

Abstract
We have generated mouse models of non-Hodgkin lymphoma (NHL) that rely on the cooperation between MYC overexpression and B-cell antigen receptor (BCR) signaling for the initiation and maintenance of B-cell lymphomas. Using these mouse models of NHL, we have focused on the identification of BCR-derived signal effectors that are important for the maintenance of NHL tumors. In the present study, we concentrate on Spleen tyrosine kinase (Syk), a nonreceptor tyrosine kinase required to transduce BCR-dependent signals. Using a genetic approach, we showed that Syk expression is required for the survival of murine NHL-like tumors in vitro and that tumor cells deficient in Syk fail to expand in vivo. In addition, a pharmacologic inhibitor of Syk was able to induce apoptosis of transformed B cells in vitro and led to tumor regression in vivo. Finally, we show that genetic or pharmacologic inhibition of Syk activity in human NHL cell lines are generally consistent with results found in the mouse models, suggesting that targeting Syk may be a viable therapeutic strategy.
Introduction
Non-Hodgkin lymphoma (NHL) is a heterogeneous group of malignancies principally composed of mature B cells expressing surface B-cell receptor (BCR).1 These tumors can involve the overexpression of MYC that may result from a variety of mechanisms, including chromosomal translocations, gene amplification and mutations in the transcriptional regulatory regions.2 In the majority of Burkitt lymphomas (BL), the t(8;14) chromosomal translocation places the MYC gene under the control of the immunoglobulin heavy chain (IgH) gene enhancer3,4 resulting in constitutive overexpression of MYC. The Eμ-MYC mouse strain was generated to model this chromosomal translocation.5 However, unlike in BL tumors, Eμ-MYC mice develop lymphoid tumors consisting of pre-pro–B cells lacking BCR expression that do not resemble NHL, but rather acute lymphoblastic leukemia (ALL).6,7 In contrast, circumstantial evidence links chronic antigenic stimulation through the BCR to the development of certain B-cell NHLs,2,8 and our own work has demonstrated direct contributions of antigen and the antigen receptor in a transgenic mouse model.6
We have developed novel murine models of NHL that demonstrate the cooperation between BCR-derived signals and MYC overexpression during B-cell lymphomagenesis. We introduced a transgenic BCR onto the Eμ-MYC background to generate Eμ-MYC/BCRHEL transgenic mice. These mice develop tumors composed of mature, naive B cells, resembling a subset of CD5− B-cell chronic lymphocytic leukemia (B-CLL) malignancies in humans.9 The further introduction of a transgene encoding for the cognate antigen, soluble hen egg lysosyme (sHEL), onto the Eμ-MYC/BCRHEL background results in more aggressive tumors composed of activated mature post–germinal center (GC)–like B cells. Tumors in Eμ-MYC/BCRHEL/sHEL mice strongly resemble BL by several criteria.6 Pharmacologic and genetic disruption of BCR signaling indicates that both Eμ-MYC/BCRHEL and in Eμ-MYC/BCRHEL/sHEL tumors require BCR expression for their maintenance, although the nature of the BCR signal is different and is responsible for the different tumor phenotypes.6 Our results suggest that Eμ-MYC/BCRHEL tumor B cells rely on a BCR signal analogous to a constitutive BCR survival signal,10,11 whereas Eμ-MYC/BCRHEL/sHELs depend on a chronic cognate antigen–driven signal.9
The BCR is composed of a variant extracellular antigen–interacting subunit (IgM) coupled to 2 invariant cytoplasmic signaling subunits, Igα and Igβ. These subunits become phosphorylated on immunoreceptor tyrosine-based activation motifs (ITAMs) by Src family kinases after antigen-dependent aggregation of the BCR. Spleen tyrosine kinase (Syk) is a nonreceptor kinase that is recruited to dually phosphorylated ITAMs on Igα and Igβ after BCR aggregation. Once recruited to the BCR complex, Syk is activated and can phosphorylate several targets, including BLNK and PLCγ2, to propagate BCR signaling via a robust calcium response.12 Syk-deficient mice have a near complete developmental block at pre-pro–B-cell stage.13 Other work demonstrates that Syk is necessary for the maintenance of mature naive B cells.14 These data suggest that Syk is necessary to transduce constitutive BCR signals10,11 in addition to cognate antigen–driven signals.
A variety of evidence indicates that Syk is an attractive therapeutic target to treat NHL. We have previously found that surface BCR expression is required for the maintenance of mouse NHL-like tumors.6 Although phosphorylation of Igα and Igβ by Lyn, Fyn, or other Src family kinases is generally regarded as the first biochemical step in BCR signaling, targeting this family of kinases with pharmacologic inhibitors is problematic due to their ubiquitous expression, diverse activities, and redundant nature. Syk, on the other hand, is expressed predominantly in hematopoietic cells, and only has one relative, Zap-70, not commonly expressed in most B-cell NHLs.15 We have also observed that these murine tumor cells rely heavily upon calcium-dependent signaling for their survival, which is directly regulated upstream by Syk. This BCR-activated calcium signaling pathway is required for the maintenance of murine and human BL cells.6
In the present study, we used genetic and pharmacologic approaches to define the role of Syk in the maintenance of NHL. Using murine models of NHL, we found that genetic disruption of Syk expression in B-cell lymphoma cells by RNA interference resulted in a disadvantage in an in vitro competition assay as a result of increased apoptosis without obvious effects on proliferation. Furthermore, mouse B-cell tumor cells in which Syk expression was disrupted failed to implant upon adoptive transfer in vivo. We also tested the efficacy of a novel Syk inhibitor in the murine system. Pharmacologic inhibition of Syk activity in vitro also induced apoptosis of murine B-cell lymphoma cells, and treatment of mice bearing NHL-like B-cell lymphomas resulted in regression of the tumors. Finally, we determined that Syk expression and function are necessary for the survival of human NHL cell lines. These results validate Syk as a therapeutic target in vivo, using both genetic and pharmacologic approaches. The human cell line data further support our results in the mouse models and are in accord with a published report using DLBCL cell lines.16 Our results indicate that inhibition of BCR signaling by targeting Syk is a viable therapeutic strategy to treat NHL.
