

Abstract
Cytomegalovirus (CMV) persistently infects more than 60% of the worldwide population. In immunocompetent hosts, it has been implicated in several diseases, including cardiovascular disease, possibly through the induction of inflammatory pathways. Cardiovascular risk factors promote an inflammatory phenotype in the microvasculature long before clinical disease is evident. This study determined whether CMV also impairs microvascular homeostasis and synergizes with hypercholesterolemia to exaggerate these responses. Intravital microscopy was used to assess endothelium-dependent and -independent arteriolar vasodilation and venular leukocyte and platelet adhesion in mice after injection with either mock inoculum or murine CMV (mCMV). Mice were fed a normal (ND) or high-cholesterol (HC) diet beginning at 5 weeks postinfection (p.i.), or a HC diet for the final 4 weeks of infection. mCMV-ND mice exhibited impaired endothelium-dependent vasodilation versus mock-ND at 9 and 12 weeks and endothelium-independent arteriolar dysfunction by 24 weeks. Transient mild leukocyte adhesion occurred in mCMV-ND venules at 7 and 21 weeks p.i. HC alone caused temporary arteriolar dysfunction and venular leukocyte and platelet recruitment, which were exaggerated and prolonged by mCMV infection. The time of introduction of HC after mCMV infection determined whether mCMV+HC led to worse venular inflammation than either factor alone. These findings reveal a proinflammatory influence of persistent mCMV on the microvasculature, and suggest that mCMV infection enhances microvasculature susceptibility to both inflammatory and thrombogenic responses caused by hypercholesterolemia.
Cytomegalovirus (CMV) is a β-herpesvirus that infects a majority of the world's population, primarily during early childhood. The virus establishes a lifelong infection, with periods of reactivation and latency.1 While it can cause severe disease in immunocompromised hosts, CMV infection is asymptomatic in immunocompetent individuals. However, evidence is accumulating that CMV may contribute to other diseases, including cardiovascular disease2–5 and inflammatory bowel disease.6–8 The prevailing thought is that CMV is playing a role in these other pathologies by promoting inflammation,9–11 a feature of this virus that is central to its dissemination and, therefore, survival strategy within the host.12,13 CMV can infect many different cell types that are involved in cardiovascular disease, including leukocytes and endothelial cells, and primary infection of these cells leads to cellular adhesion molecule (CAM) up-regulation,12,14–19 leukocyte18,20,21 and platelet adhesion,22 and cytokine release,15,17,20 all hallmarks of cardiovascular disease.23–29 While these cell culture and in vivo studies have significantly advanced our understanding of CMV-induced inflammatory pathways in terms of vascular responses, less is known about the specific impact of CMV infection on vascular homeostasis in intact vessels during persistent infection. Therefore we sought to characterize the responses of both the arteriolar and venular sides of the microvasculature to primary and persistent CMV infection.
The microvasculature is the site of events leading to tissue injury after exposure to inflammatory stimuli such as bacteria and ischemia/reperfusion. One of the first responses to an inflammatory signal is endothelial dysfunction, characterized by impaired endothelium-dependent vasodilation on the arteriolar side, and activation of the vascular endothelium in postcapillary venules resulting in the up-regulation of CAMs that support leukocyte and platelet recruitment. The microvessels in many organs have been shown to respond to cardiovascular risk factors in this manner.30–32 Not only are these responses evident long before large vessel disease and the associated clinical symptoms appear, but this low-grade inflammation predisposes the tissue to worse injury responses to other stimuli including ischemia-reperfusion.33–37 While little is known about the impact of CMV on the microvasculature, there is some evidence from transplanted organs that CMV infection is associated with thickening of the arteriolar walls during rejection,38 vasculopathy,39 and arteriolar dysfunction.40 The present study uses a murine model to systematically assess the microvascular responses to CMV infection over the course of 6 months (primary and persistent infection). To this end we measured arteriolar vasodilation responses to endothelium-dependent and -independent vasodilators, acetylcholine and papaverine, respectively, as well as leukocyte and platelet recruitment in postcapillary venules of mock-inoculated and murine CMV (mCMV)-infected mice. To reflect the asymptomatic infection seen in immunocompetent humans, we used a mouse strain (C57BL/6J) that is relatively resistant to CMV infection and is well characterized in terms of vascular responses to cardiovascular risk factors.
In terms of cardiovascular disease it is also noteworthy that the extent of disease is associated with risk factor burden.41 Because patients typically do not present to a cardiologist because of CMV infection, the possibility that CMV synergizes with other cardiovascular risk factors to induce microvascular inflammation should be considered. Such a scenario is supported by studies showing that mCMV infection accelerated atherosclerosis development in hyperlipidemic mice.42–45 In these studies, the use of genetically hyperlipidemic mice necessitated the introduction of mCMV when they were already becoming hypercholesterolemic, however a majority of people are infected with CMV during early childhood before circulating cholesterol levels are elevated. Therefore the second part of our study was designed to determine whether the prior presence of mCMV infection exacerbates HC-induced microvascular dysfunction and whether the timing of this hypercholesterolemia relative to mCMV infection influences this synergism.
Materials and Methods
Virus
A stock of the Smith strain of mCMV was grown on NIH3T3 cells. The supernatant and cell contents were then purified and concentrated on a sucrose gradient, and the virus was titered (as 8 × 107 plaque forming units), using a standard plaque assay on NIH3T3 cells as previously described,46 aliquoted, and stored at −80°C until required. Mock inoculum was similarly prepared using noninfected NIH3T3 cells.
Animals
Wild-type C57BL/6J mice (WT) were obtained from Jackson Laboratories (Bar Harbor, ME). Mice (3–5 weeks old) were injected with mock inoculum or 3 × 104 plaque forming units mCMV. Intravital microscopy was performed at different time points between 1 day and 24 weeks postinfection (p.i.). Mice were divided into two parallel substudies: i) to determine whether the presence of persistent mCMV infection prolonged hypercholesterolemia-induced inflammatory responses, mice were maintained on normal diet (ND) or placed on a high-cholesterol diet (HC) (Teklad 90221 containing 1.25% cholesterol, 15.8% fat and 0.125% choline chloride, Harlan Teklad, Madison, WI) at 5 weeks postinfection until observation at 7, 9, 13, 17 or 21 weeks p.i.; or ii) to determine whether the time of introduction of a fixed duration of diet altered microvascular responses, mice were either maintained on ND or placed on HC for the final 4 weeks before observation at 4, 6, 9, 12, 18, and 24 weeks p.i. n = 2–13 for all groups (groups containing <5 mice were almost exclusively mock-inoculated controls).
Surgical Protocol
At the time of experimentation, mice were anesthetized with ketamine hydrochloride (150 mg/kg body weight, i.p.) and xylazine (7.5 mg/kg body weight, i.p.). The right jugular vein was canulated for administration of platelets. Core body temperature was maintained at 35 ± 0.5°C. Animal handling procedures were approved by the LSU Health Sciences Center Institutional Animal Care and Use Committee and were in accordance with the guidelines of the American Physiological Society.
Intravital Microscopy
The cremaster muscle was prepared for intravital microscopy and superfused with bicarbonate buffered saline at a rate of 1 ml/min as described previously.32 Postcapillary venules were selected for observation. A leukocyte was considered adherent if it remained stationary for ≥30 seconds (#/mm2 vessel surface) and was measured throughout the observation period. Leukocyte emigration was measured online at the end of each 1-minute observation period. Emigrated leukocytes were expressed as the number of interstitial leukocytes per mm2 of view adjacent to the segment under observation (#/mm2 interstitium).
