

Summary
Alzheimer's disease (AD) is characterized pathologically by the abundance of senile plaques and neurofibrillary tangles in the brain. We synthesized over 1200 novel gamma-secretase modulator (GSM) compounds that reduced Abeta42 levels without inhibiting epsilon-site cleavage of APP and Notch, the generation of the APP and Notch intracellular domains, respectively. These compounds also reduced Abeta40 levels while concomitantly elevating levels of Abeta38 and Abeta37.
Immobilization of a potent GSM onto an agarose matrix quantitatively recovered Pen-2 and to a lesser degree PS-1 NTFs from cellular extracts. Moreover, oral administration (once daily) of another potent GSM to Tg 2576 transgenic AD mice displayed dose-responsive lowering of plasma and brain Abeta42; chronic daily administration led to significant reductions in both diffuse and neuritic plaques. These effects were observed in the absence of Notch-related changes (e.g. intestinal proliferation of goblet cells), which are commonly associated with repeated exposure to functional gamma-secretase inhibitors (GSIs).
Introduction
A large number of published studies, including those providing genetic, biochemical, pathological and epidemiological evidence, lend significant support to the theory that alterations in the relative levels of the Aβ42 and Aβ40 peptide species, i.e., Aβ42/40 ratio, may play a pivotal role in the pathogenesis of AD (reviewed in Tanzi and Bertram, 2005). Generation of Aβ peptides requires sequential cleavage of APP by γ-secretase-mediated proteolysis of the β-secretase-generated C-terminal APP cleavage product known as APP-C99 or β-CTF (Vassar et al., 2009). Early approaches to therapeutic intervention focused on lowering total Aβ peptide production by inhibiting the catalytic activities of either β-secretase or BACE 1 (β-amyloid cleaving enzyme 1) or γ-secretase. “γ-secretase” is a heterogeneous complex of membrane proteins (Serneels et al., 2009) which regulate intramembrane proteolysis of APP (Sisodia and St. George-Hyslop, 2002) and a multitude of other substrates (Wakabayashi and De Strooper, 2008), including Notch. γ-secretase-mediated Notch cleavage at the site 3 (S3) or epsilon (ε) site yields a large cytoplasmic peptide (Notch intracellular domain, NICD) that translocates to the nucleus and which is necessary for proper cellular differentiation and development (De Strooper et al., 1999). Typical γ-secretase inhibitors (GSIs) prevent catalysis of both the γ-sites and ε-sites, resulting in the potential for adverse effects secondary to inhibition of ε-site proteolysis of Notch, thereby preventing NICD formation (Kreft, Martone and Porte, 2009; Tomita et al., 2009).
In view of the potential problems associated with inhibiting both the γ-site and ε-site cleavages of γ-secretase, a more attractive therapeutic alternative is to identify small molecules that can preferentially lower the levels of the most fibrillogenic Aβ peptide, Aβ42, without affecting catalytic activity at the ε-site. This approach has been supported in AD animal models using an APP/Aβ-binding non-steroidal anti-inflammatory drug (NSAID)-like compounds to achieve allosteric modulation of γ-secretase activity and attenuate the Aβ42/Aβ40 ratio (Weggen et al., 2001; Kukar et al., 2008).
In order to identify potent, selective, and orally bioavailable small molecule modulators of γ-secretase, we employed cell-based assays to screen for compounds that lowered Aβ42 levels without inhibiting Notch proteolytic processing (NICD generation) or related ε-site γ-secretase-mediated cleavages. For in vivo proof-of-principle studies, the most potent orally bioavailable compounds were then tested for efficacy in Tg 2576 transgenic mice, which express the “Swedish mutant” of human APP (APPswe) and overproduce Aβ42 and Aβ40, at levels leading to neuritic plaques and cerebral amyloid angiopathy (Hsiao et al., 1996). If successful, this approach could lead to the development of therapeutic regimens capable of safely intervening in key neuropathologic processes associated with AD.
Results
High-throughput Screening
A chemical library composed of commercially available compounds was designed using computational tools to provide broad coverage of chemical space with “drug-like” chemical properties. The chemical library, comprising ∼80,000 compounds, was purchased from a variety of commercial sources and screened for the ability to suppress extracellular Aβ42 levels produced from a Chinese hamster ovary (CHO) cell line stably overexpressing APP695 (referred to as CHO-PZ3 or CHO-APPwt) using a monoclonal antibody-based homogeneous fluorescence resonance energy transfer (FRET) high-throughput screening (HTS) assay. One hit, which had an IC50 value of 15 μM for the inhibition of Aβ42, passed all subsequent screening criteria and several focused chemical libraries were then designed and synthesized based on this structure. One compound from one of the focused libraries led to Compound 1 the original compound in the 2-aminothiazole series (Series A). Lead optimization efforts led to Compound 3 and Compound 4 (Figure 1A). Subsequently, lead evolution efforts led to urea-containing analogues (Series B, e.g., Compound 7). All the compounds were subsequently tested in vitro for their ability to inhibit the production of Aβ42 from a human neuroblastoma cell line (SH-SY5Y) stably overexpressing human APP751 (SH-SY5Y-APP cells). Several compounds from Series A exhibited impressive Aβ42 lowering potencies (low nanomolar IC50s) comparable to some of the most potent GSIs (e.g., BMS-299897, LY-411575 and GSI-953) in similar cell-based in vitro assay systems (Martone et al., 2009). This cell-based assay served to generate structure-activity relationships (SAR) for modulators of γ-secretase activity and was the key parameter used to evaluate potencies for the selection and prioritization of compounds pursued in primary and secondary pharmacological studies, including in vivo efficacy testing (Figure 1B).
Figure 1.

Diarylaminothiazoles (Series A) and Diarylureas (Series B) are Potent Modulators of γ-Secretase Activity. (A) Chemical structures of key molecules from Series A and Series B GSMs, including Compound 6, the ethylene amino derivative of Compound 3 that was immobilized onto an Affigel matrix and used as an affinity chromatography ligand. (B) Concentration response curves (CRCs) for lowering of Aβ42 levels produced by SH-SY5Y-APP cells. IC50 values were derived using four parameter fit non-linear regression analyses.
Differentiation of GSMs Versus GSIs
The Series A GSM, Compound 3, differentially inhibited Aβ42 and Aβ40 with greater potency on reducing Aβ42 levels, but had no effect on Aβtotal peptide levels in either the CHO-APPswe or SH-SY5Y-APP cell-based assays (Figure 2A&B). This is in contrast to the arylsulfonamide-containing GSI, BMS-299897, which equivalently inhibited Aβ42, Aβ40 and Aβtotal peptide production in these same two cell-based assay systems (Figure 2C&D). To further characterize the effects of GSM compounds on Aβ peptide species (Aβ37, Aβ38, Aβ40, Aβ42 and Aβtotal), several different methods were employed to quantitate these peptides from both in vitro and in vivo samples. GSMs and GSIs exhibited differential inhibition profiles for several Aβ peptide variants in CHO-APPswe cells using immunoprecipitation in combination with Surface Enhanced Laser Desorption/Ionization Time-Of-Flight Mass Spectrometry (SELDI-TOF MS) (Figure 3A). The arylsulfonamide GSI, BMS-299897 reduced the levels of all Aβ peptide variants to baseline levels (exogenously added Aβ1-28 was used as an internal standard). In contrast, three different Series A GSMs (Compound 2, Compound 5 and Compound 3) reduced Aβ40 levels while increasing the levels of Aβ37 and Aβ38; Aβ42 was not detectable in this assay. Similar results were observed when comparing the effects of the arylsulfonamide GSI, BMS-299897, with the Series A GSM Compound 2, on various Aβ peptide variants secreted from Tg 2576-derived primary mixed brain cultures (MBCs) analyzed using tris-glycine gel electrophoresis in combination with anti-Aβ peptide immunoprecipitation and immunoblotting (Figure 3B). In good agreement with the SELDI-TOF MS experiments depicted in Figure 3A, BMS-299897 reduced the levels of all Aβ variants (Aβ37, Aβ38, Aβ40 and Aβ42) to undetectable levels, while the Series A GSM Compound 2 reduced the levels of Aβ42 and Aβ40 while concomitantly increasing the levels of Aβ38 and Aβ37. We next attempted to extend the observations on the differential effects of Compound 2 on the various Aβ peptide variants to an in vivo experimental paradigm (Figure 3C). Plasma and brain samples from Tg 2576 mice dosed orally with Compound 2 (100 mg/kg for five consecutive days) showed a similar profile with respect to effects on the various Aβ peptide variants (decreased levels of Aβ42 and Aβ40; increased levels of Aβ38 and Aβ37). Interestingly, plasma levels of Aβ39 were also reduced in the Compound 2-treated versus the vehicle-treated mice, as were the levels of Aβ40 and Aβ42, while the levels of Aβ37 and Aβ38 were increased, in concordance with the in vitro studies as well as data from the various brain extracts (Figure 3C).
Figure 2.

Differential Effects of a Series A GSM, on Levels of Aβ42, Aβ40 and Aβtotal Peptide Variants. Concentration-response curves showing effects of the Series A GSM, Compound 3 (A and B) or the functional GSI, BMS-299897 (C and D) on levels of Aβ42, Aβ40 and Aβtotal peptide variants produced by CHO-APPswe cells (A and C) or SH-SY5Y-APP cells (B and D).
