

Abstract
A novel iminium ion cascade reaction has been developed that allows for the stereoselective synthesis of a variety of substituted aza-fused bicycles. The combination of amino allylsilanes and aldehydes (or ketones) was used to synthesize a number of quinolizidines and indolizidines in an one-pot reaction sequence. This technology has been used to effect the facile syntheses of several indolizidine and quinolizidine natural products including, (±)-epilupinine, (±)-tashiromine, and (−)-epimyrtine. Substrate scope has been examined varying the type of amino allylsilanes (primary, secondary and conjugated) and carbonyl compounds (aldehydes and ketones) to give a variety of fused ring structures. Varying the components chosen allows for the inclusion of synthetically useful functional groups at different positions on the core structure. The methodology has been used to construct the tricyclic core structures present in the cylindricine family and halichlorine.
Keywords: Iminium ion, cascade reaction, alkaloid synthesis, stereoselective
1. Introduction
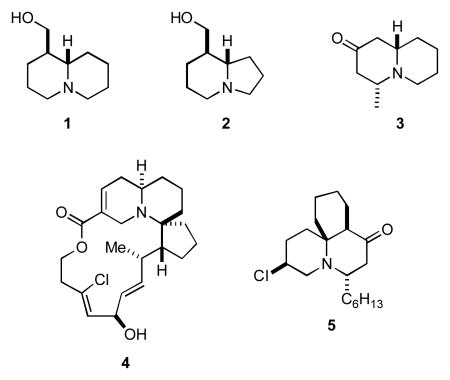
The quinolizidine and indolizidine ring systems comprise core structural subunits found in a large number of natural products ranging from simple bicyclic alkaloids such as epilupinine (1),i tashiromine (2),ii and epimyrtine (3)iii to more complex multicyclic molecules like halichlorine (4)iv and cylindricine B (5).v The diverse structures of these compounds coupled with the biological activity exhibited by many members of these families of alkaloids have made natural products containing these ring systems popular targets for synthesis. Consequently it does not occasion surprise that numerous methods and strategies have been developed for the stereoselective construction of substituted quinolizidine and indolizidines.vi
We have had a longstanding interest in developing useful and general entries to substituted quinolizidine and indolizidines during the normal course of our general programs in alkaloid synthesis.ive,vii In that context, we recognized that imines and their activated derivatives play pivotal roles in the synthesis of nitrogen heterocycles via a diverse array of bond forming reactions.viii We therefore became intrigued by the challenge of combining some of these different constructions and inventing new cascade reactions involving imines and allylsilanes as the key reaction partners to rapidly access the quinolizidine and indolizidine frameworks. Cascade reactions are becoming increasingly attractive as synthetic tools because they involve multiple reactants and sequential bond-forming events in which the product of one reaction is preprogrammed to be the starting material for the next one in a domino-like process.ix Such methods have gained popularity because they may enable the rapid assembly of complex molecular architectures in one-pot operations that can be highly efficient.x
Our design strategy was guided by evaluating various possibilities in which the different reactivity modes of imines as electrophiles and nucleophiles might be combined in tandem with the nucleophilic reactivity profile of allylsilanes to prepare nitrogen heterocycles having bridgehead nitrogen atoms. The plan that eventuated is outlined in general terms in Scheme 1 and enables the synthesis of a diverse array of nitrogen heterocycles 7–9 from the cascade reactions of monoprotected dicarbonyl compounds of the general form 6 with the amino allylsilanes 10–13, followed by a terminating step in which the penultimate intermediate iminium ion is trapped with a nucleophile. The ring sizes in 7–9 could be controlled by simply varying the number of carbon atoms in the tethers linking the reacting functional groups in 6 and 10–13.xi An attractive feature of this entry to quinolizidines and indolizidines is the high level of convergency wherein three components may be integrated into a product in a single chemical operation.
Scheme 1.

The putative mechanistic underpinnings for forming bicyclic nitrogen heterocycles related to 7 are exemplified in Scheme 2 for the formation of 15 from the acid-catalyzed reaction of the amino allylsilane 11 with the monoprotected dialdehyde 14, which corresponds to 6 (R = H, n = 1), followed by quenching the penultimate iminium ion intermediate with a generic nucleophile, Nu−. Thus, the sequence commences with the condensation of 11 with 14 to give the acyclic imine 16. Although several reaction manifolds are available to 16, acid-catalyzed expulsion of methanol would form an intermediate oxocarbenium ion, cyclization of which onto the imine nitrogen atom would furnish 17 that could in turn undergo cyclization via addition of the allylsilane moiety to the iminium ion to provide 18.viiia,xii Ionization of the N,O-acetal moiety of 18 in situ would generate another iminium ion that would be trapped with the nucleophile, Nu−, to deliver the fused bicyclic amine 15. Based upon principles of stereoelectronic control,xiii we anticipated that the nucleophile would add from an axial direction. Notably, four new bonds and two rings are formed from three different components in a single chemical operation. Similar mechanistic pathways may be set forth for the reactions of 14 with the branched allylsilane 12 and the pentadienyl silane 13 to give 19 and 20, respectively.
Scheme 2.

2. Results and Discussion
Having conceived of the iminium cascade processes outlined in Schemes 1 and 2, the first task was to establish the feasibility of such constructions. Toward that end, we first condensed the known aminosilane 11xiv with the monoacetal aldehyde 14, which was prepared by the procedure of Schreiber,xv and the resulting imine was treated directly with a number of different Brønsted and Lewis acids to induce the desired cascade. After rather extensive experimentation using different temperatures, solvents, and acids, we eventually discovered that cyclization of the intermediate imine proceeded best at −40 °C in acetonitrile in the presence of trifluoroacetic acid. After adding triethylsilane to reduce the putative iminium ion generated in situ, the quinolizidine 22 was isolated in 75% yield as a single diastereomer (Scheme 3).
Scheme 3.

Using the standardized conditions developed for the optimized preparation of 22, the known allyl aminosilane 10xvi was allowed to condense with the aldehyde 14, and the resultant imine was treated sequentially with TFA and then Et3SiH to provide the indolizidine 23 as a single stereoisomer in 45% yield.ic Similarly, reaction of the allylic aminosilane 11 with the monoprotected dialdehyde 21,xv followed by addition of TFA and then Et3SiH furnished the indolizidine 24 in 36% yield as a single stereoisomer. The relative stereochemistry between the two newly created stereocenters at C(1) and C(9a) of 22 was tentatively assigned based upon the observed coupling constant of 12.0 Hz of the trifluoroacetate salt of 22. This assignment as well as the structural assignments for 23 and 24 were confirmed by the conversion of these intermediates into known compounds (vide infra). Although the syntheses of indolizidines via this cascade reaction were less efficient than for quinolizidines, the levels of stereoselectivity were high. Unfortunately, preliminary efforts to extend such processes to the preparation of pyrrolizidines have proven unsuccessful.
