

Abstract
During the first trimester of pregnancy, the uterine spiral arteries are remodeled, creating heavily dilated conduits that lack maternal vasomotor control but allow the placenta to meet an increasing requirement for nutrients and oxygen. To effect permanent vasodilatation, the internal elastic lamina and medial elastin fibers must be degraded. In this study, we sought to identify the elastolytic proteases involved in this process. Primary first-trimester cytotrophoblasts (CTBs) derived from the placenta exhibited intracellular and membrane-associated elastase activity; membrane-associated activity was primarily attributable to matrix metalloproteinases (MMP). Indeed, Affymetrix microarray analysis and immunocytochemistry implicated MMP-12 (macrophage metalloelastase) as a key mediator of elastolysis. Cultured human aortic smooth muscle cells (HASMCs) exhibited constitutive membrane-associated elastase activity and inducible intracellular elastase activity; these cells also expressed MMP-12 protein. Moreover, a specific inhibitor of MMP-12 significantly reduced CTB- and HASMC-mediated elastolysis in vitro, to 31.7 ± 10.9% and 23.3 ± 8.7% of control levels, respectively. MMP-12 is expressed by both interstitial and endovascular trophoblasts in the first-trimester placental bed and by vascular SMCs (VSMCs) in remodeling spiral arteries. Perfusion of isolated spiral artery segments with CTB-conditioned medium stimulated MMP-12 expression in medial VSMCs. Our data support a model in which trophoblasts and VSMCs use MMP-12 cooperatively to degrade elastin during vascular remodeling in pregnancy, with the localized release of elastin peptides and CTB-derived factors amplifying elastin catabolism.
Transformation of the uterine spiral arteries during the first 20 weeks of gestation ensures that a constant supply of blood is delivered to the developing placenta, at an optimal rate of flow.1–3 This allows the placenta to meet an increasing requirement for nutrients and oxygen and enables the developing fetus to attain its growth potential. The remodeling process leads to vessel dilatation, loss of spirality, and decreased vasoactivity, allowing a nonpulsatile low-pressure supply of blood to be delivered to placental villi at the maternofetal interface. Early alterations in arterial structure include endothelial vacuolation, hypertrophy of vascular smooth muscle cells (VSMCs), and disruption of medial smooth muscle layers, which occur in the absence of fetal-derived trophoblast and correlate with perivascular accumulation of macrophages and uterine natural killer (uNK) cells.4,5 After colonization of the uterine decidua and myometrium by extravillous cytotrophoblast (EVT), endothelial cells and VSMCs are lost from the arterial wall and replaced by trophoblast embedded in a fibrinoid matrix. Remodeling is regulated in a spatial and temporal manner, such that the successive steps of trophoblast adherence, intravasation, fibrinoid deposition, and mural incorporation are effected without any loss in vessel integrity. A complex and highly orchestrated combination of vascular cell apoptosis, dedifferentiation, and matrix breakdown is probably required to achieve this alteration in vessel wall structure.5–9
Two distinct populations of EVT originate from anchoring placental villi and contribute to vessel transformation.10,11 Interstitial EVT invade the uterine wall, migrating through the decidua and myometrium to adopt a perivascular position. Endovascular EVT enter the lumen of the spiral arteries and migrate as far as the first third of the myometrium, colonizing the arterial wall from within. Impaired arterial remodeling is distinguished by shallow EVT invasion, decreased numbers of EVT, and the persistence of muscular, narrow-bore arteries, and is associated with second trimester miscarriage,12 preterm labor,13 pre-eclampsia,14 and fetal growth restriction.15
To effect a permanent increase in vessel diameter it is crucial that elastin fibers within each artery are catabolized, eliminating their capacity for stretch and recoil. Myometrial segments of the spiral arteries possess an internal elastic lamina (IEL), and the musculo-elastic media of both decidual and myometrial arteries is rich in elastic fibers.16,17 During pregnancy, EVT traverse the IEL during mural incorporation,18 thus it is highly likely that they possess elastase activity: indeed, first-trimester EVT synthesize and secrete the elastolytic proteases matrix metalloproteinase-2 (MMP-2), MMP-7, MMP-9, cathepsin B, and cathepsin L.19,20 Although both uNK cells and macrophages produce enzymes capable of elastolysis,5 uNK cells are not abundant in myometrium,21 and elastin breakdown is associated with the presence of endovascular EVT17 rather than macrophages.22 Previous studies have demonstrated that the availability of nitric oxide (NO) can influence protease expression and activity,23–26 and we have shown NO to be an important regulator of trophoblast function.27–29 As dysregulation of NO production has been implicated in the pathogenesis of pre-eclampsia and intrauterine growth restriction (IUGR),30–32 NO availability may regulate the process of arterial remodeling by controlling trophoblast elastolysis.
Rodent models of atherosclerosis have highlighted a role for VSMC-derived cathepsins as mediators of IEL breakdown during lesion formation,33 demonstrating that the arterial wall may be a potential source of elastases. Similarly, caspase-2, −3, and −7 derived from apoptotic VSMCs have been implicated as mediators of elastin breakdown.34 Thus, during the process of spiral artery transformation, resident VSMCs may also be stimulated to produce elastase(s) in response to pregnancy hormones, trophoblast invasion, or soluble factors released by cells within the placental bed. In this study we have investigated the origin and identity of the proteases involved in mediating elastin breakdown during spiral artery remodeling.
Materials and Methods
Reagents
Caspase inhibitor zVAD-fmk and cathepsin inhibitor Z-Phe-Gly-NHO-Bz-pOMe were from Calbiochem (San Diego, CA); MMP inhibitor N-Isobutyl-N-(4-methoxyphenylsulfonyl)-glycyl hydroxamic acid (NNGH) was from Biomol International (Exeter, UK); urokinase plasminogen activator (uPA) inhibitor uPA-STOP was from American Diagnostica Inc. (Stamford, CT); in situ cell death detection kit (TUNEL) was from Roche (Lewes, UK); CellTracker CM-Dil was from Molecular Probes (distributed by Invitrogen Ltd, Paisley, UK); recombinant human TRAIL was from PeproTech (London, UK); inducible NOS (iNOS) inhibitor 1400W was from Axxora (Nottingham, UK); porcine pancreatic elastase, N-succinyl-(L-alanine)3-p-nitroanilide, elastin from bovine neck ligament, Congo-Red labeled elastin, Nω-Nitro-L-arginine methyl ester hydrochloride (L-NAME hydrochloride), phenylmethanesulfonyl fluoride (PMSF) were from Sigma-Aldrich (Poole, UK); tissue culture medium, glutamine, antibiotics were from Cambrex (Wokingham, UK); fetal bovine serum (FBS) was from Gibco (distributed by Invitrogen Ltd, Paisley, UK); OCT embedding medium was from Raymond A. Lamb (London, UK); Matrigel was from BD Discovery Labware (Bedford, MA); mouse anti-human cytokeratin-7 monoclonal antibody (clone OV-TL 12/30), mouse anti-human α-smooth muscle actin monoclonal antibody (clone 1A4), mouse anti-human CD31 monoclonal antibody, universal negative control IgG (IgG1, IgG2a, IgG2b, IgG3, and IgM; 20 μg/ml of each isotype, 100 μg/ml total concentration), biotinylated rabbit anti-mouse, goat anti-mouse and swine anti-rabbit secondary antibodies, streptavidin-FITC were from Dako (Glostrup, Denmark); mouse anti-human MMP-12 monoclonal antibody (catalytic domain) was from R&D Systems (Minneapolis, MN); rabbit anti-human MMP-12 monoclonal antibody (carboxyterminal; EP1261Y) and mouse anti-human HLA-G monoclonal antibody were from Abcam (Cambridge, UK); Vectashield mounting medium was from Vector Laboratories Inc. (Burlingame, CA); Elastolux intracellular elastase activity assay was from OncoImmunin (Gaithersburg, MD). U133A/B GeneChips were obtained from Affymetrix (Affymetrix Inc., Santa Clara, CA). Labeling reagents for GeneChips were purchased from Enzo Life Sciences (Enzo, Farmingdale, NY). The specific MMP-12 inhibitor 470.1 (Ki 0.19 nmol/L; purity >98%) was kindly donated by Dr. V. Dive (Compound 1 in Ref. 35).
Cell Culture
Primary cytotrophoblasts (CTBs) were isolated as previously described36 and were cultured in a 1:1 ratio DMEM: Ham's F12 medium supplemented with FBS (10%), glutamine (2 mmol/L), penicillin (100 units/ml), and streptomycin (0.1 mg/ml). CTBs were grown on Matrigel-coated flasks (diluted 1:10 in serum-free medium) to promote a more advanced extravillous phenotype. Cells were also seeded onto Matrigel-coated coverslips and cultured for 48 hours for immunostaining. After 48 hours in culture, 91.1 ± 4.2% (n = 5) of cells were immunopositive for cytokeratin-7. Cells were also positive for HLA-G. To prepare CTB-conditioned medium, 1.5 × 106 CTBs were seeded into a Matrigel-coated T25 flask and left for 3 hours to adhere. The medium was removed, the cells were washed with PBS, and 5 ml of fresh serum-free DMEM/F12 was added to the flask. After 48 hours the medium was removed, centrifuged to eliminate cell debris, and stored at −20°C. The mean protein concentration of the CTB-conditioned medium was 193.45 ± 5.6 μg/ml (n = 8). Conditioned medium was diluted 1:1 with unconditioned medium before use.
Human aortic SMCs (HASMCs) were cultured in Kaighn's modification of Ham's F12 medium and SGHPL-4 cells were cultured in Ham's F10 medium. Media was supplemented with FBS (10%), glutamine (2 mmol/L), penicillin (100 units/ml), and streptomycin (0.1 mg/ml). To generate HASMC- and SGHPL-4-conditioned medium, 0.5 × 106 cells were seeded into a T25 flask. After 24 hours the medium was removed, centrifuged to eliminate cell debris, and stored at −20°C. The mean protein concentrations of the HASMC- and SGHPL-4–conditioned media were 244.36 ± 10.0 μg/ml (n = 8) and 261.31 ± 6.7 μg/ml (n = 6), respectively. Conditioned medium was diluted 1:1 with unconditioned medium before use.