Figure 3.
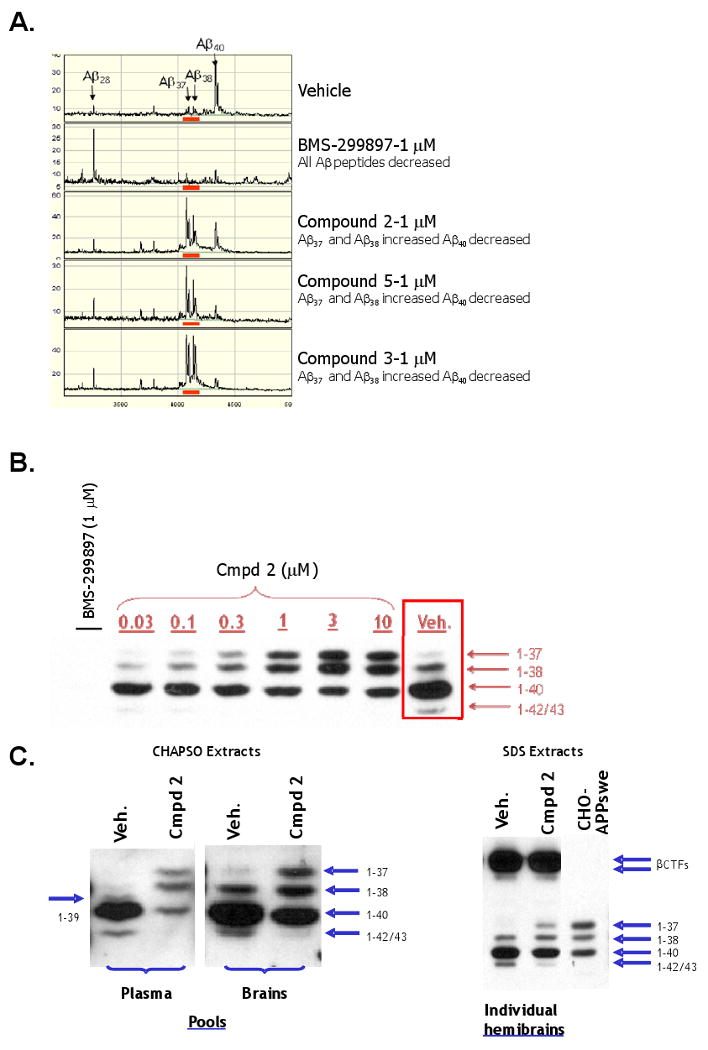
Opposing Effects of Series A GSMs on Levels of Aβ42 and Aβ40 Versus Effects on Levels of Aβ38 and Aβ37 Peptide Variants. (A) SELDI-TOF mass spectroscopic analyses of anti-Aβ17-24 mAb (4G8) immunoprecipitates of conditioned medium from CHO-APPswe cells treated with either vehicle (top panel), the functional GSI BMS-299897 (1 μM), also referred to as SIB-3520 (second panel), or the allosteric GSMs (each at 1 μM), Compound 2, Compound 5 or Compound 3 (bottom three panels, respectively). Equal amounts of synthetic Aβ1-28 peptide were spiked into aliquots of vehicle- and the various compound-treated media samples prior to performing the immunoprecipitations and was used as an internal standard. (B) Immunoblots of anti-Aβ1-12 (mAb-B436) immunoprecipitates (IP/westerns) of conditioned medium of Tg 2576 MBCs treated with vehicle, the functional GSI BMS-299897 (1 μM), also referred to as SIB-3520, or the indicated concentrations of the allosteric GSM, Compound 2. (C) Immunoblots of anti-Aβ1-12 (mAb-B436) immunoprecipitates (IP/westerns) of CHAPSO-solubilized extracts of plasma or brain pools (left panel) from either vehicle or Compound 2-treated (100 mg/kg p.o for five consecutive days) Tg 2576 mice; (right panel) SDS-solubilized extracts of representative individual hemibrains from either vehicle or Compound 2-treated Tg 2576 mice. Immunoprecipitates of conditioned medium of CHO-APPswe cells following treatment with Compound 2 (1 μM) were used as Aβ peptide variant standards.
APP-C99 is cleaved by γ-secretase at two distinct processing sites; one near the middle of the transmembrane domain to release Aβ peptides (γ-cleavage sites), and another near the membrane-cytosol interface releasing an intracellular domain termed AICD (ε-cleavage sites). GSMs not only differ dramatically from GSIs with respect to their effects on γ-secretase processing at γ-cleavage sites but also at ε-cleavage sites (Figure 4). Upon binding to certain ligands such as Delta and Jagged, the Notch receptor is rendered a γ-secretase substrate (LaVoie and Selkoe 2003) that is cleaved at a site analogous to the ε-site of APP. This generates the Notch intracellular domain (NICD), a signaling fragment that translocates to the nucleus and ultimately affects cell fate (Kopan and Goate, 2000). In order to compare the effects of GSIs and GSMs on γ-secretase-mediated ε-site processing of APP and Notch, we employed doubly-transfected human embryonic kidney 293 (HEK-APP-NΔE_7-4 cells) harboring both APP and NΔE cDNA constructs. The dipeptidic carboxamide GSI, N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester or DAPT (Dovey et al., 2001), inhibited NICD generation in a dose-dependent manner, at concentrations equivalent to those required to inhibit Aβ peptide generation (∼10-30 nM). In contrast, the Series A GSMs (Compound 5, Compound 3 and Compound 2) had no detectable effect on NICD formation in this very same cell-based assay (Figure 4A), even at concentrations ≥ 1000-fold higher (30 μM) than their IC50,s for inhibiting Aβ42 (∼5-30 nM).
Figure 4.
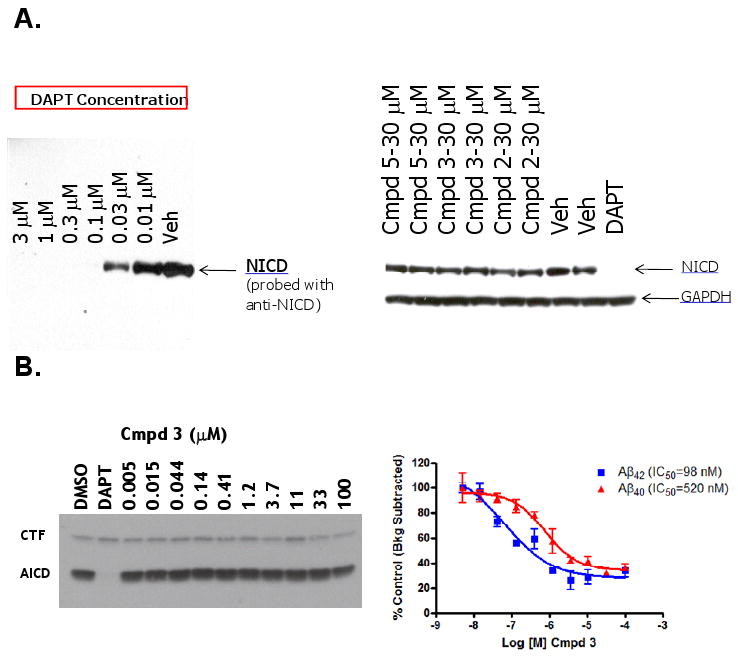
Series A GSMs do not Inhibit NICD Formation from Notch (NΔE) or AICD Formation from APP-CTFs. (A) Immunoblots (anti-NICD) of conditioned medium from HEK-APP-NΔE cells treated with the indicated concentrations of either DAPT or the designated allosteric GSMs. (B) Parallel analysis of AICD production (left panel) and Aβ42 and Aβ40 peptide formation (right panel) from CHO-APPswe membranes treated with either vehicle (0.2% DMSO), DAPT (1μM) or the indicated concentrations of the allosteric GSM, Compound 3. Data are expressed as mean ± SEM (n ≥ 3).
To further confirm that GSM compounds can modulate γ-secretase cleavages at γ-sites without inhibiting cleavage at ε-sites, a cell-free assay system capable of detecting Aβ42, Aβ40 and AICD was employed (Figure 4B; for experimental details, see Supplemental Information). Utilization of this cell-free assay allowed for a semi-quantitative measurement of AICD formation (ε-site activity) and Aβ peptide formation (γ-site activity) from identical cell-free preparations. Unlike DAPT (1 μM), which totally inhibited the formation of AICD and Aβ (Aβ data not shown), the Series A GSM Compound 3 had no effect on AICD levels when tested over a broad range of concentrations (Figure 4B). This same Series A GSM compound demonstrated dose-dependent inhibition of both Aβ42 and Aβ40 with a similar preference for lowering Aβ42 as previously shown in the various cell-based assays (see Figure 2). Although Compound 3 displayed significantly higher Aβ42 and Aβ40 IC50s in this cell-free assay as compared to the cell-based assays (perhaps due to increased non-specific protein binding and/or decreased effective drug concentrations), it did not affect ε-site activity as evidenced by the unchanged levels of AICD, even at concentrations 200- to 1000-times higher than those required for 50% inhibition of Aβ40 and Aβ42, respectively (Figure 4B).