The ultimate test of any new synthetic method or strategy lies in its applicability to the preparation of targets that may be of interest, and it did not escape attention that compounds 22 and 24 might be readily transformed into natural quinolizidines and indolizidines. Indeed, although compound 22 was a known intermediate in a previous synthesis of (±)-epilupinine (1),ic we developed an improved protocol for effecting this conversion. Namely, ozonolysis of the trifluoroacetate salt of 22, followed by reduction of the intermediate ozonide with LiAlH4 furnished synthetic (±)-epilupinine (1), which gave 1H and 13C NMR spectral data consistent with those reported, in 88% yield (Scheme 3).if Compound 24 was converted into the natural product (±)-tashiromine (2) in 56% yield via ozonolysis of its trifluoroacetate salt followed by hydride reduction of the ozonide thus obtained. The synthetic 2 gave 1H and 13C NMR spectral data consistent with those reported.iia,d Similarly, compound 23 was converted into the known indolizidine 25.ic, xvii
Having succeeded in preparing simple vinyl-substituted quinolizidines and indolizidines, we turned our attention to trapping the penultimate iminium ion with nucleophiles other than a hydride ion. Perhaps not unexpectedly, several initial attempts to trap the iminium ion generated from the reaction of with organometallic reagents such as alkyllithium and alkyl Grignard reagents formed an unstable enamine as the major product. However, we discovered that the intermediate iminium ion 26 could be readily trapped by the nucleophilic addition of cyanide ion under phase-transfer conditions to give the aminonitrile 27 as a single diastereomer in 79% overall yield (Scheme 4). As will be demonstrated in discussions that follow, it is noteworthy that α-aminonitriles such as 27 are versatile synthetic intermediates that may serve as precursors of nucleophiles (via deprotonation)xviii or electrophiles via Bruylants reactions.xix
Scheme 4.

We had thus established the viability of using cascade reactions of dialdehyde monoacetals and the linear amino allylsilanes 10 and 11 for the rapid formation of indolizidine and quinolizidine alkaloids and their precursors. At this juncture, we wished to explore related processes involving branched amino allylsilanes and keto aldehyde monoacetals. Toward this end, we first applied this chemistry to the branched allylsilane 12 and the monoprotected dialdehyde 14. In the event, condensation 12 with 14 followed by sequential treatment of the imine thus generated in situ with TFA and then aqueous NaCN furnished an excellent yield of an epimeric mixture (88:12) of the aminonitrile 31, favoring the α-CN isomer (Scheme 5).xx The presence of Bohlmann bands (2800-2700) in the IR spectrum of 31 was consistent with a trans-ring fused quinolizidine ring. The axial orientation of the cyano group, which is consistent with the preferred conformation of cyano groups in quinolizidine systems,xxi in the major isomer was assigned based upon examination of the 1H NMR spectrum of 31. The 1H NMR spectrum of the minor stereoisomer, which was assigned as having a β-CN group, the hydrogen at C(6) exhibited a large coupling constant (J = 10.4 Hz) indicative of an axial-axial interaction and suggesting that the cyano group was equatorial.
Scheme 5.

The nature of the monoprotected dicarbonyl component was then extended to include the ketal aldehydes 28–30, which were generally prepared according to the plan outlined in Scheme 6. These aldehydes were each condensed with the branched allylsilane 12, and the intermediate imines were treated with trifluoroacetic acid to initiate cyclization; addition of cyanide ion in the last step then afforded the aminonitriles 32–34 (Scheme 5). Because there was no observed nOe interaction between the bridgehead hydrogen atom and those on the R group at C(6), these groups were tentatively assigned as being trans to each other.xxii
Scheme 6.

It is noteworthy that the quinolizidines 31–34 each bear functionality that might be further elaborated to give a variety of derivatives. In order to illustrate the utility of this process, it was applied to a concise, enantioselective synthesis of the quinolizidine alkaloid (−)-epimyrtine (3) (Scheme 7). Thus, the known amino silane 39xxiii was condensed with the dialdehyde monoacetal 14 to give an imine that was then treated sequentially with trifluoroacetic acid and aqueous NaCN to give a mixture of diastereomeric amino nitriles 40 in 90% yield. Subsequent reduction of 40 with NaCNBH3 then provided a mixture (95:5) of epimeric quinolizidines 41a,b. We were also able to reduce the intermediate bicyclic iminium ion generated from the cascade reaction of 39 with 14 directly, but the yield of 41a,b was only about 60%. Formation of the trifluoroacetate salt of 41a,b followed by ozonolysis of the exocyclic olefin gave an inseparable mixture (95:5) of (−)-epimyrtine (3) and (+)-myrtine (42) was obtained. The 1H and 13C NMR spectral data for synthetic 3 and the specific rotation for 3 were consistent with those reported.xxiv
Scheme 7.

The potential utility of the α-aminonitriles formed in these sequences did not escape our attention. For example, α-aminonitriles may be deprotonated with strong bases to generate stabilized carbanions that may undergo reactions with a number of different electrophiles, and they may also serve as iminium ion equivalents in Bruylants and related processes.xxv In order to exemplify the utility of α-aminonitriles derived from our cascade reaction as precursors of interesting heterocyclic targets, a mixture of epimeric α-aminonitriles 31 was readily transformed into the tricycle 45, which possesses a skeletal framework related to the core of halichlorine (4). Thus, deprotonation of 31 with LDA followed by alkylation of the carbanion with the known tosylate 43xxvi gave 44 in good yield. Subsequent exposure of 44 to AgOTf generated an iminium ion in situ that underwent efficient cyclization via addition of the allylsilane to give 45 as a single diastereomer.
That the cyclization to give 45 proceeded in accord with the principles of stereoelectronic control was verified by NMR spectroscopy.xiii,xxvii In particular, the relative stereochemistry of the spiro and bridgehead centers at C(9a) and C(6) in 45 was assigned on the basis of nOe correlations observed in a GOESY experiment performed on the TFA salt in which a strong interaction was observed between the bridgehead proton at C(9a) and one of the hydrogen atoms on the C(10) methylene group. This observation is consistent with the cis-relationship between these protons in 45.
In order to extend this chemistry to the preparation of more highly substituted spirocyclic quinolizidines that might better serve as intermediates in syntheses of halichlorine and derivatives thereof, we identified 47 as a potential target. Toward this end, quinolizidine 34 was allowed to react with 3-butenylmagnesium bromide to deliver quinolizidine 46 as a single diastereomer in virtually quantitative yield (Scheme 9). The stereochemistry of 46 was assigned on the basis of a GOESY experiment in which NOE interactions were observed between the protons at C(9a) and C(13). The hydrochloride salt of 46 underwent facile enyne ring-closing metathesis in the presence of Grubbs II catalyst (48) to provide 47 in 97% yield. The synthesis of 47, which comprises the tricyclic core of halichlorine (4) is remarkable for its brevity, only three steps from acyclic starting materials, and efficiency of 72% overall yield.
Scheme 9.

The versatility of this cascade approach to substituted quinolizidines may be further illustrated by the synthesis of the C(6) epimer of 46 in an unoptimized, two-step sequence of reactions from 31 (Scheme 10). Thus, deprotonation of 31 followed by alkylation with 4-bromobutane led to aminonitrile 49, which underwent a Bruylants reaction with propynylmagnesium bromide to deliver quinolizidine 50 as a single diastereomer.xxiiia,xxv The stereochemistry of 50 was assigned on the basis of a GOESY experiment that revealed a nOe interaction between the equatorial proton on C(2) with the C(10) proton. There was no nOe between the protons at C(9a) and C(10).
Scheme 10.