Methods
Transgenic mice and cell lines
Eμ-MYC mice were previously described.5 Eμ-MYC/BCRHEL and Eμ-MYC/BCRHEL/sHEL, and their derivative murine B-cell lymphoma cells, DBL-114, DBL-120, TBL-1, TBL-5, and TBL-90, were previously described.6 BL cell lines, RAJI, BJAB, and DAUDI were obtained from Dr John Cambier (National Jewish Medical Research Center [NJMRC], Denver, CO). DLBCL cell lines OCI-LY1, OCI-LY3, and Val were kindly provided by Dr Hilda Ye (Albert Einstein College of Medicine, New York, NY). DLBCL cell lines SudHL-4 and SudHL-6, and MCL cell lines GRANTA and NCEB were a gift of Drs Craig T. Jordan and Randall M. Rossi (University of Rochester, Rochester, NY). All cells were maintained in C10 media,6 except DAUDI, OCI-LY1, OCI-LY3, and Val cells, which were maintained in Iscove modified Dulbecco medium (IMDM) supplemented with 10% fetal bovine serum (FBS) and 100 U/mL penicillin/streptomycin. Approval for the use of deidentified human lymphoid cells was obtained from the NJMRC Institutional Review Board for these studies. All experiments with mice also received approval from the NJMRC Institutional Review Board.
Western blot analysis
Cells were lysed with 1% Triton X-100 as previously described.17 Protein concentration of the postnuclear supernatants (PNSs) was determined as previously described.17 PNSs were analyzed by SDS-PAGE as previously described.15 Anti-Syk rabbit serum was kindly provided by Dr John Cambier. Anti-phospho (Y525/Y526) Syk rabbit serum from Cell Signaling Technologies (Danvers, MA) and anti–β-actin mouse antibody (8226) was obtained from Abcam (Cambridge, MA). Secondary horseradish peroxidase (HRP)–linked anti–rabbit and anti–mouse were purchased from Cell Signaling Technologies. Blots were stripped by incubation with 0.2 M NaOH for 5 minutes, quenched with Tris-buffered saline Tween-20 (TBST; 50 mM Tris, pH 7.6, 150 mM NaCl2 and 0.1% [vol/vol] Tween-20), and then reblocked. Blots were developed on Kodak BioMax Light film (Kodak, Rochester, NY) using a X-OMAT 2000 developer (Kodak).
Disruption of gene expression by shRNA in tumor cells
The generation and cloning of small RNA hairpins (shRNAs) into lentiviral vectors was previously described.6 Target sequences are as follows: shmSyk TAGTGAAGCCACAGATGTA; shLuc CTTACGCTGAGTACTTCGA; shhSyk.1 GTCGAGCATTATTCTTATA; shhSyk.2 GGAATAATCTCAAGAATCA. Lentiviral infection protocols were previously described.6
To validate knockdown of Syk, infected DBL-114 or BJAB cells were sorted to greater than 95% green fluorescent protein (GFP) expression on a MoFlo cell sorter (Dako, Glostrup, Denmark) in the NJMRC flow cytometry core facility. Sorted cells were lysed and subjected to Western blot analysis with anti-Syk rabbit antisera. Blots were stripped and reprobed with anti–β-actin to determine relative loading. Western blots were scanned from film with a Perfection 1670 digital scanner (Epson, Long Beach, CA) and densitometry of bands was determined using UnScanit software (Silk Scientific, Orem, UT). The density of each band corresponding to Syk protein was normalized to the density of bands corresponding to β-actin, and this ratio was normalized to the control lane (no transduction).
The in vitro and in vivo competition assays were described previously.6 Tumor spleens and nodules were imaged in brightfield and epifluorescence by a Zeiss Discovery V08 Stereo microscope (Carl Zeiss, Peabody, MA).
Pharmacologic inhibitors of Syk
R406 and R78818 were provided by Rigel Pharmaceuticals (South San Francisco, CA). R788 was administered to mice intraperitoneally (IP) at 80 mg/kg per day.
Cell viability and apoptosis assays
MTS cell viability assays were performed using the Celltiter 96 reagent (Promega, Madison, WI) as previously described.19 Briefly, a 2-fold titration of R406 was made and 2.5 × 104 live cells added per well in a 96-well plate and allowed to incubate overnight before addition of MTS reagent. For data analysis, the value for media alone was subtracted from all wells as background, and then all samples were normalized to the value of the well without R406.
Apoptosis was measured using either 7-amino-actinomycin D (7-AAD) or propidium iodide (PI). 7-AAD (EMD, Gibbstown, NJ) was performed as previously described.20 The protocol for PI staining was previously described.21 Cells for PI labeling were obtained through 2 methods. First, cells were infected with a virus possessing a GFP reporter gene and sorted for greater than 95% GFP expression, allowed to recover for 2 days, and then stained. Second, cells were infected with virus containing a Thy1.1 reporter gene. These cells would have a mix of infected (Thy1.1+) and uninfected (Thy1.1−) cells, and infected cells were determined by a fluorescein isothiocyante (FITC)–conjugated anti-Thy1.1 antibody (Becton Dickinson, Franklin Lakes, NJ). For both 7-AAD and PI analysis, necrotic cells were excluded based on their forward and side light scattering properties.
Pharmacologic inhibition of Syk in vivo
Tumors were transplanted into cohorts of age- and sex-matched C57/BL6 mice as previously described.6 Mice in one group were injected IP with 250 μL R788 at 4 mg/mL once daily (∼80 mg/kg per day), while mice in the other group were injected with 250 μL vehicle daily. Pharmacokinetic data for R788 administered IP revealed detectable levels of R406 in the plasma of mice given a dose of 10 mg/kg (Figure S1; available on the Blood website; see the Supplemental Materials link at the top of the online article). Injections were carried out for a total of 7 days. The following day, between 3 and 5 mice were taken from each cohort, plus 2 or 3 wild-type control mice, for analysis of tumor burden. Those mice were killed and their lymph nodes were harvested as previously described.6 The remaining mice were left in the vivarium and survival kinetics were determined in accordance with NJMRC Institutional Animal Care and Use Committee (IACUC) guidelines.
Results
Syk expression is retained in murine B-cell lymphomas
We hypothesized that Syk expression and activity are required for the maintenance of NHL. We initially examined the levels of Syk expression and function in our murine B-cell lymphomas to ensure the validity of our hypothesis. We prepared detergent lysates of cells obtained from Eμ-MYC, Eμ-MYC/BCRHEL, and Eμ-MYC/BCRHEL/sHEL mice, and from primary, wild-type B cells purified from lymph nodes (LN) and spleens of C57/BL6 mice, without or with acute BCR stimulation by anti-IgM to crosslink surface BCR and activate Syk kinase. We loaded 15 μg of each lysate on to an SDS-PAGE gel for subsequent Western blot analysis. All samples had Syk expression as shown by Western blot with anti-Syk rabbit antibody (Figure 1 top panel). Relative to naive B cells, we found that Eμ-MYC and Eμ-MYC/BCRHEL tumors have reduced Syk expression, whereas Eμ-MYC/BCRHEL/sHEL tumors have modestly enhanced Syk expression when normalized to levels of β-actin expression (Figure 1 bottom panel). By blotting with anti–phospho-Syk Y525/526 rabbit antibodies, we observed activation of Syk in wild-type B cells stimulated with anti-IgM (Figure 1 middle panel). In contrast, we found no comparable activation in any of the transformed B-cell lysates. These results indicate that Syk expression is maintained in transformed B cells, but that expressed Syk is not acutely activated (see “Discussion”).