Affinity Chromatography
To further investigate the mechanism of action of Series A GSMs, we employed affinity chromatography using a highly active compound (Compound 6) as a ligand. We observed a quantitative retention of Pen-2 and partial retention of PS1-NTFs (N-terminal fragments) from Triton X-100/Tween-20 (1.0%/0.2%;v/v) detergent-solubilized cellular lysates to the Compound 6 affinity column (Figure 5). There was only minor recovery of the PS1-CTFs (C-terminal fragments) and no recovery of APP-CTFs. One possibility is that under these conditions some PS1-NTF remains associated with compound 6-bound Pen-2 (perhaps via Pen-2/PS1-NTF heterodimers), especially when considering the known association of these two essential subunits (Kim and Sisodia, 2005), under very similar conditions (Fraering, et al., 2004). Another γ-secretase subunit, nicastrin, as well as a control protein, PLC-γ, showed no detectable binding to the Compound 6-derivatized affinity column. Aph-1a subunits were not probed. Elution with GSM compounds and competition experiments were not possible due to the core aqueous solubility of the compounds.
Figure 5.
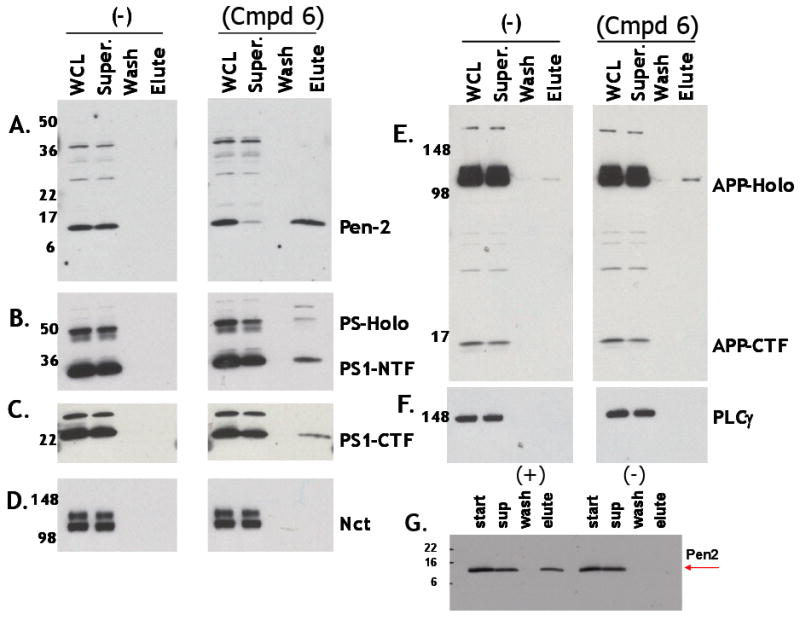
Affinity Chromatography Using an Immobilized Series A GSM Binds Isolated Pen-2 and Quantitatively Recovers Pen-2 from Nonionic Detergent-solubilized Cellular Extracts. (A-F) Compound 6 specifically retains Pen-2 from Triton X-100/Tween-20 (1.0%/0.2%)-solubilized CHO-APPswe whole cell lysates. Following gentle agitation of solubilzed whole cell lysates (WCL) with either the non-modified Affigel matrix (-) or Compound 6-immobilized Affigel matrix, flow thru supernatants (Super) were collected and columns were washed with PBS (Wash) and eluted with sample dilution buffer containing 1.0 % SDS (Elute). Equivalent aliquots of each fraction were electrophoresed using 4-20% gradient gels, immunoblotted/probed with the following antibodies; (A) anti-Pen-2, (B) anti- PS1-NTF, (C) anti-PS1-CTF, (D) anti-nicastrin (NCT), (E) anti-APP-CTF and (F) anti-phospholipase C-γ (PLCγ). (G) Affigel 10 with immobolized Compound 6 (+) retains purified recombinant Pen-2 and Affigel 10 alone (-) does not. PS-Holo and APP-Holo correspond to to PS1-holoprotein and APP-holoprotein, respectively.
An identical binding pattern to the Compound 6 resin: Pen-2 > PS1-NTFs > >PS1-CTFs was also observed using the same cellular extracts when solubilized with another non-denaturing, non-ionic detergent, dodecyl β-D-maltoside (DDM; 1.0%, v/v) (Figure S1), but not when using the zwitterionic CHAPSO detergent (data not shown). It is expected that these non-ionic, non-denaturing detergent solutions (DDM and Triton X-100/Tween-20) would solubilize the γ-secretase complexes in a more dissociating manner than would the zwitterionic, non-denaturing CHAPSO detergent, that is known to preserve intact γ-secretase complexes (Li et al., 2000). Steric hindrance due to immobilization of compound 6 using a short 10 atom spacer (Affigel 10) may have prevented access to Pen-2/PS1-NTF when localized within the fully assembled complex, since affinity capture of CHAPSO-solubilized catalytically active gamma-secretase with derivatized GSIs requires spacers of far greater length (> 25 atoms; Placanica, et al., 2009). Considering the ability of the immobilized Compound 6 to bind to isolated recombinant Pen-2 (Figure 5G) and the ability of a structurally-related Series A GSM analogue, Compound 4, to inhibit Aβ42 (as well as Aβ40) formation at nanomolar concentrations in membranes and in a reconstituted γ-secretase assay system utilizing highly purified requisite γ-secretase subunits (Figure 6C), suggest that Series A GSMs essentially shift the preferred γ-cleavage sites, increasing Aβ37 and Aβ38 and decreasing Aβ42 and Aβ40. Together with the lack of inhibition of NICD or AICD formation (ε-site cleavage products), these data begin to illustrate a mechanism of action distinct from all known subclasses of GSIs (Kreft, Martone and Porte, 2009) including the “allosteric GSIs” (AGSIs), coumarin dimer-based non-competitive inhibitors of γ-secretase that preferentially block the γ-site cleavage responsible for generating Aβ42 without altering the levels of Aβ37, Aβ38 or Aβ40 (Shelton et al., 2009). These data also serve to further differentiate these Series A GSMs from NSAIDs, such as sulindac sulfide, that have been shown to differentially and independently affect Aβ42 and Aβ38 generation without affecting production of either Aβ40 or Aβ37 (Page et al., 2008).
Figure 6.
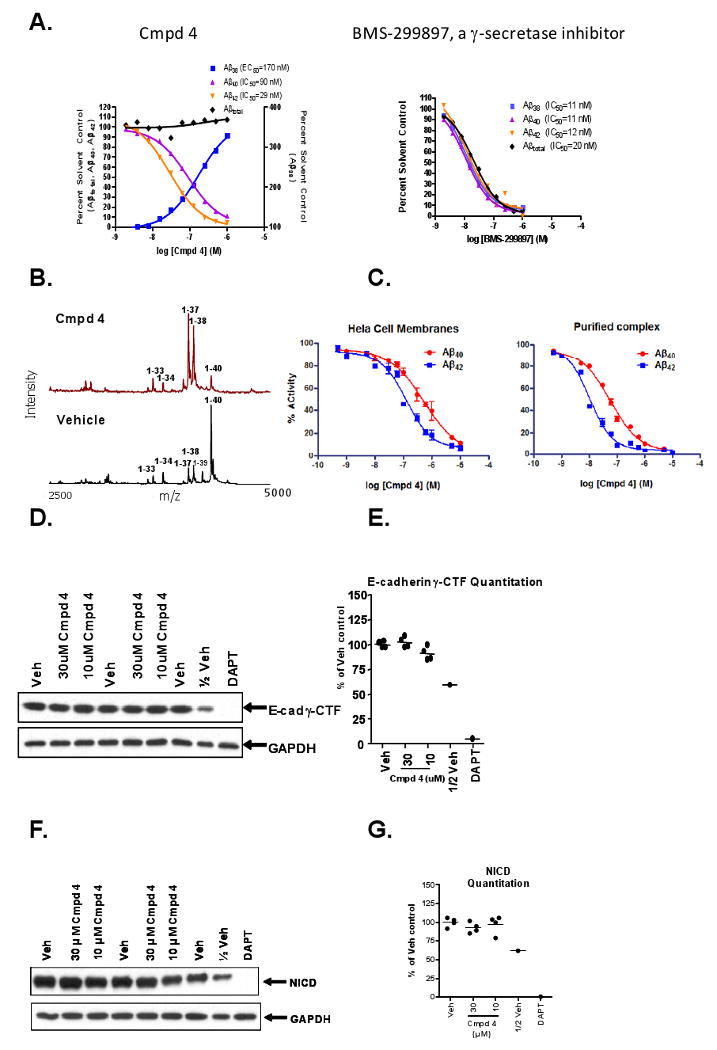
Compound 4, a Prototypical Series A GSM that Potently Lowers Levels of Aβ42 and Aβ40, Concomitantly Increases Levels of Aβ38 and Aβ37 and does not Inhibit γ-Secretase-Mediated Generation of E-Caderin γ-CTFs (E-Cad/CTF-γ) or NICD Generation from Notch (NΔE). (A) CRCs for Compound 4 (left panel) or BMS-299897 (right panel) on various Aβ peptide levels in conditioned medium following treatment (18 h) of Tg 2576 MBCs. Data from each specific sandwich ELISA or MesoScale assay were plotted as percent solvent control (0.2% v/v DMSO). IC50 or EC50 values were determined using four parameter fit non-linear regression analysis. (B) MALDI-TOF mass spectrometry of anti-Aβ17-24 mAb (4G8) immunoprecipitates of conditioned medium from CHO-APPswe cells after treatment (18 h) with either Compound 4 (1μM) or vehicle (0.2% v/v DMSO). (C) Concentration response curves for inhibition of Aβ42 and Aβ40 production in HeLa cell membrane (left panel) or TAP-purified protein reconstituted γ-secretase complex (right panel) in vitro γ-secretase assays. IC50 values were derived using four parameter fit non-linear regression analyses. (D) Immunoblots of E-cad γ-CTFs and the GAPDH control protein following treatment of human A431 cells with either DAPT (1 μM) or the indicated concentrations of Compound 4 for 18 h. Proteolysis of E-cadherin is induced by staurosporine (1 μM) treatment of A431 cells for 6 h. (F) Immunoblots of NICD and the GAPDH control protein following treatment of HEK-APP-NΔE cells with either DAPT (1 μM) or the indicated concentrations of Compound 4. DMSO (0.2%v/v) is the vehicle control (Veh) and 0.5× sample volume from vehicle treated cells (1/2 Veh) was analyzed to quantify relative levels of E-cad γ-CTF immunoreactivity (E) or NICD immunoreactivity (G) using laser scanning densitometry.