Platelets
Approximately 0.9 ml of blood was collected from a donor mouse via the carotid artery into 0.1 ml acid-citrate-dextrose (Sigma Chemicals Co., St. Louis, MO). Platelet donors matched the source of the recipients in terms of infection and diet. Platelets were isolated by a series of centrifugation steps and fluorescently labeled as described previously. This technique does not lead to platelet activation, as assessed by P-selectin expression.47 Platelets were considered saltating if they paused transiently (for ≥2 seconds but <30 s) on the vessel wall and firmly adherent if they arrested for ≥30 seconds. Total adhesion was calculated as the sum of saltation and firm adhesion, and all platelet parameters were expressed as # platelets/mm2.
Experimental Protocol
Postcapillary venules (20–40 μm diameter) with a wall shear rate of ≥500/s were studied. This threshold was selected based on previous reports describing a propensity for leukocytes and platelets to adhere in venules at low wall shear rates.48 To avoid bias and inflammation due to surgical trauma, the venule with the least number of adherent and emigrated leukocytes at the end of 30 minutes stabilization was chosen for the study. Platelets (108, in a volume of 120 μl) were infused via the jugular vein over 5 minutes and allowed to circulate for a further 5 minutes. One-minute recordings of the leukocytes (light microscopy) followed by 1-minute recordings of the platelets (fluorescent microscopy) were made of the first 100 μm of every 300 μm along the length of the unstimulated vessel, beginning as near to the source of the venule as possible. The mean value of each variable within a single venule was calculated, and comparisons were made between the experimental groups.
Once the venular data had been collected, the animals were allowed to stabilize for 20–30 minutes, and arterioles with diameters between 15–40 μm and wall shear rate of ≥500/s were chosen for study. Diameter and Vrbc were measured in the chosen sections before and after superfusion with 10−5 M of the endothelium-dependent vasodilator, acetylcholine (ACh), for 5 minutes. Arteriolar diameters were allowed to return to baseline with bicarbonate buffered saline superfusion, before papaverine (papav) was applied to test for maximal endothelium-independent vasodilation. Arteriolar vasorelaxation responses were expressed as the percentage diameter change versus baseline. Arterioles that were not responsive to papav were excluded from the study.
Serum Cholesterol Levels
Serum was frozen for subsequent measurement of cholesterol levels using a spectrophotometric assay (Sigma Chemicals Co.).
Blood Cell Counts
Blood was drawn from the heart at the end of the experiment for leukocyte (using crystal violet stain) and platelet counts (using the unopette system; BD Biosciences, San Jose, CA) with the aid of a hemocytometer.
Plaque Assays
NIH3T3 cells were propagated in the DMEM medium with 10% fetal bovine serum and 1:100 penicillin/streptomycin and passaged at multiplicity of 5–10. For plaque assays, cells were grown to 90% confluency in 48-well tissue culture plates. Samples of salivary glands, liver, spleen (primary organs of dissemination), and cremaster muscle (used for microvasculature observation) from mice infected with mCMV ± HC were analyzed using a standard plaque assay (4–7 per group). Tissues were weighed and homogenized in the sterile Hanks' balanced salt solution with 0.1% fetal bovine serum (Volume [μl] = tissue weight [mg] × 10), debris was removed by centrifugation, and three 10× serial dilutions of the supernatants from each sample were added to 3T3 monolayers (in duplicate). Plates were incubated at 37°C for 1–1.5 hours, overlayed with methylcellulose, and plaques were counted at 4–5 days after infection. Mock medium and mock organ homogenates were used as negative controls. Serial virus dilutions with known titer, and known virus titers added to mock liver samples before homogenizing were used as positive controls (virus titers corresponded to the amount of virus added to the tissue/well).
PCR for Viral DNA
PCR for viral DNA was performed on liver, spleen, and salivary glands from mCMV-ND and mCMV-HC mice at 9 weeks p.i., which coincided with the maximal overall microvascular response to infection. In addition, salivary glands and/or livers from other time points (6, 7, 13, and 21 weeks p.i.), corresponding to each individual peak response, were tested. Briefly, total DNA was isolated using a DNeasy Blood and Tissue Kit (Qiagen Inc., Valencia, CA). It was then analyzed by PCR using a GoTaq Green MasterMix (Promega Corp., Madison, WI) and primers for mCMV immediate early gene (Accession No. M11788) 5′-tca gcc atc aac tct gct acc aac-3′ and 5′-gtg cta gat tgt atc tgg tgc tcc tc-3′, and mouse β-actin (Accession No. M12481) 5′-gct gta ttc ccc tcc atc gtg-3′ and 5′-cac ggt tgg cct tag ggt tca-3′.49 Positive controls included organs from BALB/c mice at 4 days p.i., in which viral DNA was detected by this method.
Statistical Analysis
All values are reported as mean ± SEM. Analysis of variance with Fisher posthoc test was used for statistical comparison of experimental groups, with statistical significance set at P < 0.05. Please note that figures comparing Mock-ND and mCMV-ND groups are only presented for ease of comparison; the data in these graphs are derived from the combination of all ND groups in subsequent figures and therefore contain all time points. In one substudy, HC was introduced at 5 weeks postinfection until observation at 7, 9, 13, 17, or 21 weeks p.i. In the second substudy, mice were placed on HC for the final 4 weeks before observation at 4, 6, 9, 12, 18, and 24 weeks p.i. Data for the 9 weeks groups are common to all graphs. Because of the different time points tested, the patterns of microvascular responses in the CMV-ND group are distinct between the two substudies, therefore the effect of CMV alone on the microvasculature is interpreted from the first three figures.
Results
Plasma cholesterol levels were increased in HC-fed mice to almost twofold over normocholesterolemic values after 2 weeks of cholesterol-enriched diet and appeared to plateau by 4 weeks of HC (∼2.5-fold higher than ND). The presence of mCMV did not significantly alter cholesterol levels. No significant differences were observed for blood leukocyte counts between mock and mCMV-infected mice or ND and HC treatments (data not shown).
The Impact of mCMV on the Microvasculature
mCMV-Induced Arteriolar Dysfunction
Mock-inoculated mice exhibited comparable levels of vasodilation in response to acetylcholine at all time points examined. mCMV infection did not induce any discernable arteriolar dysfunction during primary infection (1–6 days p.i., data not shown), however by 6 weeks p.i., endothelium-dependent vasodilation responses were beginning to decrease when compared with mock-inoculated counterparts, and arteriolar dysfunction was most evident at 12 weeks p.i. This vasodilation response then recovered before arteriolar dysfunction was observed again at 24 weeks p.i. (Figure 1A). In contrast to the first few months p.i., when papav-induced dilation was comparable between mock and mCMV groups, these vasodilation responses were also reduced at 24 weeks p.i. (Figure 1B), to approximately the same extent as the ACh responses (shown in Figure 1A), suggesting that primarily endothelium-independent vasodilation was responsible for the arteriolar dysfunction induced by mCMV infection 6 months after initial exposure.
Figure 1.
