produce greater amounts of VEGF than more differentiated human fetal RPE and were chosen for this experiment to mimic the conditions of neovascular AMD, in which the RPE produces more VEGF than normal.26 ARPE-19 cells were pretreated for 6 hours with 500 μmol/L apocynin, 2 μmol/L DPI, or PBS in serum-free medium. Immediately thereafter, cells were washed with PBS, retreated, and placed into room air (21% O2) for 16 hours. Cells were then washed with PBS, and total RNA was purified using the RNeasy Mini kit (Qiagen, Valencia, CA).
Real-Time Quantitative PCR
Assays were performed using the Applied Biosystems 7900HT Real-Time PCR System. In brief, 1 μg of total RNA was reverse-transcribed into cDNA using a TaqMan Reverse Transcription reagent (Applied Biosystems, Foster City, CA), according to the manufacturer's protocol. One-twentieth of the total cDNA (50 ng of equivalent RNA) was used in each amplification reaction. Each TaqMan reaction (16 μl) contained 5 μl of cDNA, 8 μl of TaqMan PCR MasterMix (Applied Biosystems), 1 μl of forward primer (5 μmol/L), 1 μl of reverse primer (5 μmol/L), and 1 μl of probe (5 μmol/L) (see below for primer sequences). PCR cycles consisted of denaturation at 95°C for 10 minutes, followed by 40 cycles at 95°C for 15 seconds and at 60°C for 60 seconds. To confirm amplification specificity, PCR products from each primer pair were subjected to a melting curve analysis. Amplification of the 18S RNA (Eukaryotic 18S rRNA, Hs99999901_s1, Applied Biosystems) was performed in parallel as a control for sample loading and to allow normalization between samples. Each sample was run in duplicate, and each experiment included three nontemplate control wells. Relative expression levels were determined by normalization to 18S rRNA using REST 2009.27 Results are expressed as the mean ± SE. Primers and probes were as follows: for human VEGF121: 5′-CATAGGAGAGATGAGCTTCC-3′ (forward), 5′-CCTCGGCTTGTCACATTTTTCT-3′ (reverse), and FAM-CAGCACAACAAATGTGAATGCAGACCA-TAMRA (probe); for human VEGF165: 5′-CATAGGAGAGATGAGCTTCC-3′ (forward), 5′-AAGGCCCACAGGGATTTTCT-3′ (reverse), and FAM-CAGCACAACAAATGTGAATGCAGACCA-TAMRA (probe); and for human VEGF189: 5′-CCAAAGAAAGATAGAGCAAGAC-3′ (forward) and 5′-AGGACTTATACCGGGATTTCT-3′ (reverse), FAM-TGCCCCTTTCCCTTTCCTCGAACTG-TAMRA (probe).
ROS Assay
Formation of ROS was monitored by the conversion of nonfluorescent 6-carboxy-2′7′-dichlorodihydrofluorescin diacetate, di(acetoxymethyl ester) (H2DCF-DA) (Invitrogen) to fluorescent 6-carboxy-2′,7′-dichlorofluorescein diacetate di(acetoxymethyl ester) (DCF) (Invitrogen Molecular Probes, San Diego, CA). Cells were preincubated with apocynin, NAC, or DPI for 2 hours before loading with 5 μmol/L H2DCF-DA in serum-free medium for 30 minutes at 37°C. After loading, cells were washed twice with PBS and incubated for an additional 20 minutes at 37°C to allow for dye de-esterification. Cells were stimulated with VEGF165 for the described times as indicated in the figure legends. Fluorescence was determined using a fluorometer with an excitation of 485 nm and an emission of 520 nm.
Subcellular Fractionation and Western Blot Analysis
Membrane-bound and cytosolic proteins were separated from choroidal ECs using the Subcellular Protein Fractionation Kit (Thermo Scientific, Rockford, IL). In brief, cells were trypsinized with trypsin-EDTA (Sigma-Aldrich), washed with PBS, and centrifuged at 400g for 5 minutes. Cells were then incubated in cytoplasmic extraction buffer (Thermo Scientific) for 10 minutes at 4°C. The lysed cells were centrifuged, and the supernatant containing cytoplasmic proteins was removed. Likewise, membrane-bound proteins were extracted using membrane extraction buffer. Equal amounts of protein samples were resolved by SDS-polyacrylamide gel electrophoresis and then transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA), which were blocked with 5% blocking grade dry milk (Bio-Rad Laboratories) for 1 hour before incubation with the primary antibody. Membranes were probed with anti-p47phox and anti-p22phox antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) at a 1:1000 dilution. The membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) and visualized by enhanced chemiluminescence using Immobilon Western (Millipore Corporation, Billerica, MA). Digitized images of Western blots were quantified using UN-SCAN-IT (Silk Scientific, Orem, Utah) and normalized to β-actin. The densitometric values were displayed as a ratio of p47phox/p22phox.
Rac1 Assay
After stimulation, choroidal ECs were kept on ice, washed with ice-cold PBS, and assayed for Rac1 activation.23 Choroidal ECs were lysed in 300 μl of Rac1 assay buffer B (50 mmol/L Tris, pH 7.6, 150 mmol/L NAC, 1% Triton X-100, and 0.5 mmol/L MgCl2) containing protease inhibitors and 1 mmol/L sodium vanadate. Lysates were clarified by centrifugation at 13,000 rpm for 5 minutes at 4°C. Lysates were then incubated for 30 minutes at 4°C with gentle rocking with 30 μg of GST-PBD (Rac1-binding domain of glutathione S-transferase p21-activating kinase) fusion protein immobilized on glutathione beads to specifically pull down active (GTP-bound) Rac1. The beads were washed four times with the same lysis buffer and then were resuspended in 2× gel sample buffer. Bead-bound active Rac1 and total Rac1 from reserved cell lysates were then analyzed by SDS-polyacrylamide gel electrophoresis. Proteins were transferred to nitrocellulose membranes (Schleicher & Schuell BioScience, Inc., Keene, NH) and processed for Western analysis using a monoclonal antibody directed against Rac1. Bound antibodies were detected by enhanced chemiluminescence. For quantification of Western blots, intensity values of bands were measured from three different repeats for each experiment using ImageJ software (National Institutes of Health, Bethesda, MD).
Small Interfering RNA Transfection
Choroidal ECs plated at approximately 50% confluence were transfected with small interfering RNA (siRNA) to Rac1 (Dharmacon RNA Technologies, Lafayette, CO) at a concentration of 25 nmol/L using OligofectAMINE (Invitrogen) according to the manufacturer's instructions. A nontargeting siRNA (Dharmacon RNA Technologies) was used as a control. Cells were transfected for 4 hours in serum-free medium, after which 1.5 ml of EGM-2MV was added. Cells were harvested after 72 hours.
Migration Assay
Transwell polycarbonate plates with tissue culture inserts containing a membrane with 8-μm pores were coated with 20 μg/ml PureCol (INAMED Biomaterials, Fremont, CA). Choroidal ECs were suspended in serum-free EBM and seeded into each insert. The cells were then incubated with 10 ng/ml of VEGF added to each of the outer chambers. After 4 hours of incubation, ECs adhering to the top surface of the tissue culture inserts were removed with a cotton-tipped applicator, and ECs adhering to the bottom surface of the inserts were trypsinized and counted using a hemocytometer.
In Vivo Laser-Induced CNV Model
Wild-type mice (C57BL/6) were purchased from Charles River. p47phox knockout mice have been shown to lack p47phox by Southern analysis.28 Three-month-old mice were used in laser-induced CNV experiments. All animals were cared for in accordance with the University of North Carolina's Institute for Laboratory Animal Research (Guide for the Care and Use of Laboratory Animals) and the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research.