In Vitro and In Vivo Primary Pharmacology
Comparisons between several different Series A GSMs (data not shown) demonstrated that Compound 4 had the most favorable PK properties (Tmax p.o.= 3 ± 0.6 h; Cl i.v. = 0.2 ± 0.03 l/h/kg; t½ p.o. = 2.0 ± 0.2 h; F = 49%) and was therefore selected for further in vitro and in vivo studies, including single dose, subchronic, and chronic long-term efficacy studies. Using Meso Scale Aβ alloform specific ELISAs, in vitro characterization of Compound 4 in MBCs from Tg 2576 mice showed a concentration-dependent decrease in Aβ42 as well as Aβ40, markedly increased levels of Aβ38, and no significant change in Aβtotal levels (as defined by Aβ1-x, where x is ≥ 24). In contrast, BMS-299897 induced a dose-dependent decrease in Aβ42, Aβ40, Aβ38 and Aβtotal levels (Figure 6A). To confirm the differential effect on various Aβ peptides including Aβ37 by Compound 4, MALDI-TOF analysis of conditioned medium of the CHO-APPswe cells following treatment with Compound 4 was conducted (Figure 6B). In addition, when using either a HeLa cell membrane in vitro γ-secretase assay system (Li et al., 2000) or a reconstituted γ-secretase assay composed of tandem affinity purified (TAP) γ-secretase complex subunits (Figure 6C; see also Figure S2 and Supplemental Information for experimental details) Compound 4 inhibited Aβ42 production (IC50 = 131 nM in the HeLa cell membrane in vitro assay; IC50 = 12 nM in the reconstituted TAP-isolated protein complex γ-secretase assay) and Aβ40 production (IC50 = 531 nM in the HeLa cell membrane assay; IC50 = 56 nM in the reconstituted TAP-isolated protein complex γ-secretase assay) and did not inhibit NICD formation from the NΔE Notch/APP co-transfected cell line, in contrast to DAPT (1 μM), which prevented NICD production entirely (Figure 6F&G). In order to assess the effects of Compound 4 on another known γ-secretase substrate, E-cadherin, an assay similar to one described previously (Marambaud et al. 2002) was employed using a human epithelial cell line (A431) that endogenously expresses measureable levels of E-cadherin proteolytic peptide products. Proteolysis of the full length E-cadherin protein is induced upon treatment of the cells with staurosporin, followed by generation of the E-cad CTF-γ by γ-secretase. Results of A431 cells treated with Compound 4 in the E-cadherin γ-secretase assay are shown in Figure 6D&E, and demonstrate no inhibition of γ-secretase-mediated proteolysis of E-cadherin peptides at concentrations up to 1000-fold above the IC50 values for lowering Aβ42 levels in SH-SY5Y-APP (Figure 6A) and NΔE Notch/APP (data not shown) cell-based assays. These results strongly support the conclusion that Compound 4 behaves like a prototypical Series A GSM.
Single and Repeat-Dose Efficacy Studies
Prior to testing for efficacy in vivo, brain and plasma concentrations following oral dosing (50 mg/kg) were measured to assess brain: plasma ratios of Compound 4 in C57Bl/6 mice. Plasma and brain concentration vs. time data from a representative exposure study following oral dosing with Compound 4 are shown in Figure 7A, and the mean data from two studies showed plasma exposure (AUC) at 0-24 hours was 70,475 ng·h/ml while in brain was 65,545 ng·h/ml, giving an average brain: plasma ratio of 0.93. Compound 4 was then tested for efficacy for lowering concentrations of brain Aβ42 and Aβ40 peptides in 3-4 month old (non-plaque-bearing) Tg 2576 female mice (5-100 mg/kg p.o. in a PEG 400:water vehicle) following once daily dosing for three consecutive days with samples being collected 6 h after the last dose. This in vivo experimental paradigm was established based on data from numerous pharmacodynamic experiments confirming that 6 h post-dose is an appropriate collection time to capture optimal Compound 4 compound levels (Figure 7A) and maximal effects for lowering both the plasma and brain levels of Aβ42 (data not shown). Brain and plasma Aβ42 concentrations were measured by ELISA and compound concentrations were measured in drug-treated animals by LC-MS/MS. Compound 4 elicited dose-ordered lowering of Aβ42 levels in both plasma and brain following three days of consecutive daily dosing (Figure 7B). The compound appeared to be more efficacious in plasma than in brain due to a combination of possibilities: (1) compound exposures were possibly slightly greater in plasma than in brain; (2) plasma Aβ42 turns over much more rapidly than does brain Aβ42 (Cirrito et al., 2005); (3) nonspecific protein binding of Compound 4 in brain may be greater than in plasma, thus lowering the effective Compound 4 concentration to a greater extent in brain than in plasma. Figure 7 shows that following treatment of young female Tg 2576 mice with increasing doses of Compound 4 (5-100 mg/kg, p.o.), Aβ42 levels were significantly lower relative to vehicle-treated mice, with a minimum effective dose of 25-50 mg/kg, and the extent of reduction in brain Aβ42 levels correlated with brain concentrations of Compound 4 (Figure 7C). The brain concentrations of Compound 4 achieved following these dosing regimens were in excess of those necessary to reduce Aβ42 levels based on the IC50 (29 nM) obtained from cell-based assays in MBCs (Figure 6A).
Figure 7.
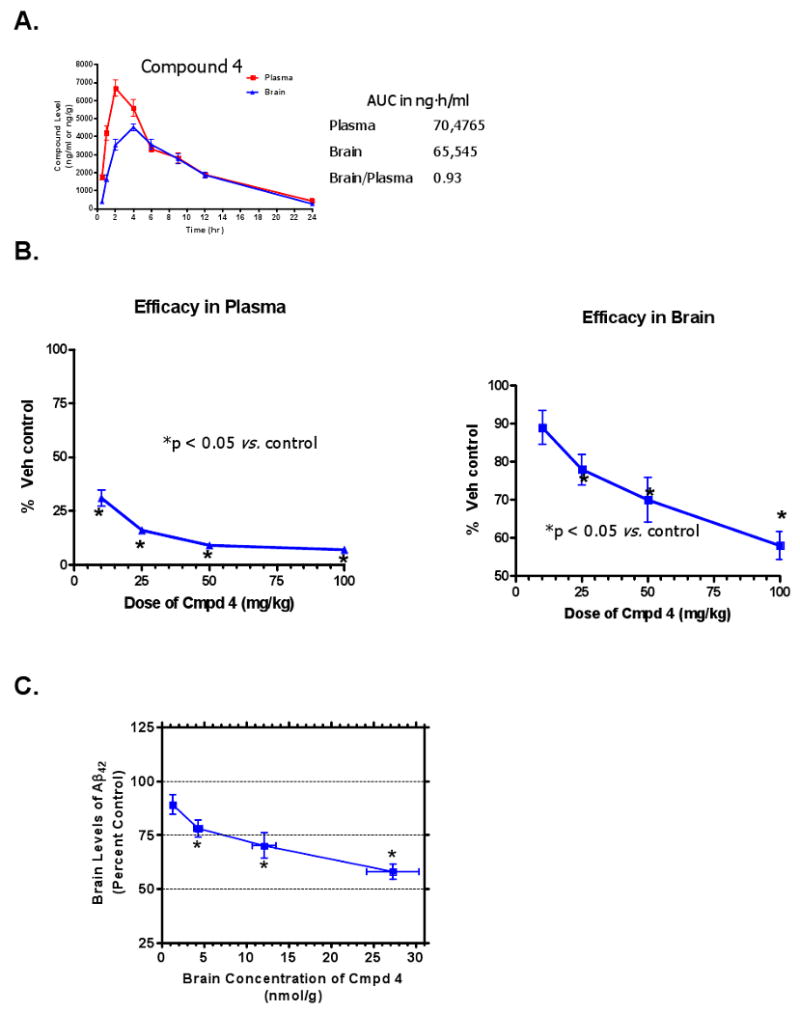
Dose-dependent Reduction of Aβ42 Levels in Plasma and Brain in Tg 2576 Mice Following Oral Administration of the Series A GSM Compound 4. (A) Time course of plasma and brain exposures of Compound 4 in female C57Bl/6 mice (2-3 mo of age, n=6 per group) following once daily oral administration for 3 consecutive days of Compound 4 (50 mg/kg, p.o.) in F110 vehicle. Plasma and brains were collected at the indicated times post-dose on day 3 and concentrations of Compound 4 were measured in plasma and brain extracts by LC-MS/MS. Exposure is given as the integrated area under the curve (AUC) of the compound concentration vs. time curve in ng·hr/ml. Brain/plasma is the brain:plasma ratio of Compound 4 exposure. (B) Dose-dependent efficacy of Compound 4 (p.o.) for lowering Aβ42 in plasma (left panel) and brain (right panel) in Tg 2576 mice (3-4 mo of age, n=10/dose) following three days of once-daily dosing expressed as a percentage of vehicle (80% PEG-400 v/v) control. (C) Brain concentrations of Compound 4 attained following each dose. Data are mean ± SEM. *p < 0.05 by t-test for brain measurements or ANOVA with Dunnett's post-hoc analysis for plasma measurements.