Tissue
Informed consent was obtained for all myometrial and placental tissue used in this study, and local ethics committee approval was in place. First-trimester placenta (n = 21) and decidua (n = 8; 7–12 weeks gestation) was obtained after surgical or medical termination of pregnancy for psychosocial reasons. Term decidual/myometrial biopsies (n = 6) taken from non-placental bed tissue were obtained from women with normal pregnancies at elective caesarean section.
Vessel Explant Model
Dissection and perfusion of spiral arteries was performed as previously described.36 Non-placental bed decidual/myometrial biopsies were obtained at Caesarean section from healthy pregnant women at term. Unmodified spiral arteries were dissected under sterile conditions and mounted on glass cannulae in a pressure myography perfusion chamber (Living Systems Instrumentation, Burlington, VT). A subset of vessels were fixed immediately, and the remaining arteries were denuded of endothelium by passing a column of air through the lumen; removal of the endothelium was confirmed by CD31 immunostaining. Denuded arteries were perfused with unconditioned CTB-medium (1:1 DMEM: Ham's F12; control) or CTB-conditioned medium (diluted 1:1 with unconditioned DMEM: Ham's F12). The ends of each vessel were tied off to prevent the medium from leaking out, and the arteries were cultured in 1:1 DMEM: Ham's F12 culture medium for 24 hours.
Immunocytochemistry
Arteries were fixed (2% [vol/vol] paraformaldehyde in PBS, 30 minutes) and incubated with sucrose (0.5 mol/L in PBS; 1 hour). Tissue was embedded in OCT, frozen, and cut into 10-μm transverse sections. For immunostaining of VSMCs, cells were cultured on glass coverslips. Cells/tissue sections were fixed (4% [vol/vol] paraformaldehyde in PBS; 20 minutes), permeabilized (0.1% [vol/vol] Triton-X in PBS; 5 minutes), and washed in PBS. Antibodies were applied for 1 hour, and slides were washed (3 × 5 minutes; PBS) after each antibody incubation. Working concentrations of antibodies were as follows: cytokeratin-7 (0.9 μg/ml), CD31 (5.15 μg/ml), MMP-12 (catalytic domain; 10 μg/ml), biotinylated goat anti-mouse (7 μg/ml), and swine anti-rabbit secondary antibodies (4.05 μg/ml), streptavidin-FITC (10 μg/ml), control IgG (20 μg/ml of each isotype, 100 μg/ml final concentration). Sections and coverslips were mounted using Vectashield mounting medium containing propidium iodide or DAPI and imaged using either an Olympus IX70 inverted fluorescence microscope or a Biorad Radiance 2100 confocal microscope with a ×10 or a ×40 oil immersion objective lens and LaserSharp 2000 image analysis software.
Immunohistochemistry
Wax-embedded decidual tissue sections (5 μm) were deparaffinized in Histoclear and alcohol and microwaved for 10 minutes in sodium citrate buffer (0.01 mol/L; containing 0.05% [vol/vol] Tween 20, pH 6.0) to facilitate antigen unmasking. After cooling, endogenous peroxide activity was blocked by placing the slides in methanol containing 0.4% (vol/vol) HCl and 0.5% (vol/vol) hydrogen peroxide for 30 minutes. Tissue sections were washed three times in 0.05 mol/L Tris buffered saline (TBS) and blocked with 5% (wt/vol) BSA in TBS for 30 minutes. Primary antibodies, diluted to working concentration with 0.05 mol/L TBS (cytokeratin-7, 0.9 μg/ml; MMP-12 [carboxyterminal], 4 μg/ml; HLA-G, 2 μg/ml; α-smooth muscle actin, 0.18 μg/ml; control IgG [20 μg/ml of each isotype, final concentration 100 μg/m]) were applied to the tissue sections, which were incubated overnight at 4°C in a humidity chamber. Slides were washed (3× TBS), and the secondary antibodies, diluted in TBS (biotinylated swine anti-rabbit IgG, 4.05 μg/ml; biotinylated goat anti-mouse IgG, 7 μg/ml), were applied for 30 minutes at room temperature. Slides were washed again (3× TBS) and incubated with avidin peroxidase (5 μg/ml in 0.125M TBS) for 30 minutes at room temperature. Slides were washed in TBS and incubated for 1–5 minutes with 0.05% (wt/vol) DAB and 0.015% (vol/vol) hydrogen peroxide. Slides were washed in dH2O, counterstained with hematoxylin, rehydrated in alcohol and Histoclear, and mounted in DPX mountant containing distyrene, a plasticizer, and xylene.
Intracellular Elastase Activity Assay
Intracellular elastase activity was quantified using an Elastolux assay kit. Cells were cultured alone or with protease inhibitors as described. After trypsinization, cell pellets were incubated in culture medium containing a cell-permeable elastase substrate (9 μmol/L) and 10% FBS, for 1 hour at 37°C. On cleavage by intracellular elastases, the substrate becomes fluorescent and is retained within the cell. Cells were washed once in PBS, and the percentage of fluorescent cells was quantified using a Coulter Epics Elite flow cytometer.
Total Elastase Activity Assay
Cell extracts were generated by incubating CTBs, SGHPL-4, or HASMCs with 0.1% (vol/vol) Triton X100 in PBS for 30 minutes. This process induced cell lysis, release of intracellular elastases, and dissociation of membrane-associated elastases. Extracts were centrifuged to remove any cellular debris, and the resulting supernatant, which contained both intracellular and membrane-associated elastases, was retained for analysis. No elastase activity was detected in the cell pellets. Supernatants or conditioned culture medium (50 μl) were incubated with 200 μl of N-succinyl-(L-alanine)3-p-nitroanilide (1 mmol/L) dissolved in Tris-HCl (200 mmol/L; pH 8.0) for 2 hours at 37°C in a 96-well plate. On cleavage by elastase this substrate absorbs at 405 nm. After addition of 5 μl of glacial acetic acid, the A405 of each sample was determined and compared to a calibration curve prepared using porcine pancreatic elastase. The protein concentration of each sample was measured using a Biorad protein assay with a standard curve prepared using bovine serum albumin. Elastase activity data were expressed as activity per mg of protein.
Time Lapse Microscopy
Proliferating SGHPL-4 and HASMCs (0.25 × 106 cells) were seeded into six-well plates and left to adhere. After addition of elastin (1 mg/ml), NNGH (50 μmol/L), and L-NAME (5 mmol/L), elastin uptake was monitored by time-lapse microscopy using an Olympus IX70 inverted fluorescence microscope with a motorized stage and cooled CCD camera (Hamamatsu model C4742–95) enclosed in a humidified chamber at 37°C with 5% CO2 in air, as described previously.37 Images were taken at 15-minute intervals, and time-lapse sequences were analyzed using ImagePro Plus (Media Cybernetics, Silver Spring, MD). Fifteen cells were scored from each field of view (wells were analyzed in triplicate), and the time at which a cell took up a fragment of elastin was recorded.
RNA Extraction, cRNA Synthesis, and Gene Expression Profiling
EVT outgrowths from villous explants cultured on Matrigel (obtained from six different first-trimester placentas, 7–10 weeks gestation) were isolated and total RNA was extracted as previously described.38,39 RNA integrity was checked by agarose gel electrophoresis and with the Agilent Bioanalyzer 2100 (Agilent Technologies, Inc., Palo Alto, CA). Total RNA (5 μg) was then used for GeneChip analysis. Preparation of cRNA, hybridization to human U133 GeneChips, and scanning of the arrays were performed according to the manufacturer's protocols (Affymetrix Inc., Santa Clara, CA), as previously described.40
Statistics
Statistical analyses were performed using GraphPad Prism software version 4 (GraphPad Software, San Diego, CA). Nonparametric data were represented by median and range and was analyzed using a Kruskal–Wallis tests with appropriate post hoc analysis. Data represent at least three independent experiments. Significance was taken as P < 0.05.
Results
Elastase Activity in Trophoblasts Is Attributed to Caspases and Matrix Metalloproteinases
To investigate the contribution of trophoblast-derived elastases to arterial remodeling, we analyzed intracellular and total elastase activity of human first-trimester CTBs. When cultured with Congo red-labeled elastin, CTBs were observed to engulf and degrade elastin fibers (Figure 1A). CTBs exhibited detectable intracellular elastase activity, which was significantly reduced by a broad spectrum caspase inhibitor, but only when CTBs were isolated from placentas of less than 9 weeks of gestation (P < 0.05; Figure 1B). None of the broad spectrum protease inhibitors tested reduced the intracellular elastase activity of CTBs at later gestations, when added individually, or in combination (Figure 1C). Cell supernatants containing both cytoplasmic and membrane-associated proteases were prepared by incubating CTBs (8–10 weeks gestation) or the first-trimester extravillous trophoblast cell line SGHPL-4 with 0.1% (vol/vol) Triton X-100. Supernatants were incubated with the elastase substrate N-succinyl-(L-alanine)3-p-nitroanilide to quantify elastase activity. Both SGHPL-4 cells and CTB supernatants demonstrated marked elastase activity, which was completely inhibited by the broad spectrum MMP inhibitor NNGH (P < 0.05, Figure 1, D and E). As the MMP inhibitor had no effect on intracellular elastase activity, the MMP activity in the CTB supernatants must be attributable to membrane-associated elastases. Elastase activity in CTB-conditioned medium was below the level of detection of this assay (data not shown).
Figure 1.