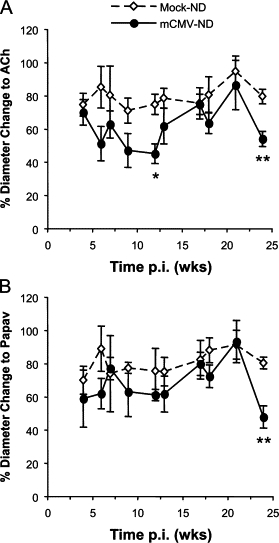
Arteriolar vasodilation responses to acetylcholine (ACh) (A) or papaverine (papav) (B) in mock-inoculated or mCMV-infected mice maintained on a normal diet (ND). Measurements (expressed as % change in diameter from baseline) were performed at different time points between 4 and 24 weeks p.i. The data in this figure are the combined ND data from both substudies. *P < 0.01 versus corresponding Mock-ND; **P < 0.005 versus corresponding Mock-ND.
mCMV-Induced Venular Blood Cell Recruitment
During the primary infection period (1, 2, 4, or 6 days p.i.), mCMV did not cause an increase in leukocyte recruitment when compared with corresponding mock controls (data not shown). Leukocyte adhesion was transiently elevated by mCMV infection at 7 weeks p.i., after which time this inflammation resolved before briefly increasing above mock levels again at 21 weeks p.i. (Figure 2A). Emigration followed a similar pattern, with a highly variable increase at 7 weeks p.i. and a significant rise in infiltrating leukocytes at 21 weeks of mCMV infection. Although there was a significant difference also observed at 13 weeks, this appeared to be primarily due to low emigration in the Mock-ND controls (Figure 2B). Platelet adhesion in postcapillary venules was not significantly raised by mCMV at any time point during primary (data not shown) or persistent infection, although there was a tendency for higher platelet adhesion at 7 and 13 weeks p.i. (Figure 3C). This was primarily due to increased brief interactions (≥2 s, <30 s) of the platelets with the vessel wall (Figure 3A) rather than firm adhesion (Figure 3B).
Figure 2.
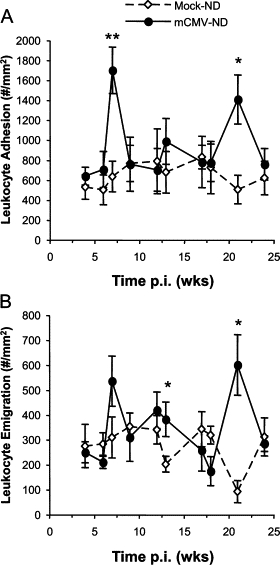
Leukocyte adhesion in postcapillary venules (#/mm2 vessel surface) (A) and emigration into the interstitium (#/mm2 interstitium) (B) measured in mock-inoculated or mCMV-infected mice maintained on a normal diet (ND) between 4 and 24 weeks p.i. The data shown are the combined data from all ND mice at all time points from the two substudies, which are represented separately in Figures 5 and 7. *P < 0.05 versus corresponding Mock-ND; **P < 0.01 versus corresponding Mock-ND.
Figure 3.
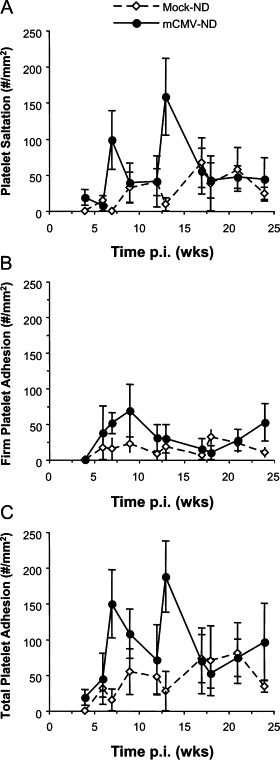
Time course of platelet recruitment in postcapillary venules of mice injected with mock inoculum or mCMV at wk 0. Platelet interactions are expressed as saltation (transient; A), firm adhesion (≥30 s; B), or total adhesion (C; #/mm2 vessel surface). The ND data collected at all time points in the two substudies are shown jointly here
mCMV Infection Exacerbates and Prolongs the Impact of Hypercholesterolemia on Microvascular Responses (Mice Placed on HC at 5 weeks p.i.)
Arteriolar Function
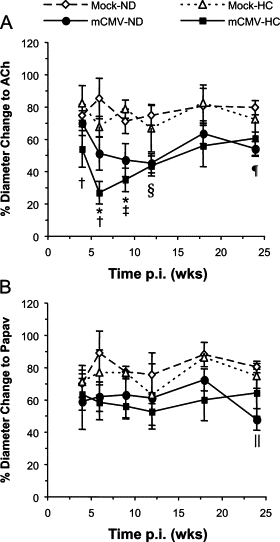
Focusing on the first substudy (7, 9, 13, 17, or 21 weeks p.i. only), in mock-inoculated mice, the introduction of HC at 5 weeks p.i. led to a mild transient impairment of vasodilation at 2 weeks of diet (7 weeks p.i.), which had corrected itself by 4 weeks of HC feeding (Figure 4A). Such dysfunction was apparent again at 8 and 12 weeks HC (13 and 17 weeks p.i.), although this did not reach significance. While mCMV infection did not alter the vasodilation responses to HC at 7 weeks p.i., the combination of mCMV and HC led to the worst impairment of all groups at 9 and 13 weeks p.i., which was significantly different from Mock-ND controls but not the CMV-only groups. However, by 21 weeks p.i. (16 weeks HC), arteriolar function in both mCMV-ND and Mock-HC groups had returned to normal levels, whereas the combination of mCMV+HC continued to cause impairment of vasodilation, which was significant versus all other groups, suggesting that mCMV and HC can indeed synergize to induce arteriolar dysfunction. Interestingly, although both the mCMV and mCMV+HC mediated impairment of dilation was endothelium-dependent at 9 weeks pi., this developed into endothelium-independent dysfunction at the later time points in the mCMV-HC groups (Figure 4B). Because papav acts directly on the vascular smooth muscle, this suggests that the combination of mCMV and HC accelerates the appearance of vascular smooth muscle dysfunction.
Figure 4.
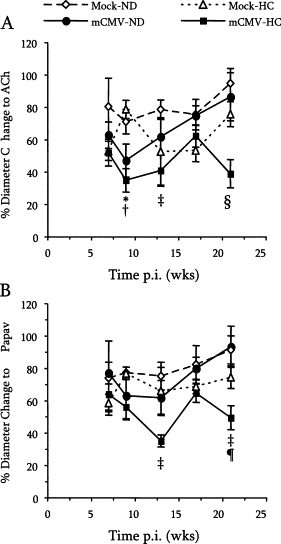
Endothelium-dependent (A) and -independent (B) vasodilatory responses to ACh or Papav, respectively in arterioles of mice exposed to mock inoculum or mCMV and maintained on a normal diet (ND) or placed on a high-cholesterol diet (HC) at 5 weeks p.i. before observation at 7, 9, 13, 17, or 21 weeks p.i. Data here are also represented in Figure 1. *P < 0.05 mCMV-ND and mCMV-HC versus Mock-ND; †P < 0.01 mCMV-ND and mCMV-HC versus Mock-HC; ‡P < 0.05 mCMV-HC versus Mock-ND; §P < 0.05 versus all other groups; ¶P < 0.05 mCMV-HC versus mCMV-ND (comparisons made within each time point).