We had thus verified that the cascade processes generally outlined in Scheme 1 could be applied to the syntheses of quinolizidines and indolizidines lacking substituents at the bridgehead position. However, there are a number of quinolizidine alkaloids such as cylindricine (5) that bear alkyl substituents on the bridgehead carbon atom. In order to probe whether cascade iminium ion reactions could be implemented to fabricate the tricyclic core of cylindricine, the known keto acetal 51xxviii was first condensed with the amino allylsilane 12. Trifluoroacetic acid was then added to the reaction, and the resulting tricyclic iminium was trapped with cyanide ion to provide a mixture (2.8:1.0:1.7:1.7) of diastereomers 52a,b and 53a,b in 66% combined yield (Scheme 11). Compound 52a was isolated and crystallized, and x-ray crystallographic analysis enabled its unambiguous stereochemical assignment. When the mixture of 52a,b and 53a,b was treated with sodium borohydride, an inseparable mixture (1.1:1.0) of the two diastereomeric tricyclic amines 54 and 55 was obtained.
Scheme 11.

As foreshadowed in the introductory discussion of our general approach to polycyclic nitrogen heterocycles via the iminium ion cascade reactions outlined in Scheme 1, we were also intrigued by the possibility of using amino dienylsilanes as 13 as reacting partners to allow access to more highly functionalized products. Because 13 was unknown, it was necessary to devise a means for its synthesis. Toward this end propargyl alcohol (56) was treated with allyl chlorodimethylsilane (57) in the presence of base to give silyloxy ether 58 (Scheme 12). Ring-closing enyne metathesis of 58 using 3 mol % of the Grubbs II Hoveyda catalyst 59 under an ethylene atmosphere gave the volatile cyclic silyloxy ether 60, which was not isolated by rather treated directly with methylmagnesium bromide to yield the hydroxy allylsilane 61 in 58% yield over two steps.xxix The hydroxyl group of the allylsilane 61 was converted into an azide function via a Mitsunobu reaction that surprisingly proceeded to give a mixture (3:1) of E-and Z-isomers. Preliminary efforts to avoid forming a mixture of geometric isomers were unsuccessful. Because the obtention of a mixture of geometric isomers was presumably inconsequential for the purpose at hand, we did not attempt to separate the isomers. Subsequent reduction of the azide gave the desired amino E-/Z-dienylsilanes 13.
Scheme 12.

Gratifyingly, we found that condensation of 13 with the monoprotected dialdehyde 14, followed by the addition of trifluoroacetic acid and subsequent nucleophilic trapping with cyanide furnished a separable mixture (18:82) of aminonitriles 62a,b in 32% combined yield (Scheme 13). The relative chemistry between C(5a) and C(9) was assigned based upon the observation of nOe interactions between the protons at these positions in a GOESY experiment with the minor diastereoisomer 62a. Despite the modest yield in this reaction, the result is noteworthy as it represents the first example of the use of a conjugated dienylsilane in an intramolecular Mannich-like reaction.xxx
Scheme 13.

3. Conclusion
In summary we have developed a number of related cascade reactions in which iminium ions are generated and trapped by allylsilanes and other nucleophiles such as cyanide ion and hydride donors to give functionalized quinolizidines and indolizidines according to the general plan set forth in Scheme 1. This novel cascade sequence features a one-pot process involving the formation of two rings and four new bonds from simple acyclic starting materials. A variety of amino allylsilanes and monoprotected dicarbonyl compounds may serve as inputs in these reactions. The practical utility of this new entry to polycyclic nitrogen heterocycles was convincingly demonstrated by its application to the facile syntheses of a number of quinolizidine and indolizidine alkaloids, including (±)-epilupinine, (−)-epimyrtine, and (±)-tashiromine as well as the tricyclic core structures of the more complex alkaloids halichlorine and cylindricine. Significantly, the amino nitrile function that may be obtained in some of these cascade reactions serves as a convenient functional handle for the introduction of a variety of other substituents onto the heterocyclic framework. Further applications of these processes to the preparation of targets of biological interest are under active investigation, and the results will be reported in due course.
4. Experimental
4.1 General
Tetrahydrofuran, dimethylformamide, acetonitrile, and toluene were dried according to the procedure described by Grubbs.xxxi All solvents were determined to contain less than 50 ppm H2O by Karl Fischer coulometric moisture analysis. Triethylamine was distilled from calcium hydride prior to use. Reactions involving air or moisture sensitive reagents or intermediates were performed under an inert atmosphere of nitrogen or argon in glassware that was flame dried. Thin layer chromatography was run on pre-coated silica gel plates with a 0.25 mm thickness containing 60F-254 indicator (Merck), and the plates were visualized by staining with AMCAN (ammonium molybdate/cerium ammonium nitrate), potassium permanganate, or p-anisaldehyde. Flash chromatography was performed using the indicated solvent system on 230–400 mesh silica gel (E. Merck reagent silica gel 60) according to Still’s protocol.xxxii Melting points are uncorrected. Infrared (IR) spectra were obtained as solutions in the solvent indicated. Proton nuclear magnetic resonance spectra (1H NMR) were obtained in CDCl3 solutions, and chemical shifts are reported in parts per million (ppm) referenced to the solvent. Coupling constants (J) are reported in Hz and the splitting abbreviations used are: s, singlet; d, doublet; t, triplet; app t, apparent triplet; q, quartet; m, multiplet; comp, complex multiplet; br, broad. Carbon nuclear magnetic resonance spectra (13C NMR) were obtained using CDCl3 as the internal reference.
4.2.1 Representative Procedure for the Reductive Cascade Reaction: Synthesis of (1S*, 9aS*)-1-vinyloctahydro-1H-quinolizine (22)
Freshly prepared amine 11 (363 mg, 2.12 mmol) was added dropwise to a suspension of freshly prepared aldehyde 14 (310 mg, 2.12 mmol) and molecular sieves (4 Å, 0.50 mg) in CH3CN (2.0 mL) at 0°C. The ice bath was removed, and the reaction was stirred for 2 h at which time the solution was transferred via syringe to a flame dried round-bottom flask. Additional CH3CN (30 mL) was added, and the mixture was cooled to 0°C with stirring. Freshly distilled CF3CO2H (2.4 g, 1.6 mL, 21.2 mmol) was added dropwise, and the reaction mixture was stirred for 2 h. The ice bath was removed, and the solution was stirred at room temperature for 12 h. Neat Et3SiH (2.5 g, 3.2 mL, 21.2 mmol) was then added dropwise, and the mixture was heated under reflux for 24 h. The reaction mixture was cooled to room temperature, and the solvents were removed under reduced pressure. The crude residue was dissolved in Et2O (15 mL), and a solution of 2 N HCl (15 mL) was added dropwise. The layers were separated, and the aqueous layer was extracted with Et2O (3 × 15 mL). The aqueous layer was made basic with a solution of 5% NaOH saturated with NaCl (15 mL) and extracted with Et2O (3 × 15 mL). The combined organic layers were dried (K2CO3) and concentrated under reduced pressure (300 mm Hg). The crude residue was purified by flash chromatography eluting with pentane/EtOH (60:1). An aliquot of the fractions containing pure 22 was concentrated under reduced pressure (300 mm Hg) to give an analytical sample for characterization. The remaining fractions containing pure 22 were combined, CF3CO2H (1.5 mL) was added, and the mixture was concentrated under reduced pressure to give 444 mg (75%) of the trifluoroacetate salt of 22 as a single diastereomer as a pale yellow oil; 1H NMR (500 MHz, CD3CN) δ5.60 (ddd, J = 17.5, 10.0, 9.0 Hz, 1 H), 4.99 (ddd, J = 17.5, 2.0, 1.0 Hz, 1 H), 4.94 (dd, J = 10.0, 2.0 Hz, 1 H), 2.76-2.69 (comp, 2 H), 2.00-1.93 (comp, 2 H), 1.85 (app tdd, J = 11.0, 9.0, 3.0 Hz, 1 H), 1.74 (app dp, J = 13.0, 2.5 Hz, 1 H), 1.74-1.64 (comp, 2 H), 1.63-1.54 (comp, 4 H), 1.54-1.46 (m, 1 H), 1.23-1.13 (comp, 2 H), 1.04 (app dq, J = 10.5, 3.5 Hz, 1 H); 13C NMR (125 MHz)δ142.2, 114.8, 66.5, 57.2, 56.9, 48.3, 32.5, 31.3, 26.4, 25.6, 25.2; IR (neat) 3400, 3079, 2953, 2869, 2723, 2627, 2584, 1738, 1671, 1448, 1201 cm−1; mass spectrum (CI) m/z 166.1591 [C11H20N (M+1) requires 166.1596], 164, 194, 248, 327, 329, 341, 399, 401.