Wild-type mice were injected intraperitoneally with 10 mg/kg/day apocynin (Sigma-Aldrich, St. Louis, MO) dissolved in PBS each day for 5 days preceding and 5 days after laser treatment. The treatment and time points were chosen based on other interventional studies for laser-induced CNV29 and based on peak recruitment of macrophages.30,31 PBS-injected animals were used as controls.
Mice were anesthetized with 10 mg/kg ketamine and 100 mg/kg xylazine, and the pupils of each eye were dilated with 1% topical Tropicamide (Alcon Pharmaceuticals, Ft. Worth, TX). The retina was visualized via a glass coverslip held against the cornea with Gonak (Akorn, Lake Forest, IL) as a coupling agent using a Zeiss SL30 slit lamp biomicroscope. Laser photocoagulation (200 mW, 100 ms, 50 μm) was induced in both eyes of adult female mice using a 532-nm laser from Iridex (Irvine, CA). Four to six laser spots were delivered, each approximately 2 disk diameters from the optic nerve with care to avoid major vessels. Disruption of Bruch's membrane was confirmed by the appearance of a cavitation bubble.
Three and 5 days after injury, eyes were removed and fixed in 4% paraformaldehyde for 1 hour. The lens, globe, and retina were removed, and the choroid/sclera segment was cut into a cloverleaf pattern and mounted onto a microscope slide for choroidal flat mounts. The tissue was treated with cold 70% methanol for 20 minutes, washed with PBS, treated with 0.1% Triton X-100 (Sigma-Aldrich) in PBS, and washed three more times with PBS. The segments were then incubated in Isolectin Alexa Fluor 568 (Invitrogen) overnight at 4°C and flat-mounted with a 1:1 PBS/glycerol mix.
Images of the flat mounts were taken with a Leica SP2 upright confocal microscope (Leica, Wetzlar, Germany). All images were taken with a ×20 objective. Multiple horizontal sections of the choroid/sclera were taken at 1-μm increments. Laser burns that were too deep (indicated by dark areas in the center of the lesion), associated with hemorrhage, or co-joined with other lesions were excluded from the analysis. The stacked images of the lesions were measured and expressed as total volumes (μm3) using Volocity 5.0 (Improvision/PerkinElmer, Waltham, MA). For each experiment, multiple spots from both eyes of four to five animals per condition were analyzed. All volumes calculated were averaged as one value per animal. A maximum projection of the z-stack was created for each CNV lesion for qualitative comparisons.
Immunohistochemical Labeling
Whole eyes were placed in Hanks' buffered salt solution, and the anterior segment and retina were removed. The eye cup, consisting of RPE-choroid-sclera, was flat mounted and dissected into small, rectangular laser burn and nonlaser burn sections, which were fresh frozen in Optimal Cutting Temperature (O.C.T.) compound (Sakura Finetek USA, Inc., Torrance, CA) and serially cut into 10-μm sections. For comparisons, labeled serial sections were placed onto the same microscope slide to assure equal handling and antibody labeling conditions. Sections were fixed for 15 minutes in 2% paraformaldehyde, washed three times in PBS, and incubated overnight at 4°C with Alexa Fluor 568-isolectin B4 in PBS (1:100) to stain the vasculature. After being washed in PBS three times, retinas were incubated for 30 minutes in 5% bovine serum albumin to block nonspecific binding of the primary antibody. All sections were rinsed three times in PBS and mounted in PBS/glycerol (2:1 with Vectashield, Vector Laboratories, Burlingame, CA). On all slides, the coverslips were sealed with nail polish, and images of the sections were captured using a Leica SP2 scanning laser confocal microscope and digitally stored for analysis. Control sections were labeled with secondary antibodies only to assess for background staining due to nonspecific binding.
ROS Detection in Retina/Choroid Sections
ROS generation was detected using dihydroethidium (DHE) fluorescence measurement as described previously.32 Fresh-frozen sections of RPE-choroid were prepared from eye cups as described above. Control and experimental sections were placed on the same slide and processed under the same conditions. Sections were thawed, washed in Tris-buffered saline and incubated with 10 μmol/L DHE (Invitrogen) in the dark at 37°C for 15 minutes. Slides were rinsed and mounted in PBS/glycerol (2:1 with Vectashield, Vector Laboratories). Confocal images were acquired with an Olympus FluoView FV500 laser scanning confocal microscope using a ×20 objective and 530 nm excitation/615 nm emission filters at the same photomultiplier tube voltage, gain, and offset. Images were converted to grayscale using Adobe Photoshop CS3. The area of the RPE and choroid was measured, including the lesion area and neovascular tissue in laser-burned sections. The sclera and extraocular tissues were excluded from analysis. The sum of the gray values of all pixels (integrated density) per image area of interest was measured using ImageJ 1.43. The integrated densities of sections of CNV from several mice were analyzed for each condition and time point. Sections for DHE analysis were fresh-frozen and obtained within 3 hours of staining.
Statistical Assays
For in vitro assays, experiments were performed in triplicate with four to five samples per condition. For in vivo experiments, at least four animals (eight eyes) per condition were used. Statistics were determined with analysis of variance and a post hoc Bonferroni t-test. For real-time PCR analyses, the REST was used as described earlier.27
Results
VEGF-Induced Rac1 Activation in Choroidal ECs
VEGF was previously reported to activate Rac1 through VEGF receptor 2 in human umbilical vein endothelial cells.24 We therefore examined whether VEGF165 also caused Rac1 activation in choroidal ECs. Rac1 GTP loading was measured by pulldown assays with GST-PBD, followed by blotting for Rac1, as described previously.23 We determined that Rac1 was activated within 5 minutes of treatment with VEGF165 and remained active for 60 minutes in the choroidal ECs (Figure 1, A and B).
Figure 1.
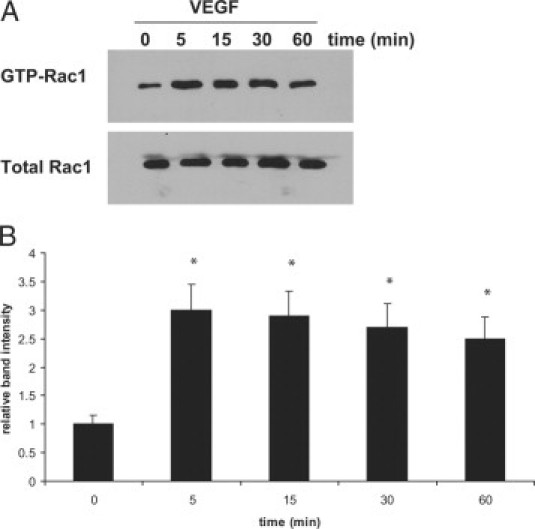
VEGF treatment activates Rac1 in choroidal ECs. Choroidal ECs were treated with 10 ng/ml VEGF for the indicated times, and the activation of Rac1 was measured using a GST-PBD pulldown assay as described in Materials and Methods. Total Rac1 was used as a loading control. A: Representative blot showing increased active Rac1 within five minutes of treatment with VEGF. B: Densitometry analysis of blots performed as described in Materials and Methods. Band intensity is expressed for each time point as a relative number compared with the 0-minute time point. Rac1 activation at the zero time point is statistically different from that of cells treated with VEGF for 5, 15, 30, or 60 minutes (*P < 0.05).
VEGF-Induced ROS in Choroidal ECs
It has been demonstrated that Rac1 activation can lead to increased generation of ROS in an NADPH oxidase-dependent manner.33,34 Because NADPH oxidase is an effector for Rac1, we tested whether exposure of choroidal ECs to different concentrations of VEGF would stimulate the generation of ROS. VEGF165 increased ROS production in choroidal ECs in a dose-dependent manner, as measured using the H2DCF-DA assay (Figure 2A). The production of VEGF-induced ROS at 15 minutes was inhibited by pretreatment of ECs with either of two NADPH oxidase inhibitors, apocynin or DPI, to a similar degree as with a broad antioxidant, NAC (Figure 2B).