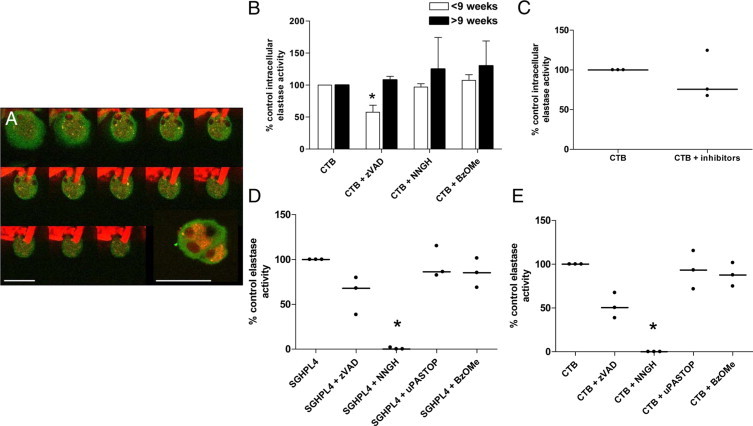
Elastase activity in first-trimester cytotrophoblasts. A: First-trimester cytotrophoblasts (CTBs) were cultured with Congo Red-labeled elastin (1 mg/ml) for 48 hours and stained with an antibody to cytokeratin-7 (green). Serial images at 2-μm intervals along the z axis were captured by confocal microscopy; inset depicts intracellular elastin fragments. Scale bars = 50 μm (n = 3). B: CTBs were isolated from placentas of fewer than 9 weeks gestation or greater than 9-weeks gestation. Cells were cultured with a vehicle control (DMSO), the broad-spectrum caspase inhibitor zVADfmk (50 μmol/L), the broad-spectrum MMP inhibitor NNGH (50 μmol/L), the broad-spectrum cathepsin inhibitor Z-FG-NHO-BzOMe (10 μmol/L), or (C) with all three inhibitors for 18 hours. B and C: Intracellular elastase activity was determined using a cell-permeable elastase substrate. Median + range (n = 3). B: *P < 0.05; Kruskal-Wallis test. D and E: Membrane-associated elastase activity in SGHPL4 (D) and first-trimester CTBs (E) was assessed using the substrate N-succinyl-(L-alanine)3-p-nitroanilide. Cell supernatants were assayed for elastase activity in the presence of vehicle control (DMSO), zVADfmk (50 μmol/L), NNGH (50 μmol/L), Z-FG-NHO-BzOMe (10 μmol/L), or the uPA inhibitor uPA-STOP (10 μmol/L). Median, (n = 3); *P < 0.05; Kruskal-Wallis test.
To investigate the kinetics of elastin breakdown, SGHPL-4 cells were incubated with elastin and monitored at 15-minute intervals for 24 hours by time-lapse video microscopy. Over time, cells began to engulf and degrade these fibers, retaining small pieces of elastin within their cytoplasm (Figure 2A). After 24 hours, approximately 85% of control cells contained one or more elastin fragments (Figure 2B). Addition of the broad-spectrum MMP inhibitor zVAD-fmk to the cultures significantly reduced the percentage of SGHPL-4 cells containing elastin to 66% of controls after 6 hours and 81% after 24 hours (P < 0.05; Figure 2C). It has previously been shown that in some cell types, the availability of NO can influence the expression and activity of certain proteases,23–26 and we have previously identified MMP-9 as a potential target for S-nitrosylation in trophoblast.29 Thus, we investigated whether elastin uptake was NO-dependent, using the nitric oxide synthase (NOS) inhibitor L-NAME. In the presence of L-NAME, the percentage of cells containing elastin fragments was significantly reduced after 6 hours (P < 0.01) but not at any other time points, implying that NO availability is not a prerequisite for elastin uptake.
Figure 2.
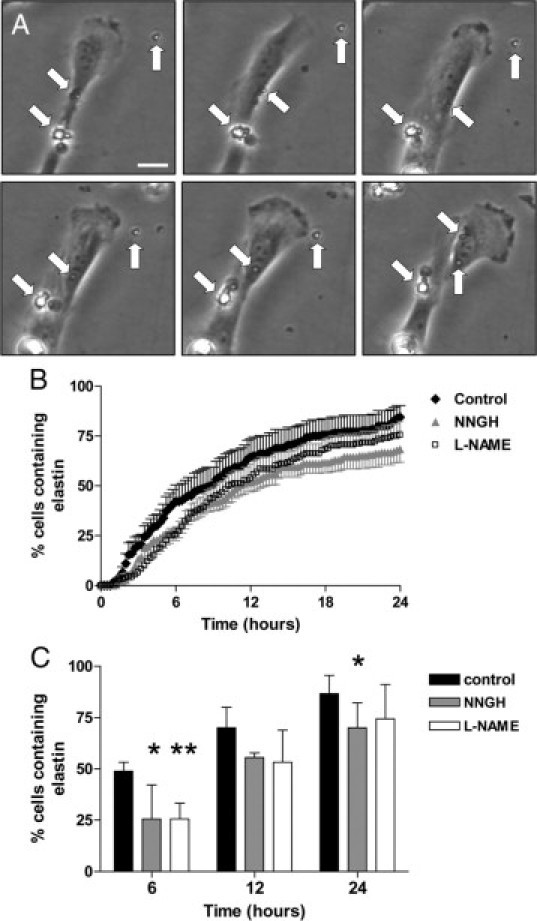
Elastin uptake and MMP expression in first-trimester trophoblasts. SGHPL-4 were cultured with elastin fragments (1 mg/ml) (closed diamonds) in the presence of vehicle control (DMSO), the broad-spectrum MMP inhibitor NNGH (50 μmol/L; closed triangles), or L-NAME (5 mmol/L; open squares) for 24 hours. Cultures were monitored by time-lapse video-microscopy, and the time point at which an elastin fragment appeared within a cell was recorded; 45 cells were scored for each treatment. A: Selected images taken at hourly intervals, showing accumulation of elastin fragments within a cell. Arrows denote elastin fragments. Scale bar = 10 μm. B: Time course of elastin uptake. Mean ± SEM (n = 4). C: Percentage of SGHPL-4–containing elastin fragments at 6, 12, and 24 hours. Black bars represent control cells, gray bars represent NNGH-treated cells, and white bars represent L-NAME–treated cells. Median + range (n = 4); *P < 0.05, **P < 0.01; two-way analysis of variance.
Identification of Candidate Elastases in First-Trimester Trophoblasts
To determine the identity of candidate trophoblast elastases, a transcriptomic approach was used. Affymetrix gene chip analysis of two separate pools of first-trimester EVT mRNA (7–10 weeks gestation) demonstrated that caspase-2, MMP-2, and MMP-12 were the most highly expressed protease transcripts (Table 1). The role of MMP-2 in migration and invasion of first-trimester EVT has been well characterized previously20; however, the role of MMP-12 (macrophage metalloelastase) in trophoblast function has not been investigated.
Table 1.
Affymetrix Analysis of Protease Expression in First Trimester EVT
| Protease | Affymetrix reference number | Relative expression 1 | Relative expression 2 |
|---|---|---|---|
| MMP-2 | 201069_at | 19,976.2* | 25,128.7* |
| MMP-3 | 205828_at | 793.2 | 1557.3 |
| MMP-9 | 203936_s_at | 193.3 | 258.7 |
| MMP-11 | 203878_s_at | 1330.8 | 1031 |
| MMP-12 | 204580_at | 18,953.8* | 33,777.5* |
| MMP-14 | 160020_at | 980 | 859.1 |
| MMP-15 | 203365_s_at | 6427 | 2102.7 |
| MMP-19 | 204575_s_at | 1151.6 | 950.2 |
| Caspase-1 | 211366_x_at | 129.4 | 244.9 |
| Caspase-2 | 226032_at | 4885.5* | 5775.1* |
| Caspase-3 | 202763_at | 467.8 | 352.6 |
| Caspase-4 | 209310_s_at | 370.7 | 406.5 |
| Caspase-4 | 213596_at | 58.5 | 143.1 |
| Caspase-6 | 209790_s_at | 382.7 | 250.3 |
| Caspase-6 | 211464_x_at | 210.4 | 203.6 |
| Caspase-7 | 207181_s_at | 801.6 | 797 |
| Caspase-8 | 213373_s_at | 167.8 | 210.8 |
| Caspase-9 | 203984_s_at | 490.6 | 449.2 |
Relative expression of protease transcripts in two pools of first trimester EVT mRNA. Asterisks denote the most highly expressed transcripts.
MMP-12 Mediates Elastolysis in First-Trimester Trophoblasts
To validate our transcriptomic data, isolated first-trimester CTBs were immunostained with an antibody to MMP-12, confirming protein expression (Figure 3, A and B). As previously shown, pretreatment of individual CTB supernatants (7–12 weeks gestation) with a broad-spectrum MMP inhibitor abolished elastase activity (P < 0.001; Figure 3C). Pretreatment of the same supernatants with a specific inhibitor of MMP-12 (470.1) significantly reduced elastase activity to a mean value of 31.7 ± 10.9% (P < 0.05) of control activity, implicating MMP-12 as an important membrane-associated elastase in trophoblast.
Figure 3.