Compound 4 was further examined for in vivo efficacy following either one day (50 or 200 mg/kg p.o.) or fourteen days (50 mg/kg p.o.) of once daily administration. Compound 4 produces consistent efficacy in lowering both plasma and brain levels of Aβ42 and Aβ40 peptides when given acutely or for up to two weeks of oral dosing (Table S1).
Chronic Administration Efficacy Study
We next asked whether chronic administration, and presumably sustained reduction of Aβ42 and Aβ40 brain concentrations, of Compound 4 could affect the extent of pathology (i.e., amyloid plaques) in the brains of Tg 2576 mice. To examine this, 8 month old female (pre-plaque-bearing) Tg2576 mice were dosed daily with an estimated 50 mg/kg/day (p.o.) of Compound 4 for seven consecutive months. For this purpose Compound 4 was milled into the standard rodent diet (Harlan Teklad 8664) at a concentration of 0.3125 g Compound 4/kg chow and repelleted and the mice (n=19-20 per group) were allowed to feed ad lib either Compound 4-containing (treated) or standard (vehicle) rodent chow. Compound 4-fed and vehicle-fed mice were carefully observed and food consumption and body weights were recorded weekly. Compound 4 was well tolerated based upon behavioral observations and the lack of differences in food consumption (data not shown) or rate of body weight gain between the groups throughout the 29 week period of exposure (Figure 8A). In addition, plasma concentrations of Compound 4 were measured in a subset of treated mice at the end of the chronic study and were found to be 3.95 ± 0.49 nmol/ml (n=12). This level of exposure, which presumably reflects steady-state plasma concentration under this dosing regimen, was roughly equal to peak plasma concentrations seen following three days of once-daily dosing at 25 mg/kg given via oral gavage. Data from the three-day dosing paradigm has shown that this plasma concentration is sufficient to significantly reduce levels of brain Aβ42 in Tg 2576 mice (Figure 7C).
Figure 8.
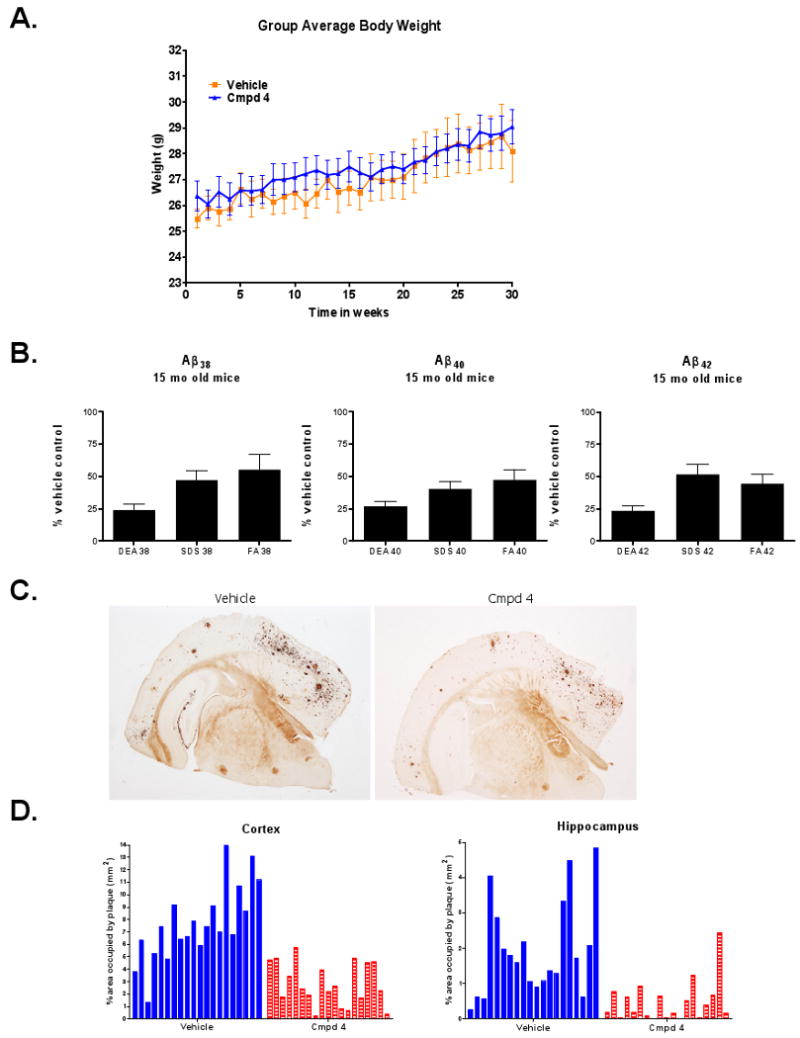
Chronic Daily Exposure of Tg 2576 Mice to the Series A GSM Compound 4 for Seven Consecutive Months is Well Tolerated and Significantly Reduces Overall Amyloid Load and Aβ Deposition in Neuritic Plaques. (A) Weekly group averaged body weights of Tg 2576 mice (8 mo – 15 mo of age) fed (ad libitum) either normal rodent chow (Vehicle; n=20) or rodent chow containing an estimated dose of 50 mg/kg/day of Compound 4 (Compound 4; n=19) for 29 consecutive weeks. Data are mean ± SEM. (B) Brain Aβ peptide (Aβ42, Aβ40 and Aβ38) concentrations (measured using Meso Scale triplex kits) in aqueous buffer soluble (DEA), denaturing detergent-extractable (SDS) and formic acid-extractable (FA) brain fractions from Tg 2576 mice fed either normal rodent chow or chow containing Compound 4. Data are expressed as mean (± SEM) of percentage of control levels in normal chow-fed mice (p ≤ 0.002 for each of the Aβ peptide variants in all extracts except for Aβ38 in FA extract; p = 0.0441). (C) Representative photomicrographs of Campbell-Switzer silver stained sections showing neuritic plaque staining in the cerebral cortex and hippocampus from a normal chow-fed 15-mo old Tg 2576 mouse (left panel) and a 15-mo old Tg 2576 mouse fed Compound 4 (50 mg/kg/day)-containing chow (right panel) from 8 to 15 months of age. (D) Percentage of cerebral cortical area (left panel) and hippocampal area (right panel) occupied by neuritic plaques, quantified from 4 brain levels. Data are expressed as percent area (mm2) of individual mice from normal chow-fed (Vehicle) and Compound 4-containing chow-fed (Compound 4) Tg 2576 mice.
Evaluation of the gastrointestinal tract and semi-quantitative analysis of GI goblet cell densities in both the small and large intestine of treated and untreated mice showed no treatment-related differences between vehicle- and Compound 4-fed mice (Table S2). GI lesions, such as those previously reported after repeated exposure to GSI's (Searfoss et al., 2003; Wong et al., 2004; Hyde et al., 2006) are typically associated with inflammation, erosion and goblet cell hyperplasia, which was not evidenced after chronic Compound 4 treatment. These data strongly support the conclusion that allosteric modulation of γ-secretase is sufficient to significantly reduce Aβ42-dependent pathological endpoints without induction of gastrointestinal changes associated with inhibiting Notch proteolytic processing.
Serial extraction of one brain hemisphere for each study animal was done to capture soluble (DEA-extractions), detergent-soluble (SDS) and formic acid-extracted (FA) Aβ peptides. Biochemical analyses of these extracts using the Meso Scale Aβ triplex kit revealed that the levels of all three Aβ peptides measured (Aβ38, Aβ40, Aβ42) were dramatically and significantly reduced in all three brain fractions of Compound 4-treated mice, relative to control mice. Figure 8B shows percentage of each Aβ peptide, relative to control levels, in each brain extract of treated mice. In DEA extracts, Compound 4 treatment, relative to control mice, resulted in 24 ± 4% (mean ± SEM) concentration of Aβ38 (p<0.0001), 27 ± 4% of Aβ40 (p<0.0001) and 24 ± 3% of Aβ42 (p<0.0001) concentrations. In the SDS extracts, Compound 4-treated mice had 47 ± 7% of Aβ38 (p=0.0016), 41 ± 6% of Aβ40 (p<0.0001) and 52 ± 8% of Aβ42 (p=0.0003). In the FA extracts, the Compound 4-treated mice had 55 ± 11% of Aβ38 (p=0.0441), 48 ± 8% of Aβ40 (p=0.0014) and 45 ± 7% of Aβ42 (p=0.002). Somewhat surprisingly, levels of Aβ38 were significantly reduced in each of the various brain extracts analyzed, in contrast to Compound 4's effects of increasing Aβ38 in APPswe-expressing cell cultures and in brain and plasma with acute or short-term in vivo dosing. This could be due to the inability to detect this modest increase in aged plaque-bearing Tg 2576 mice under conditions of exceptionally high amyloid loads.