Figure 2.
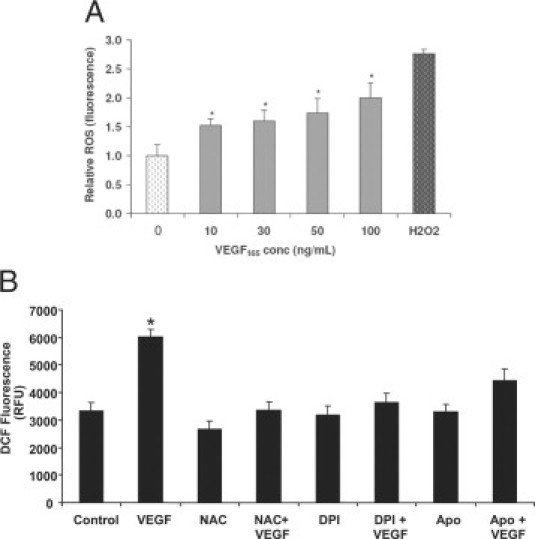
VEGF-induced ROS generation in choroidal ECs. A: Cells were incubated with different concentrations of VEGF165 for 15 minutes and then ROS generation was determined by DCF fluorescence measurement (*P < 0.05); 200 μmol/L H2O2 was used as a positive control. B: Choroidal ECs were pretreated with 1 mmol/L NAC, 500 μmol/L apocynin (Apo), or 1 μmol/L DPI for two hours. The choroidal ECs were loaded with H2DCF-DA and then treated with 10 ng/ml VEGF165 for ten minutes. The generation of ROS was measured by the fluorescence intensity of DCF. Data are expressed in relative fluorescent units (RFU) as given by the plate reader. *P < 0.05 when VEGF is compared with control choroidal ECs. Treatment with NAC, DPI, or Apo abrogated VEGF-induced generation of ROS.
To determine whether Rac1 was required for VEGF-induced ROS generation in the choroidal ECs, we knocked down Rac1 protein expression with siRNA (Figure 3A). We then compared the ability of the ECs to generate ROS after VEGF treatment in both control and Rac1 siRNA-treated cells. In cells treated with control siRNA, VEGF led to ROS generation, whereas in ECs treated with Rac1 siRNA, VEGF was no longer capable of inducing ROS production (Figure 3B). These data suggest that Rac1 is required for VEGF-induced generation of ROS in choroidal ECs and is upstream of ROS production. To further examine the relationship between Rac1 and ROS in response to VEGF treatment, we tested whether ROS inhibition had an effect on VEGF-induced Rac1 activation. We found that pretreatment with either the antioxidant NAC (1 mmol/L) or the NADPH oxidase inhibitors, apocynin (500 μmol/L) or DPI (1 μmol/L), did not reduce Rac1 activation in choroidal ECs (Figures 4, A–C). Collectively, these data indicate that VEGF causes Rac1-dependent generation of ROS in choroidal ECs and that Rac1 activation is required upstream of NADPH oxidase in this pathway.
Figure 3.
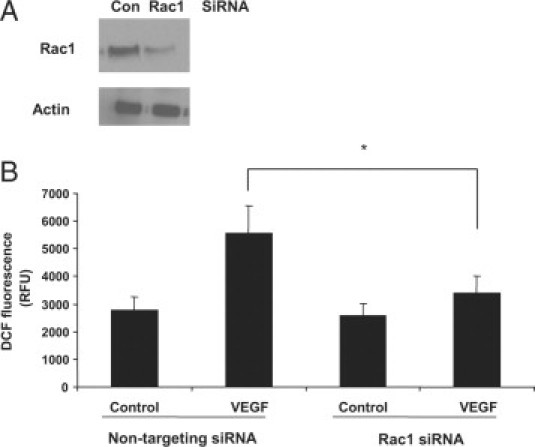
Rac1-dependent VEGF-induced ROS in choroidal ECs. A: Choroidal ECs were transfected with Rac1 siRNA or nontargeting siRNA for 72 hours. Cells were then lysed in sample buffer, electrophoresed, and immunoblotted with an antibody against Rac1 or actin. B: Choroidal ECs were transfected with Rac1 siRNA or a nontargeting siRNA for 72 hours. Cells were then treated with 10 ng/ml VEGF, and ROS generation was monitored by DCF fluorescence. Data are expressed as relative fluorescent units (RFU) as given by the plate reader. ROS generation in the nontargeting siRNA cells treated with VEGF is statistically different from that in the Rac1 siRNA cells treated with VEGF at 15 minutes (*P < 0.05). Con, control.
Figure 4.
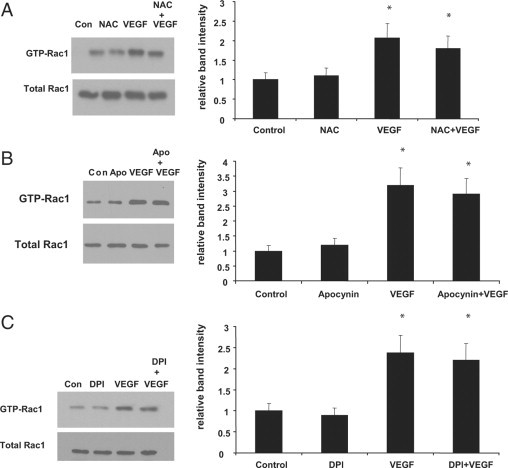
Rac1 activated upstream of NADPH oxidase in VEGF-stimulated choroidal ECs. A: Choroidal ECs were pretreated with 1 mmol/L NAC for two hours before treatment with VEGF165 (10 ng/ml). The activation of Rac1 was measured through a GST-PBD pulldown assay. Representative blots are shown for each. B: Choroidal ECs were pretreated with 500 μmol/L apocynin for 2 hours before treatment with VEGF165 (10 ng/ml). The activation of Rac1 was measured through a GST-PBD pulldown assay. C: Choroidal ECs were pretreated with 1 μmol/L DPI for two hours before treatment with VEGF165 (10 ng/ml). The activation of Rac1 was measured through a GST-PBD pulldown assay. Representative blots are shown for each. Densitometry analysis of blots was performed as described in Materials and Methods. Band intensity is expressed for each treatment as a relative number compared with the control. Rac1 activation in the control cells is statistically different from that for cells treated with VEGF or VEGF + drug. (*P < 0.05). Con, control.
VEGF-Induced Activation of NADPH Oxidase
The above data demonstrate that inhibition of NADPH oxidase activity attenuates VEGF-induced ROS production. We next sought to determine whether VEGF treatment led to the activation of NADPH oxidase in choroidal ECs. Activation of NADPH oxidase occurs through assembly of cytosolic with membrane-bound subunits.25 Because isoforms of the subunits can vary in different species and even tissues,35 we confirmed expression of the p47phox subunit of NADPH oxidase by Western blot in human choroidal ECs (data not shown). We then examined the recruitment of p22phox and p47phox to the membrane compartment as a measure of NADPH oxidase activation25,36 in choroidal ECs exposed to VEGF165. Compared with the control, VEGF treatment led to an increase in the p47phox/p22phox subunit ratio (Figure 5). This provides support that VEGF treatment leads to the activation of NADPH oxidase in choroidal ECs.
Figure 5.