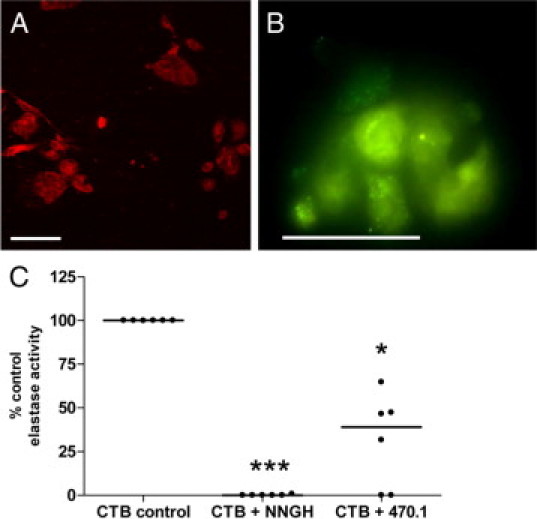
Membrane-associated MMP-12 mediates elastolysis in first-trimester CTBs. First-trimester CTBs cultured for 48 hours on Matrigel-coated coverslips were immunostained with control IgG (A) or an antibody to MMP-12 (B) (catalytic domain; green); nuclei were counterstained with propidium iodide (red). Scale bar = 50 μm. Pictures are representative of three independent CTB isolations. C: Membrane-associated elastase activity in first-trimester CTBs was assessed using the substrate N-succinyl-(L-alanine)3-p-nitroanilide. Cell supernatants were assayed for elastase activity in the presence of vehicle control (DMSO), the broad-spectrum MMP inhibitor NNGH (50 μmol/L), or the specific MMP-12 inhibitor 470.1 (100 μmol/L). Median, (n = 6); *P < 0.05; ***P < 0.001; Kruskal–Wallis test.
Intracellular Elastase Activity of HASMCs Is Stimulated by Elastin Fragments
As VSMC-derived elastases have been shown to mediate elastin breakdown during the progression of atherosclerosis and aneurysm, we investigated whether VSMCs could contribute to the elastolysis observed during arterial remodeling in pregnancy. Only a small proportion of cultured HASMCs exhibited exogenous intracellular elastase activity (4.81% ± 1.4; n = 7); this was unchanged after culture on Matrigel, a collagen IV and laminin-rich extracellular matrix preparation similar in composition to the basement membrane on which VSMCs reside in vivo (Figure 4A). Similarly, HASMCs cultured with CTB-conditioned medium did not exhibit increased elastase activity, suggesting that soluble factors released from trophoblast do not modulate intracellular elastase activity in vitro. As VSMCs may be exposed to elastin fragments generated by trophoblast-mediated elastolysis during remodeling in vivo, HASMCs were cultured with elastin fibers. This treatment significantly enhanced intracellular elastase activity (P < 0.05), suggesting that the presence of elastin fragments in vivo may stimulate elastase production in the arterial wall.
Figure 4.
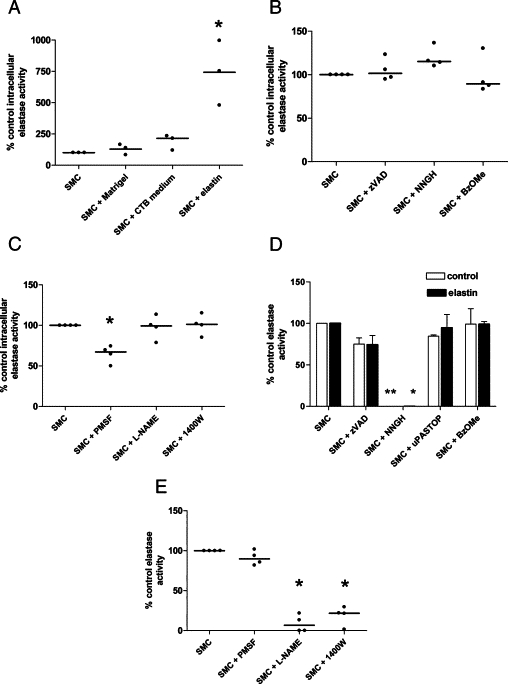
HASMCs exhibit elastase activity attributable to serine proteases and MMP. A: HASMCs were cultured on uncoated plastic dishes (control), Matrigel-coated dishes, or with first-trimester CTB-conditioned medium (50% [vol/vol]), or elastin fibers (1 mg/ml) for 48 hours. Intracellular elastase activity was quantified using a cell-permeable elastase substrate and flow cytometry. Median (n = 3); *P < 0.05; Kruskal-Wallis test. B: HASMCs were cultured on plates coated with elastin fragments for 48 hours and were incubated with vehicle control (DMSO), the broad-spectrum caspase inhibitor zVADfmk (50 μmol/L), the MMP inhibitor NNGH (50 μmol/L), or the cathepsin inhibitor Z-FG-NHO-BzOMe (10 μmol/L) for a further six hours. Intracellular elastase activity was determined as above. Median (n = 4). C: HASMCs were cultured on elastin coated flasks for 48 hours then incubated with vehicle control (DMSO), the serine protease inhibitor PMSF (10 mg/ml; 6 hours), the NOS inhibitor L-NAME (5 mmol/L; 24 hours), or the iNOS inhibitor 1400W (5 μmol/L; 24 hours). Intracellular elastase activity was determined as above. Median (n = 4); *P < 0.05; Kruskal-Wallis test. D and E: Membrane-associated elastase activity in HASMCs was assessed using the substrate N-succinyl-(L-alanine)3-p-nitroanilide. Cell supernatants were assayed for elastase activity in the presence of vehicle control (DMSO), the caspase inhibitor zVADfmk (50 μmol/L), the MMP inhibitor NNGH (50 μmol/L), the uPA inhibitor uPA-STOP (10 μmol/L), or the cathepsin inhibitor Z-FG-NHO-BzOMe (10 μmol/L). D: HASMCs were cultured in uncoated flasks (white bars) or in flasks coated with elastin fragments (black bars) for 48 hours. Median + range (n = 4); *P < 0.05, **P < 0.01; Kruskal–Wallis test. E: HASMCs were cultured on elastin coated flasks, in the absence or presence of the NOS inhibitor L-NAME (5 mmol/L; 24 hours) or the iNOS inhibitor 1400W (5 μmol/L; 24 hours). Control supernatants were analyzed in the presence of vehicle control (DMSO) or the serine protease inhibitor PMSF (10 mg/ml). Median (n = 4); *P < 0.05; Kruskal–Wallis test.
Intracellular Elastase Activity of HASMCs Is Partially Mediated by Serine Proteases
To identify the intracellular proteases responsible for the observed elastase activity, while minimizing the amount of elastin introduced into the flow cytometer, HASMCs were cultured on elastin-coated plates for 48 hours and incubated with cell-permeable protease inhibitors for a further 6 hours. Broad-spectrum caspase, MMP, and cathepsin inhibitors had no effect on intracellular elastase activity (Figure 4B); however, the serine protease inhibitor PMSF reduced activity by approximately 35% (P < 0.05; Figure 4C). Pretreatment of HASMCs with the broad-spectrum NOS inhibitor L-NAME or the specific iNOS inhibitor 1400W for 24 hours had no effect on intracellular elastase activity, suggesting that it was not NO-dependent.
Membrane-Associated Elastase Activity in HASMCs Is MMP-Dependent
To identify membrane-associated, pericellular, or secreted elastases, HASMC supernatants or conditioned medium were incubated with N-succinyl-(L-alanine)3-p-nitroanilide. HASMCs cultured on plastic exhibited constitutive membrane-associated elastase activity, which was completely inhibited by a broad spectrum MMP inhibitor (P < 0.01; Figure 4D). HASMCs cultured on plates coated with elastin did not exhibit an enhancement of elastase activity (0.435 ± 0.16 μg elastase/mg protein [plastic] versus 0.316 ± 0.09 μg elastase/mg protein [elastin], mean ± SEM; n = 4) and again, activity was completely inhibited by a broad spectrum MMP inhibitor (P < 0.05). The broad spectrum caspase inhibitor zVADfmk, the cathepsin inhibitor Z-FG-NHO-BzOMe, and uPA-STOP had no effect. As the MMP inhibitor had no effect on intracellular elastase activity of HASMCs, the activity detected in the cell supernatants must be attributable to membrane-associated elastases. Elastase activity present in the conditioned medium of HASMCs was below the level of detection (data not shown).
Membrane-Associated Elastase Activity in HASMCs Is NO-Dependent
To investigate whether the activity of elastolytic MMP was NO-dependent, HASMCs were pretreated with L-NAME or the iNOS inhibitor 1400W for 24 hours before preparation of cell supernatants. Elastase activity was significantly decreased by both inhibitors, indicating that NO availability is necessary for effective MMP function in HASMCs (P < 0.05; Figure 4E).
Elastin Uptake Is MMP- and NO-Dependent
To study elastin fragment uptake over time, HASMCs were incubated with elastin and monitored by time lapse video-microscopy (Figure 5A). When HASMCs were cultured with elastin alone, approximately 80% of control cells contained elastin fragments after 24 hours (Figure 5B). Addition of NNGH or L-NAME to the cultures had a marked effect on the kinetics of elastin uptake. Uptake was significantly decreased at 6 hours, 12 hours, and 24 hours in the presence of the MMP inhibitor (P < 0.01, P < 0.001), and at 6 hours and 12 hours by L-NAME (P < 0.05, P < 0.001; Figure 5C). These data suggest that MMP and NO are required for effective elastin uptake by HASMCs.
Figure 5.