4.2.2 (1S*, 8aS*)-1-Vinyloctahydroindolizine (23)
Prepared in 45% yield in accordance with the representative procedure. 1H NMR (500 MHz, CD3CN) δ5.70 (ddd, J = 17.0, 10.5, 8.0 Hz, 1 H), 5.18 (ddd, J = 17.0, 1.5, 1.0 Hz, 1 H), 5.18 (ddd, J = 10.5, 1.5, 1.0 Hz, 1 H), 3.62-3.56 (comp, 2 H), 3.05-2.97 (m, 1 H), 2.86-2.74 (comp, 2 H), 2.64 (app p, J = 8.5 Hz, 1 H), 2.20 (app ddt, J = 11.5, 9.5, 7.5 Hz, 1 H), 2.04-1.94 (m, 1 H), 1.90-1.80 (comp, 3 H), 1.73-1.64 (m, 1 H), 1.55-1.40 (comp, 2 H); 13C NMR (125 MHz) δ137.9, 118.7, 70.7, 53.4, 52.7, 47.2, 27.6, 27.1, 23.8, 22.8. The spectral data are in accordance with reported spectra for 23.ic
4.2.3 (8S*, 8aS*)-8-Vinyloctahydroindolizine (24)
Prepared in 36% yield in accordance with the representative procedure. 1H NMR (500 MHz, CD3CN) δ5.71 (ddd, J = 17.0, 10.0, 8.0 Hz, 1 H), 5.23 (d, J = 17.0 Hz, 1 H), 5.13 (d, J = 10.0 Hz, 1 H), 3.70-3.60 (comp, 2 H), 3.07-2.88 (comp, 2 H), 2.39-2.33 (m, 1 H), 2.28-2.22 (m, 1 H), 2.06-2.00 (comp, 3 H), 1.92-1.82 (comp, 2 H), 1.75-1.67 (m, 1 H), 1.52-1.44 (m, 1 H); 13C NMR (125 MHz) δ138.3, 117.7, 70.4, 54.1, 52.3, 45.7, 30.1, 28.4, 24.0, 20.0; IR (neat) 2923, 1454, 1376, 725; mass spectrum (CI) m/z 152.143559 [C10H17N (M+1) requires 152.143925], 371, 222, 152 (base), 97.
4.2.4 (±)–Epilupinine (1)
The trifluoroactetate salt of 22 (75 mg, 0.27 mmol) was dissolved in Et2O (3 mL) and ozone was passed through the stirred solution at −78 °C until the solution turned blue. Air was then passed through the reaction mixture for 5 min. A solution of LiAlH4 (51 mg, 1.5 mmol) in THF (2 mL) was added, the dry ice bath was removed, and the suspension was stirred at room temperature for 12 h. H2O (0.05 mL), 5% NaOH (0.05 mL), and H2O (0.15 mL) were added dropwise sequentially with stirring, and the mixture was stirred until the suspension turned white. The solids were removed by vacuum filtration and washed with Et2O. The combined filtrate and washings were concentrated under reduced pressure to give 40 mg (88%) of 1 as a colorless oil. Recrystallization of the crude product from pentane provided a white solid; (mp 79–80°C; lit.if 78–79°C); 1H NMR (500 MHz, MeOD) δ3.55 (dd, J = 11.0, 3.5 Hz, 1 H), 3.50 (dd, J = 11.0, 5.5 Hz, 1 H), 2.83-2.74 (comp, 2 H), 2.10-2.01 (comp, 2 H), 1.98-1.94 (m, 1 H), 1.84-1.54 (comp, 7 H), 1.37-1.23 (comp, 3 H), 1.16 (app tdd, J = 13.5, 11.0, 4.0 Hz, 1H); 13C NMR (125 MHz) δ65.8, 64.4, 57.9, 57.7, 44.9, 30.4, 29.3, 26.3, 25.8, 25.4; IR (neat) 3382, 2940, 2850, 2800, 2750, 2681, 2540, 2252, 2070, 1819, 1795, 1467, 1379, 1298, 1121, 1094 cm−1; mass spectrum (CI) m/z 170.1537 [C10H19NO (M+1) requires 170.1545], 170 (base), 168, 152. These spectral data are in accordance with reported spectra for (±)-epilupinine.if
4.2.5 (±)–Tashiromine (2)
The trifluoroactetate salt of 24 (142 mg, 0.57 mmol) was dissolved in Et2O (5 mL) and ozone was passed through the stirred solution at −78°C until the solution turned blue. Air was then passed through the reaction mixture for 5 min. A solution of LiAlH4 (215 mg, 4.46 mmol) in THF (3 mL) was added, the dry ice bath was removed, and the suspension was stirred at room temperature for 12 h. H2O (0.2 mL), 5% NaOH (0.2 mL), and H2O (0.6 mL) were added dropwise sequentially with stirring, and the mixture was stirred until the suspension turned white. The solids were removed by vacuum filtration and washed with Et2O. The combined filtrate and washings were concentrated under reduced pressure to give 44 mg (56%) of 2 as a colorless oil; 1H NMR (500 MHz, CD3CN) δ3.46 (dd, J = 11.0, 5.0 Hz, 1 H), 3.30 (dd, J = 11.0, 7.0 Hz, 1 H), 2.98 (app dp, J = 11.0, 2.0 Hz, 1 H), 2.93 (app dt, J = 9.0, 2.5 Hz, 1 H), 1.98 (app q, J = 9.0 Hz, 1 H), 1.91-1.78 (comp, 3 H), 1.70-1.48 (comp, 5 H), 1.40-1.28 (comp, 2 H), 1.04-0.90 (m, 1 H); 13C NMR (125 MHz)δ67.3, 65.4, 54.8, 53.4, 45.6, 29.9, 28.6, 26.1, 21.5; IR (neat) 3622, 3100 (broad peak), 2928, 2876, 2789, 2760, 2755, 2241, 2171, 1459, 1442, 1386, 1329 cm−1; mass spectrum (CI) m/z 156.1391 [C9H18NO (M+1) requires 156.1388], 156 (base). The spectral data are in accordance with reported spectra for (±)-tashiromine.iia,d
4.3.1 Representative Procedure for the Cascade Reaction with Cyanide-Trapping: Synthesis of (4R*, 9aR*)-8-methyleneoctahydro-1H-quinolizine-4-carbonitrile (31)