Figure 1.
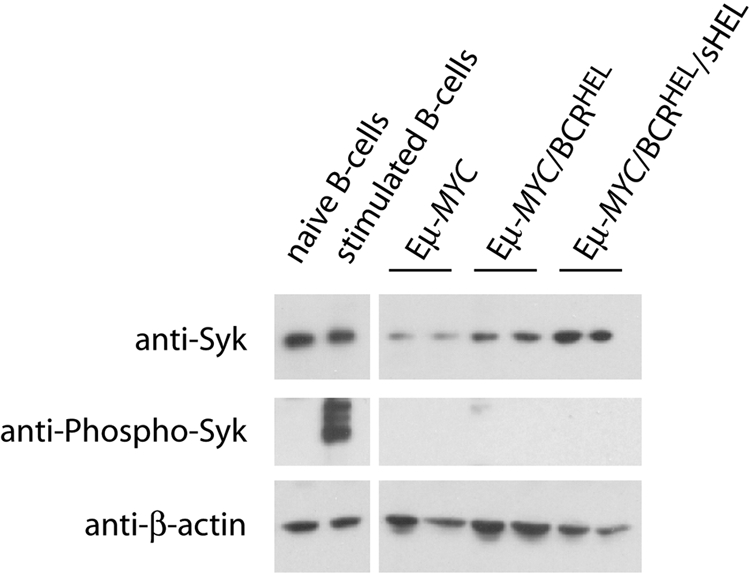
Syk expression and activity in NHL-like murine tumor B cells. Representative Western blot of naive and anti-IgM stimulated (5 minutes) wild-type B cells from spleen compared with 2 separate tumor spleens each from Eμ-MYC, Eμ-MYC/BCRHEL, and Eμ-MYC/BCRHEL/sHEL mice. Cells were lysed with 1% Triton X-100 in the presence of phosphatase and protease inhibitors, and blotted for anti–phospho-Syk (human Y525/526) (middle panel), stripped and reprobed for anti-Syk (top panel), and then stripped a second time and probed for anti–β-actin loading control (bottom panel).
Genetic disruption of Syk expression in transformed B cells results in apoptosis
To evaluate the role of Syk expression in the maintenance of B-cell lymphomas, we used RNA interference to reduce Syk expression. A shRNA targeting murine Syk was designed and designated shmSyk, using a previously described algorithm22 and software, and then cloned into a lentiviral expression vector that also encodes a GFP reporter.23 To evaluate this shRNA, we used polyclonal B-cell tumor lines derived from either Eμ-MYC/BCRHEL tumor cells, named DBL-114 cells, or Eμ-MYC/BCRHEL/sHEL tumors, named TBL-1 cells (see “Methods”). DBL-114 cells were transduced with empty vector, a control shRNA targeting firefly luciferase (shLuc), or the shRNA specific to Syk (shmSyk). Transduced cells were sorted to enrich for GFP expression to greater than 95%, using a high-speed cell sorter (data not shown). Sorted cells were next lysed in 1% Triton X-100, and 15 μg of each lysate was subjected to Western blot analysis with anti-Syk rabbit polyclonal antibody. Blots were then stripped and reprobed with anti–β-actin mouse monoclonal antibody to determine relative protein loading between samples (Figure 2A). Quantitation of band densities on 2 independent blots showed that the Syk-specific shRNA reduced Syk expression by 80% relative to controls (see “Methods”).
Figure 2.

Knockdown Syk expression confers a competitive disadvantage through the induction of apoptosis. (A) Semiquantitative Western blot analysis of Syk knockdown in murine tumor B cells (DBL-114) with anti-Syk (top panel) and anti–β-actin (bottom panel) of the stripped blot. Cells were infected with empty vector (pLL3.7), control shRNA (shLuc), or Syk-specific shRNA (shmSyk). Percent knockdown determined from 2 separate blots. (B) Plot of in vitro competition assay in TBL-1 or DBL-114 cells infected with either control shLuc or shmSyk. Competition between infected (GFP+) and uninfected cells (GFP−) cells is shown. All samples were normalized to percent of GFP+ cells on day 2 after infection and shown relative to empty vector pLL3.7 thereafter (n = 4). (C) Plot of 7-AAD–positive cells normalized to levels of 7-AAD expression in cells expressing shLuc relative to 7-AAD expression in shmSyk (n = 3). (D) PI staining of DBL-114 tumor B cells expressing shLuc (gray filled, top number) or shmSyk (black trace, bottom number) 2 days after sorting for greater than 95% GFP-expressing cells.
We next performed an in vitro competition assay to evaluate whether targeted knockdown of Syk expression affected NHL-like mouse tumor cells. DBL-114 and TBL-1 cells were transduced with empty vector, shLuc, or shmSyk. Initial transduction efficiency was determined by flow cytometry 2 days after transduction, and the frequency of GFP-positive cells relative to the empty vector control was monitored every other day for 14 days. Tumor cell lines harboring shmSyk showed a time-dependent reduction in the frequency of GFP-positive cells relative to uninfected cells in the same well (Figure 2B), suggesting they had a competitive disadvantage. Transduction of shLuc had no significant effect on the frequency of GFP-positive cells over time, demonstrating that engagement of the Dicer pathway by a double-stranded RNA substrate does not affect tumor B cells (Figure 2B). Similar rates for loss of shmSyk-expressing cells were observed for Eμ-MYC/BCRHEL and Eμ-MYC/BCRHEL/sHEL tumor cells, indicating that both types of BCR signals rely on Syk expression (Figure 2B).
The in vitro competition assay used in Figure 2B is unable to differentiate between changes in cellular proliferation and survival. To determine whether Syk knockdown resulted in decreased cell proliferation, we transduced DBL-114 cells with a lentivirus encoding shmSyk and a Thy1.1 reporter gene. Transduced DBL-114 cells were labeled with carboxyfluoroscein succinimidyl ester (CFSE) 4 days after the last day of transduction. Cell division can be tracked by following the dilution of CFSE over time using flow cytometry, which is diluted among the daughter cells after cell division. Analysis of CFSE-labeled cells 24, 48, and 72 hours later did not reveal a significant difference in proliferation between the control shLuc and the Syk-specific shRNA (data not shown).