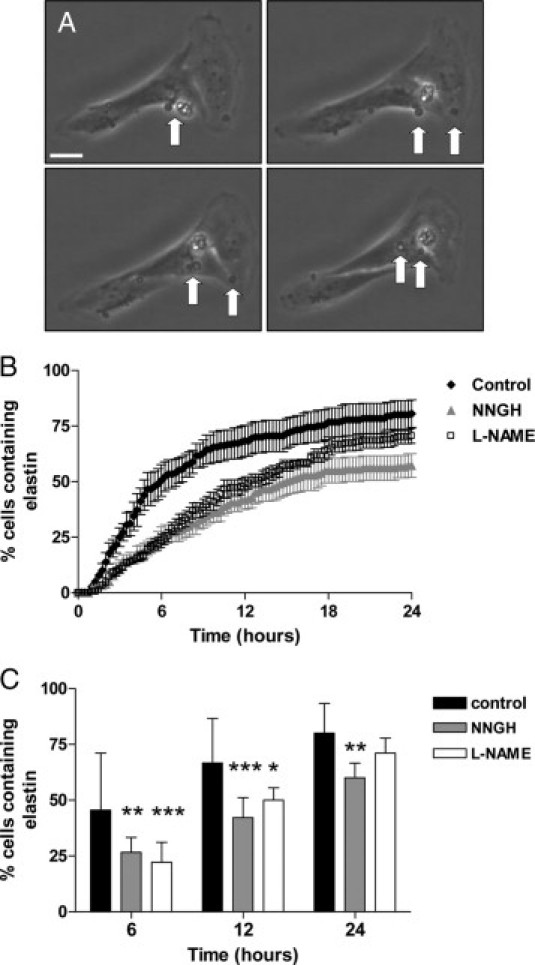
Elastin uptake in HASMCs is regulated by MMP and NO. HASMCs were cultured with elastin fragments (1 mg/ml) in the presence of vehicle control (DMSO), the broad-spectrum MMP inhibitor NNGH (50 μmol/L), or L-NAME (5 mmol/L) for 24 hours. Cultures were monitored by time-lapse video-microscopy, and the time point at which an elastin fragment appeared within a cell was recorded. Forty-five cells were scored for each treatment. A: Selected images taken at hourly intervals, showing accumulation of elastin fragments within a cell. Arrows denote elastin fragments. Scale bar = 10 μm. B: Time course of elastin uptake. Mean ± SEM (n = 4). C: Percentage of HASMC-containing elastin fragments at 6, 12, and 24 hours. Black bars represent control cells, gray bars represent NNGH-treated cells, and white bars represent L-NAME-treated cells. Median + range (n = 4); *P < 0.05, **P < 0.01, ***P < 0.001; two-way analysis of variance.
VSMCs Express Elastolytic MMP in Vitro and in Vivo
To investigate whether cultured HASMCs express MMP-12, immunostaining was used to confirm expression of the active form (Figure 6, A and B). During pregnancy, invading EVT release soluble factors including FasL and TRAIL, which trigger VSMC apoptosis.41,42 To assess whether these factors can alter MMP-12 expression in spiral artery VSMCs ex vivo, unremodeled spiral arteries were dissected from myometrial biopsies obtained from healthy pregnant women undergoing caesarean section at term. Vessels were denuded of endothelium to facilitate access of factors to the underlying VSMCs then perfused with unconditioned medium (control), CTB-conditioned medium (which contains detectable soluble FasL but not TRAIL),41,42 or recombinant human TRAIL (rhTRAIL), and cultured for 24 hours. CD31 immunostaining confirmed removal of the endothelium, while morphological analysis of the arterial wall verified the integrity of the underlying VSMC layers (Figure 6, C and D). No MMP-12 expression was observed in VSMCs of freshly isolated arteries with intact endothelium (Figure 6E) or in denuded vessels perfused with unconditioned (control) medium (Figure 6F), indicating that removal of the endothelium does not up-regulate MMP-12 expression in the underlying VSMCs. Introduction of CTB-conditioned medium or rhTRAIL into the lumen of denuded arteries induced expression of active MMP-12 in VSMCs, although the extent of expression was heterogeneous, with only 30% of tissue sections examined containing MMP-12–positive cells (Figure 6, G and H). These data suggest that factors produced by EVT during colonization and remodeling of the spiral arteries have the ability to influence MMP-12 expression in a proportion of mural VSMCs. Membrane-associated elastase activity of HASMCs was reduced to a mean value of 23.3 ± 8.7% of control activity after pretreatment of HASMC supernatants with a specific inhibitor of MMP-12 (P < 0.05; Figure 6I). As previously observed, the broad spectrum MMP inhibitor NNGH abolished elastase activity (P < 0.001). These data implicate MMP-12 as an important mediator of HASMC elastolysis in vitro.
Figure 6.
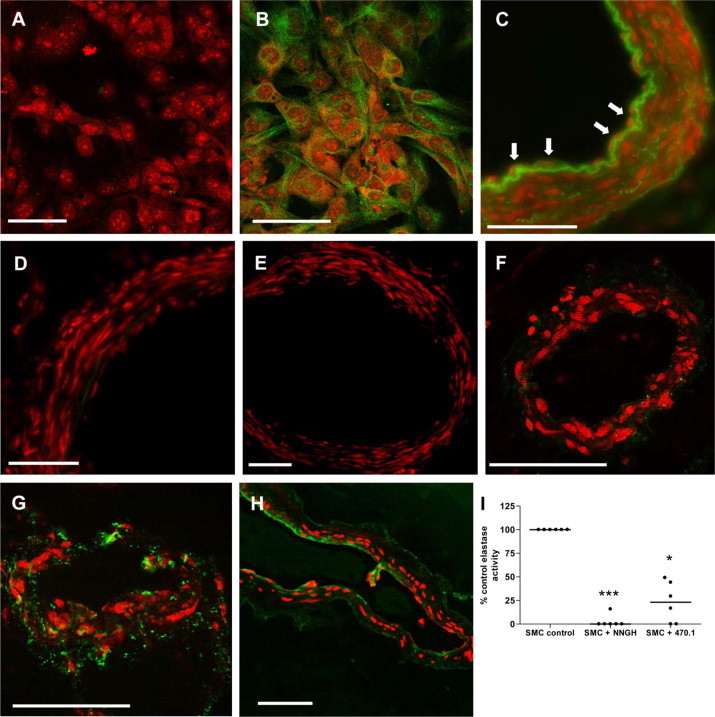
MMP-12 expression in cultured HASMCs and excised human spiral arties. A and B: Cultured HASMCs were immunostained with control IgG (A) or an antibody to the catalytic domain of MMP-12 (B) (green). Nuclei were counterstained with propidium iodide (red). Scale bar = 50 μm. Intact human spiral arteries (C) and arteries denuded of endothelium (D) were fixed immediately and stained with an antibody to CD31 (green). Nuclei were counterstained with propidium iodide (red). Scale bar = 100 μm. C: Arrows denote endothelial cells. Note autofluorescence of elastin. E–H: Spiral arteries were denuded of endothelium and were fixed immediately (E) or perfused with unconditioned first-trimester CTB medium (1:1 DMEM: Ham's F12; control) (F), CTB-conditioned medium (diluted 1:1 with DMEM: Ham's F12) (G), or rhTRAIL (0.5 μg/ml in DMEM: Ham's F12) (H) and cultured for 24 hours. Sections were stained with an antibody to the catalytic domain of MMP-12 (green). Scale bar = 100 μm. Nuclei were counterstained with propidium iodide (red). Images are representative of six independent experiments; however, MMP-12–positive VSMCs were only observed in seven or eight (∼30%) of the 24 serial tissue sections examined from each vessel. I: HASMC supernatants were assayed for membrane-associated elastase activity in the presence of vehicle control (DMSO), the broad-spectrum MMP inhibitor NNGH (50 μmol/L), or the specific MMP-12 inhibitor 470.1 (100 μmol/L). Median (n = 6); *P < 0.05; ***P < 0.001; Kruskal–Wallis test.
MMP-12 Is Expressed by Trophoblast and VSMCs in the First Trimester of Pregnancy
Sections of first-trimester placenta and decidua (8–12 weeks gestation) were stained with an antibody to MMP-12; villous CTBs, trophoblast cell columns and EVT were immunopositive (Figure 7, A, B, D, and E). A subset of villous stromal cells, most likely placental macrophages (Hofbauer cells), also expressed MMP-12 (Figure 7D). Medial VSMCs in unremodeled arteries did not express MMP-12, consistent with our ex vivo observations; however, decidual stromal cells were weakly positive for MMP-12 and strong expression was noted in the vascular endothelium (EC; Figure 7, C and F). MMP-12 expression was also observed in endovascular EVT (eEVT; identified by HLA-G immunostaining) and heterogeneously in disrupted VSMCs (identified by α-smooth muscle actin immunostaining) in remodeling spiral arteries (Figure 7, G–L).
Figure 7.
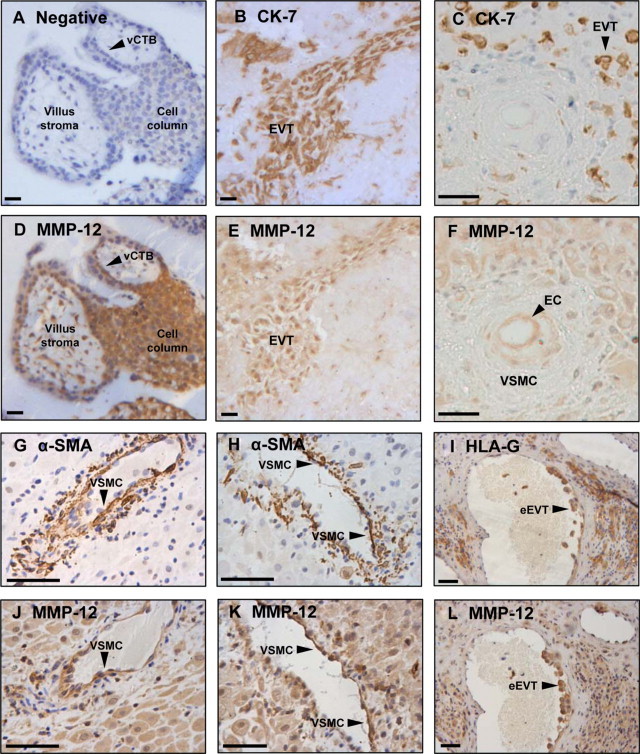
MMP-12 expression in cultured VSMCs and excised human spiral arties. Serial sections of first-trimester placenta (n = 5) and decidua basalis (n = 8) were immunostained with control IgG (A), an antibody to cytokeratin-7 (CK-7; B, C) or HLA-G (I) to detect trophoblasts, an antibody to α-smooth muscle actin (α-SMA) to detect VSMCs (G and H), or an antibody to MMP-12 (carboxyl-terminal) (D–F, J–L). Sections were counterstained with hematoxylin. Scale bar = 50 μm. Serial sections: (A and D), (B and E), (C and F), (G and J), (H and K), (I and L). EC indicates endothelial cells; vCTB, villous cytotrophoblasts; VSMC, vascular smooth muscle cell; eEVT, endovascular extravillous trophoblasts.