Freshly prepared amine 12 (696 mg, 4.4 mmol) was added dropwise to a mixture of freshly prepared 14 (642 mg, 4.4 mmol) and molecular sieves (4 Å, 0.70 mg) in CH3CN (2.0 mL) at 0 °C. The ice bath was removed, and the mixture was stirred at room temperature for 2 h. The supernatant was transferred via syringe to another round-bottom flask, and additional CH3CN (50 mL) was added. The mixture was cooled to −40°C, and freshly distilled CF3CO2H (2.5 g, 1.7 mL, 22.0 mmol) was added dropwise. The reaction mixture was stirred at −40°C for 4 h, whereupon the cooling bath was removed and the mixture stirred at room temperature for 2 h. The solvents were removed under reduced pressure and the residue was dissolved in CH2Cl2 (2.2 mL) at 0°C. A solution of NaCN (1.0 g, 20.4 mmol) in H2O (3 mL) was added to the reaction, the ice bath was removed and the reaction mixture stirred at room temperature for 12 h. CH2Cl2 (10 mL) and 0.5 M NaOH saturated with NaCl were added, and the layers were separated. The aqueous layer was extracted with CH2Cl2 (3 × 25 mL), and the combined organic layers were dried (K2CO3) and concentrated under reduced pressure (300 mmHg). The crude residue was purified by flash chromatography eluting with pentane/Et2O (10:1). The fractions were combined and concentrated under reduced pressure (300 mm Hg) to give 690 mg (89%) of an epimeric mixture (88:12) of 31 as a pale yellow oil: For αH: 1H NMR (500 MHz, CD3CN) δ4.71-4.67 (comp, 2 H), 3.90 (app t, J = 3.4 Hz, 1 H), 2.82-2.78 (m, 1 H), 2.30-2.20 (comp, 5 H), 2.07 (app tt, J = 11.0, 3.0 Hz, 1 H), 1.92-1.85 (m, 1 H), 1.78 (app ddt, J = 11.0, 4.5, 4.0 Hz, 1 H), 1.72-1.62 (comp, 2 H), 1.54 (app qt, J = 13.5, 3.5, 1 H), 1.50–1.28 (m, 1 H); 13C NMR (125 MHz) δ146.0, 117.0, 107.8, 58.2, 55.1, 54.9, 41.7, 34.3, 32.8, 28.8, 20.4 4. IR (neat) 3061, 2925, 2812, 2223, 1653, 1438, 1279, 1172, 1115, 889 cm−1; mass spectrum (CI) m/z 177.1400 [C11H16N2 (M+1) requires 177.1391], 150 (base), 177.
For β-H: 1H NMR (500 MHz, CD3CN) δ4.71-4.67 (comp, 2 H), 3.40 (app dt, J = 10.4, 3.2 Hz, 1 H), 3.06 (app dd, J = 11.6, 3.2, 1 H), 2.34-1.20 (comp, 13 H); 13C NMR (100 MHz) δ146.8, 121.1, 108.3, 63.6, 56.2, 55.8, 42.4, 34.8, 33.2, 31.7, 23.5 cm−1; mass spectrum (CI) m/z 177.1400 [C11H16N2 (M+1) requires 177.1391], 150 (base), 177.
4.3.2 (1R*, 6R*, 9aS*)-1-Vinyl-6-cyanoquinolizidine (27)
Prepared in 79% yield in accordance with the representative procedure. 1H NMR (500 MHz) δ5.64 (ddd, J = 17.0, 10.0, 9.0 Hz, 1 H), 5.02 (ddd, J = 17.0, 2.0, 1.0 Hz, 1 H), 4.97 (dd, J = 10.0, 2.0 Hz, 1 H), 3.88 (app t, J = 3.0 Hz, 2 H), 2.67 (app dp, J = 11.0, 2.0 Hz, 2 H), 2.33 (app dt, J = 11.0, 1.0 Hz, 1 H), 1.86-1.44 (comp, 10 H), 1.28-1.20 (m, 1 H), 1.07-0.99 (m, 1 H); 13C NMR (125 MHz) δ142.1, 115.9, 99.2, 61.2, 56.2, 54.7, 49.0, 32.3, 31.2, 25.7, 21.5; IR (neat) 3075, 2935, 2858, 2814, 2747 (Bohlmann Bands), 2221, 1641, 1442 cm−1; mass spectrum (CI) m/z 191.1539 [C12H19N2 (M+1) requires 191.1548], 164 (base), 191, 220, 236, 263.
4.3.3 (4R*, 9aR*)-4-Methyl-8-methyleneoctahydro-1H-quinolizine-4-carbonitrile (32)
Prepared in 65% yield in accordance with the representative procedure. 1H NMR (500 MHz, C6H6) δ 4.57-4.55 (m, 2 H), 3.10-3.07 (m, 1 H), 2.18-2.05 (comp, 5 H), 1.91-1.86 (m, 1 H), 1.82-1.79 (m, 1 H), 1.61-1.51 (comp, 4 H), 1.40 (s, 1 H), 1.20-1.15 (m, 1 H); 13C NMR (125 MHz) 144.9, 119.1, 107.7, 59.2, 49.4, 42.1, 38.5, 34.4, 33.2, 21.2; IR (neat) 2976, 2938, 2860, 2814, 1659, 1444, 1371, 1281, 1228, 1198, 1122, 1096, 1036, 890 cm−1; mass spectrum (CI) m/z 191.1544 [C12H19N2 (M+1) requires 191.1548], 191, 164 (base).
4.3.4 (4R*, 9aR*)-4-Ethyl-8-methyleneoctahydro-1H-quinolizine-4-carbonitrile (33)
Prepared in 60% yield in accordance with the representative procedure. 1H NMR (500 MHz) δ 4.64-4.63 (m, 2 H), 3.10 (app dt, J = 10.7, 3.8 Hz, 1 H), 2.26-2.17 (m, 4 H), 2.12-2.06 (m, 1 H), 1.98 (app t, J = 1.4 Hz, 1 H), 1.88-1.81 (m, 3 H), 1.72-1.60 (m, 4 H), 1.28-1.18 (m, 2 H), 0.97 (t, J = 7.5 Hz, 3 H); 13C NMR (125 MHz) 145.1, 119.1, 107.5, 62.2, 59.2, 48.9, 42.3, 34.5, 33.8, 33.2, 30.8, 21.0, 7.4; IR (neat) 2880, 2838, 2817, 1660, 1444, 1382, 1281, 1186, 1098, 891 cm−1; mass spectrum (CI) m/z 205.1707 [C13H21N2 (M+1) requires 205.1705], 205, 178 (base).