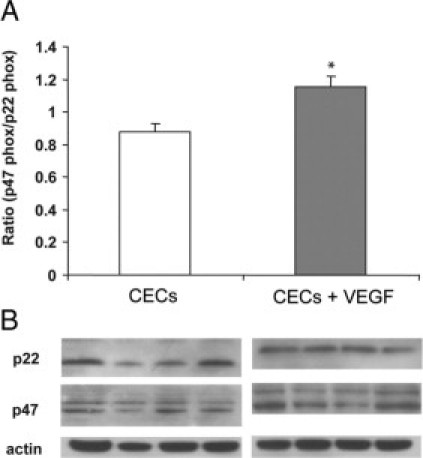
p47phox/p22phox increased in VEGF-stimulated choroidal ECs. A: Choroidal ECs (CECs) were incubated with 30 ng/ml VEGF165 for 15 minutes, and isolated membrane protein was separated by SDS-polyacrylamide gel electrophoresis. Membranes were then blotted for p47phox or p22phox. B: Figure is representative of densitometry analyses of Western blot of membrane protein probed with anti-p47phox and anti-p22phox antibodies. β-Actin was used as a loading control. The ratio in control CECs is statistically different from that in CECs treated with VEGF (*P < 0.05).
Inhibition of NADPH Oxidase Activity on VEGF-Induced Migration of Choroidal ECs
The ability of ECs to migrate is a critical component of both angiogenesis and CNV. To determine whether NADPH oxidase might be required for VEGF-induced choroidal EC migration, the inhibitors DPI and apocynin and the broad spectrum antioxidant NAC were used. Treatment with VEGF caused an approximate twofold increase in the number of choroidal ECs that migrated across a transwell insert compared with untreated control ECs. However, treatment with apocynin, DPI, or NAC significantly reduced VEGF-induced EC migration (Figure 6). These data suggest that NADPH oxidase activity and ROS generation play an important role in regulation of choroidal EC migration.
Figure 6.
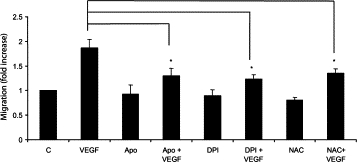
NADPH oxidase required for VEGF-induced migration of choroidal ECs. Endothelial cells were seeded onto collagen-coated transwell tissue culture inserts in the presence or absence of NAC, DPI, or apocynin (Apo) for two hours. After this initial pretreatment with inhibitors, 10 ng/ml VEGF was added to the outer chamber, and cells were allowed to migrate for two hours. After two hours, cells remaining on the top of the chamber were removed, and cells that had migrated to the bottom chamber were trypsinized and counted. Migration of the VEGF-treated choroidal ECs is statistically different from that of the choroidal ECs treated with VEGF + drug (*P < 0.05). All values are expressed as relative to the control, which has been set equal to 1. C, control choroidal ECs.
Production of ROS and Inhibition of NADPH Oxidase in Laser-Induced CNV
Rac1 activation is necessary for choroidal EC transmigration across the RPE,23 a critical step in vision-threatening neovascular AMD. Because VEGF-induced Rac1 activation triggered downstream activation of NADPH oxidase and contributed to choroidal EC migration, we investigated whether inhibition of NADPH oxidase would affect the development of CNV in the laser-induced CNV model. This model is a widely used animal model of neovascular AMD.30 As a measurement of superoxide radical and intracellular ROS, we assayed DHE fluorescence. We treated animals with daily 0.1-ml intraperitoneal injections of apocynin to inhibit NADPH oxidase (10 mg/kg/day) or PBS, as a control, for 5 days before and 5 days after laser treatment to induce CNV. Doses were based on previous studies showing effective and significant reductions in retinal ROS.37 Compared with unlasered areas, DHE fluorescence was greater in RPE-choroid areas that had received laser injury 3 days (P < 0.001, analysis of variance) or 5 days previously (P < 0.01, analysis of variance) (Figure 7A). Qualitative assessment of ROS determined by DHE fluorescence also revealed less DHE staining in unlasered eyes (Figure 7, B–E) compared with respective lasered eyes (Figure 7, F–I). Compared with mice that received control PBS, apocynin treatment significantly reduced ROS within the RPE-choroid of sections with laser injury at day 5 (P < 0.05, analysis of variance) (Figure 7A). Likewise, Figure 7 shows less DHE staining in CNV in apocynin-treated (Figure 7I) compared with control lasered eyes (Figure 7G), in which predominantly nonpigmented and some pigmented cells seen in the corresponding phase image in Figure 7F were DHE-positive. In unlasered RPE-choroid, no significant difference was found between apocynin- and PBS-treated mice. In addition, compared with the PBS control, apocynin significantly reduced CNV volumes measured from lectin-stained choroidal flat mounts (P = 0.007, analysis of variance) (Figure 7J) and as noted in underlying maximum projections of lectin-stained CNV in choroidal flat mounts (Figure 7K).
Figure 7.
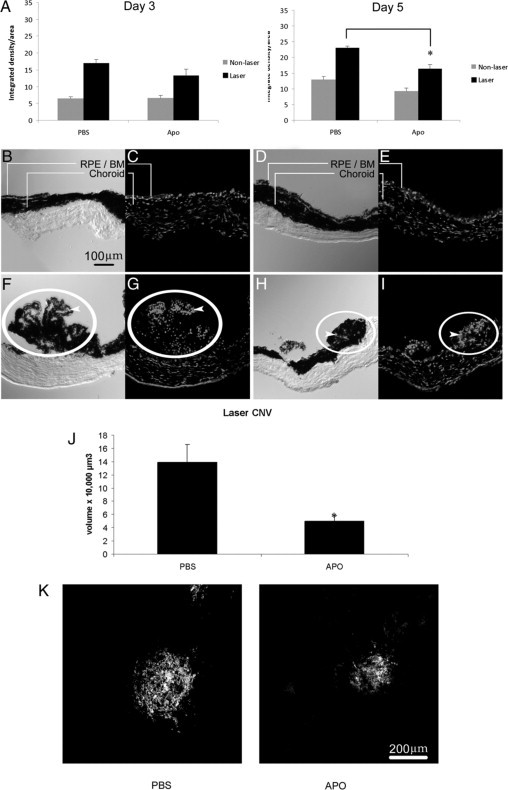
Inhibition of NADPH oxidase decreases laser-induced CNV in mice. A: ROS levels in lasered and unlasered sections of RPE-choroid in three month-old C57BL/6 mice. ROS levels were determined by measuring DHE fluorescence. Data are presented as integrated density per area (mean ± SEM [10 μmol/L]) using ImageJ 1.43 for analysis. *P < 0.05, laser-treated, PBS versus apocynin (Apo), day five. B–I: Representative images of retinal/choroidal cross sections from mice unlasered (B–E) or lasered (F–I) and administered apocynin (D, E, H and I) or PBS (B, C, F and G). Sections are oriented with CNV and/or RPE-choroid at the top and the sclera below. Phase images (B, D, F, and H) were taken of corresponding DHE-stained images (C, E, G, and I). Note the qualitatively increased number of DHE-stained cells in lasered sections (bottom row) compared with respective images (top row) and qualitatively reduced number of DHE-stained cells in lasered sections from apocynin-treated (I) compared with PBS-treated (G) mice. DHE fluorescence is found in mainly nonpigmented cells in PBS-treated lasered mice (compare arrowheads in phase [F] and DHE fluorescence [G]) and in both pigmented and nonpigmented cells in both PBS-treated (F) and apocynin-treated lasered mice (compare arrowheads in phase [H] and DHE fluorescence [I]). J: C57BL/6 mice (three months old) were treated with apocynin (10 mg/kg/day) or PBS for five days preceding and five days after photocoagulation by laser treatment. Choroidal flat mounts of lasered areas were stained with lectin and 1-μm horizontal sections were captured with confocal microscopy, measured, and analyzed as described (*P = 0.007, analysis of variance). K: Representative images of maximum projections of CNV from choroidal flat mounts are shown for each condition: left, PBS-treated; right, apocynin-treated. Scale bar = 200 μm. RPE/BM, retinal pigment epithelium/Bruch's membrane; laser CNV, choroidal neovascular lesion induced with laser injury.