Discussion
Remodeling of uterine spiral arteries during pregnancy involves not only replacement of vascular endothelial cells and VSMCs by invading trophoblast but also structural and functional transformation of the vascular extracellular matrix. To allow a permanent increase in luminal diameter, the internal elastic lamina and medial elastin fibers must be degraded. Matrix breakdown is mediated in part by the actions of trophoblast-derived proteases, and we now provide evidence that spiral artery VSMC-derived proteases add their own contribution to elastin catabolism.
Primary first-trimester CTBs can phagocytose elastin in vitro and exhibit both intracellular and membrane-associated elastase activity. Intracellular activity is partially attributable to caspases in early gestation, but we were unable to identify candidate enzymes beyond 9 weeks, possibly due to functional redundancy of the enzymes involved. Approximately 60–70% of membrane-associated elastase activity was attributable to MMP-12 (macrophage metalloelastase), which is known to act in association with the cell surface43; it is likely that the remaining proportion is mediated by a combination of MMP-2, −7, and −9, which are capable of elastolysis and are expressed by first-trimester trophoblast.19,20 MMP-12 has previously been implicated as a potential regulator of trophoblast adhesion and migration44; here we identify MMP-12 as a candidate elastase which is expressed by both interstitial and endovascular trophoblasts in close proximity to spiral arteries in first trimester. Although NO bioavailability is an important regulator of trophoblast function,27,28 including MMP-9 activity at the leading edge of migrating cells,29 phagocytosis of elastin by trophoblast was not reduced in the presence of a NOS inhibitor.
Elastin degradation appears to be a cooperative process, because VSMCs also have the capacity to engulf and degrade small particles of elastin using intracellular and membrane-associated enzymes. In vitro, HASMCs expressed low endogenous levels of intracellular elastase; however, this was significantly increased after culture with elastin and was mediated by serine proteases independently of NO bioavailability. In contrast, HASMCs exhibited constitutive membrane-associated elastase activity, which was attributable to MMP and caspases, was significantly reduced by broad-spectrum- and iNOS-specific inhibitors, and was not significantly increased after culture with elastin. Phagocytosis of elastin fragments was reduced in the presence of inhibitors of either MMP or NOS. A major fraction of membrane-associated elastase activity in VSMCs was attributable to MMP-12; moreover, expression of MMP-12 was induced in the mural VSMCs of excised spiral arteries that had been denuded of endothelium and perfused with CTB-conditioned medium. We therefore hypothesize that soluble factors secreted by endovascular EVT up-regulate MMP expression in spiral artery VSMCs in vivo, which contributes to, or is a consequence of, the alteration in VSMC phenotype observed in actively remodeling vessels.4,5 Indeed, the hypertrophy, dedifferentiation, and disruption of medial smooth muscle layers observed in vivo reflects transition from a stable contractile phenotype toward a more synthetic proliferative state, a change that has previously been linked to enhanced MMP expression.45 Furthermore, cultured HASMCs constitutively expressed MMP- 12 in vitro, whereas medial VSMCs in excised spiral arteries did not, and CTB-conditioned medium induced MMP-12 expression in spiral artery VSMCs in situ but did not enhance elastase activity of undifferentiated HASMCs in vitro. These observations suggest that induction of MMP-12 expression in VSMCs may be coincident with dedifferentiation and loss of contractile phenotype. Our finding that MMP-12 is expressed in layers of disrupted VSMCs within remodeling arteries, but is absent from the organized VSMC layers of unremodeled vessels, supports this hypothesis.
Breakdown of elastin and formation of elastin-derived peptides (EDP) plays an important role in the regulation of tissue remodeling. Macrophage-derived MMP-12 is crucial for the development of emphysema in a murine model46: release of chemotactic EDP recruits monocytes to the lung, which differentiate into macrophages, generate more EDP, and perpetuate the condition. EDP also stimulate chemotaxis and chemokinesis of leukocytes into the walls of damaged arteries, where they contribute to matrix breakdown and repair.47 It is therefore interesting to note that leukocytes adopt a perivascular location around untransformed vessels in the first-trimester placental bed48 and mediate the initial stages of vascular transformation.5 EDP can also stimulate quiescent arterial VSMCs to adopt a more proliferative phenotype49–52 and promote MMP expression in cancer cell lines.53–55 In addition, EDP may regulate NOS activity, as elastin peptides stimulated NO production in coronary artery endothelial cells and cardiomyocytes.56 Our data support a model whereby trophoblast-mediated breakdown of the elastin within the spiral arteries liberates EDP that promote MMP-12 expression in medial VSMCs. EDP may also increase NO production and MMP activity in trophoblasts, establishing a positive feedback mechanism by which EDP production and MMP-12 activity are maintained.
Elastase activity in HASMCs, but not CTBs, was dependent on NO bioavailability. This is in contrast to previous work showing that NO donors stimulated MMP expression and activity in term CTBs, whereas NOS inhibitors reduced it.57,58 These conflicting data may be explained by the later gestational age of the cells, or because we were studying elastolysis, rather than collagenolysis, which is primarily mediated by MMP-2 and −9. Nitration of a tyrosine residue in active site of MMP-2 by peroxynitrite59 or nitrosylation of a critical cysteine residue in MMP-960 by NO and other reactive nitrogen oxides has been shown to promote enzymatic activity. However, cysteine residues in the zinc-tetrathiolate cluster of endothelial NOS (eNOS) and iNOS are susceptible to NO-induced thiol modification, leading to impaired stability of NOS dimers.61–63 Hence, high concentrations of NO resulting from forced expression of iNOS may reduce NOS activity and therefore MMP activity. Cellular localization could hold the key to successful MMP regulation by NO: we and others have observed concentrated expression of eNOS64 and iNOS29 in membrane ruffles at the leading edge of migrating, but not static cells. Thus, localization of NO production to the site of MMP activation may be important in regulating enzyme activity, matrix breakdown, and cell migration.
In summary, we have identified a novel role for MMP-12 as a mediator of uterine vascular remodeling during pregnancy. Our data support a model whereby trophoblast and vascular VSMCs use MMP-12 to cooperatively degrade mural elastin during spiral artery remodeling, with the localized release of elastin peptides and CTB-derived factors amplifying elastin catabolism. The role of leukocytes in this scheme of events has yet to be addressed; however, cytokines or chemokines released by decidual macrophages and uNK cells may influence MMP-12 expression in VSMCs and EVT.44 Breakdown of elastin in decidual vessels may be initiated by resident VSMCs before the arrival of the trophoblast, facilitating the entry of both leukocytes and EVT into the arterial wall. Macrophages and uNK cells may also contribute to elastolysis in decidual arteries, via the release of MMP-7 and MMP-9.5 However, MMP-12 expression and elastolysis may not occur efficiently in the deeper myometrial vessel segments until trophoblasts reside in close proximity to the medial VSMCs. Interestingly, elastin catabolism is impaired in the myometrial spiral arteries of women with pre-eclampsia,17 and a dramatic reduction in expression of MMP-12 mRNA has been reported in the first-trimester placentas of women who subsequently develop pre-eclampsia,65 implicating MMP-12 as a potential therapeutic target. As MMP-12 is a multifunctional enzyme that is capable of degrading not only elastin, but collagen IV, laminin, fibronectin, heparan sulfate, and vitronectin,66 there may be serious consequences if MMP-12 expression is suboptimal. Pre-eclampsia is also characterized by increased circulating concentrations of the endogenous inhibitor of NOS asymmetric dimethyl arginine (ADMA).30,32 It is therefore possible that this may limit NO production, reduce elastase activity, and impair arterial vasodilatation. However, further work is needed to elucidate the complex mechanisms controlling matrix breakdown and vascular transformation and to investigate the spatiotemporal interplay between medial VSMCs, invasive trophoblast, and other cell types present within the placental bed.
Acknowledgements
We thank Dr Laurent Devel and Dr Vincent Dive (CEA, France) for kindly supplying the specific MMP-12 inhibitor, 470.1.
Footnotes
Supported by funding from the Wellcome Trust, UK (069939), a MRC studentship, and Tommy's the baby charity. The Maternal and Fetal Health Research Group is supported by the Manchester Academic Health Sciences Centre (MAHSC) and the National Institute for Health Research (NIHR) Manchester Biomedical Research Centre.