4.3.5 8-Methylene-4-(prop-1-ynyl)octahydro-1H-quinolizine-4-carbonitrile (34)
Prepared in 75% yield in accordance with the representative procedure. 1H NMR (500 MHz, CD3CN) δ 4.66 (ddt, J = 11.3, 1.8, 1.7 Hz, 2 H), 3.61 (ddd, J = 10.6, 4.7, 4.5 Hz, 1 H), 2.36-2.28 (m, 2 H), 2.27-2.17 (m, 4H), 2.10-1.95 (m, 2 H), 1.85 (s, 3 H) 1.73-1.62 (m, 3 H) 1.34-1.24 (m, 1 H); 13C NMR (125 MHz) 144.7, 115.4, 108.2, 82.2. 75.6, 58.4, 56.8, 51.8, 41.9, 38.4, 34.3, 32.5, 20.5, 3.6; IR (neat) 2939, 2868, 2819, 2360, 1658, 1442, 1284, 1207, 1106, 1018, cm−1; mass spectrum (CI) m/z 215.1552 [C14H19N2 (M+1) requires 215.1548], 219 (base), 215, 205.
4.3.6 (4R)-2-Methylenyl-4-methyl-6-cyanoquinolizidine (40)
Prepared in 90% yield in accordance with the representative procedure. 1H NMR (500 MHz, CD3CN) δ4.70 (app t, J = 2.0 Hz, 2 H), 4.35 (app t, J = 3.5 Hz, 1 H), 2.35-2.18 (comp, 4 H), 2.07-1.95 (comp, 3 H), 1.82-1.70 (comp, 3 H), 1.59 (app qt, J = 12.0, 1.5 Hz, 1 H), 1.31-1.23 (m, 1 H), 1.14 (d, J = 6.0 Hz, 1 H); 13C NMR (125 MHz) δ146.3, 117.4, 107.8, 59.3, 58.3, 50.7, 43.7, 42.8, 33.9, 29.6, 21.3, 20.3; IR (neat) 3074, 2939 2868, 2825, 2736, 2221, 1778, 1726, 1659, 1444, 1379, 1328, 1182 cm−1; mass spectrum (CI) m/z 191.1541 [C15H23N (M+1) requires 191.1548], 220, 191, 164 (base).
4.3.7 7-Vinyl-1,3,4,6,9,9a-hexahydro-2H-quinolizine-4-carbonitrile (62a, 62b)
Prepared in 79% yield in accordance with the representative procedure. (β-CN) 1H NMR (500 MHz) δ 6.27 (dd, J = 17.8, 10.9 Hz, 1 H), 5.27 (s, 1 H), 5.06 (d, J = 17.8 Hz, 1 H), 4.96 (d, J = 10.9 Hz, 1 H), 4.00 (d, J = 15.5 Hz, 1 H), 3.08 (dd, J = 12.1, 3.1 Hz, 1 H), 2.81 (dd, J = 15.1, 2.6 Hz, 1 H), 2.20-2.08 (comp, 4 H), 1.98-1.90 (m, 1 H), 1.84-1.81 (m, 1 H), 1.76-1.73 (m, 1 H), 1.37-1.29 (comp, 2 H); 13C NMR (125 MHz) 136.6, 132.2, 125.7, 119.3, 111.0, 57.0, 55.8, 52.7, 33.9, 32.2, 30.7, 22.9; IR (neat) 2867, 2817, 2787, 2356, 1661, 1611, 1440, 1370, 1310, 1249, 1165, 1119, 1044, 994, 894, 853 cm−1; mass spectrum (CI) m/z 189.1397 [C12H17N2 (M+1) requires 189.1392], 252, 217, 189, 163 (base). (α-CN) 1H NMR (500 MHz) δ 6.28 (dd, J = 11.01, 17.9 Hz, 1 H), 5.71 (s, 1 H), 5.01 (d, J = 17.3 Hz, 1 H), 4.95 (d, J = 11.0 Hz, 1 H), 3.97 (app t, J = 3.4 Hz, 1 H), 3.31 (comp, 2 H), 2.55-2.52 (m, 1 H), 2.26 (d, J = 16.7 Hz, 1 H), 2.07-1.80 (m, 4 H), 1.84-1.81 (m, 1 H), 1.77-1.71 (m, 2 H), 1.27-1.21 (comp, 1 H); 13C NMR (125 MHz) 136.7, 132.9, 125.4, 117.1, 110.8, 54.9, 52.0, 51.2, 33.9, 32.9, 28.8, 20.3; IR (neat) 2870, 2849, 2822, 2361, 1657, 1144, 1440, 1267, 1127, 852, 837, 738 cm−1; mass spectrum (CI) mz 189.1391 [C12H17N2 (M+1) requires 189.1392], 189, 162 (base).
4.4 (4R)-2-Methylenyl-4-methylquinolizidine (41)
Acetic acid (276 mg, 4.6 mmol) was added dropwise to a solution of NaBH3CN (145 mg, 2.3 mmol) in CH3CN (2 mL) with stirring, whereupon 18 (88 mg, 0.46 mmol) was added dropwise over 2 min. The solution was stirred for 24 h at rt, and then CH2Cl2 (5 mL) and a solution of 2 N HCl (5 mL) were added. The layers were separated, and the aqueous layer was made basic with a solution of 5% NaOH saturated with NaCl and extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were dried (K2CO3), CF3CO2H (60 mg) was added dropwise, and the crude mixture was concentrated under reduced pressure to give 111 mg (86%) of 40 as an inseparable mixture (95:5) of diastereomers (based on GC/MS and 1H NMR) as a pale yellow oil. Major isomer - 1H NMR (500 MHz, CD3CN) δ 4.85 (app t, J = 2.0, Hz, 2 H), 3.77-3.70 (m, 1 H), 3.06 (app ttd, J = 10.0, 6.5, 3.5 Hz, 1 H), 2.95 (app tdt, J = 12.0, 9.0, 3.0 Hz, 1 H), 2.65 (app tdd, J = 13.0, 10.0, 3.0 Hz, 1 H), 2.54-2.33 (comp, 4 H), 1.92-1.89 (comp, 2 H), 1.81-1.70 (comp, 2 H) 1.49 (app tdd, J = 17.0, 7.0, 4.0 Hz, 1 H), 1.36 (d, J = 6.5 Hz, 1 H); 13C NMR (125 MHz) δ 140.9, 111.8, 66.6, 63.7, 52.3, 40.6, 39.3, 31.7, 24.3, 22.6, 17.8; IR (CH2Cl2) 2943, 2869, 2263, 1785, 1673, 1453, 1415, 1198, 1131 cm−1; mass spectrum (CI) m/z 166.1591 [C11H20N (M+1) requires 166.1596], 107, 150, 166 (base), 224. The spectral data were in accordance with reported values.iiib
4.5 (−)-Epimyrtine (3a)
The trifluoroacetate salt of 19 (115 mg, 0.41 mmol) was dissolved in a mixture of CH2Cl2 (3 mL) and MeOH (0.5 mL) and ozone was bubbled through the solution at −78 °C until a blue color persisted (5 min). Air was passed through the reaction mixture for 5 min. Methyl sulfide (128 mg, 0.15 mL, 2.06 mmol) was added dropwise, the dry ice bath was removed and the reaction mixture stirred at room temperature for 12 h. A solution of 5% NaOH saturated with NaCl (1 mL) was added dropwise with stirring. The layers were separated, and the aqueous layer was extracted with Et2O (3 × 15 mL). The combined organic layers were dried (K2CO3) and concentrated under reduced pressure (300 mm Hg). The residue was purified by flash chromatography eluting with hexanes/EtOH (10:1) to give 40 mg (60%) of a mixture (95:5) of 3a and 3b as a pale yellow oil. [α]23D −18.0 (c 0.4, CDCl3); [lit.iiib ([α]20 D −17.4 (c 0.7, CHCl3); [α]23 D −19.0 (c 0.4, CHCl3)]; 1H NMR (500 MHz) δ 3.30-3.26 (m, 1 H), 2.40-2.12 (comp, 6 H), 1.83-1.52 (comp, 5 H), 1.41-1.33 (m, 1 H), 1.30-1.20 (m, 1 H), 1.17 (d, J = 5.5 Hz, 3 H); 13C NMR (125 MHz) δ 208.4, 62.1, 59.3, 51.0, 49.8, 48.7, 34.2, 25.9, 23.9, 20.7; IR (neat) 2929, 2846, 2788, 2741, 1718, 1442, 1336, 1283, 1166 cm−1; mass spectrum (CI) m/z 168.1388 [C10H18NO (M+1) requires 168.1388], 240, 196, 168 (base), 152. The spectral data were in accordance with reported values for (−)-epimyrtine.iii