Fixed brain sections were stained with Campbell-Switzer stain (Campbell, Switzer and Martin, 1987) to visualize both diffuse and neuritic amyloid plaques as depicted in representative photomicrographs of Campbell-Switzer-stained sections from control and drug-treated mice (Figure 8B). The percentage of cortical and hippocampal area covered by plaque was measured by quantitative image analysis through 4 levels of brain and morphological and statistical comparisons were made between treated and untreated mice (Figure 8D). While there is noticeable variability among animals within the treatment groups (which is entirely expected for this age of animals in this particular AD mouse model), there is a highly statistically significant difference between the Compound 4-treated and untreated mice. In untreated mice, the mean percent of cortex occupied by plaque was 7.6%, while in the Compound 4-treated mice the mean percent area was 2.8%, p < 0.0001. In the hippocampus, 1.9% of the hippocampal area was occupied by plaque in normal chow-fed animals, while an average of 0.5% of hippocampal area was occupied by plaque in Compound 4-treated mice, p=0.0001 (Figure 8D). These histological data show that chronic treatment (29 weeks) with Compound 4, given orally in rodent chow, substantially inhibited plaque deposition in Tg 2576 mice between the ages of 8 and 15 months as shown by significantly lower plaque density and amyloid load in both hippocampus and cortex.
Discussion
We have characterized a class of GSM compounds that display unique pharmacological properties compared to other disease-modifying therapeutic approaches (e.g., GSI's) previously tested or currently undergoing human clinical trials for AD (for review see Pissarnitski, 2007). Unlike “substrate-targeting GSMs” e.g. R-flurbiprofen or tarenflurbil (Kuklar et al., 2008), these Series A GSMs exhibit potencies and levels of brain penetration that are far superior to tarenflurbil or other NSAID-like GSMs such as sulindac sulfide or CHF5074 (Weggen et al., 2001; Imbimbo et al., 2007). In addition, Series A GSMs are approximately 1000-10,000-fold more potent than tarenflurbil, sulindac sulfide or CHF5074, based on in vitro IC50 values in comparable cell-based assays.
The Series A GSMs bind directly to the γ-secretase enzyme complex via Pen-2/PS1-NTFs and, in effect, elicit a truncation of the major secreted Aβ peptides (Aβ42 and Aβ40), without inhibiting ε-site proteolysis of APP, Notch, E-cadherin or LRP-1 (data not shown for LRP-1). Series A GSMs potently inhibit the production of Aβ42 and Aβ40 and cause a concomitant increase in the levels of Aβ38 and Aβ37. It is currently not known whether products of γ-site cleavages from other γ-secretase substrates such as Notch, i.e., Notch β, upon treatment with A GSMs, would also be similarly affected. In any event, selectively affecting the γ-site cleavage products (e.g., Aβ, Nβ, etc.) from γ-secretase-mediated proteolysis of the numerous γ-secretase substrates would appear to be a prudent therapeutic approach, especially when weighing the risks of inhibiting ε-site proteolysis. Interestingly, ε-site proteolytic processing by γ-secretase has been much more widely studied than have the γ-sites and in an ever-expanding number of type I membrane proteins, involves the generation of intracellular domains (ICDs), e.g., AICD, NICD and LICD, many of which appear to translocate to the nucleus (De Strooper et al., 1999; Cao and Sudhof, 2001; Wakabayashi and De Strooper, 2008).
A few of the functional GSIs previously described, i.e., the arylsulfonamide- or sulfonamide-containing, alternative-site binding GSIs, BMS-299897 (Milano et al., 2004), GSI-953 (Begacestat) and BMS-708163 (Martone et al., 2009; Kreft, Martone and Porte, 2009; Starrett, Gillman and Olson, 2009), as well as at least one other sulfonamide-containing GSI (Best et al., 2007) appear to be “Notch-sparing”. However, the mechanistic basis of this apparent therapeutic window is poorly understood and the effects of these “Notch-sparing” GSIs on γ-secretase-mediated ε-site proteolysis of APP and other γ-secretase substrates with the exception of Notch, have not been reported.
In the present study, we analyzed a finite number of γ-secretase (ε-site-cleaved) substrates including APP (AICD), Notch (NICD), E-cadherin (E-Cad/CTF-γ) and LRP-1 (LICD); and even when tested at concentrations far above (∼1000-fold) the in vitro IC50's for lowering the levels of Aβ42 in cell-based assays, we detected no inhibition regarding the ε-site proteolysis on any of these four known substrates of γ-secretase. Furthermore, the lack of inhibition in cell-based in vitro assays on γ-secretase-mediated ε-site proteolysis of Notch (i.e., NICD formation) is consistent with the lack of effect of Compound 4 on intestinal goblet cell densities, even after chronic daily administration to Tg 2576 transgenic mice (∼50 mg/kg/day p.o. for seven months). This is not the case for some of the more thoroughly studied GSIs, including Compound X (Searfoss et al., 2003) and LY-411,575 (Wong et al., 2004; Hyde et al., 2006) that show evidence of Notch-related toxicity even after much shorter periods of administration than those employed here.
GSIs have been recently categorized into a number of different subclasses based on their functional or structural similarities; e.g., active-site binding, substrate docking-site binding, peptidic, di-peptidic or peptidomimetic transition state mimics or transition state analogs, and alternative binding site carboxamides, sulfamides, sulfonamides, sulfones, dibenzazepines, benzodiazepines, etc., (for review see Kreft, Martone and Porte, 2009; Tomita, 2009). Thus far most, if not all, of the various subclasses of GSIs appear to interact directly with PS, the catalytic component of γ-secretase and would therefore be expected to similarly inhibit the proteolysis of other γ-secretase substrates such as Notch (Lewis et al., 2003). Interestingly, the arylsulfonamide-containing GSI, BMS- 299897 (Tian et al., 2002; Anderson et al., 2005), a non-competitive γ-secretase inhibitor that lowers the levels of all measureable Aβ peptide variants, concomitantly causes accumulation of both α- and β-CTFs, suggesting a mechanism consistent with dual inhibition of ε-site and γ-site proteolysis.
Three other GSIs, LY450139 (Semagacestat), GSI-953 (Begacestat) and BMS-708163 have recently entered into clinical trials (Fleisher et al., 2008; Martone et al., 2009). These GSIs (a carboxamide, a sulfonamide, and an arylsulfonamide, respectively) would all be expected to interact directly with the catalytic component of γ-secretase (PS), and would therefore be anticipated to similarly inhibit Notch, APP and a variety of other known γ-secretase substrates. Interestingly, recent clinical studies with LY450139 have reported various adverse events during phase I and phase II that may be interpreted as being Notch-mediated (Fleisher et al, 2008). The other two sulfonamide-containing GSIs (GSI-953 and BMS-708163) with an open IND (investigational new drug) status both have a mechanism of total Aβ peptide inhibition described as “Notch-sparing.” This classification is based primarily on the relative IC50 or EC50 values obtained from a variety of comparable in vitro cell based assays, similar to those employed here, demonstrating that these two GSIs can preferentially inhibit Aβ peptide production from APP/βCTFs versus inhibiting NICD production from Notch/NΔE with a rather impressive yet highly variable (14-fold for GSI-953 and 26- to 193-fold for BMS-708163) preference for lowering Aβ peptide levels (Martone et al., 2009; Starrett, Gillman and Olson, 2009).
However, γ-secretase complexes are known to hydrolyze a large number of type I membrane proteins (Wakabayashi and De Strooper, 2008), implying possible cooperation in major membrane protein degradation and signaling pathways. Therefore, inhibiting an enzymatic complex like γ-secretase, which has been described as the “proteosome of the membrane” (Kopan and Ilagan, 2004), may not be at all beneficial to an AD population with compromised neuronal catabolism. The aminothiazole-containing GSMs described in the present study are not inhibitors of γ-secretase activity and are thus, genuinely “Notch-sparing”. This is based on numerous in vitro and in vivo endpoints observed even after prolonged periods of exposure, throughout which the compound sustained the ability to reduce Aβ42 peptide levels and attenuate amyloid deposition without evidence of Notch-related GI toxicity. Furthermore the in vitro studies, including affinity chromatography with a biologically active Series A GSM (Compound 6), strongly suggests γ-secretase (i.e., Pen-2/PS1-NTFs) as the binding target and support these aminothiazole GSMs as being capable of potently and preferentially lowering both Aβ42and Aβ40 levels without inhibiting NICD or AICD formation. In summary, these experiments describe a class of aminothiazole γ-secretase modulators, hereafter referred to as AGSMs, which exhibit a unique pharmacological profile, in vitro and in vivo, that under steady state conditions, is consistent with a non-inhibitory mechanism of action involving direct interaction with γ-secretase. These types of AGSM compounds warrant further in vitro and in vivo investigation as a potentially safe and effective therapeutic approach for the treatment and/or prevention of AD and perhaps other related neurodegenerative proteinopathies (Ghaemmaghami et al., 2009).