Venular Blood Cell Recruitment
Feeding mock-inoculated mice a HC diet led to a significant increase in leukocyte adhesion at 2 weeks HC (7 weeks p.i.), to a level comparable to that observed in the mCMV-ND group (Figure 5A). As in the mCMV-ND group, this inflammation resolved thereafter, although it began to increase again after 12–16 weeks of HC. However, when mice were infected with mCMV and placed on HC at 5 weeks p.i., the level of leukocyte adhesion was significantly higher than Mock-ND at 7 weeks p.i., and approximately 50% higher when compared with either risk factor alone. Although the inflammation in the single-risk factor groups fell to control levels by 9 weeks, the combination of mCMV and HC led to maintenance of leukocyte adhesion above all other groups. By 17 weeks p.i., partial resolution of this inflammation was observed. Leukocyte emigration followed a similar pattern albeit it fell toward baseline earlier than the adhesion (Figure 5B). However, in both single factor groups (mCMV alone and mock with 16 weeks HC) and in CMV+HC mice, adhesion and emigration appeared to be rising again by 21 weeks p.i. reaching significance in the CMV+HC group. Platelets behaved somewhat differently to the leukocytes (Figure 5C). Two weeks HC (7 weeks post mock-inoculation) led to a moderate increase in platelet adhesion that resolved thereafter. When mice were exposed to both mCMV and HC, platelet adhesion was significantly elevated versus Mock-ND at 7 weeks p.i. (2 weeks HC), and versus all groups by 9 weeks p.i. (4 weeks HC). However, this did not last, and recruitment of platelets had returned toward baseline levels by 13 weeks p.i. (8 weeks HC).
Figure 5.
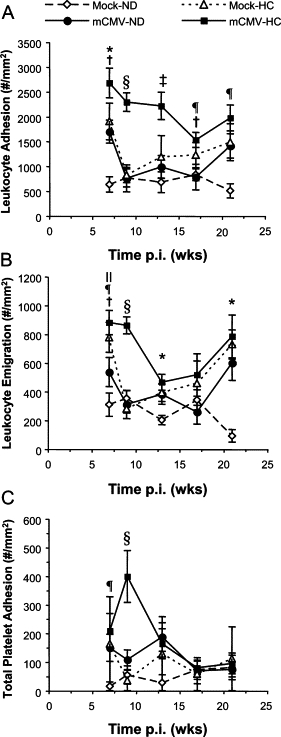
Leukocyte adhesion (#/mm2 vessel surface) (A) and emigration (#/mm2 interstitium) (B), and total platelet adhesion (#/mm2 vessel surface) (C) in postcapillary venules of mice injected with mock inoculum or mCMV at Day 0. Mice were either kept on normal chow (ND) for the entire period or changed to a high-cholesterol diet (HC) at 5 weeks p.i. before observation at 7, 9, 13, 17, or 21 weeks p.i. Data here are also represented in Figures 2 and 3. *P < 0.05 Mock-ND versus all other groups; †P < 0.05 mCMV-HC versus mCMV-ND; ‡P < 0.05 versus all other groups; §P < 0.001 versus all other groups; ¶P < 0.05 mCMV-HC versus Mock-ND; ∥P < 0.01 Mock-HC versus Mock-ND (comparisons made within each time point).
Time Post-Infection Determines the Impact of Hypercholesterolemia on Microvascular Responses
To determine whether the synergism between mCMV and HC is dependent on when the HC is introduced relative to time p.i. with mCMV, we placed mice on ND or HC during the final 4 weeks before observation at 4, 6, 9, 12, 18 and 24 weeks p.i. 4 weeks of HC was chosen because both arteriolar responses to ACh and venular blood cell recruitment were not altered in mock mice at that time of HC (versus mock-ND).
Arteriolar Function
Arteriolar dilation to ACh was comparable between Mock-ND and Mock-HC groups at all time points examined (Figure 6A). The combination of mCMV-HC resulted in the worst impairment at almost all of these time points and was significant versus Mock-ND between 6 and 12 weeks p.i. However, this never reached significance versus responses in mCMV-ND arterioles, suggesting that mCMV is the primary determinant for the changes in arteriolar function in mice early after hypercholesterolemia and infection. In addition, the reduced dilation observed in the mCMV-HC group could be largely attributed to endothelium-dependent impairment at all time points examined because vasodilation responses to papav were not significantly different from those in corresponding Mock-ND groups (Figure 6B).
Figure 6.
Vasodilation responses to ACh (endothelium-dependent) (A) or papav (endothelium-independent) (B) measured in arterioles of mice over a 6-month period after injection with mock inoculum or mCMV. Mice from both groups were maintained on a normal diet (ND) for the entire period or switched to a high-cholesterol diet (HC) for the final 4 weeks before observation at 4, 6, 9, 12, 18, and 24 weeks p.i. Data here are also represented in Figure 1. *P < 0.05 mCMV-ND and mCMV-HC versus Mock-ND; †P < 0.05 mCMV-HC versus Mock-HC; ‡P < 0.01 mCMV-ND and mCMV-HC versus Mock-HC; §P < 0.05 mCMV-HC versus Mock-ND; ¶P < 0.05 mCMV-ND versus both Mock groups; ∥P < 0.05 mCMV-ND versus Mock-ND (comparisons made within each time point).
Venular Blood Cell Recruitment
As expected, neither HC nor mCMV alone induced significant leukocyte adhesion or emigration at the time points examined (this substudy did not include 7 and 21 weeks p.i. at which mCMV alone increased leukocyte adhesion). Interestingly, in contrast to the arteriolar side, the timing of when mice began HC feeding clearly had a critical impact on the synergism between mCMV infection and HC with respect to the inflammatory response in venules, in that feeding HC for the final 4 weeks before observation led to significant leukocyte adhesion (Figure 7A) and emigration (Figure 7B) by 6 weeks, peaking at 9 weeks, and starting to fall by 12 weeks p.i., when compared to all other groups, reaching Mock-ND levels by 24 weeks p.i. Adhesion was quicker to rise and took longer to fully resolve than emigration. Platelet adhesion exhibited a muted version of the pattern of leukocyte responses, in that platelet recruitment began to rise at 6 weeks p.i., primarily due to firm adhesion being significantly higher in mCMV+HC mice than in all other groups at that time point (Figure 7C). Platelet adhesion peaked at 9 weeks, with both saltating and firm adhesion being significantly greater than mock-ND controls, or mice exposed to either risk factor alone. However, by 12 weeks p.i., platelet recruitment appeared to returning toward control values and it had reached near to Mock-ND levels by 24 weeks p.i.
Figure 7.
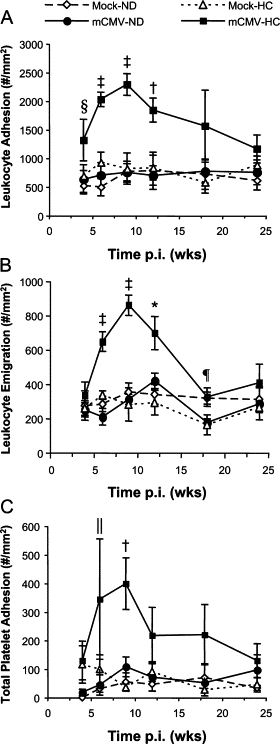
Blood cell recruitment in postcapillary venules of mice injected with mock inoculum or infected with mCMV and kept on a normal diet (ND) or placed on a high-cholesterol diet (HC) for the final 4 weeks before observation. Leukocyte adhesion (#/mm2 vessel surface) (A) and emigration (#/mm2 interstitium) (B), and platelet adhesion (#/mm2 vessel surface) (C) were measured at 4, 6, 9, 12, 18, and 24 weeks after infection. Data here are also represented in Figures 2 and 3. *P < 0.05 mCMV-HC versus all other groups; †P < 0.005 mCMV-HC versus all other groups; ‡P ≤ 0.0001 mCMV-HC versus all other groups; §P < 0.05 mCMV-HC versus Mock-ND and mCMV-ND; ¶P < 0.05 mCMV-HC versus Mock-HC and mCMV-ND; ∥P < 0.05 mCMV-HC versus Mock-ND (comparisons made within each time point).