To confirm the role of NADPH oxidase in CNV, we subjected mice lacking the p47phox subunit and hence a functional NADPH oxidase to laser-induced injury. First, we examined the morphology of untreated whole-eye cross sections of p47phox−/− and wild-type counterparts that were stained with hematoxylin and eosin at the Histology Research Core Unit at the University of North Carolina (Figure 8). Compared with a wild-type eye, the p47phox−/− eye had no discernible structural differences in the layers of the neurosensory retina, RPE, or choroid. In lasered regions of p47phox−/− mice, there was less DHE fluorescence at day 3 (P < 0.001, analysis of variance) and day 5 (P < 0.05, analysis of variance) (Figure 9A) compared with that for wild-type mice. A representative image (Figure 9) shows qualitatively increased DHE fluorescence in sections from lasered (Figure 9, F–I) versus unlasered (Figure 9, B–E) mice. Qualitatively fewer DHE-stained cells were present in sections from lasered p47phox knockout (Figure 9I) compared with wild-type (Figure 9G) mice. In wild-type lasered eyes, DHE fluorescence was seen predominantly in nonpigmented (arrowheads, compare Figure 9, F and G) and some pigmented cells, whereas in p47phox−/− eyes, DHE fluorescence was less and seen in both pigmented and nonpigmented cells (compare Figure 9, H and I). DHE fluorescence was not significantly different in unlasered sections at either time point (P > 0.05, analysis of variance). CNV volume measured from lectin-stained choroidal flat mounts was reduced in lasered p47phox−/− compared with wild-type mice at day 5 (P = 0.036, analysis of variance) (Figure 9J) and as noted in the underlying image (Figure 9K) of maximum projections of lectin-stained CNV in choroidal flat mounts).
Figure 8.
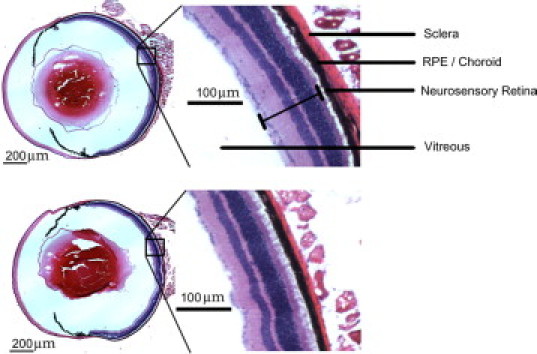
Ocular histology of p47phox−/− compared with wild-type mice. Representative images of H&E stains of wild-type (top) and p47phox−/− whole eyes (bottom). Inset: magnified retinal layers. Scale bar = 200 μm.
Figure 9.
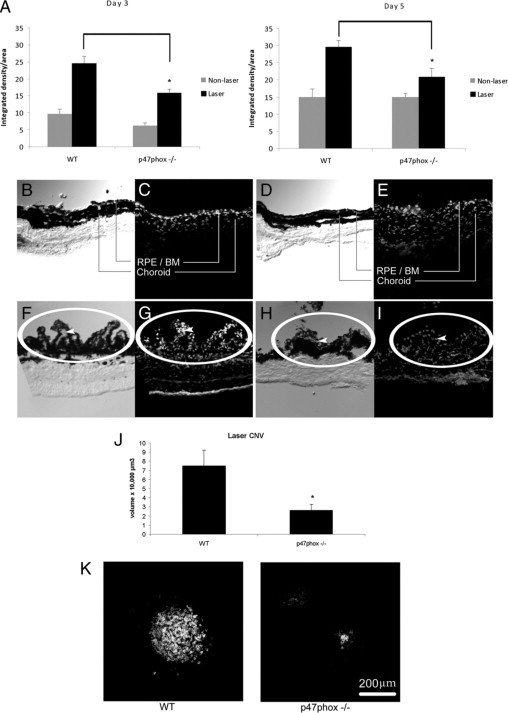
Deficiency in the p47phox subunit of NADPH oxidase decreases laser-induced CNV in mice. A: Three-month-old wild-type and p47phox−/− mice received photocoagulation by laser treatment. ROS levels in lasered and unlasered sections of RPE-choroid were determined by measuring DHE fluorescence. Data are presented as integrated density per area (mean ± SEM, DHE [10 μmol/L] using ImageJ for analysis). *Analysis of variance, P < 0.05, day three and day five, laser-treated, wild-type versus p47phox−/− mice. B–I: Representative images of retinal-choroidal cross sections from mice untreated (top row) or laser-treated (bottom row). Phase images (B, D, F, and H) were taken of corresponding DHE-stained images (C, E, G, and I). Sections from wild-type mice (B, C, F and G) or p47phox−/− mice (D, E, H and I). Sections are oriented with CNV and/or the RPE-choroid at the top and sclera (nonpigmented layer) at the bottom. An edge artifact is noted at the inferior boundary of the sclera in G. Note the qualitatively increased number of DHE-stained cells in lasered sections (F–I) compared with unlasered respective comparisons (B–E) and qualitatively fewer DHE-stained cells in lasered sections from p47phox−/− mice (I) compared with wild-type mice (G). DHE fluorescence is found in mainly nonpigmented cells in wild-type lasered mice (compare arrowheads in phase [F] and DHE fluorescence [G]) and in both pigmented and nonpigmented cells in p47phox−/− lasered mice (compare phase [H] and DHE fluorescence [I]). J: Wild-type and p47phox−/− mice received photocoagulation by laser treatment 5 days earlier. Choroidal flat mounts of lasered areas were stained with lectin and 1-μm horizontal sections were captured with confocal microscopy, measured, and analyzed as described (analysis of variance, *P = 0.036, laser treated wild-type versus p47phox−/− mice). K: Representative images of maximum projections of lectin-stained CNV in choroidal flat mounts shown below: left, wild-type; right, p47phox−/−. Scale bar = 200 μm. WT, wild-type; RPE/BM, retinal pigment epithelium/Bruch's membrane; laser CNV, choroidal neovascular lesion induced with laser injury.
NADPH oxidase can be activated in several different cell types involved in the development of CNV, including choroidal ECs, RPE, and macrophages.25 RPE express VEGF in response to age-related stimuli, including oxidative stress,38 and VEGF plays an important role in CNV in human AMD.39 To examine the role of RPE, we cultured human ARPE-19 cells in the presence of NADPH oxidase inhibitors, apocynin or DPI, and obtained purified RNA to measure VEGF splice variants by real-time PCR. Compared with control PBS treatment, both apocynin and DPI significantly reduced expression of all VEGF splice variants (Figure 10A). This result suggests that NADPH oxidase activation in RPE plays a role in the expression of VEGF.
Figure 10.
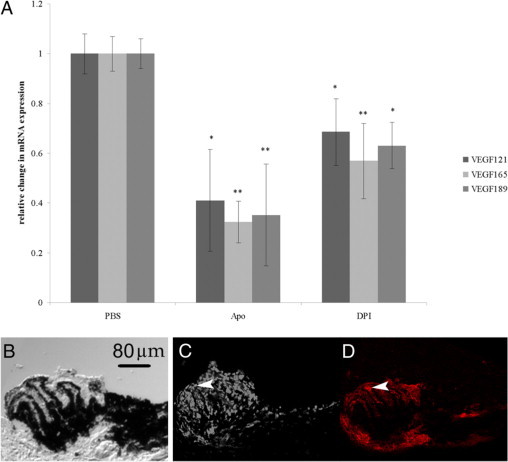
A: Inhibition of ROS generation decreases expression of VEGF splice variants in human RPE. Total RNA was extracted from human ARPE treated with PBS, apocynin (Apo), or DPI. Changes in gene expression were detected by real-time PCR. Data are presented as mean ± SEM change in gene expression normalized to 18s RNA levels and relative to PBS control normalized to 18s levels. *P < 0.01; **P < 0.001 versus PBS control, n = 10 (analyzed with REST software). Apo, apocynin. B–D: Panels of cross sections through the CNV lesion 5 days after laser treatment in wild-type mice. Sections taken in 10-μm increments are shown. B: Phase image showing pigmented and nonpigmented cells (neurosensory retina is removed and the sclera is toward the bottom of the image) of a fresh tissue section that had been stained for DHE fluorescence and imaged previously (C). D: Adjacent section through the same CNV lesion fixed and stained with lectin to visualize vascular cells. Arrowheads indicate lectin-stained tissue in D corresponding to DHE-positive cells in C. Scale bar = 80 μm.