We next determined if Syk knockdown resulted in increased apoptosis, as determined by 7-AAD (7-amino-actinomycin D) exclusion from live cells. TBL-1 and DBL-114 cells transduced with virus-expressing shLuc or shmSyk encoding a Thy1.1 reporter gene were labeled with 7-AAD 5 days after infection. Examination of Thy1.1-positive cells revealed that knockdown of Syk resulted in a 3-fold increase in the proportion of 7-AAD–positive cells compared with control shRNA (Figure 2C). We confirmed these results with PI staining for total DNA content of cells in each culture. Using this method, we observed a 2-fold increase in the population of cells harboring subdiploid DNA,24 a hallmark of apoptosis,25 in shmSyk versus shLuc-transduced cells (Figure 2D). Together, these results indicate that Syk knockdown induces apoptosis in B-cell lymphoma cells.
Syk expression is required for tumor maintenance in vivo
To validate the conclusions drawn from the in vitro competition assay we next examined how genetic ablation of Syk affects tumor maintenance in vivo. DBL-114 cells were transduced with control shLuc or Syk-specific shmSyk shRNA. Four days after transduction, a mixture of infected (GFP+) and uninfected (GFP−) cells were transplanted into Rag1−/− recipient mice. When the transplant-recipient mice developed externally evident clinical signs of disease (scruffy fur, hunched posture, dehydration, labored breathing, splenomegaly, and lymphadenopathy), they were euthanized, and their tumors were harvested. GFP fluorescence was observed in spleens and tumor masses from these mice by epifluorescence microscopy (Figure 3A); tumorous lymph nodes were not imaged because of low GFP fluorescence. The images in Figure 3A show a qualitative decrease in GFP fluorescence in tumors expressing shmSyk, relative to shLuc. Although both cohorts of mice developed tumors, GFP+ cells were observed only in those mice that received control shRNA (Figure 3A). The percentage of GFP+ tumor cells from each mouse was determined by fluorescence-activated cell sorting (FACS) analysis of single-cell suspensions obtained from tumor masses (Figure 3B). We again observed an absence of GFP+ cells in those mice that received shmSyk- transduced cells, demonstrating that loss of Syk expression confers a competitive disadvantage to established B-cell tumors. These data suggest that Syk expression is necessary for expansion of mature Eμ-MYC/BCRHEL tumors in vivo. TBL cells were not used in this assay because their transduction efficiency with lentiviral vectors is not high enough for this less sensitive in vivo assay.
Figure 3.

Syk-deficient lymphomas fail to expand in mice. DBL-114 tumor B cells were infected with either pLL3.7-shLuc or pLL3.7-shmSyk and then adoptively transferred into mice. Mice were killed when they developed externally palpable tumors (A). Representative brightfield or fluorescent images of tumor spleens and tumor nodules harvested from mice. (B) Quantitation of percent GFP+ cells left in tumors relative to the percentage of GFP+ cells transferred into the mice, as determined by FACS analysis of cells before adoptive transfer and after tumor harvest.
Pharmacologic inhibition of Syk activity induces apoptosis of B-cell lymphoma cells in vitro
Our results from genetic disruption of Syk on tumor maintenance (Figures 2,3) indicate that Syk is a potential therapeutic target to treat NHL. We sought to test this idea with a pharmacologic approach. We obtained the small molecule inhibitors of Syk function named R406 and R788, where R788 is a prodrug of R406 for use in vivo. R406 is a bioavailable Syk inhibitor shown to inhibit Syk-dependent phosphorylation of SLP-65 in anti-IgM–treated RAMOS cells.18 Furthermore, R406 treatment of primary splenocytes inhibits both CD69 up-regulation18 and calcium flux after BCR cross-linking (R.M.Y. and Y.R., unpublished results, August 2006). We decided to test R406 and R788 in our mouse models of NHL.
We performed an MTS cell viability assay on Eμ-MYC/BCRHEL and Eμ-MYC/BCRHEL/sHEL tumor cells. We found a dose-dependent decrease in cellular metabolism with increasing concentrations of R406 with the EC50 of R406 in the high nanomolar range for both types of murine tumors (Figure 4A). The MTS assay is unable to distinguish between decreases in cellular metabolism caused by changes in cell proliferation or cell death. Eμ-MYC/BCRHEL and Eμ-MYC/BCRHEL/sHEL tumor cells were stained with CFSE and then treated with 2 μM R406, the lowest dose at which we observed a maximal effect. CFSE staining did not reveal a significant defect in cell proliferation after 24 or 48 hours compared with dimethyl sulfoxide (DMSO) alone (data not shown). We next examined whether R406 treatment of these murine lymphoma cells resulted in cell death. Eμ-MYC/BCRHEL (DBL-114 and DBL-120) and Eμ-MYC/BCRHEL/sHEL (TBL-1, TBL-90) tumor cells were treated with 2 μM R406 for 24 hours before PI staining and analysis. R406 treatment resulted in a substantial increase in subdiploid apoptotic cells (black) compared with DMSO vehicle control (gray) for all cell lines tested (Figure 4B,C). Moreover, when we used a second method to detect apoptotic cells, 7-AAD, we observed that 2 μM R406 for 24 hours resulted in a marked increase in 7-AAD staining (Figure 4C). Experiments on DLBCL cell lines also found that R406 induced apoptosis via a cell-intrinsic caspase 9 and 3 pathway.16 These results confirmed our genetic data and indicate that inhibition of Syk activity by R406/788 induces apoptosis of transformed B cells.
Figure 4.

Inhibition of Syk activity induces cell death in tumor B cells. Cells were treated with R406 in vitro. (A) A representative plot of cell viability as determined by MTS assay by concentration of the Syk inhibitor R406 (n = 4). (B) Representative plots of PI stains of TBL-1 and DBL-114 tumor B cells treated with DMSO (gray shaded) or 2 μM R406 (black trace). The bar is gated on subdiploid DNA for apoptotic cells. The bottom percentage is for DMSO treatment and the top for R406. (C) A bar graph depicting percent positive 7-AAD (top panel) or PI (bottom panel) after 24-hour treatment with DMSO (■) or 2 μM R406 (□).