Assessment of Infectious Virus
Salivary glands, liver, spleen, and cremaster samples from mice infected with mCMV and fed with normal and high cholesterol diets were analyzed in plaque assays at most of the time points used for intravital microscopy, including those where microvascular responses were maximum (ie, 9 weeks post-mCMV infection [4 weeks HC]). No replicating virus was detected in any of the samples analyzed (data not shown). We also tested multiple organs at 6, 7, 9, 13, and 21 weeks p.i. for the presence of viral DNA using PCR and were unable to detect virus at any of the times examined in either normocholesterolemic or hypercholesterolemic mCMV-infected groups.
Discussion
Cytomegalovirus infects a majority of the population and has been implicated in many inflammatory diseases, including cardiovascular disease.2–5 One way in which CMV may contribute to cardiovascular disease is through the activation of inflammatory pathways (eg, CAM up-regulation, leukocyte and platelet adhesion, and cytokine release).12,14,15,17–20 Many of these responses have been shown to participate in the pathogenesis of cardiovascular disease. Furthermore they are evident in the microvasculature, manifesting as a dysfunctional phenotype, in response to cardiovascular risk factors long before clinical signs of disease are seen.29–32 However little is known about the impact of CMV on arterioles and venules in vivo. We have characterized the microvascular responses to primary and persistent mCMV for the first time and now show that mCMV can impair vasodilation responses primarily to endothelium-dependent factors, but also to endothelium-independent stimuli. In addition, mCMV has a mild transient inflammatory impact on postcapillary venules that appears to show some periodicity during persistent infection. Because higher risk factor burden is associated with worse outcome in cardiovascular disease,41 we also sought to determine whether mCMV synergized with another cardiovascular risk factor to exacerbate microvascular dysfunction. Our findings indicate that mCMV infection together with hypercholesterolemia produces slightly worse arteriolar dysfunction and prolonged and exaggerated leukocyte and platelet recruitment in venules, and that in the case of venules, the timing of the hypercholesterolemia relative to mCMV infection was critical in generating the strongest inflammatory and thrombogenic responses.
While information about the impact of CMV on the microvasculature is limited, in particular in vivo, more is known about its effect on larger vessels. Gombos et al (2009) reported that methacholine-induced vasodilation of mesenteric and uterine arteries was accentuated in the first few weeks following mCMV infection.50 Our new data presented here appear to be in contrast to this published study. The differences may stem from the use of different strains of virus, different virus doses, female versus male mice, and/or different time points p.i. to assess dilation in isolated arteries versus intact arterioles. On the other hand, our findings showing that persistent mCMV infection led to impaired arteriolar dysfunction are largely in agreement with human studies (Figure 1). For example, in transplanted organs, microvascular endothelial function is compromised in patients with a history of CMV disease.40 Grahame-Clarke et al found that CMV seropositivity was associated with impaired endothelium-dependent arterial function in healthy or diabetic individuals,51 albeit they found that dilation to bradykinin, but not ACh, was reduced. Two other results from our study also support what they found. First, at most time points examined, CMV infection did not synergize with another cardiovascular risk factor (diabetes in their case and hypercholesterolemia in our case) to significantly exacerbate the vasodilation dysfunction (Figures 4 and 6), although in the human study the diabetes would have been at different stages, whereas our study could be controlled for that issue and only at 21 weeks p.i. with prolonged HC was a significantly worse impairment of vasodilation observed versus either risk factor alone (Figure 4). Second, there was evidence for endothelium-independent arteriolar dysfunction (tested with glycerol trinitrite in the human study), which we found at 24 weeks p.i. using papav in CMV-ND mice (Figure 1) and at 13 and 21 weeks in the mCMV-infected prolonged HC mice (Figure 4). Thus our findings offer the first direct evidence for a deleterious effect of persistent mCMV infection on arteriolar responses, which supports several studies in humans showing an association between arteriolar/arterial impairment and CMV infection.
There are some data showing that leukocytes and platelets exhibit enhanced adhesion to venous and microvascular endothelial cells during primary CMV infection,12,16,18,22 and in fact in normocholesterolemic rats, CMV promotes a low level of leukocyte recruitment in the aorta during primary infection.52,53 Although we did not find significant blood cell adhesion in either postcapillary venules or arterioles in our model during the first week after CMV infection, perhaps due to the strain of mouse, or vascular bed examined, we were able to detect low levels of leukocyte recruitment in postcapillary venules over the observed period of persistent infection. These elevations in leukocyte adhesion and emigration at 7 and 21 weeks p.i. were transient in nature (Figure 2). Whether they represent different leukocyte populations remains to be elucidated. The appearance of these inflammatory infiltrates did not seem to correspond to reactivation of the virus in the organs we tested. Although we cannot discount that the replication was below the detection limit of our assay (which we determined to be 500 plaque forming units/g tissue), or that organs other than the salivary gland, liver, spleen, and cremaster harbored replicating virus, CMV can also induce inflammatory pathways in the absence of replication. For example, replication-deficient CMV (UV-inactivated virus) was shown to enhance the adhesion of infected monocytes to endothelial cells, via an NF-κB pathway,54 and CMV also increases the generation of inflammatory cytokines from monocytes by binding toll-like receptor 2 on the monocyte surface.55 Therefore it is plausible that reactivation of the virus is not necessary for the responses obtained in our study. Furthermore, in light of our lack of detection of viral DNA in these mice at the time points where microvascular responses were observed, an alternative possibility is that primary mCMV infection initiates a series of epigenetic changes and/or immune responses that in turn are responsible for the subsequent microvascular dysfunction and inflammation, and do not require prolonged detectable presence of virus. These potential pathways require further investigation. Nonetheless, the inflammatory response to CMV infection that we observed in immunocompetent mice supports the prevailing thought that this virus may contribute to chronic diseases through the induction of inflammatory pathways (either directly or indirectly), and provides a model for testing the underlying mechanisms of these actions.
CMV has been associated with enhanced CAM expression and inflammation after transplantation,56 suggesting this virus can exacerbate responses to other inflammatory stimuli. In individuals with cardiovascular disease, CMV nucleic acids have been localized to atherosclerotic lesions.57,58 While this does not indicate whether CMV is simply a bystander or is actively contributing to disease, the latter possibility is supported by animal studies where the progression and size of atherosclerotic lesions in hyperlipidemic animals were increased after infection with mCMV.42–45 In atherosclerosis-prone ApoE−/− mice, mCMV infection was accompanied by an early elevation in serum tumor necrosis factor-α and interferon-γ levels, which was followed by enhanced local cytokine generation and T cell infiltration in early lesions of infected mice when compared with mock controls.43,44 Our findings in postcapillary venules of mCMV-infected hypercholesterolemic mice that leukocyte recruitment was higher in mice exposed to both risk factors when compared with either alone at many time points (both substudies, Figures 5 and 7) are in agreement with the notion that CMV and HC may synergize to exacerbate inflammatory responses. While much of the evidence supporting the ability for CMV to promote CAM up-regulation and leukocyte adhesion is derived from studies of primary CMV infection, the fact that persistent mCMV infection can exacerbate responses to hypercholesterolemia could have important implications in cardiovascular disease, where the virus may represent a therapeutic target. An additional novel finding in our study was that, like leukocytes, the significant platelet adhesion induced by mCMV+HC at 6 and 9 weeks p.i. (Figure 7) was more than the sum of platelet adhesion to either stimulus alone. Interestingly, the peak platelet recruitment coincided with peak leukocyte adhesion, but the platelet response was more short-lived. Whether this reflects an inability of CMV infection to directly activate platelets, as shown by Rahbar et al22 or whether the platelets require a higher threshold of, or different, circulating factors to induce their recruitment remains to be elucidated. Alternatively, the population of recruited leukocytes may change over time, which in turn may impact the thrombogenic responses. Nonetheless, in light of the overwhelming evidence that platelets can promote inflammation and inflammation can promote platelet activation and adhesion,59–61 this represents an additional component that might explain the influence of CMV on microvascular inflammation and dysfunction and perhaps cardiovascular disease.