We also labeled tissue sections of CNV from wild-type mice that had received laser injury 5 days previously. Fresh sections were stained with DHE to detect ROS. Phase images of the same sections revealed mainly nonpigmented and some pigmented cells that were DHE-positive. In adjacent fixed sections, lectin-stained vascular cells seemed to correspond to nonpigmented DHE positive cells (Figure 10, B–D).
Discussion
Previous studies have reported that external stimuli leading to ROS generation15,16,40,41 contribute to the pathophysiology in AMD. However, much of the evidence in vivo has supported a role of ROS in early or atrophic forms of AMD or retinal degeneration.16,42 Our data provide evidence that activation of NADPH oxidase in vivo is also important in neovascular AMD and that subunits p47phox and Rac1 may play complementary roles in the complex development of CNV.
Laboratory research using the well accepted laser-induced CNV model provided evidence that VEGF was essential in the development of CNV, and clinical experience has since confirmed the importance of VEGF in human neovascular AMD.43 We found that VEGF165 stimulated the generation of ROS from cultured choroidal ECs. This occurred through the activation of Rac1, which triggered downstream activation of NADPH oxidase. A pathway has been characterized in human umbilical vein endothelial cells, in which engagement of the VEGF receptor 2 stimulates the Rac1 guanine nucleotide exchange factor, Vav2.24,44 In pulmonary microvascular ECs, it was demonstrated that VEGF led to Rac1 activation, stimulating the subsequent activation of NADPH oxidase.45 Active Rac1 facilitates EC migration through the formation of lamellipodia and directed cell motility.46 Further, activation of Rac1 in choroidal ECs was found to be essential for choroidal ECs to migrate across a monolayer of RPE toward a VEGF gradient.23 In this study, we showed that Rac1-induced NADPH oxidase activation and ROS generation enhanced migration of choroidal ECs toward a VEGF gradient. This result suggests that Rac1 uses both its well known effects on the cytoskeleton and its ability to induce ROS-dependent signaling to maximally regulate migration.
The in vivo evidence using the laser-induced model in the current study provides support that activation of NADPH oxidase contributes to the formation of CNV. NADPH oxidase becomes activated as a result of aggregation of membrane bound and cytosolic subunits.25 Heumüller et al47 have reported that apocynin works mainly as an antioxidant in cultured ECs, because it requires ROS to become active, and ECs lack myeloperoxidases that activate apocynin. However, in vivo, apocynin is a strong NADPH oxidase inhibitor, believed in part to be due to its activation by myeloperoxidases in cells adjacent to ECs, such as macrophages.25 We found that inhibition of NADPH oxidase with apocynin or knockout of subunit p47phox was associated with reduced DHE fluorescence in association with a reduction in CNV volume after laser injury.
Activation of NADPH oxidase can occur in multiple cells involved in the development of CNV including choroidal ECs, RPE, and macrophages.48–50 VEGF is released from RPE and macrophages and has been localized in CNV lesions.51 ROS exposure to cultured RPE enhanced VEGF release.52 Li et al50 reported that knockdown of the NADPH oxidase subunit p22phox in RPE cells inhibited laser-induced CNV in mice. These investigators proposed that p22phox knockdown reduced ROS generation from RPE-derived NADPH oxidase and therefore led to reduced VEGF production. In agreement with this study, we found significant inhibition of VEGF mRNA expression with two different inhibitors of NADPH oxidase, suggesting that NADPH oxidase does stimulate enhanced VEGF production from RPE cells. Investigators have also reported an influx of F4/80+ macrophages in laser-induced CNV that peaked at day 3 and then were reduced when CNV developed.30 We found that DHE fluorescence appeared in both pigmented and nonpigmented cells within CNV lesions, and some cells also appeared to stain with lectin. Qualitatively, fewer DHE-stained cells appeared in CNV lesions of both apocynin-treated mice and p47phox−/− mice compared with the respective controls, and this result corresponded to measured DHE fluorescence in these groups. Collectively, these data suggest roles for multiple cells in the development of CNV. Further studies are necessary to determine the roles of each individual cell type in the regulation of CNV.
The pathology of AMD is complex and occurs in the outer retina in the region of the RPE, Bruch's membrane, and the choroid. Neovascular AMD accounts for 90% of the legal blindness that occurs in AMD.38 Vision loss from neovascular AMD occurs with CNV, in which blood vessels originating from the choroidal vasculature and choriocapillaris grow into the neurosensory retina. Activated choroidal ECs must migrate through Bruch's membrane toward the RPE. There is evidence that VEGF released from the basal RPE is an important chemoattractant for migrating choroidal ECs53,54 and that the RPE cell junctions break down with RPE-EC contact.55 It is believed that breakdown of the RPE barrier permits VEGF to enter the neurosensory retinal space and attract choroidal ECs to form neurosensory retinal CNV. Neurosensory retinal CNV is associated with disruption of the architecture of the neurosensory retina and leakage of fluid and blood substances into the neurosensory retina, all of which reduce visual acuity. Clinically, vision is restored to some degree by reduction of fluid leakage into the neurosensory retinal layers, and the current leading pharmacological intervention is inhibition of the permeability and growth factor, VEGF.56 It is known that VEGF is increased in laser-induced injury models and in human specimens of CNV.39 Treatment with anti-VEGF agents has improved the management of neovascular AMD but is only effective in approximately 40% of eyes and requires repeated intraocular injections. There is also concern of possible toxicity from inhibiting the beneficial57–61 while treating the pathological functions of VEGF. Intervening downstream of VEGF, through NADPH oxidase or Rac1, may provide more effective and safer therapies.
In summary, we provide evidence that NADPH oxidase plays a role in neovascular AMD and may affect several cell types involved in the development of CNV. A plausible scenario is that age-related stresses induce RPE to express VEGF. Soluble VEGF activates and acts as a chemoattractant to direct migration of choroidal ECs.23 The activation of Rac1 in choroidal ECs is followed by downstream activation of NADPH oxidase and generation of ROS. The ROS from the choroidal ECs may then induce RPE or macrophages to express VEGF, thereby leading to a positive feedback loop and contributing to the pathology of neovascular AMD.
Acknowledgements
We acknowledge Sarah Moyer, C.R.A., C.O.T., and Erika Wittchen, Ph.D., for help with images and the Histology Research Core Unit (University of North Carolina) for staining of sections from p47phox−/− and wild-type mice. We acknowledge the University of North Carolina Michael Hooker Microscopy Facility for acquisition of choroidal flat mounts and the University of North Carolina Microscopy Services Laboratory for measurement of dihydroethidium fluorescence.
Footnotes
Supported by the National Institutes of Health (grants R01-EY015130 to M.E.H., principle investigator [PI]; R01-EY017011 to M.E.H., PI; P01-HL080166 to K.B., PI; HL-57352 to M.S.R. and N.M., PIs; and AG-024282 to M.S.R.), Research to Prevent Blindness, and the American Cancer Society (grant PF-09-119-01-CSM to E.M.-B.).
E.M.-B. and J.H. contributed equally to this work as first authors.