References
- 1.Burton GJ, Woods AW, Jauniaux E, Kingdom JC. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta. 2009;30:473–482. doi: 10.1016/j.placenta.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aplin JD. Implantation, trophoblast differentiation and haemochorial placentation: mechanistic evidence in vivo and in vitro. J Cell Sci. 1991;99:681–692. doi: 10.1242/jcs.99.4.681. [DOI] [PubMed] [Google Scholar]
- 3.Aplin JD. The cell biology of human implantation. Placenta. 1996;17:269–275. doi: 10.1016/s0143-4004(96)90050-8. [DOI] [PubMed] [Google Scholar]
- 4.Craven CM, Morgan T, Ward K. Decidual spiral artery remodeling begins before cellular interaction with cytotrophoblasts. Placenta. 1998;19:241–252. doi: 10.1016/s0143-4004(98)90055-8. [DOI] [PubMed] [Google Scholar]
- 5.Smith SD, Dunk CE, Aplin JD, Harris LK, Jones RL. Evidence for immune cell involvement in decidual spiral arteriole remodeling in early human pregnancy. Am J Pathol. 2009;174:1959–1971. doi: 10.2353/ajpath.2009.080995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brosens I, Robertson WB, Dixon HG. The physiological response of the vessels of the placental bed to normal pregnancy. J Pathol Bacteriol. 1967;93:569–579. doi: 10.1002/path.1700930218. [DOI] [PubMed] [Google Scholar]
- 7.Pijnenborg R, Bland JM, Robertson WB, Brosens I. Uteroplacental arterial changes related to interstitial trophoblast migration in early human pregnancy. Placenta. 1983;4:397–413. doi: 10.1016/s0143-4004(83)80043-5. [DOI] [PubMed] [Google Scholar]
- 8.Pijnenborg R, Vercruysse L, Hanssens M. The uterine spiral arteries in human pregnancy: facts and controversies. Placenta. 2006;27:939–958. doi: 10.1016/j.placenta.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 9.Harris LK, Aplin JD. Vascular remodeling and extracellular matrix breakdown in the uterine spiral arteries during pregnancy. Reprod Sci. 2007;14:28–34. doi: 10.1177/1933719107309588. [DOI] [PubMed] [Google Scholar]
- 10.Kaufmann P, Black S, Huppertz B. Endovascular trophoblast invasion: implications for the pathogenesis of intrauterine growth retardation and preeclampsia. Biol Reprod. 2003;69:1–7. doi: 10.1095/biolreprod.102.014977. [DOI] [PubMed] [Google Scholar]
- 11.Vicovac L, Aplin JD. Epithelial-mesenchymal transition during trophoblast differentiation. Acta Anat (Basel) 1996;156:202–216. doi: 10.1159/000147847. [DOI] [PubMed] [Google Scholar]
- 12.Ball E, Bulmer JN, Ayis S, Lyall F, Robson SC. Late sporadic miscarriage is associated with abnormalities in spiral artery transformation and trophoblast invasion. J Pathol. 2006;208:535–542. doi: 10.1002/path.1927. [DOI] [PubMed] [Google Scholar]
- 13.Kim YM, Bujold E, Chaiworapongsa T, Gomez R, Yoon BH, Thaler HT, Rotmensch S, Romero R. Failure of physiologic transformation of the spiral arteries in patients with preterm labor and intact membranes. Am J Obstet Gynecol. 2003;189:1063–1069. doi: 10.1067/s0002-9378(03)00838-x. [DOI] [PubMed] [Google Scholar]
- 14.Kadyrov M, Schmitz C, Black S, Kaufmann P, Huppertz B. Pre-eclampsia and maternal anaemia display reduced apoptosis and opposite invasive phenotypes of extravillous trophoblast. Placenta. 2003;24:540–548. doi: 10.1053/plac.2002.0946. [DOI] [PubMed] [Google Scholar]
- 15.Khong TY, De Wolf F, Robertson WB, Brosens I. Inadequate maternal vascular response to placentation in pregnancies complicated by pre-eclampsia and by small-for-gestational age infants. Br J Obstet Gynaecol. 1986;93:1049–1059. doi: 10.1111/j.1471-0528.1986.tb07830.x. [DOI] [PubMed] [Google Scholar]
- 16.Metaxa-Mariatou V, McGavigan CJ, Robertson K, Stewart C, Cameron IT, Campbell S. Elastin distribution in the myometrial and vascular smooth muscle of the human uterus. Mol Hum Reprod. 2002;8:559–565. doi: 10.1093/molehr/8.6.559. [DOI] [PubMed] [Google Scholar]
- 17.Pijnenborg R, Vercruyssse L, Verbist L, Van Assche FA. Relative contribution of interstitial and endovascular trophoblast to elastica breakdown in placental bed spiral arteries. Am J Obstet Gynecol. 1999;180:S43. [Google Scholar]
- 18.Crocker IP, Wareing M, Ferris GR, Jones CJ, Cartwright JE, Baker PN, Aplin JD. The effect of vascular origin, oxygen, and tumour necrosis factor alpha on trophoblast invasion of maternal arteries in vitro. J Pathol. 2005;206:476–485. doi: 10.1002/path.1801. [DOI] [PubMed] [Google Scholar]
- 19.Laszlo A, Sohar I, Falkay G, Kovacs A, Halmos V, Szabo J. Physiological values of cysteine and metalloproteinase activities in chorionic villi. Acta Obstet Gynecol Scand. 1990;69:397–398. doi: 10.3109/00016349009013300. [DOI] [PubMed] [Google Scholar]
- 20.Staun-Ram E, Goldman S, Gabarin D, Shalev E. Expression and importance of matrix metalloproteinase 2 and 9 (MMP-2 and -9) in human trophoblast invasion. Reprod Biol Endocrinol. 2004;2:59–71. doi: 10.1186/1477-7827-2-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pijnenborg R. Implantation and immunology: maternal inflammatory and immune cellular responses to implantation and trophoblast invasion. Reprod Biomed Online. 2002;4(Suppl 3):14–17. doi: 10.1016/s1472-6483(12)60110-2. [DOI] [PubMed] [Google Scholar]
- 22.Reister F, Frank HG, Heyl W, Kosanke G, Huppertz B, Schroder W, Kaufmann P, Rath W. The distribution of macrophages in spiral arteries of the placental bed in pre-eclampsia differs from that in healthy patients. Placenta. 1999;20:229–233. doi: 10.1053/plac.1998.0373. [DOI] [PubMed] [Google Scholar]
- 23.Cui X, Chen J, Zacharek A, Li Y, Roberts C, Kapke A, Savant-Bhonsale S, Chopp M. Nitric oxide donor upregulation of SDF1/CXCR4 enhances BMSC migration into ischemic brain after stroke. Stem Cells. 2007;25:2777–2785. doi: 10.1634/stemcells.2007-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Genis L, Gonzalo P, Tutor AS, Galvez BG, Martinez-Ruiz A, Zaragoza C, Lamas S, Tryggvason K, Apte SS, Arroyo AG. Functional interplay between endothelial nitric oxide synthase and membrane type 1 matrix metalloproteinase in migrating endothelial cells. Blood. 2007;110:2916–2923. doi: 10.1182/blood-2007-01-068080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gurjar MV, Sharma RV, Bhalla RC. eNOS gene transfer inhibits smooth muscle cell migration and MMP-2 and MMP-9 activity. Arterioscler Thromb Vasc Biol. 1999;19:2871–2877. doi: 10.1161/01.atv.19.12.2871. [DOI] [PubMed] [Google Scholar]
- 26.Marcet-Palacios M, Graham K, Cass C, Befus AD, Mayers I, Radomski MW. Nitric oxide and cyclic GMP increase the expression of matrix metalloproteinase-9 in vascular smooth muscle. J Pharmacol Exp Ther. 2003;307:429–436. doi: 10.1124/jpet.103.050385. [DOI] [PubMed] [Google Scholar]
- 27.Dash PR, Cartwright JE, Baker PN, Johnstone AP, Whitley GS. Nitric oxide protects human extravillous trophoblast cells from apoptosis by a cyclic GMP-dependent mechanism and independently of caspase 3 nitrosylation. Exp Cell Res. 2003;287:314–324. doi: 10.1016/s0014-4827(03)00156-3. [DOI] [PubMed] [Google Scholar]
- 28.Dash PR, Whitley GS, Ayling LJ, Johnstone AP, Cartwright JE. Trophoblast apoptosis is inhibited by hepatocyte growth factor through the Akt and beta-catenin mediated up-regulation of inducible nitric oxide synthase. Cell Signal. 2005;17:571–580. doi: 10.1016/j.cellsig.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 29.Harris LK, McCormick J, Cartwright JE, Whitley GS, Dash PR. S-nitrosylation of proteins at the leading edge of migrating trophoblasts by inducible nitric oxide synthase promotes trophoblast invasion. Exp Cell Res. 2008;314:1765–1776. doi: 10.1016/j.yexcr.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 30.Holden DP, Fickling SA, Whitley GS, Nussey SS. Plasma concentrations of asymmetric dimethylarginine, a natural inhibitor of nitric oxide synthase, in normal pregnancy and preeclampsia. Am J Obstet Gynecol. 1998;178:551–556. doi: 10.1016/s0002-9378(98)70437-5. [DOI] [PubMed] [Google Scholar]
- 31.Rutherford RA, McCarthy A, Sullivan MH, Elder MG, Polak JM, Wharton J. Nitric oxide synthase in human placenta and umbilical cord from normal, intrauterine growth-retarded and pre-eclamptic pregnancies. Br J Pharmacol. 1995;116:3099–3109. doi: 10.1111/j.1476-5381.1995.tb15111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pettersson A, Hedner T, Milsom I. Increased circulating concentrations of asymmetric dimethyl arginine (ADMA), an endogenous inhibitor of nitric oxide synthesis, in preeclampsia. Acta Obstet Gynecol Scand. 1998;77:808–813. [PubMed] [Google Scholar]
- 33.Cheng XW, Kuzuya M, Sasaki T, Arakawa K, Kanda S, Sumi D, Koike T, Maeda K, Tamaya-Mori N, Shi GP, Saito N, Iguchi A. Increased expression of elastolytic cysteine proteases, cathepsins S and K, in the neointima of balloon-injured rat carotid arteries. Am J Pathol. 2004;164:243–251. doi: 10.1016/S0002-9440(10)63114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cowan KN, Leung WC, Mar C, Bhattacharjee R, Zhu YH, Rabinovitch M. Caspases from apoptotic myocytes degrade extracellular matrix: a novel remodeling paradigm. FASEB J. 2005;13:1848–1850. doi: 10.1096/fj.05-3706fje. [DOI] [PubMed] [Google Scholar]