In our model, the timing of the introduction of HC had a major impact on the likelihood of a synergism being found between CMV infection and acute (4 weeks) hypercholesterolemia in postcapillary venules. Although we could not determine whether peak inflammation correlated with virus reactivation, the impact of this timing difference could make it more difficult to detect synergism between CMV infection and other inflammatory stimuli in studies with just a few time points, or in human studies. This could in turn affect our interpretation of whether or how CMV infection exacerbates responses to other stimuli. While we are cautious about extrapolating this dependence on time postinfection to chronic diseases such as cardiovascular disease, which takes decades to develop, and in which each risk factor persists for long periods, this observation may have bearing on the susceptibility of the microvasculature, and therefore organ(s), to more acute injuries such as ischemia or sepsis.
In conclusion we provide the first evidence for arteriolar dysfunction and mild transient venular inflammatory and thrombogenic responses during persistent mCMV infection in the absence of other factors. The peak responses to virus did not coincide with detectable viral replication. Whether intermittent low-level reactivation of virus, low-grade continuation of the initial immune response to mCMV, or early mCMV-induced epigenetic changes are responsible for these microvascular responses to mCMV remains to be elucidated. We also provide evidence that mCMV not only enhances but also prolongs hypercholesterolemia-induced inflammation in postcapillary venules and this peaks alongside a transient platelet recruitment. Furthermore, the timing of the introduction of HC diet during mCMV infection was critical for the initial prolongation of the HC-induced inflammation, although it is unclear whether the influence of this timing continues over the course of chronic exposure to both stimuli. Taken together these data support epidemiological studies showing an association between CMV and vascular dysfunction or cardiovascular disease and suggest this virus may be a feasible therapeutic target in the treatment of chronic disease in infected individuals.
Acknowledgements
We acknowledge the assistance of Eunice Johnson and Candiss Hamric in performing the work contained in this manuscript.
Footnotes
Supported by grants from the American Heart Association (0565285B and 0730294N to K.Y.S.) and National Institutes of Health (P20RR018724 to K.Y.S.; AI-056077 and HD-051998 to A.D.Y.).
References
- 1.Bruggeman CA. Cytomegalovirus and latency: an overview. Virchows Arch B Cell Pathol Incl Mol Pathol. 1993;64:325–333. doi: 10.1007/BF02915131. [DOI] [PubMed] [Google Scholar]
- 2.Chiu B, Viira E, Tucker W, Fong IW. Chlamydia pneumoniae, cytomegalovirus, and herpes simplex virus in atherosclerosis of the carotid artery. Circulation. 1997;96:2144–2148. doi: 10.1161/01.cir.96.7.2144. [DOI] [PubMed] [Google Scholar]
- 3.Blum A, Giladi M, Weinberg M, Kaplan G, Pasternack H, Laniado S, Miller H. High anti-cytomegalovirus (CMV) IgG antibody titer is associated with coronary artery disease and may predict post-coronary balloon angioplasty restenosis. Am J Cardiol. 1998;81:866–868. doi: 10.1016/s0002-9149(98)00019-8. [DOI] [PubMed] [Google Scholar]
- 4.Espinola-Klein C, Rupprecht HJ, Blankenberg S, Bickel C, Kopp H, Victor A, Hafner G, Prellwitz W, Schlumberger W, Meyer J. Impact of infectious burden on progression of carotid atherosclerosis. Stroke. 2002;33:2581–2586. doi: 10.1161/01.str.0000034789.82859.a4. [DOI] [PubMed] [Google Scholar]
- 5.Grattan MT, Moreno-Cabral CE, Starnes VA, Oyer PE, Stinson EB, Shumway NE. Cytomegalovirus infection is associated with cardiac allograft rejection and atherosclerosis. JAMA. 1989;261:3561–3566. [PubMed] [Google Scholar]
- 6.Kishore J, Ghoshal U, Ghoshal UC, Krishnani N, Kumar S, Singh M, Ayyagari A. Infection with cytomegalovirus in patients with inflammatory bowel disease: prevalence, clinical significance and outcome. J Med Microbiol. 2004;53:1155–1160. doi: 10.1099/jmm.0.45629-0. [DOI] [PubMed] [Google Scholar]
- 7.Papadakis KA, Tung JK, Binder SW, Kam LY, Abreu MT, Targan SR, Vasiliauskas EA. Outcome of cytomegalovirus infections in patients with inflammatory bowel disease. Am J Gastroenterol. 2001;96:2137–2142. doi: 10.1111/j.1572-0241.2001.03949.x. [DOI] [PubMed] [Google Scholar]
- 8.Soderberg-Naucler C. HCMV microinfections in inflammatory diseases and cancer. J Clin Virol. 2008;41:218–223. doi: 10.1016/j.jcv.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 9.Vliegen I, Duijvestijn A, Stassen F, Bruggeman C. Murine cytomegalovirus infection directs macrophage differentiation into a pro-inflammatory immune phenotype: implications for atherogenesis. Microbes Infect. 2004;6:1056–1062. doi: 10.1016/j.micinf.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 10.Freeman RB., Jr The ‘indirect’ effects of cytomegalovirus infection. Am J Transplant. 2009;9:2453–2458. doi: 10.1111/j.1600-6143.2009.02824.x. [DOI] [PubMed] [Google Scholar]
- 11.Zhu J, Quyyumi AA, Norman JE, Csako G, Epstein SE. Cytomegalovirus in the pathogenesis of atherosclerosis: the role of inflammation as reflected by elevated C-reactive protein levels. J Am Coll Cardiol. 1999;34:1738–1743. doi: 10.1016/s0735-1097(99)00410-6. [DOI] [PubMed] [Google Scholar]
- 12.Bentz GL, Jarquin-Pardo M, Chan G, Smith MS, Sinzger C, Yurochko AD. Human cytomegalovirus (HCMV) infection of endothelial cells promotes naive monocyte extravasation and transfer of productive virus to enhance hematogenous dissemination of HCMV. J Virol. 2006;80:11539–11555. doi: 10.1128/JVI.01016-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith MS, Bentz GL, Alexander JS, Yurochko AD. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J Virol. 2004;78:4444–4453. doi: 10.1128/JVI.78.9.4444-4453.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burns LJ, Pooley JC, Walsh DJ, Vercellotti GM, Weber ML, Kovacs A. Intercellular adhesion molecule-1 expression in endothelial cells is activated by cytomegalovirus immediate early proteins.[see comment] Transplantation. 1999;67:137–144. doi: 10.1097/00007890-199901150-00023. [DOI] [PubMed] [Google Scholar]