Pharmacologic inhibition of Syk activity in murine models of NHL resulted in tumor remission
We took advantage of our ability to transplant tumors, from either tumor-bearing mice or cell lines, into cohorts of recipient mice. This approach allows us to test the effects of a specific compound on a statistically relevant number of independent animals that harbor the same tumor in a synchronized manner. We transplanted Eμ-MYC/BCRHEL and Eμ-MYC/BCRHEL/sHEL (TBL-1) tumors into cohorts of syngeneic age- and sex-matched C57BL6 recipient mice. In addition, we also transplanted Eμ-MYC tumors as a control, because these tumors are not dependent on BCR-derived signals for their initiation or maintenance. After the cohorts of recipient mice developed externally evident clinical signs of disease, we administered 80 mg/kg per day of the Syk inhibitor R788, or an equal volume of vehicle as a control, for 7 consecutive days. Pharmacokinetic data for R788 administered IP revealed detectable levels of R406 in the plasma of mice given a dose of 10 mg/kg (Figure S1). Subsets of mice from the R788-treated or vehicle-treated cohorts were euthanized 24 hours after the last day of treatment. At that time, their spleens and lymph nodes were harvested and used to generate single-cell suspensions that were then enumerated. The remaining mice in the cohorts stayed in the vivarium for long-term observation to determine whether any remission induced by the transient treatment resulted in an extension of their lifespan.
R788 treatment markedly reduced tumor burden relative to vehicle in Eμ-MYC/BCRHEL (Figure 5A) and Eμ-MYC/BCRHEL/sHEL (TBL-1) tumors (Figure 5B). In addition, R788 treatment extended the survival of mice transplanted with either Eμ-MYC/BCRHEL (Figure 5D) and Eμ-MYC/BCRHEL/sHEL (Figure 5E). After an initial remission, mice with Eμ-MYC/BCRHEL and Eμ-MYC/BCRHEL/sHEL (TBL-1) tumors that were transiently treated with R788 relapsed and uniformly died as a result of their tumors. In contrast, mice transplanted with Eμ-MYC tumors had no significant reduction in tumor burden (Figure 5C), and treatment of these tumors did not affect the survival of mice transplanted with Eμ-MYC tumors relative to treatment with vehicle alone (Figure 5F), suggesting R788 is not a general mitostatic agent. One difference of tumors arising in Eμ-MYC mice is that they do not express surface BCR and developed in the absence of BCR signaling. These data suggest that R788 is inhibiting Syk activity in the context of BCR signaling, although we cannot rule out differences in R788 sensitivity due to the different developmental state of the Eμ-MYC tumors. These data also validate the notion of acute dependence of established B-cell lymphomas on continuous BCR signaling.
Figure 5.

Inhibition of Syk activity by R788 reduces tumor burden and increases survival in mouse models of NHL. Cohorts of mice were adoptively transferred with primary tumors or tumor cell lines. At the first external sign of tumors, mice were treated with either 80 mg/kg per day of R788 or control vehicle for 7 days. One day after treatment, 3 or more mice were harvested from R788 or control groups to determine tumor burden relative to wild-type mice. Representative plots of tumor burden as assessed by total number of live cells in the lymph nodes of mice are shown for (A) primary Eμ-MYC/BCRHEL tumors, (B) Eμ-MYC/BCRHEL/sHEL (TBL-1) tumors, or (C) primary Eμ-MYC tumors (n = 2). The remaining mice were left in the vivarium to determine whether R788 treatment affected survival. Representative plots of tumor survival are shown for (D) primary Eμ-MYC/BCRHEL tumors, (E) Eμ-MYC/BCRHEL/sHEL (TBL-1) tumors, or (F) primary Eμ-MYC tumors. No AT and no treatment (○), AT tumor and treatment with control vehicle (□), or AT tumor and treatment with 80 mg/kg per day R788 (▲) are shown (n = 3). P values for vehicle versus R788 are less than .002 for Eμ-MYC/BCRHEL and Eμ-MYC/BCRHEL/sHEL tumors.
Human B-cell lymphomas require Syk activity for survival in vitro
Results from our mouse models of NHL demonstrate that Syk expression and activity are required for tumor maintenance. We wanted to validate our observations obtained in the mouse model of NHL by testing the requirement for Syk in human cells to rule out any potential differences between the 2 species. We examined whether human B-cell lymphomas were also dependent on Syk expression for their survival in vitro. For these experiments, we used the human BL tumor cell lines RAJI and BJAB. We found robust Syk expression in both of these cell lines (data not shown). We generated an shRNA to disrupt expression of human Syk. BJAB cells were transduced with empty vector (pLL3.7), shLuc, or shRNAs specific for human Syk, shhSyk.1 and shhSyk.2. Transduced cells were sorted for GFP expression and then lysed with 1.0% Triton X-100 as described above for knockdown of murine Syk (Figure 2A). Semiquantitative Western blot analysis indicated that we had near 90% knockdown of Syk protein expression with the Syk-specific shhSyk.2 shRNA (Figure 6A).
Figure 6.

Genetic disruption of Syk expression in human NHL cell lines. Syk expression was disrupted in human BL cell lines. (A) Semiquantitative Western blot analysis of Syk knockdown in BJAB cells with anti-Syk (top panel); blots were then stripped and reprobed with anti–β-actin (bottom panel). Cells were infected with empty vector (pLL3.7), control shRNA (shLuc), or Syk-specific shRNA (shSyk). Percent knockdown was determined by densitometry of the Syk bands relative to the β-actin bands. (B) Plot of in vitro tumor competition assay in RAJI and BJAB cells infected with either control shLuc or shhSyk, as described in Figure 4 (n = 4).
Using these constructs, we performed an in vitro competition assay to determine whether RAJI or BJAB cells are dependent on Syk expression for their survival. Cells were transduced with empty vector, shLuc or shhSyk.2. Both shLuc and shhSyk.2 infections were normalized to the initial transduction efficiency determined 2 days after transduction, and the frequency of GFP-positive cells relative to empty vector were monitored by FACS analysis every other day for 14 days. RAJI cells harboring shhSyk.2 had a reduction in the frequency of GFP+ cells over time, whereas cells expressing shLuc did not, suggesting knockdown of Syk results in a competitive disadvantage relative to RAJI cells expressing shLuc (Figure 6B). In contrast, disruption of Syk expression in BJAB cells did not result in competitive disadvantage compared with cells expressing the control shRNA.