References
- 1.Tielsch JA. Vision Problems in the U.S.: A Report on Blindness and Vision Impairment in Adults Age 40 and Older. Prevent Blindness America; Schaumburg, IL: 1994. pp. 1–20. [Google Scholar]
- 2.Augood CA, Vingerling JR, de Jong PTVM, Chakravarthy U, Seland J, Soubrane G, Tomazzoli L, Topouzis F, Bentham G, Rahu M, Vioque J, Young IS, Fletcher AE. Prevalence of age-related maculopathy in older Europeans: the European Eye Study (EUREYE) Arch Ophthalmol. 2006;124:529–535. doi: 10.1001/archopht.124.4.529. [DOI] [PubMed] [Google Scholar]
- 3.Mitchell P, Smith W, Attebo K, Wang JJ. Prevalence of age-related maculopathy in Australia. The Blue Mountains Eye Study. Ophthalmology. 1995;102:1450–1460. doi: 10.1016/s0161-6420(95)30846-9. [DOI] [PubMed] [Google Scholar]
- 4.Friedman DS, O'Colmain BJ, Muñoz B, Tomany SC, McCarty C, de Jong PT, Nemesure B, Mitchell P, Kempen J, Eye Diseases Prevalence Research Group Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–572. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- 5.Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration—emerging pathogenetic and therapeutic concepts. Ann Med. 2006;38:450–471. doi: 10.1080/07853890600946724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maller J, George S, Purcell S, Fagerness J, Altshuler D, Daly MJ, Seddon JM. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet. 2006;38:1055–1059. doi: 10.1038/ng1873. [DOI] [PubMed] [Google Scholar]
- 8.Li M, Atmaca-Sonmez P, Othman M, Branham KEH, Khanna R, Wade MS, Li Y, Liang L, Zareparsi S, Swaroop A, Abecasis GR. CFH haplotypes without the Y402H coding variant show strong association with susceptibility to age-related macular degeneration. Nat Genet. 2006;38:1049–1054. doi: 10.1038/ng1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 10.Edwards AO, Ritter RI, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 11.Rivera A, Fisher SA, Fritsche LG, Keilhauer CN, Lichtner P, Meitinger T, Weber BHF. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet. 2005;14:3227–3236. doi: 10.1093/hmg/ddi353. [DOI] [PubMed] [Google Scholar]
- 12.Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch's membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–732. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- 13.Reynolds R, Hartnett ME, Atkinson JP, Giclas PC, Rosner B, Seddon JM. Plasma complement components and activation fragments are associated with age-related macular degeneration genotypes and phenotypes. Invest Ophthalmol Vis Sci. 2009;50:5818–5827. doi: 10.1167/iovs.09-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seddon JM, Francis PJ, George S, Schultz DW, Rosner B, Klein ML. Association of CFH Y402H and LOC387715 A69S with progression of age-related macular degeneration. JAMA. 2007;297:1793–1800. doi: 10.1001/jama.297.16.1793. [DOI] [PubMed] [Google Scholar]
- 15.Margrain TH, Boulton M, Marshall J, Sliney DH. Do blue light filters confer protection against age-related macular degeneration? Prog Retin Eye Res. 2004;23:523–531. doi: 10.1016/j.preteyeres.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 16.Espinosa-Heidmann DG, Suner IJ, Catanuto P, Hernandez EP, Marin-Castano ME, Cousins SW. Cigarette smoke-related oxidants and the development of sub-RPE deposits in an experimental animal model of dry AMD. Invest Ophthalmol Vis Sci. 2006;47:729–737. doi: 10.1167/iovs.05-0719. [DOI] [PubMed] [Google Scholar]
- 17.Ni J, Yuan X, Gu J, Yue X, Gu X, Nagaraj RH, Crabb JW, Clinical Genomic and Proteomic AMD Study Group Plasma protein pentosidine and carboxymethyllysine, biomarkers for age-related macular degeneration. Mol Cell Proteomics. 2009;8:1921–1933. doi: 10.1074/mcp.M900127-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antioxidant status and neovascular age-related macular degeneration. Eye Disease Case-Control Study Group. Arch Ophthalmol. 1993;111:104–109. doi: 10.1001/archopht.1993.01090010108035. [DOI] [PubMed] [Google Scholar]
- 19.Age-Related Eye Disease Study Research Group A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, β carotene, and zinc for age-related macular degeneration and vision loss: AREDS Report No. 8. Arch Ophthalmol. 2001;119:1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sreekumar PG, Kannan R, de Silva AT, Burton R, Ryan SJ, Hinton DR. Thiol regulation of vascular endothelial growth factor-A and its receptors in human retinal pigment epithelial cells. Biochem Biophys Res Commun. 2006;346:1200–1206. doi: 10.1016/j.bbrc.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 21.Imamura Y, Noda S, Hashizume K, Shinoda K, Yamaguchi M, Uchiyama S, Shimizu T, Mizushima Y, Shirasawa T, Tsubota K. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: a model of age-related macular degeneration. Proc Natl Acad Sci USA. 2006;103:11282–11287. doi: 10.1073/pnas.0602131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou J, Jang YP, Kim SR, Sparrow JR. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci USA. 2006;103:16182–16187. doi: 10.1073/pnas.0604255103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peterson LJ, Wittchen ES, Geisen P, Burridge K, Hartnett ME. Heterotypic RPE-choroidal endothelial cell contact increases choroidal endothelial cell transmigration via PI 3-kinase and Rac1. Exp Eye Res. 2007;84:737–744. doi: 10.1016/j.exer.2006.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garrett TA, Van Buul JD, Burridge K. VEGF-induced Rac1 activation in endothelial cells is regulated by the guanine nucleotide exchange factor Vav2. Exp Cell Res. 2007;313:3285–3297. doi: 10.1016/j.yexcr.2007.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ushio-Fukai M. Redox signaling in angiogenesis: role of NADPH oxidase. Cardiovasc Res. 2006;71:226–235. doi: 10.1016/j.cardiores.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 26.Geisen P, McColm JR, King BM, Hartnett ME. Characterization of barrier properties and inducible VEGF expression of several types of retinal pigment epithelium in medium-term culture. Curr Eye Res. 2006;31:739–748. doi: 10.1080/02713680600837408. [DOI] [PubMed] [Google Scholar]
- 27.Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST©) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucl Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jackson SH, Gallin JI, Holland SM. The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med. 1995;182:751–758. doi: 10.1084/jem.182.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Samtani S, Amaral J, Campos MM, Fariss RN, Becerra SP. Doxycycline-mediated inhibition of choroidal neovascularization. Invest Ophthalmol Vis Sci. 2009;50:5098–5106. doi: 10.1167/iovs.08-3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakurai E, Anand A, Ambati BK, van Rooijen N, Ambati J. Macrophage depletion inhibits experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:3578–3585. doi: 10.1167/iovs.03-0097. [DOI] [PubMed] [Google Scholar]
- 31.Espinosa-Heidmann DG, Caicedo A, Hernandez EP, Csaky KG, Cousins SW. Bone marrow-derived progenitor cells contribute to experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:4914–4919. doi: 10.1167/iovs.03-0371. [DOI] [PubMed] [Google Scholar]
- 32.Vendrov AE, Hakim ZS, Madamanchi NR, Rojas M, Madamanchi C, Runge MS. Atherosclerosis is attenuated by limiting superoxide generation in both macrophages and vessel wall cells. Arterioscler Thromb Vasc Biol. 2007;27:2714–2721. doi: 10.1161/ATVBAHA.107.152629. [DOI] [PubMed] [Google Scholar]