- 15.Dengler TJ, Raftery MJ, Werle M, Zimmermann R, Schonrich G. Cytomegalovirus infection of vascular cells induces expression of pro-inflammatory adhesion molecules by paracrine action of secreted interleukin-1beta. Transplantation. 2000;69:1160–1168. doi: 10.1097/00007890-200003270-00022. [DOI] [PubMed] [Google Scholar]
- 16.Knight DA, Waldman WJ, Sedmak DD. Cytomegalovirus-mediated modulation of adhesion molecule expression by human arterial and microvascular endothelial cells. Transplantation. 1999;68:1814–1818. doi: 10.1097/00007890-199912150-00030. [DOI] [PubMed] [Google Scholar]
- 17.Ricotta D, Alessandri G, Pollara C, Fiorentini S, Favilli F, Tosetti M, Mantovani A, Grassi M, Garrafa E, Dei Cas L, Muneretto C, Caruso A. Adult human heart microvascular endothelial cells are permissive for non-lytic infection by human cytomegalovirus. Cardiovasc Res. 2001;49:440–448. doi: 10.1016/s0008-6363(00)00258-3. [DOI] [PubMed] [Google Scholar]
- 18.Shahgasempour S, Woodroffe SB, Garnett HM. Alterations in the expression of ELAM-1. ICAM-1 and VCAM-1 after in vitro infection of endothelial cells with a clinical isolate of human cytomegalovirus. Microbiol Immunol. 1997;41:121–129. doi: 10.1111/j.1348-0421.1997.tb01177.x. [DOI] [PubMed] [Google Scholar]
- 19.Span AH, Mullers W, Miltenburg AM, Bruggeman CA. Cytomegalovirus induced PMN adherence in relation to an ELAM-1 antigen present on infected endothelial cell monolayers. Immunology. 1991;72:355–360. [PMC free article] [PubMed] [Google Scholar]
- 20.Craigen JL, Yong KL, Jordan NJ, MacCormac LP, Westwick J, Akbar AN, Grundy JE. Human cytomegalovirus infection up-regulates interleukin-8 gene expression and stimulates neutrophil transendothelial migration. Immunology. 1997;92:138–145. doi: 10.1046/j.1365-2567.1997.00310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knight DA, Briggs BR, Bennett CF, Harindranath N, Waldman WJ, Sedmak DD. Attenuation of cytomegalovirus-induced endothelial intercellular adhesion molecule-1 mRNA/protein expression and T lymphocyte adhesion by a 2′-O-methoxyethyl antisense oligonucleotide. Transplantation. 2000;69:417–426. doi: 10.1097/00007890-200002150-00019. [DOI] [PubMed] [Google Scholar]
- 22.Rahbar A, Soderberg-Naucler C. Human cytomegalovirus infection of endothelial cells triggers platelet adhesion and aggregation. J Virol. 2005;79:2211–2220. doi: 10.1128/JVI.79.4.2211-2220.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dansky HM, Barlow CB, Lominska C, Sikes JL, Kao C, Weinsaft J, Cybulsky MI, Smith JD. Adhesion of monocytes to arterial endothelium and initiation of atherosclerosis are critically dependent on vascular cell adhesion molecule-1 gene dosage. Arterioscler Thromb Vasc Biol. 2001;21:1662–1667. doi: 10.1161/hq1001.096625. [DOI] [PubMed] [Google Scholar]
- 24.Kitagawa K, Matsumoto M, Sasaki T, Hashimoto H, Kuwabara K, Ohtsuki T, Hori M. Involvement of ICAM-1 in the progression of atherosclerosis in APOE-knockout mice. Atherosclerosis. 2002;160:305–310. doi: 10.1016/s0021-9150(01)00587-1. [DOI] [PubMed] [Google Scholar]
- 25.Huo Y, Ley KF. Role of platelets in the development of atherosclerosis. Trends Cardiovasc Med. 2004;14:18–22. doi: 10.1016/j.tcm.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 26.Nageh MF, Sandberg ET, Marotti KR, Lin AH, Melchior EP, Bullard DC, Beaudet AL. Deficiency of inflammatory cell adhesion molecules protects against atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 1997;17:1517–1520. doi: 10.1161/01.atv.17.8.1517. [DOI] [PubMed] [Google Scholar]
- 27.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 28.Collins RG, Velji R, Guevara NV, Hicks MJ, Chan L, Beaudet AL. P-Selectin or intercellular adhesion molecule (ICAM)-1 deficiency substantially protects against atherosclerosis in apolipoprotein E-deficient mice. J Exp Med. 2000;191:189–194. doi: 10.1084/jem.191.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stokes K, Granger DN. The microcirculation: a motor for the systemic inflammatory response and large vessel disease induced by hypercholesterolemia? J Physiol. 2005;562:647–653. doi: 10.1113/jphysiol.2004.079640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishikawa M, Stokes KY, Zhang JH, Nanda A, Granger DN. Cerebral microvascular responses to hypercholesterolemia: roles of NADPH oxidase and P-selectin. Circ Res. 2004;94:239–244. doi: 10.1161/01.RES.0000111524.05779.60. [DOI] [PubMed] [Google Scholar]
- 31.Nellore K, Harris NR. Inhibition of leukocyte adherence enables venular control of capillary perfusion in streptozotocin-induced diabetic rats. Microcirculation. 2004;11:645–654. doi: 10.1080/10739680490517668. [DOI] [PubMed] [Google Scholar]
- 32.Stokes KY, Calahan L, Russell JM, Gurwara S, Granger DN. Role of platelets in hypercholesterolemia-induced leukocyte recruitment and arteriolar dysfunction. Microcirculation. 2006;13:377–388. doi: 10.1080/10739680600745877. [DOI] [PubMed] [Google Scholar]
- 33.Granger DN. Ischemia-reperfusion: mechanisms of microvascular dysfunction and the influence of risk factors for cardiovascular disease. Microcirculation. 1999;6:167–178. [PubMed] [Google Scholar]
- 34.Harris AG, Skalak TC, Hatchell DL. Leukocyte-capillary plugging and network resistance are increased in skeletal muscle of rats with streptozotocin-induced hyperglycemia. Int J Microcirc Clin Exp. 1994;14:159–166. doi: 10.1159/000178824. [DOI] [PubMed] [Google Scholar]
- 35.Kurose I, Wolf RE, Grisham MB, Granger DN. Hypercholesterolemia enhances oxidant production in mesenteric venules exposed to ischemia/reperfusion. Arterioscler Thromb Vasc Biol. 1998;18:1583–1588. doi: 10.1161/01.atv.18.10.1583. [DOI] [PubMed] [Google Scholar]
- 36.Mori N, Horie Y, Gerritsen ME, Granger DN. Ischemia-reperfusion induced microvascular responses in LDL-receptor -/- mice. Am J Physiol. 1999;276:H1647–H1654. doi: 10.1152/ajpheart.1999.276.5.H1647. [DOI] [PubMed] [Google Scholar]
- 37.Panes J, Kurose I, Rodriguez-Vaca D, Anderson DC, Miyasaka M, Tso P, Granger DN. Diabetes exacerbates inflammatory responses to ischemia-reperfusion. Circulation. 1996;93:161–167. doi: 10.1161/01.cir.93.1.161. [DOI] [PubMed] [Google Scholar]