4.6 6-Cyano-6-(3′-methyltrimethylsilyl-3′-butenyl)-2-methylenylquinolizidine (44)
A solution of n-butyllithium in hexanes (0.21 mL of 2.5 M, 0.44 mmol) was added dropwise to a solution of diisopropylamine (101 mg, 0.07 mL, 0.44 mmol) in Et2O (3 mL) at 0 °C. The solution was stirred for 0.5 h at which time 31 (60 mg, 0.35 mmol) was added dropwise. The reaction mixture was stirred for 0.5 h, and 43 (215 mg, 0.69 mmol) was added dropwise. The ice bath was removed, and the reaction was stirred for 1 h. A solution of 0.5 M NaOH saturated with NaCl (5 mL) was added, and the layers were separated. The aqueous layer was extracted with Et2O (3 × 10 mL), and the combined organic layers were dried (K2CO3) and concentrated under reduced pressure. The crude residue was purified by flash chromatography eluting with pentane/Et2O (95:5) to give 67 mg (62%) of 44 as a pale yellow oil. 1H NMR (500 MHz, CD3CN) δ 4.67-4.54 (comp, 4 H), 3.14 (app dq, J = 11.0, 2.5 Hz, 1 H), 2.29-1.15 (comp, 16 H), 1.57 (s, 3 H), 0.02 (s, 9 H); 13C NMR (125 MHz) δ 148.0, 146.9, 119.8, 107.8, 107.8, 62.6, 60.5, 50.2, 27.4, −1.3, 42.9, 37.0, 35.2, 35.0, 33.8, 31.5, 21.9; IR (neat) 3067, 2939, 2820, 2726, 2358, 2213, 1658, 1632, 1440, 1239 cm−1; mass spectrum (CI) m/z 317.2408 [C19H32NSi (M+1) requires 317.2413], 290 (base), 154, 218, 274, 317.
4.7 6-(2′-Methenylcyclopentyl)-2-methylenylquinolizidine (45)
Silver triflate (55 mg, 0.22 mmol) was added to a suspension of 4 Å molecular sieves (0.10 mg) in CH3CN (0.5 mL) at room temperature. The suspension was wrapped in tin foil and a solution of cyano amine 44 (62 mg, 0.20 mmol) in CH3CN (0.5 mL) was added dropwise. The resulting mixture was stirred at room temperature for 1 h, at which time the suspension was filtered through a cotton plug and washed with CH3CN (2 mL). The combined supernatant was heated at 120 °C (oil bath) with stirring for 24 h. The reaction was cooled to room temperature, and Et2O (10 mL) was added. A solution of 2 N HCl (5 mL) was added, and the layers were separated. The aqueous layer was washed with Et2O (3 × 5 mL) and then made basic with a solution of 5% NaOH saturated with NaCl. The mixture was then extracted with Et2O (3 × 5 mL). The combined organic layers were dried (K2CO3) and concentrated under reduced pressure to give 34 mg (81%) of 45 as a single diastereomer (based on 1H NMR) as a colorless oil. 1H NMR (500 MHz, CD3CN) δ 4.77 (app p, J = 2.0 Hz, 1 H), 4.74 (app p, J = 2.0 Hz, 1 H), 4.59-4.56 (m, 1 H), 4.56 (br s, 1 H), 3.00 (app dt, J = 9.0, 3.0 Hz, 1 H), 2.48 (d, J = 16.5 Hz, 1 H), 2.31-2.11 (comp, 6 H), 2.10-1.98 (comp, 2 H), 1.86 (dddd, J = 11.5, 9.5, 2.0, 1.5 Hz, 1 H), 1.55-1.40 (comp, 5 H), 1.32-1.25 (m, 1 H); 13C NMR (125 MHz) δ 153.7, 148.1, 107.0, 106.0, 66.9, 58.9, 47.9, 43.5, 39.2, 37.9, 36.0, 35.6, 33.0, 31.6, 21.8; IR (CH2Cl2) 2943, 2872, 2249, 1672, 1443, 1414, 1196 cm−1; mass spectrum (CI) m/z 218.1904 [C15H23N (M+1) requires 218.1908], 250, 218 (base), 152, 109.
4.8 2-Methylenyl-6-cyano-6-butenylquinolizidine (49)
A solution of n-butyllithium in hexanes (0.74 mL of 2.5 M, 1.40 mmol) was added dropwise to a solution of i-Pr2NEt (0.17 mL, 1.40 mmol) in Et2O (5 mL) at 0 °C. The solution was stirred for 0.5 h and then was cooled to −78 °C. Cyanoamine 31 (211 mg, 1.20 mmol) was added dropwise with stirring and the dry-ice/acetone bath was replaced with an ice-water bath. The mixture was stirred for 0.5 h at 0 °C then cooled to −78 °C. 4-Bromobutene (186 mg, 0.14 mL, 1.4 mmol) was added dropwise, and the dry-ice/acetone bath was again replaced with an ice-water bath. The reaction was stirred for 1 h at 0 °C, whereupon a solution of 0.5 M NaOH (10 mL) was added dropwise. The ice bath was removed, and the mixture was stirred for 15 min. The layers were separated, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic layers were dried (K2CO3) and concentrated under reduced pressure (300 mm Hg). The crude residue was purified by flash chromatography eluting with pentane/Et2O (9:1) to give 137 mg (50%) of 49 as a single diastereomer of a pale yellow oil; 1H NMR (500 MHz, CD3CN) δ 5.84 (ddt, J = 17.0, 10.0, 6.5 Hz, 1 H), 5.07 (dq, J = 17.0, 2.0 Hz, 1 H), 4.97 (ddt, J = 10.0, 2.0, 1.5 Hz, 1 H), 4.65 (dp, J = 8.0, 2.0 Hz, 2 H), 3.15 (ddd, J = 10.0, 4.5, 2.5 Hz, 1 H), 2.27-2.06 (comp, 6 H), 2.10 (app tt, J = 11.0, 2.5 Hz, 1 H), 2.00-1.91 (comp, 3 H), 1.86-1.80 (comp, 2H), 1.75 (app dt, J = 13.5, 4.0 Hz, 1 H), 1.68-1.61 (comp, 2 H), 1.56 (app tdd, J = 17.5, 8.0, 4.0 Hz, 1 H), 1.20 (app tdd, J = 15.0, 11.0, 4.0 Hz, 1H); 13C NMR (125 MHz) δ 146.9, 138.8, 119.7, 115.6, 107.8, 62.6, 60.5, 50.2, 42.9, 37.7, 35.1, 35.0, 33.8, 27.6, 21.9 cm−1; mass spectrum (CI) m/z 204 (base), 231.