- 35.Devel L, Rogakos V, David A, Makaritis A, Beau F, Cuniasse P, Yiotakis A, Dive V. Development of selective inhibitors and substrate of matrix metalloproteinase-12. J Biol Chem. 2006;281:11152–11160. doi: 10.1074/jbc.M600222200. [DOI] [PubMed] [Google Scholar]
- 36.Cartwright JE, Kenny LC, Dash PR, Crocker IP, Aplin JD, Baker PN, Whitley GS. Trophoblast invasion of spiral arteries: a novel in vitro model. Placenta. 2002;23:232–235. doi: 10.1053/plac.2001.0760. [DOI] [PubMed] [Google Scholar]
- 37.Ashton SV, Whitley GS, Dash PR, Wareing M, Crocker IP, Baker PN, Cartwright JE. Uterine spiral artery remodeling involves endothelial apoptosis induced by extravillous trophoblasts through Fas/FasL interactions. Arterioscler Thromb Vasc Biol. 2005;25:102–108. doi: 10.1161/01.ATV.0000148547.70187.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pollheimer J, Loregger T, Sonderegger S, Saleh L, Bauer S, Bilban M, Czerwenka K, Husslein P, Knofler M. Activation of the canonical wingless/T-cell factor signaling pathway promotes invasive differentiation of human trophoblast. Am J Pathol. 2006;168:1134–1147. doi: 10.2353/ajpath.2006.050686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bauer S, Pollheimer J, Hartmann J, Husslein P, Aplin JD, Knofler M. Tumor necrosis factor-alpha inhibits trophoblast migration through elevation of plasminogen activator inhibitor-1 in first-trimester villous explant cultures. J Clin Endocrinol Metab. 2004;89:812–822. doi: 10.1210/jc.2003-031351. [DOI] [PubMed] [Google Scholar]
- 40.Bilban M, Haslinger P, Prast J, Klinglmuller F, Woelfel T, Haider S, Sachs A, Otterbein LE, Desoye G, Hiden U, Wagner O, Knofler M. Identification of novel trophoblast invasion-related genes: heme oxygenase-1 controls motility via peroxisome proliferator-activated receptor gamma. Endocrinology. 2009;150:1000–1013. doi: 10.1210/en.2008-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harris LK, Keogh RJ, Wareing M, Baker PN, Cartwright JE, Aplin JD, Whitley GS. Invasive trophoblasts stimulate vascular smooth muscle cell apoptosis by a fas ligand-dependent mechanism. Am J Pathol. 2006;169:1863–1874. doi: 10.2353/ajpath.2006.060265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keogh RJ, Harris LK, Freeman A, Baker PN, Aplin JD, Whitley GS, Cartwright JE. Fetal-derived trophoblast use the apoptotic cytokine tumor necrosis factor-alpha-related apoptosis-inducing ligand to induce smooth muscle cell death. Circ Res. 2007;100:834–841. doi: 10.1161/01.RES.0000261352.81736.37. [DOI] [PubMed] [Google Scholar]
- 43.Cobos-Correa A, Trojanek JB, Diemer S, Mall MA, Schultz C. Membrane-bound FRET probe visualizes MMP12 activity in pulmonary inflammation. Nat Chem Biol. 2009;5:628–630. doi: 10.1038/nchembio.196. [DOI] [PubMed] [Google Scholar]
- 44.Hannan NJ, Salamonsen LA. CX3CL1 and CCL14 regulate extracellular matrix and adhesion molecules in the trophoblast: potential roles in human embryo implantation. Biol Reprod. 2008;79:58–65. doi: 10.1095/biolreprod.107.066480. [DOI] [PubMed] [Google Scholar]
- 45.Fitzgerald M, Hayward IP, Thomas AC, Campbell GR, Campbell JH. Matrix metalloproteinase can facilitate the heparanase-induced promotion of phenotype change in vascular smooth muscle cells. Atherosclerosis. 1999;145:97–106. doi: 10.1016/s0021-9150(99)00019-2. [DOI] [PubMed] [Google Scholar]
- 46.Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, Mecham RP, Senior RM, Shapiro SD. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest. 2006;116:753–759. doi: 10.1172/JCI25617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hinek A. Nature and the multiple functions of the 67-kD elastin-/laminin binding protein. Cell Adhes Commun. 1994;2:185–193. doi: 10.3109/15419069409004436. [DOI] [PubMed] [Google Scholar]
- 48.Reister F, Frank HG, Kingdom JC, Heyl W, Kaufmann P, Rath W, Huppertz B. Macrophage-induced apoptosis limits endovascular trophoblast invasion in the uterine wall of preeclamptic women. Lab Invest. 2001;81:1143–1152. doi: 10.1038/labinvest.3780326. [DOI] [PubMed] [Google Scholar]
- 49.Faury G. Role of the elastin-laminin receptor in the cardiovascular system. Pathol Biol (Paris) 1998;46:517–526. [PubMed] [Google Scholar]
- 50.Mochizuki S, Brassart B, Hinek A. Signaling pathways transduced through the elastin receptor facilitate proliferation of arterial smooth muscle cells. J Biol Chem. 2002;277:44854–44863. doi: 10.1074/jbc.M205630200. [DOI] [PubMed] [Google Scholar]
- 51.Varga Z, Jacob MP, Csongor J, Robert L, Leovey A, Fulop T., Jr Altered phosphatidylinositol breakdown after K-elastin stimulation in PMNLs of elderly. Mech Ageing Dev. 1990;52:61–70. doi: 10.1016/0047-6374(90)90144-5. [DOI] [PubMed] [Google Scholar]
- 52.Faury G, Ristori MT, Verdetti J, Jacob MP, Robert L. Effect of elastin peptides on vascular tone. J Vasc Res. 1995;32:112–119. doi: 10.1159/000159084. [DOI] [PubMed] [Google Scholar]
- 53.Brassart B, Randoux A, Hornebeck W, Emonard H. Regulation of matrix metalloproteinase-2 (gelatinase A. MMP-2), membrane-type matrix metalloproteinase-1 (MT1-MMP) and tissue inhibitor of metalloproteinases-2 (TIMP-2) expression by elastin-derived peptides in human HT-1080 fibrosarcoma cell line. Clin Exp Metastasis. 1998;16:489–500. doi: 10.1023/a:1006550503612. [DOI] [PubMed] [Google Scholar]
- 54.Hornebeck W, Derouette JC, Brechemier D, Adnet JJ, Robert L. Elastogenesis and elastinolytic activity in human breast cancer. Biomedicine. 1977;26:48–52. [PubMed] [Google Scholar]
- 55.Ntayi C, Labrousse AL, Debret R, Birembaut P, Bellon G, Antonicelli F, Hornebeck W, Bernard P. Elastin-derived peptides upregulate matrix metalloproteinase-2-mediated melanoma cell invasion through elastin-binding protein. J Invest Dermatol. 2004;122:256–265. doi: 10.1046/j.0022-202X.2004.22228.x. [DOI] [PubMed] [Google Scholar]
- 56.Robinet A, Millart H, Oszust F, Hornebeck W, Bellon G. Binding of elastin peptides to S-Gal protects the heart against ischemia/reperfusion injury by triggering the RISK pathway. FASEB J. 2007;21:1968–1978. doi: 10.1096/fj.06-6477com. [DOI] [PubMed] [Google Scholar]
- 57.Novaro V, Colman-Lerner A, Ortega FV, Jawerbaum A, Paz D, Lo Nostro F, Pustovrh C, Gimeno MF, Gonzalez E. Regulation of metalloproteinases by nitric oxide in human trophoblast cells in culture. Reprod Fertil Dev. 2001;13:411–420. doi: 10.1071/rd01036. [DOI] [PubMed] [Google Scholar]
- 58.Nakatsuka M, Asagiri K, Noguchi S, Habara T, Kudo T. Nafamostat mesilate, a serine protease inhibitor, suppresses lipopolysaccharide-induced nitric oxide synthesis and apoptosis in cultured human trophoblasts. Life Sci. 2000;67:1243–1250. doi: 10.1016/s0024-3205(00)00716-5. [DOI] [PubMed] [Google Scholar]
- 59.Rajagopalan S, Meng XP, Ramasamy S, Harrison DG, Galis ZS. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability J Clin Invest. 1996;98:2572–2579. doi: 10.1172/JCI119076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- 61.Erwin PA, Lin AJ, Golan DE, Michel T. Receptor-regulated dynamic S-nitrosylation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem. 2005;280:19888–19894. doi: 10.1074/jbc.M413058200. [DOI] [PubMed] [Google Scholar]
- 62.Erwin PA, Mitchell DA, Sartoretto J, Marletta MA, Michel T. Subcellular targeting and differential S-nitrosylation of endothelial nitric-oxide synthase. J Biol Chem. 2006;281:151–157. doi: 10.1074/jbc.M510421200. [DOI] [PubMed] [Google Scholar]
- 63.Mitchell DA, Erwin PA, Michel T, Marletta MA. S-Nitrosation and regulation of inducible nitric oxide synthase. Biochemistry. 2005;44:4636–4647. doi: 10.1021/bi0474463. [DOI] [PubMed] [Google Scholar]
- 64.Bulotta S, Cerullo A, Barsacchi R, Palma CD, Rotiroti D, Clementi E, Borgese N. Endothelial nitric oxide synthase is segregated from caveolin-1 and localizes to the leading edge of migrating cells. Exp Cell Res. 2006;312:877–889. doi: 10.1016/j.yexcr.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 65.Founds SA, Conley YP, Lyons-Weiler JF, Jeyabalan A, Hogge WA, Conrad KP. Altered global gene expression in first-trimester placentas of women destined to develop preeclampsia. Placenta. 2009;30:15–24. doi: 10.1016/j.placenta.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gronski TJ, Jr, Martin RL, Kobayashi DK, Walsh BC, Holman MC, Huber M, Van Wart HE, Shapiro SD. Hydrolysis of a broad spectrum of extracellular matrix proteins by human macrophage elastase. J Biol Chem. 1997;272:12189–12194. doi: 10.1074/jbc.272.18.12189. [DOI] [PubMed] [Google Scholar]