- 38.Helantera I, Koskinen P, Tornroth T, Loginov R, Gronhagen-Riska C, Lautenschlager I. The impact of cytomegalovirus infections and acute rejection episodes on the development of vascular changes in 6-month protocol biopsy specimens of cadaveric kidney allograft recipients. Transplantation. 2003;75:1858–1864. doi: 10.1097/01.TP.0000064709.20841.E1. [DOI] [PubMed] [Google Scholar]
- 39.Koskinen PK, Nieminen MS, Krogerus LA, Lemstrom KB, Mattila SP, Hayry PJ, Lautenschlager IT. Cytomegalovirus infection accelerates cardiac allograft vasculopathy: correlation between angiographic and endomyocardial biopsy findings in heart transplant patients. Transpl Int. 1993;6:341–347. doi: 10.1007/BF00335972. [DOI] [PubMed] [Google Scholar]
- 40.Petrakopoulou P, Kubrich M, Pehlivanli S, Meiser B, Reichart B, von Scheidt W, Weis M. Cytomegalovirus infection in heart transplant recipients is associated with impaired endothelial function. Circulation. 2004;110:II207–II212. doi: 10.1161/01.CIR.0000138393.99310.1c. [DOI] [PubMed] [Google Scholar]
- 41.Marma AK, Lloyd-Jones DM. Systematic examination of the updated Framingham heart study general cardiovascular risk profile. Circulation. 2009;120:384–390. doi: 10.1161/CIRCULATIONAHA.108.835470. [DOI] [PubMed] [Google Scholar]
- 42.Burnett MS, Gaydos CA, Madico GE, Glad SM, Paigen B, Quinn TC, Epstein SE. Atherosclerosis in apoE knockout mice infected with multiple pathogens. J Infect Dis. 2001;183:226–231. doi: 10.1086/317938. [DOI] [PubMed] [Google Scholar]
- 43.Vliegen I, Duijvestijn A, Grauls G, Herngreen S, Bruggeman C, Stassen F. Cytomegalovirus infection aggravates atherogenesis in apoE knockout mice by both local and systemic immune activation. Microbes Infect. 2004;6:17–24. doi: 10.1016/j.micinf.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 44.Vliegen I, Stassen F, Grauls G, Blok R, Bruggeman C. MCMV infection increases early T-lymphocyte influx in atherosclerotic lesions in apoE knockout mice. J Clin Virol. 2002;25(Suppl 2):S159–S171. doi: 10.1016/s1386-6532(02)00095-1. [DOI] [PubMed] [Google Scholar]
- 45.Hsich E, Zhou YF, Paigen B, Johnson TM, Burnett MS, Epstein SE. Cytomegalovirus infection increases development of atherosclerosis in Apolipoprotein-E knockout mice. Atherosclerosis. 2001;156:23–28. doi: 10.1016/s0021-9150(00)00608-0. [DOI] [PubMed] [Google Scholar]
- 46.Brune W, Hengel H, Koszinowski UH. A mouse model for cytomegalovirus infection. Curr Protoc Immunol. 2001 doi: 10.1002/0471142735.im1907s43. Chapter 19:Unit 19. [DOI] [PubMed] [Google Scholar]
- 47.Cooper D, Chitman KD, Williams MC, Granger DN. Time-dependent platelet-vessel wall interactions induced by intestinal ischemia-reperfusion. Am J Physiol Gastrointest Liver Physiol. 2003;284:G1027–G1033. doi: 10.1152/ajpgi.00457.2002. [DOI] [PubMed] [Google Scholar]
- 48.Russell J, Cooper D, Tailor A, Stokes KY, Granger DN. Low venular shear rates promote leukocyte-dependent recruitment of adherent platelets. Am J Physiol Gastrointest Liver Physiol. 2003;284:G123–G129. doi: 10.1152/ajpgi.00303.2002. [DOI] [PubMed] [Google Scholar]
- 49.Wheat RL, Clark PY, Brown MG. Quantitative measurement of infectious murine cytomegalovirus genomes in real-time PCR. J Virol Methods. 2003;112:107–113. doi: 10.1016/s0166-0934(03)00197-6. [DOI] [PubMed] [Google Scholar]
- 50.Gombos RB, Wolan V, McDonald K, Hemmings DG. Impaired vascular function in mice with an active cytomegalovirus infection. Am J Physiol Heart Circ Physiol. 2009;296:H937–H945. doi: 10.1152/ajpheart.01027.2008. [DOI] [PubMed] [Google Scholar]
- 51.Grahame-Clarke C, Chan NN, Andrew D, Ridgway GL, Betteridge DJ, Emery V, Colhoun HM, Vallance P. Human cytomegalovirus seropositivity is associated with impaired vascular function. Circulation. 2003;108:678–683. doi: 10.1161/01.CIR.0000084505.54603.C7. [DOI] [PubMed] [Google Scholar]
- 52.Span AH, Grauls G, Bosman F, van Boven CP, Bruggeman CA. Cytomegalovirus infection induces vascular injury in the rat. Atherosclerosis. 1992;93:41–52. doi: 10.1016/0021-9150(92)90198-p. [DOI] [PubMed] [Google Scholar]
- 53.Span AH, Frederik PM, Grauls G, Van Boven GP, Bruggeman CA. CMV induced vascular injury: an electron-microscopic study in the rat. In Vivo. 1993;7:567–573. [PubMed] [Google Scholar]
- 54.Smith MS, Bivins-Smith ER, Tilley AM, Bentz GL, Chan G, Minard J, Yurochko AD. Roles of phosphatidylinositol 3-kinase and NF-kappaB in human cytomegalovirus-mediated monocyte diapedesis and adhesion: strategy for viral persistence. J Virol. 2007;81:7683–7694. doi: 10.1128/JVI.02839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Compton T, Kurt-Jones EA, Boehme KW, Belko J, Latz E, Golenbock DT, Finberg RW. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol. 2003;77:4588–4596. doi: 10.1128/JVI.77.8.4588-4596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yilmaz S, Koskinen PK, Kallio E, Bruggeman CA, Hayry PJ, Lemstrom KB. Cytomegalovirus infection-enhanced chronic kidney allograft rejection is linked with intercellular adhesion molecule-1 expression. Kidney Int. 1996;50:526–537. doi: 10.1038/ki.1996.345. [DOI] [PubMed] [Google Scholar]
- 57.Hendrix MG, Dormans PH, Kitslaar P, Bosman F, Bruggeman CA. The presence of cytomegalovirus nucleic acids in arterial walls of atherosclerotic and nonatherosclerotic patients. Am J Pathol. 1989;134:1151–1157. [PMC free article] [PubMed] [Google Scholar]
- 58.Melnick JL, Hu C, Burek J, Adam E, DeBakey ME. Cytomegalovirus DNA in arterial walls of patients with atherosclerosis. J Med Virol. 1994;42:170–174. doi: 10.1002/jmv.1890420213. [DOI] [PubMed] [Google Scholar]
- 59.Langer HF, Gawaz M. Platelet-vessel wall interactions in atherosclerotic disease. Thromb Haemost. 2008;99:480–486. doi: 10.1160/TH07-11-0685. [DOI] [PubMed] [Google Scholar]
- 60.Semple JW, Freedman J: Platelets and innate immunity. Cell Mol Life Sci 67:499–511 [DOI] [PMC free article] [PubMed]
- 61.von Hundelshausen P, Koenen RR, Weber C. Platelet-mediated enhancement of leukocyte adhesion. Microcirculation. 2009;16:84–96. doi: 10.1080/10739680802564787. [DOI] [PubMed] [Google Scholar]