- 33.Bokoch GM, Diebold BA. Current molecular models for NADPH oxidase regulation by Rac GTPase. Blood. 2002;100:2692–2695. doi: 10.1182/blood-2002-04-1149. [DOI] [PubMed] [Google Scholar]
- 34.Irani K, Goldschmidt-Clermont PJ. Ras, superoxide and signal transduction. Biochem Pharmacol. 1998;55:1339–1346. doi: 10.1016/s0006-2952(97)00616-3. [DOI] [PubMed] [Google Scholar]
- 35.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 36.Ushio-Fukai M, Alexander RW. Reactive oxygen species as mediators of angiogenesis signaling. Role of NAD(P)H oxidase. Mol Cell Biochem. 2004;264:85–97. doi: 10.1023/b:mcbi.0000044378.09409.b5. [DOI] [PubMed] [Google Scholar]
- 37.Saito Y, Geisen P, Uppal A, Hartnett ME. Inhibition of NAD(P)H oxidase reduces apoptosis and avascular retina in an animal model of retinopathy of prematurity. Mol Vis. 2007;13:840–853. [PMC free article] [PubMed] [Google Scholar]
- 38.Penn JS, Madan A, Caldwell RB, Bartoli M, Caldwell RW, Hartnett ME. Vascular endothelial growth factor in eye disease. Prog Retin Eye Res. 2008;27:331–371. doi: 10.1016/j.preteyeres.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frank RN. Growth factors in age-related macular degeneration: pathogenic and therapeutic implications. Ophthalmic Res. 1997;29:341–353. doi: 10.1159/000268032. [DOI] [PubMed] [Google Scholar]
- 40.Dunaief JL. Iron induced oxidative damage as a potential factor in age-related macular degeneration: the Cogan Lecture. Invest Ophthalmol Vis Sci. 2006;47:4660–4664. doi: 10.1167/iovs.06-0568. [DOI] [PubMed] [Google Scholar]
- 41.Fujihara M, Nagai N, Sussan TE, Biswal S, Handa JT. Chronic cigarette smoke causes oxidative damage and apoptosis to retinal pigmented epithelial cells in mice. PLoS One. 2008;3:e3119. doi: 10.1371/journal.pone.0003119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haruta M, Bush RA, Kjellstrom S, Vijayasarathy C, Zeng Y, Le YZ, Sieving PA. Depleting Rac1 in mouse rod photoreceptors protects them from photo-oxidative stress without affecting their structure or function. Proc Natl Acad Sci USA. 2009;106:9397–9402. doi: 10.1073/pnas.0808940106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferrara N, Damico L, Shams N, Lowman H, Kim R. Development of ranibizumab, an anti-vascular endothelial growth factor antigen binding fragment, as therapy for neovascular age-related macular degeneration. Retina. 2006;26:859–870. doi: 10.1097/01.iae.0000242842.14624.e7. [DOI] [PubMed] [Google Scholar]
- 44.Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the β-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol. 2006;8:1223–1234. doi: 10.1038/ncb1486. [DOI] [PubMed] [Google Scholar]
- 45.Monaghan-Benson E, Burridge K. The regulation of VEGF-induced microvascular permeability requires Rac and ROS. J Biol Chem. 2009;284:25602–25611. doi: 10.1074/jbc.M109.009894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 47.Heumüller S, Wind S, Barbosa-Sicard E, Schmidt HHHW, Busse R, Schroder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 48.Espinosa-Heidmann DG, Reinoso MA, Pina Y, Csaky KG, Caicedo A, Cousins SW. Quantitative enumeration of vascular smooth muscle cells and endothelial cells derived from bone marrow precursors in experimental choroidal neovascularization. Exp Eye Res. 2005;80:369–378. doi: 10.1016/j.exer.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 49.Ushio-Fukai M. VEGF signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2007;9:731–739. doi: 10.1089/ars.2007.1556. [DOI] [PubMed] [Google Scholar]
- 50.Li Q, Dinculescu A, Shan Z, Miller R, Pang J, Lewin AS, Raizada MK, Hauswirth WW. Downregulation of p22phox in retinal pigment epithelial cells inhibits choroidal neovascularization in mice. Mol Ther. 2008;16:1688–1694. doi: 10.1038/mt.2008.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nozaki M, Raisler BJ, Sakurai E, Sarma JV, Barnum SR, Lambris JD, Chen Y, Zhang K, Ambati BK, Baffi JZ, Ambati J. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci USA. 2006;103:2328–2333. doi: 10.1073/pnas.0408835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thurman JM, Renner B, Kunchithapautham K, Ferreira VP, Pangburn MK, Ablonczy Z, Tomlinson S, Holers VM, Rohrer B. Oxidative stress renders retinal pigment epithelial cells susceptible to complement-mediated injury. J Biol Chem. 2009;284:16939–16947. doi: 10.1074/jbc.M808166200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blaauwgeers HGT, Holtkamp GM, Rutten H, Witmer AN, Koolwijk P, Partanen TA, Alitalo K, Kroon ME, Kijlstra A, van Hinsbergh VWM, Schlingemann RO. Polarized vascular endothelial growth factor secretion by human retinal pigment epithelium and localization of vascular endothelial growth factor receptors on the inner choriocapillaris: evidence for a trophic paracrine relation. Am J Pathol. 1999;155:421–428. doi: 10.1016/S0002-9440(10)65138-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Geisen P, McColm JR, Hartnett ME. Choroidal endothelial cells transmigrate across the retinal pigment epithelium but do not proliferate in response to soluble vascular endothelial growth factor. Exp Eye Res. 2006;82:608–619. doi: 10.1016/j.exer.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 55.Hartnett ME, Lappas A, Darland D, McColm JR, Lovejoy S, D'Amore PA. Retinal pigment epithelium and endothelial cell interaction causes retinal pigment epithelial barrier dysfunction via a soluble VEGF-dependent mechanism. Exp Eye Res. 2003;77:593–599. doi: 10.1016/s0014-4835(03)00189-1. [DOI] [PubMed] [Google Scholar]
- 56.Rosenfeld PJ, Moshfeghi AA, Puliafito CA. Optical coherence tomography findings after an intravitreal injection of bevacizumab (avastin) for neovascular age-related macular degeneration. Ophthalmic Surg Lasers Imaging. 2005;36:331–335. [PubMed] [Google Scholar]
- 57.Nishijima K, Ng YS, Zhong L, Bradley J, Schubert W, Jo N, Akita J, Samuelsson SJ, Robinson GS, Adamis AP, Shima DT. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007;171:53–67. doi: 10.2353/ajpath.2007.061237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oosthuyse B, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, Dorpe JV, Hellings P, Gorselink M, Heymans S, Theilmeier G, Dewerchin M, Laudenbach V, Vermylen P, Raat H, Acker T, Vleminckx V, Bosch LVD, Cashman N, Fujisawa H, Drost MR, Sciot R, Bruyninckx F, Hicklin DJ, Ince C, Gressens P, Lupu F, Plate KH, Robberecht W, Herbert JM, Collen D, Carmeliet P. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet. 2001;28:131–138. doi: 10.1038/88842. [DOI] [PubMed] [Google Scholar]
- 59.Maharaj ASR, Saint-Geniez M, Maldonado AE, D'Amore PA. Vascular endothelial growth factor localization in the adult. Am J Pathol. 2006;168:639–648. doi: 10.2353/ajpath.2006.050834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Korte GE, Reppucci V, Henkind P. RPE destruction causes choriocapillary atrophy. Invest Ophthalmol Vis Sci. 1984;25:1135–1145. [PubMed] [Google Scholar]
- 61.Marneros AG, Fan J, Yokoyama Y, Gerber HP, Crouch RK, Olsen BR. Vascular endothelial growth factor expression in the retinal pigment epithelium is essential for choriocapillaris development and visual function. Am J Pathol. 2005;167:1451–1459. doi: 10.1016/S0002-9440(10)61231-X. [DOI] [PMC free article] [PubMed] [Google Scholar]