To test the requirement for Syk function in the human NHL cell lines, we treated cells with R406 in vitro. We tested the affect of R406 treatment on a panel of human NHL cell lines in vitro, including BL, DLBCL, and mantle cell lymphoma (MCL) cell lines. We evaluated the effect of R406 on the human NHL cell lines by MTS cell viability assays, as described for the murine cell lines in Figure 3C. We found that the human NHL cell lines were generally less sensitive to R406 compared with the murine cell lines (Figure 7A). For the BL cell lines tested, we found that RAJI cells were highly sensitive to R406, but that only a fraction of the BJAB cells were sensitive (see “Discussion”). This mirrors the genetic results shown in Figure 6B. We found that the DLBCL cell lines were generally more resistant to R406 than the BL cell lines, whereas MCL cell lines were the most resistant to R406. To ascertain the induction of apoptosis by R406 in the human NHL cell lines, RAJI and BJAB cells were treated with 2 μM R406 or an equivalent volume of vehicle (DMSO) for 24 hours, fixed and permeabilized with ethanol, and labeled with PI. FACS analysis revealed that R406 treatment (black) led to a dramatic increase in apoptotic cells with subdiploid DNA compared with DMSO control (gray) in RAJI cells, and to a lesser extent in BJAB cells (Figure 7B), in agreement with results obtained from the MTS assay. These data demonstrate that inhibition of Syk activity induces apoptosis in human NHL cell lines, suggesting Syk is an attractive therapeutic target for treating human NHL.
Figure 7.

Inhibition of Syk activity in a panel of human NHL cell lines. The R406 Syk inhibitor was tested against several human NHL cell lines. (A) Representative cell viability plots as determined by MTS assay by concentration of the Syk inhibitor R406. Plots are separated by cell type: BL (left), DLBCL (center), and MCL (right). (B) Representative plots of PI stains of RAJI and BJAB human tumor B cells treated with DMSO (gray) or 2 μM R406 (black). The bar shows apoptotic cells staining for subdiploid DNA. The top percentage is for DMSO treatment and the bottom for R406.
Discussion
Current treatment approaches to certain NHLs have raised the possibility that targeting the antigen receptor pathway in lymphomas may be a viable approach for the development of novel therapeutics. In the present study, we used complementary genetic and pharmacologic approaches to show that Syk expression and function are necessary for the survival of mature B-cell neoplasias. We found that genetic disruption of Syk in both murine B-cell lymphoma cell lines and the human RAJI B-cell lymphoma line conferred a competitive disadvantage, relative to untransduced cells in vitro, as a result of increased apoptosis. Moreover, Syk expression is required for the expansion of murine B-cell lymphomas in vivo. This genetic data suggested that inhibition of Syk activity might be a possible therapeutic strategy for NHL. Inhibition of Syk activity with R406 generally induced apoptosis in both murine and human B-cell lymphoma cell lines. We used the mouse models of NHL to test the efficacy R788 (in vivo R406) in vivo on transplanted tumors. We found that treatment of mice with Eμ-MYC/BCRHEL or Eμ-MYC/BCRHEL/sHEL NHL-like tumors with the Syk inhibitor led to acute tumor remission and extended mouse survival. In contrast, treatment of mice with Eμ-MYC tumors, which are not dependent on BCR signaling for their survival, did not result in tumor remission. These results show the value of our murine model systems to first, identify potential therapeutic targets, and then to perform preclinical testing of novel therapeutic agents against the targets.
We have described 2 types of BCR-derived signals that cooperate with MYC in our murine models of NHL.6 Both types of BCR-dependent signals are required for the survival of the resultant lymphoma, but they are qualitatively distinct and differentially affect lymphoma development. Eμ-MYC/BCRHEL tumors require a constitutive BCR signal, which occurs in the absence of a cognate antigen. We hypothesize that this signal is analogous to the constitutive BCR signals essential to B-cell development and the survival of mature naive B cells.26,27 On the other hand, Eμ-MYC/BCRHEL/sHEL tumor B cells develop with cognate antigen to their transgenic BCR.6 In this case, MYC overexpression breaks self-tolerance, and B cells continue to proliferate in response to chronic stimulation by self-antigens.9 This signal is less characterized, as it does not have a wild-type homologue. To our surprise, we observed no difference in Syk phosphorylation between Eμ-MYC/BCRHEL and Eμ-MYC/BCRHEL/sHEL tumor B cells. In contrast, we have previously reported that Eμ-MYC/BCRHEL and Eμ-MYC/BCRHEL/sHEL tumors are sensitive to distinct immunosuppressant drugs acting downstream of the BCR.6 We also have genetic data indicating that these 2 BCR signals are differentially reliant on nuclear factor kappa B (NF-κB) and Akt signaling (R.M.Y. and Y.R., unpublished results, March 2007) Therefore, although 2 distinct signals are present, it is not clear whether they are different at the level of Syk expression and phosphorylation. It is, therefore, possible that chronic cognate-antigen stimulation of a BCR on a B-cell lymphoma could be confused with a constitutive BCR signal, although antigen may, in fact, be driving the BCR-derived signal.
Previous reports have examined the role of constitutively active or hyperactive Syk in lymphoma. For example, Syk was found to be constitutively phosphorylated in RAJI and follicular lymphoma cell lines.28 Likewise, expression of a constitutively active mutant of Syk, Tel-Syk, in pre-B cells induced a novel lymphoma.29 In contrast, our results suggest that basal Syk activity, as observed through the absence of elevated Y525/526 phosphorylation, is enough to maintain NHL-like lymphomas, in agreement with findings in DLBCL cell lines.16 Moreover, it has been demonstrated that a dominant positive form of Syk cannot substitute for BCR ligation alone.30 Therefore, it is normal BCR-dependent signals that cooperate with deregulated MYC expression to drive lymphomagenesis. In this scenario, BCR signals transduce a survival signal, and the overexpression of MYC substitutes for cytokine signaling to promote cellular proliferation and survival.9 This suggests that wild-type B cells could also be equally affected by R406/R788 treatment since they also rely on basal Syk activity for their survival.13 In support of this hypothesis, toxicity studies show that R788-treated rats have mild lymphopenia.18
Our models demonstrate a role for BCR signaling in promoting lymphoma survival is conserved across a spectrum of B-cell NHLs.6 This suggests that targeting Syk activity may be a viable therapeutic strategy these malignancies. We have focused on BL, but our R406 cell viability data on human cells in Figure 7 shows that other human NHL cell lines tested had some response to R406. We found that MCL cell lines tested were somewhat resistant to R406 treatment. A recent study also found that inhibition of Syk activity with piceatannol in MCL cell lines had mixed results in inhibiting cell growth.31 On the other hand, we generally found a high efficacy of R406 on DLBCL cell lines, in agreement with results from Chen et al,16 which reported that surface BCR expression directly correlated to R406 sensitivity. Likewise, all DLBCL cell lines we tested were sensitive to R406 and reported to be BCR+, with the exception of Val, which was not examined in that study. The work by Chen et al also examined the tyrosine phosphorylation of state of Syk by Western blot and FACS analysis. They found no Syk phosphorylation at Y525/526 before BCR crosslinking, in agreement with our results. However, resting levels of Y352 correlated with DLBCL cell sensitivity to R406 treatment, suggesting that a basal level of Syk activity exists downstream of the BCR. Taken as a whole, these results indicate that targeting BCR-dependent signals by the inhibition of Syk activity may be a potential therapeutic strategy for a spectrum of NHLs that rely on BCR-derived survival signals.