Experimental Procedures
Stable Cell lines and Cell-Based Assays for Measuring Extracellular Levels of Aβ42 and Aβ40
The CHO-PZ3 (CHO-APPwt) cell line was derived by transfecting a Chinese hamster ovary (CHO) cell line with a plasmid expressing wild type human APP695 cDNA and selecting for stable expression of human APP and human Aβ. The CHO-APPswe cell line was derived by transfecting the same CHO cell line with a plasmid expressing the “Swedish” (swe) double mutant (670/671 K/M to N/L) cDNA of human APP695; the SH-SY5Y-APP cell line was derived by transfecting a human neuroblastoma (SH-SY5Y) cell line with a plasmid expressing wild type human APP751 cDNA. In each case, the levels of Aβ42 and Aβ40 secreted into the media were measured using either a two-site monoclonal antibody (mAb)-based homogeneous FRET high-throughput screen (HTS) or a comparable HTS sandwich ELISA assay. Cytotoxicity was determined using an Alamar Blue cell-based assay at the same compound concentrations used to determine compound potency and efficacy up to a maximum concentration of 30 μM. For complete experimental details, see Supplemental Information.
Cell-Based Assays for Extracellular Aβ42 and Aβ40 in Tg 2576 Mixed Brain Cultures (MBCs)
Neuronal cultures were prepared from brains of Tg 2576 embryos (E17) and were plated onto 96-well tissue culture dishes. Cells were grown for 6 days and then treated in triplicate wells for 18 h with compound at 10 concentrations with 3-fold dilution steps. Vehicle (PEG-400:H2O 80:20 v/v) and BMS-299897 were included as negative and positive controls, respectively. For complete experimental details, see Supplemental Information.
Aβ38, Aβ40, Aβ42 and Aβtotal Sandwich ELISAs
The Aβ38 peptide was quantified using either the Meso Scale Aβ38,40,42 triplex kit or the Meso Scale Aβ38 kit alone with the Meso Scale Sector Imager 6000. Aβ40, Aβ42, and Aβtotal levels were also measured using mAb-specific sandwich ELISA assays.
Mass Spectrometry
Ciphergen chips prepared according to manufacturer-recommended protocols from GSI- or GSM-treated CHO-APPswe conditioned medium were analyzed in the SELDI mass spectrometer (Ciphergen). MALDI-TOF mass spectrometry of anti-Aβ peptide immunoprecipitates of conditioned medium from compound-treated CHO-APPswe cells was performed by Dr. Rong Wang (Mount Sinai School of Medicine, New York, N.Y.) as described previously (Wagner and Munoz, 1999).
Cell-Based Assays for Measuring Notch and E-Cadherin Proteolytic Processing
Doubly-transfected human embryonic kidney 293 (HEK-APP-NΔE_7-4 cells) harboring both APP and Notch (NΔE) cDNA constructs (A gift of Dr. Edward H. Koo, University of California, San Diego) were treated with compound overnight, conditioned medium was collected and cell lysates were prepared. Cell lysates were analyzed for levels of NICD by western blot, and immunopositive protein bands were quantified by laser scanning densitometry. As a means of verifying compound activity, the conditioned medium was concomitantly analyzed for levels of secreted Aβ40 by sandwich ELISA, as described above. Measurement of inhibition of E-cadherin proteolytic processing was similar to that described previously (Marambaud et al., 2002).
Affinity Chromatography and Immunoblotting
Compound 6 was coupled to Affigel 10 agarose for affinity chromatography. Whole cell lysates (WCL) were prepared from CHO-APPswe cells and were incubated with 100 μl of resin for 2 hrs at 4 °C. Bound proteins were eluted with 1% SDS sample dilution buffer and were separated on 4-20% SDS polyacrylamide gels, transferred to nitrocellulose filters, and probed with a panel of antibodies. For complete experimental details, see Supplemental Information.
Brain and Plasma Compound Concentration Measurements
Blood and brains were collected at the indicated interval (after the last dose), and quantitative measurements of compound concentrations in the corresponding plasma and brain extracts were made by LC-MS/MS (Tandem Labs, West Trenton, NJ).
Rat Pharmacokinetics
Male Sprague-Dawley rats (∼300 g) were instrumented with jugular vein or jugular and femoral vein catheters and allowed several days to recover then fasted overnight and compounds were injected into the femoral vein catheter at 2 mg/kg or administered by oral gavage at 10 mg/kg (n = 3 - 4/group). Compound concentrations in plasma were measured by LC-MS/MS (Tandem Labs, West Trenton, NJ). Pharmacokinetic parameters were calculated from non-compartmental modeling using WinNonlin software (Pharsight Corp.).
In Vivo Efficacy Testing
All procedures conducted on animals were approved by TorreyPines Therapeutics' IACUC and conform to current animal welfare guidelines. Briefly, 3-4 mo old female Tg 2576 mice were used for short term (≤14 days) efficacy studies (generally n=10/group), and 8 mo old female Tg 2576 mice were used for the chronic efficacy study (n=19-20/group). Dosing was done by daily oral gavage in an 80% PEG 400 (v/v) vehicle for the short-term studies and compound was milled into the rodent chow (0.3125g Compound 4/kg chow; Harlan Teklad 8664) for oral delivery in the chronic study. For complete experimental details, see Supplemental Information.
Chronic Study GI evaluation
Slides containing tissue from the small and large intestine were trimmed, processed and stained with the Periodic Acid Schiff stain. Tissues were evaluated for goblet cell numbers and density and results were recorded separately for small and large intestines. Goblet cell density was graded on a 3 point scale with 1 having the least and 3 having the greatest goblet cell density. Evaluation of goblet cell densities in the intestinal tracts of Compound 4-treated and untreated Tg 2576 mice was performed in a blinded fashion by Midwest ToxPath Sciences, Inc., Chesterfield, MO.
Histopathology
For complete experimental details, see Supplemental Information. Briefly, brain hemispheres were processed and stained with the Campbell-Switzer stain (Campbell, Switzer and Martin, 1987) to visualize both diffuse and neuritic amyloid plaques at NeuroScience Associates, Knoxville, TN. Four coronal levels were used for image analysis to determine the percent area of the brain section occupied by plaques. Image analysis of plaques in the cortex and hippocampus was done using NIH Image/Image J. An area of interest (cortex or hippocampus) in the tissue sections was traced and was converted into two other images, one used for obtaining the area (mm2) of the region of interest, and the second for performing a particle analysis to obtain individual plaque areas and total plaque number. The area of each plaque and the total number of plaques was determined by using NIH Image's Particle Analysis. All sections were evaluated blind as to treatment.
Statistical Analysis
Values are mean ± standard error of the mean (SEM) and expressed as the % of the mean of vehicle-treated control groups. Differences between treatment and control groups were analyzed by one-way analysis of variance and by Dunnett's t-test when a significant (P< 0.05) F-value is attained. Correlation analyses are performed using Pearson Product Moment correlation analysis and IC50 and ED50 values estimated with non-linear regression (GraphPad Prism).