BCR expression alone may not account for sensitivity to R406. We hypothesize that bypass mutations resulting in R406 resistance may be a result of Zap-70 expression. Zap-70 is a relative of Syk primarily expressed in T-cell lineages, but it is functionally quite different from Syk. Zap-70 is 100-fold less active than Syk, and Zap-70 also requires Src family kinases for its own activation.32 Interestingly, Zap-70 expression has been implicated in the pathogenesis of B-CLL,33 and patients with B-CLL tumors expressing Zap-70 generally have a poor prognosis.34 We speculate that tumors that do not respond to R406 treatment express Zap-70, which could be activated independently of the BCR. The specific contributions of Zap-70 to murine BL tumors that lack Syk can be directly examined in our mouse models of NHL.
Therapeutic targeting of the BCR pathway in NHL is currently being explored in the clinic. For example, mucosa-associated lymphoid tissue (MALT) lymphoma is treated with a course of 3 antibiotics to eliminate the antigen that propels those tumors.35,36 It is not clear, however, that the specific antigen that drives MALT lymphomas is encoded by the Helicobacter pylori genome, or a self-antigen whose expression on epithelial cells is induced by the bacteria. This is an unusual case where the instigating antigen can be removed, and may not easily translate into cases where the specificity of the B-cell lymphoma cells is directed to a self-antigen. Another example involves the targeting of immunoglobulin complexes on the surface of B-cell lymphomas, by specifically generating monoclonal antibodies to an idiotype,37,38 and is, thus, a tumor-specific approach. Currently, R788 is in clinical trials to treat NHL in relapsed refractory cases as part of a phase 1/2 clinical trial. The key advantage of a small molecule inhibitor of downstream signals from the BCR, like R788, is that it moves away from specific instances and can be applied more readily across a wide spectrum of tumors. Our results using mouse models of NHL indicate that R788 will be effective against NHLs that rely on BCR signaling for their survival.
Acknowledgments
The authors thank Drs Polly Pine and Elliot Grossband at Rigel Pharmaceuticals for providing R406 and R788, and Dianne Ashby and the Biological Resource Center at NJRMC for care and maintenance of the mice used in this study.
R.M.Y. was supported by a National Institute of Allergy and Infectious Diseases (NIAID; Bethesda, MD) postdoctoral training grant to the Integrated Department of Immunology at the University of Colorado Health Science Center (T32-07 405-16) and a Translational Research Award from the Leukemia & Lymphoma Society (White Plains, NY); I.R.H. was supported by a Cancer Research Institute (New York, NY) predoctoral training grant to NJMRC; B.C.T. was funded by a translational award from the Leukemia & Lymphoma Society and a postdoctoral fellowship from the American Cancer Society (Atlanta, GA); T.A.P. and R.L.C. were supported by NIAID grant R01 AI055701; Y.R. was supported by United States Public Health Service grant CA-117802 from the National Cancer Institute (Bethesda, MD), a Translational Research Award from the Leukemia & Lymphoma Society, and startup funds from NJMRC.
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: R.M.Y. designed and performed research, contributed vital new reagents, collected data, analyzed and interpreted data, performed statistical analysis, and wrote the manuscript; I.R.H. contributed vital new reagents, analyzed and interpreted data, and edited the manuscript; R.L.C. and N.L. contributed vital new reagents; P.P. contributed PK data for R788 and R406; B.C.T. performed research; T.A.P. contributed vital new reagents and Y.R. designed and performed research and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yosef Refaeli, National Jewish Medical and Research Center, Departments of Pediatrics, Program in Cell Biology, 1400 Jackson Street, Denver, CO 80206; e-mail: refaeliy@njc.org.
References
- 1.Dameshek W, Schwartz RS. Leukemia and auto-immunization: some possible relationships. Blood. 1959;14:1151–1158. [PubMed] [Google Scholar]
- 2.Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer. 2005;5:251–262. doi: 10.1038/nrc1589. [DOI] [PubMed] [Google Scholar]
- 3.Yustein JT, Dang CV. Biology and treatment of Burkitt's lymphoma. Curr Opin Hematol. 2007;14:375–381. doi: 10.1097/MOH.0b013e3281bccdee. [DOI] [PubMed] [Google Scholar]
- 4.Kuppers R, Dalla-Favera R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene. 2001;20:5580–5594. doi: 10.1038/sj.onc.1204640. [DOI] [PubMed] [Google Scholar]
- 5.Adams JM, Harris AW, Pinkert CA, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- 6.Refaeli Y, Young RM, Turner BC, Duda J, Field KA, Bishop JM. The B-cell antigen receptor and overexpression of MYC can cooperate in the genesis of B-cell lymphomas. PLoS Biol. 2008;6:e152. doi: 10.1371/journal.pbio.0060152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galler GR, Mundt C, Parker M, Pelanda R, Martensson IL, Winkler TH. Surface mu heavy chain signals down-regulation of the V(D)J-recombinase machinery in the absence of surrogate light chain components. J Exp Med. 2004;199:1523–1532. doi: 10.1084/jem.20031523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rui L, Goodnow CC. Lymphoma and the control of B-cell growth and differentiation. Curr Mol Med. 2006;6:291–308. doi: 10.2174/156652406776894563. [DOI] [PubMed] [Google Scholar]
- 9.Refaeli Y, Field KA, Turner BC, Trumpp A, Bishop JM. The protooncogene MYC can break B-cell tolerance. Proc Natl Acad Sci U S A. 2005;102:4097–4102. doi: 10.1073/pnas.0409832102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lam KP, Kuhn R, Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 1997;90:1073–1083. doi: 10.1016/s0092-8674(00)80373-6. [DOI] [PubMed] [Google Scholar]