Supplementary Material
Acknowledgments
The authors wish to thank Nicolas Patch for technical support, Pat Baskin for critical review of the manuscript and Eisai, Co. Ltd. and the NIH grant RO1 AG026660 (to YML) and the John Adler Foundation (to SSS) for financial support. Drs. Comer and Kounnas are employees of Neurogenetics Pharmaceuticals (NGP), Inc. Drs. Comer, Tanzi, and Wagner are shareholders and founders of NGP. Dr. Tanzi and Dr. Wagner were co-founders at TorreyPines Therapeutics. Dr. Tanzi is a co-founder, shareholder, and consultant in Prana Biotechnology and Eisai.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson JJ, Holtz G, Baskin PB, Turner M, Rowe BR, Wang B, Kounnas MZ, Lamb BT, Barten D, Felsenstein K, et al. Reductions in β-amyloid concentrations in vivo with the γ-secretase inhibitors BMS-289948 and BMS-299897. Biochem Pharmacol. 2005;69:689–698. doi: 10.1016/j.bcp.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Best JD, Smith DW, Reilly MA, O'Donnell R, Lewis HD, Ellis S, Wilkie N, Rosahl TW, Laroque PA, Boussiquet-Leroux C, et al. The γ-secretase inhibitor N-[cis-4-[(4-Chlorophenyl)sulfonyl]-4-(2,5-difluorophenyl)cyclohexyl]-1,1,1-trifluoromethanesulfonamide (MRK-560) reduces amyloid plaque deposition without evidence of notch-related pathology in the Tg2576 mouse. J Pharmacol Exper Ther. 2007;320:552–558. doi: 10.1124/jpet.106.114330. [DOI] [PubMed] [Google Scholar]
- Campbell SK, Switzer RC, Martin TL. Alzheimer's plaques and tangles: A controlled and enhanced silver staining method. Soc Neurosci Abst. 1987;13:189.9. [Google Scholar]
- Cao X, Sudof TC. A transcriptionally active complex of APP with Fe65 and histone acetyl-transferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, May PC, O'dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JE, Bales KR, Paul SM, et al. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid beta metabolism and half–life. J Neurosci. 2003;23:8844–8853. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Annart W, Cupers P, Saftig P, Craessarts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WI, et al. A presenilin-1-dependent γ -secretase-like protease mediates release of notch intracellular domain. Nature. 1999;358:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- Dovey HF, John V, Anderson JP, Chen LZ, de Saint Andrieu P, Fang LY, Freedman SB, Folmer B, Goldbach E, Holsztynska EJ. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem. 2001;76:173–181. doi: 10.1046/j.1471-4159.2001.00012.x. [DOI] [PubMed] [Google Scholar]
- Fraering PC, LaVoie MJ, Ye W, Ostaszewski BL, Kimberly WT, Selkoe DJ, Wolfe MS. Detergent-dependent dissociation of active gamma-secretase reveals an interaction between Pen-2 and PS1-NTF and offers a model for subunit organization within the complex. Biochemistry. 2004;43:323–333. doi: 10.1021/bi035748j. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S, May BCH, Rensio AR, Pruisner SB. Discovery of 2-aminothiazoles as potent antiprion compounds. J Virol. 2009 doi: 10.1128/JVI.0214509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsaio K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hyde LA, McHugh NA, Chen J, Zhang Q, Manfra D, Nomeir AA, Josien H, Bara T, Clader JW, Zhang L, et al. Studies to investigate the in vivo therapeutic window of the γ-secretase inhibitor N2-[(2S)-2-(3,5-difluorophenyl)-2-hydroxethanoyl]-N1-[(7S)-5-methyl-6-oxo-6,7-dihydro-5H-dibenzo[b,d]azepin-7-L-alaninamide (LY411,575) in the CRND8 mouse. J Pharmacol Exp Ther. 2006;319:1133–1143. doi: 10.1124/jpet.106.111716. [DOI] [PubMed] [Google Scholar]
- Imbimbo BP, Giudice ED, Colavito D, D'Arrigo A, Carbonare MD, Villetti G, Facchinetti F, Volta R, Pietrini V, Baroc MF, et al. 1-(3′, 4′-Dichloro-2-fluoro[1,1′–biphenyl]-4-yl)-cyclopropranecarboxylic acid (CHF5074), a γ-secretase modulator, reduces brain β-amyloid pathology in a transgenic mouse model of Alzheimer's disease without causing peripheral toxicity. J Pharmacol Exp Ther. 2007;323:822–830. doi: 10.1124/jpet.107.129007. [DOI] [PubMed] [Google Scholar]
- Kim SH, Sisodia SS. A sequence within the first transmembrane domain of PEN-2 is critical for PEN-2-mediated endoproteolysis of presenilin 1. J Biol Chem. 2005;280:1992–2001. doi: 10.1074/jbc.M412404200. [DOI] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX. Gamma-secretase: Proteosome of the membrane? Nat Rev Mol Cell Biol. 2004;5:486–488. doi: 10.1038/nrm1406. [DOI] [PubMed] [Google Scholar]
- Kopan R, Goate A. A Common Enzyme Connects Notch Signaling and Alzheimer's Disease. Genes and Development. 2000;14:2799–2806. doi: 10.1101/gad.836900. [DOI] [PubMed] [Google Scholar]
- Kreft AF, Martone R, Porte A. Recent advances in the identification of γ-secretase inhibitors to clinically test the Aβ oligomer hypothesis of Alzheimer's disease. J Med Chem. 2009;52:6169–6188. doi: 10.1021/jm900188z. [DOI] [PubMed] [Google Scholar]
- Kukar TL, Ladd TB, Bann MA, Fraering PC, Narlawar R, Maharvi GM, Healy B, Chapman R, Welzel AT, Price RW, et al. Substrate-targeting γ-secretase modulators. Nature. 2008;453:925–930. doi: 10.1038/nature07055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVoie MJ, Selkoe DJ. The Notch ligands, Jagged and Delta are sequentially processed by α-secretase and presenilin/γ-secretase and release signaling fragments. J Biol Chem. 2003;278:34427–34437. doi: 10.1074/jbc.M302659200. [DOI] [PubMed] [Google Scholar]
- Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, et al. Photo-activated gamma-secretase inhibitors directed to the active site covalently label presenilin. Nature. 2000;405:689–694. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- Martone RL, Zhou H, Atchison K, Comery T, Xu JZ, Huang X, Gong X, Jin M, Kreft A, Harrison B, et al. Begacestat (GSI-953): A novel, selective thiophene sulfonamide inhibitor of amyloid precursor protein γ-secretase for treatment of Alzheimer's disease. J Pharmacol Exp Ther. 2009;331:598–608. doi: 10.1124/jpet.109.152975. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Shioi GS, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shoa, Wisniewski T, et al. A presenilin-1/γ-secretase cleavage releases the e-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO. 2002;21:1948–1956. doi: 10.1093/emboj/21.8.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milano J, McKay J, Daenais C, Foster-Brown L, Pognan F, Gadient R, Jacobs RT, Zacco A, Breenberg B, Ciaccio PJ. Modulation of notch processing by γ-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci. 2004;82:341–358. doi: 10.1093/toxsci/kfh254. [DOI] [PubMed] [Google Scholar]
- Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, Ray WJ, Kopan R. A ligand-induced extracellular cleavage regulates γ-secretase-like proteolytic activation of notch. Mol Cell. 2000;5:197–206. doi: 10.1016/s1097-2765(00)80416-5. [DOI] [PubMed] [Google Scholar]
- Page RM, Baumann K, Tomioka M, Perez-Reveulta B, Fukumori A, Jacobsen H, Flohr A, Luebbers T, Ozmen L, Steiner H, et al. Generation of Aβ38 and Aβ42 is independent and differentially affected by familial Alzheimer disease-associated presenilin mutations and γ-secretase modulation. J Biol Chem. 2008;283:677–683. doi: 10.1074/jbc.M708754200. [DOI] [PubMed] [Google Scholar]
- Pissarnitski D. Advances in γ-secretase modulation. Cur Opin Drug Discov Dev. 2007;10:392–402. [PubMed] [Google Scholar]
- Searfoss GH, Jordan WH, Calligaro DO, Galbreath EJ, Schirtzinger LM, Berridge BR, Gao H, Higgins MA, May PC, Ryan TP. Adipsin, a biomarker of gastrointestinal toxicity mediated by a functional γ-secretase inhibitor. J Biol Chem. 2003;278:46107–46116. doi: 10.1074/jbc.M307757200. [DOI] [PubMed] [Google Scholar]
- Serneels L, Van Biervliet J, Craessaerts K, Dejaegere T, Horre K, van Houtvin T, Esselmann H, Schafer MK, Berezovska O, Hyman BT, et al. Gamma-Secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer's disease. Science. 2009;324:639–642. doi: 10.1126/science.1171176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton CC, Zhu L, Chau D, Yang L, Wang R, Djaballah H, Zheng H, Li YM. Modulation of γ-secretase specificity using small molecule allosteric inhibitors. Proc Natl Acad Sci USA. 2009;106:20228–20233. doi: 10.1073/pnas.0910757106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemers ER, Quinn JF, Kaye J, Farlow MR, Porsteinsson A, Tariot P, Zoulnouni P, Galvin JE, Holtzman DM, Knopman DS, et al. Effects of a γ-secretase inhibitor in randomized study of patients with Alzheimer's disease. Neurology. 2006;66:602–604. doi: 10.1212/01.WNL.0000198762.41312.E1. [DOI] [PubMed] [Google Scholar]
- Sisodia SS, St George-Hyslop Gamma-secretase, notch, A-beta and Alzheimer's disease: where do the presenilins fit in? Nat Rev Neurosci. 2002;3:281–290. doi: 10.1038/nrn785. [DOI] [PubMed] [Google Scholar]
- Starrett JR, J E, Gillman KW, Olson RE. A alpha-(N-sulfonamido)acetamide compound as an inhibitor of beta amyloid peptide production. WO 2009/058552 A1 PCT Publication. 2009
- Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: A genetic perspective. Cell. 2005;120:545–555. 2005. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Tian G, Sobotka-Briner CD, Zysk J, Liu X, Birr C, Sylvester MA, Edwards PD, Scott CD, Goldberg BD. J Biol Chem. 2002;277:31499–31505. doi: 10.1074/jbc.M112328200. [DOI] [PubMed] [Google Scholar]
- Tomita T. Secretase inhibitors and modulators for Alzheimer's disease treatment. Expert Rev Neurother. 2009;9:661–679. doi: 10.1586/ern.09.24. [DOI] [PubMed] [Google Scholar]
- Vassar R, Kovacs DM, Yan R, Wong PC. The beta-secretase enzyme BACE in health and Alzheimer's disease: regulation, cell biology, function, and therapeutic potential. J Neurosci. 2009;29:12787–12794. doi: 10.1523/JNEUROSCI.3657-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner SL, Munoz B. Modulation of amyloid β protein precursor processing as a means of retarding progression of AD. J Clin Invest. 1999;104:1329–1332. doi: 10.1172/JCI8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi T, De Strooper B. Presenilins: Members of the γ-secretase quartets, but part time soloists too. Physiol. 2008;23:194–204. doi: 10.1152/physiol.00009.2008. [DOI] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrik CU, Findlay KA, Smith TE, Murphy MP, Butler T, et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414:159–160. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- Wong GT, Manfra D, Poulet FM, Zhang Q, Josien H, Bara T, Engstrom L, Pinzon-Ortiz M, Fine JS, Lee HJ, et al. Chronic treatment with the γ-secretase inhibitor LY-411,575 inhibits β-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem. 2004;279:12876–12882. doi: 10.1074/jbc.M311652200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.