- 11.Kraus M, Pao LI, Reichlin A, et al. Interference with immunoglobulin (Ig)alpha immunoreceptor tyrosine-based activation motif (ITAM) phosphorylation modulates or blocks B-cell development, depending on the availability of an Igbeta cytoplasmic tail. J Exp Med. 2001;194:455–469. doi: 10.1084/jem.194.4.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dal Porto JM, Gauld SB, Merrell KT, Mills D, Pugh-Bernard AE, Cambier J. B-cell antigen receptor signaling 101. Mol Immunol. 2004;41:599–613. doi: 10.1016/j.molimm.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Turner M, Mee PJ, Costello PS, et al. Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk. Nature. 1995;378:298–302. doi: 10.1038/378298a0. [DOI] [PubMed] [Google Scholar]
- 14.Cheng AM, Rowley B, Pao W, Hayday A, Bolen JB, Pawson T. Syk tyrosine kinase required for mouse viability and B-cell development. Nature. 1995;378:303–306. doi: 10.1038/378303a0. [DOI] [PubMed] [Google Scholar]
- 15.Orchard J, Ibbotson R, Best G, Parker A, Oscier D. ZAP-70 in B-cell malignancies. Leuk Lymphoma. 2005;46:1689–1698. doi: 10.1080/09638280500260079. [DOI] [PubMed] [Google Scholar]
- 16.Chen L, Monti S, Juszczynski P, et al. SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphoma. Blood. 2008;111:2230–2237. doi: 10.1182/blood-2007-07-100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Young RM, Zheng X, Holowka D, Baird B. Reconstitution of regulated phosphorylation of FcepsilonRI by a lipid raft-excluded protein-tyrosine phosphatase. J Biol Chem. 2005;280:1230–1235. doi: 10.1074/jbc.M408339200. [DOI] [PubMed] [Google Scholar]
- 18.Braselmann S, Taylor V, Zhao H, et al. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J Pharmacol Exp Ther. 2006;319:998–1008. doi: 10.1124/jpet.106.109058. [DOI] [PubMed] [Google Scholar]
- 19.Van Parijs L, Refaeli Y, Lord JD, Nelson BH, Abbas AK, Baltimore D. Uncoupling IL-2 signals that regulate T-cell proliferation, survival, and Fas-mediated activation-induced cell death. Immunity. 1999;11:281–288. doi: 10.1016/s1074-7613(00)80103-x. [DOI] [PubMed] [Google Scholar]
- 20.Refaeli Y, Van Parijs L, Alexander SI, Abbas AK. Interferon gamma is required for activation-induced death of T lymphocytes. J Exp Med. 2002;196:999–1005. doi: 10.1084/jem.20020666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Refaeli Y, Van Parijs L, London CA, Tschopp J, Abbas AK. Biochemical mechanisms of IL-2-regulated Fas-mediated T-cell apoptosis. Immunity. 1998;8:615–623. doi: 10.1016/s1074-7613(00)80566-x. [DOI] [PubMed] [Google Scholar]
- 22.Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- 23.Ventura A, Meissner A, Dillon CP, et al. Cre-lox-regulated conditional RNA interference from transgenes. Proc Natl Acad Sci U S A. 2004;101:10380–10385. doi: 10.1073/pnas.0403954101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- 25.Earnshaw WC. Nuclear changes in apoptosis. Curr Opin Cell Biol. 1995;7:337–343. doi: 10.1016/0955-0674(95)80088-3. [DOI] [PubMed] [Google Scholar]
- 26.Smith SH, Reth M. Perspectives on the nature of BCR-mediated survival signals. Mol Cell. 2004;14:696–697. doi: 10.1016/j.molcel.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 27.Monroe JG. Ligand-independent tonic signaling in B-cell receptor function. Curr Opin Immunol. 2004;16:288–295. doi: 10.1016/j.coi.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 28.Leseux L, Hamdi SM, Al Saati T, et al. Syk-dependent mTOR activation in follicular lymphoma cells. Blood. 2006;108:4156–4162. doi: 10.1182/blood-2006-05-026203. [DOI] [PubMed] [Google Scholar]
- 29.Wossning T, Herzog S, Kohler F, et al. Deregulated Syk inhibits differentiation and induces growth factor-independent proliferation of pre-B cells. J Exp Med. 2006;203:2829–2840. doi: 10.1084/jem.20060967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsueh RC, Hammill AM, Lee JA, Uhr JW, Scheuermann RH. Activation of the Syk tyrosine kinase is insufficient for downstream signal transduction in B lymphocytes. BMC Immunol. 2002;3:16. doi: 10.1186/1471-2172-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rinaldi A, Kwee I, Taborelli M, et al. Genomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphoma. Br J Haematol. 2006;132:303–316. doi: 10.1111/j.1365-2141.2005.05883.x. [DOI] [PubMed] [Google Scholar]
- 32.Latour S, Veillette A. Proximal protein tyrosine kinases in immunoreceptor signaling. Curr Opin Immunol. 2001;13:299–306. doi: 10.1016/s0952-7915(00)00219-3. [DOI] [PubMed] [Google Scholar]
- 33.Gobessi S, Laurenti L, Longo PG, Sica S, Leone G, Efremov DG. ZAP-70 enhances B-cell-receptor signaling despite absent or inefficient tyrosine kinase activation in chronic lymphocytic leukemia and lymphoma B cells. Blood. 2007;109:2032–2039. doi: 10.1182/blood-2006-03-011759. [DOI] [PubMed] [Google Scholar]
- 34.Efremov DG, Gobessi S, Longo PG. Signaling pathways activated by antigen-receptor engagement in chronic lymphocytic leukemia B-cells. Autoimmun Rev. 2007;7:102–108. doi: 10.1016/j.autrev.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 35.Casella G, Buda CA, Maisano R, Schiavo M, Perego D, Baldini V. Complete regression of primary gastric MALT-lymphoma after double eradication Helicobacter pylori therapy: role and importance of endoscopic ultrasonography. Anticancer Res. 2001;21:1499–1502. [PubMed] [Google Scholar]
- 36.Montalban C, Santon A, Boixeda D, Bellas C. Regression of gastric high grade mucosa associated lymphoid tissue (MALT) lymphoma after Helicobacter pylori eradication. Gut. 2001;49:584–587. doi: 10.1136/gut.49.4.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L, French RR, Chan HT, et al. The development of anti-CD79 monoclonal antibodies for treatment of B-cell neoplastic disease. Ther Immunol. 1995;2:191–202. [PubMed] [Google Scholar]
- 38.Polson AG, Yu SF, Elkins K, et al. Antibody-drug conjugates targeted to CD79 for the treatment of non-Hodgkin lymphoma. Blood. 2007;110:616–623. doi: 10.1182/blood-2007-01-066704. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}