4.9 6-(1′-Propynyl)-6-(3′-butenyl)-2-methylenylquinolizidine (50)
A solution of propynylmagnesium chloride in Et2O (3.4 mL of 0.5 M, 1.7 mmol) was added dropwise to a solution of cyanoamine 49 (137 mg, 0.60 mmol) in THF (3.0 mL) at room temperature. The solution was stirred for 5 h, at which time a solution of 2 N HCl (5 mL) was added dropwise. The layers were separated, and the aqueous layer was extracted with Et2O (3 × 5 mL). The aqueous layer was made basic with a solution of 5% NaOH saturated with NaCl and extracted with Et2O (3 × 5 mL). The combined organic layers were dried (K2CO3) and concentrated under reduced pressure to give 116 mg (80%) of 50 as a single diastereomer (based on 1H NMR) as a colorless oil; 1H NMR (500 MHz, CD3CN) δ 5.84 (ddt, J = 17.0, 10.5, 6.5 Hz, 1 H), 5.02 (dq, J = 17.0, 1.5 Hz, 1 H), 4.92 (dq, J = 9.0, 1.0 Hz, 1 H), 4.58 (dd, J = 4.0, 2.5 Hz, 2 H), 3.00 (ddd, J = 11.0, 4.5, 3.0 Hz, 1 H), 2.21-2.06 (comp, 6 H), 2.00 (app td, J = 11.0, 4.0 Hz, 1 H), 1.87 (app t, J = 12.5 Hz, 1 H), 1.79 (s, 3 H), 1.76 (ddd, J = 16.5, 14.0, 5.5 Hz, 1 H), 1.70-1.60 (comp, 2 H), 1.59-1.49 (comp, 4 H), 1.14 (app dtd, J = 15.5, 13.5, 4.0 Hz, 1 H); 13C NMR (125 MHz) δ 148.4, 140.2, 114.6, 106.8, 82.0, 80.1, 59.4, 59.2, 49.3, 43.5, 40.6, 37.1, 35.6, 34.8, 30.9, 28.3, 21.8 cm−1; mass spectrum (CI) m/z 244.2074 [C17H25N (M+1) requires 244.2065], 244 (base), 204, 188.
4.10 (6R,9aR)-6-(But-3-enyl)-2-methylene-6-(prop-1-ynyl)octahydro-1H-quinolizine (46)
4-Bromo-butene (0.23 mL, 2.27 mmol) was added to a suspension of Mg (48 mg, 1.98 mmol) in THF (3 mL), and the solution was heated to 70 °C for 1.5 h. A portion of the solution (1 mL) was added to 34 (14 mg, 0.07 mmol) in CH2Cl2 (0.40 mL) at −78 °C. The reaction was allowed to warm to room temperature with stirring over 8 h. The reaction was cooled to 0 °C, and 2.9 N HCl (3 mL) was added. The mixture was washed with Et2O (3 × 4 mL). The aqueous layer was cooled to 0 °C, 6 N NaOH (5 mL.) was added, and the mixture was extracted with Et2O (3 × 3 mL). The combined organic extracts were concentrated under reduced pressure to yield 17 mg (100%) of 46 as a single diastereomer of a clear oil. 1H NMR (500 MHz) δ 5.89-5.81 (comp, 1 H), 5.04-5.00 (ddt, J = 17.2, 1.8, 1.7 Hz, 1 H), 4.94-4.91 (ddt, J = 10.2, 1.4, 1.0 Hz, 1 H), 4.59-4.56 (m, 2 H), 3.50-3.48 (m, 1 H), 2.34-2.29 (app tt, J = 10.9, 3.1 Hz, 1 H) 2.25-2.12 (m, 5 H) 2.03-1.86 (m, 3 H) 1.81 (s, 3 H) 1.76-1.70 (app td, J = 12.8, 5.4 Hz, 1 H) 1.64-1.55 (m, 5 H), 1.50-1.45 (m, 1 H) 1.40-1.32 (app qt, J = 13.3, 3.3 Hz, 1 H) 1.28-1.20 (m, 1 H); 13C NMR (125 MHz) 147.9, 139.5, 114.2, 106.5, 84.0, 78.6, 58.5, 55.9, 49.6, 43.7, 35.8, 35.7, 34.5, 30.0, 26.7, 19.3, 3.6; IR (neat) 2934, 2863, 2812, 1656, 1450, 1091, 885 cm−1; mass spectrum (CI) m/z 244.2066 [C17H26N (M+1) requires 244.2065], 269, 244 (base), 243.
4.11 Spirocycle 47
A solution of 46 (121 mg, 0.50 mmol) in 3% HCl/MeOH (5 mL) was stirred at room temperature for 15 min. and then concentrated. The resulting salt was dissolved in benzene (10 mL) that had been sparged with ethylene, and a solution of Grubbs II catalyst (34 mg, 0.04 mmol) in benzene (2 mL) was added. The reaction was stirred at room temperature under ethylene for 36 h, whereupon 3 M NaOH in saturated brine (10 mL) was added. The mixture was extracted with Et2O (3 × 20 mL), and the combined organic layers were washed with brine (15 mL), dried (MgSO4), filtered and concentrated. The crude material was purified by silica gel column chromatography eluting with a 5–10% gradient of ether/pentane to yield 118 mg (97%) of 47 as a colorless oil. 1H NMR (500 MHz) δ 6.19 (d, J = 3.3 Hz, 1 H), 5.71 (t, J = 2.7 Hz, 1 H), 4.92 (s, 1 H), 4.56-4.55 (m, 2 H), 2.74 (ddd, J = 11.3, 4.1, 3.0 Hz, 1 H), 2.26-2.22 (m, 2 H) 2.16-2.01 (m, 6 H) 1.96 (td, J = 10.7, 4.5 Hz, 1 H) 1.90-1.86 (m, 1 H) 1.89 (s 3 H) 1.69-1.50 (m, 5 H), 1.34-1.29 (m, 1 H), 1.29-1.24 (m, 1 H); 13C NMR (125 MHz) 148.6, 148.3, 138.1, 128.8, 113.4, 105.9, 74.2, 57.8, 48.9, 43.9, 35.6, 35.2, 35.0, 29.9, 24.6, 23.3, 21.3; IR (neat) 2934, 2859, 1655, 1444, 1374, 1100, 1026, 907, 884 cm−1; mass spectrum (CI) m/z 244.2066 [C17H26N (M+1) requires 244.2065], 244, 243 (base), 242.
4.12 2-Methylenedodecahydropyrido[1,2-j]quinoline-6-carbonitrile (52a, 52b, 53a, 53b)
A solution of 2-(3,3-dimethoxypropyl)cyclohexanone (76 mg, 0.38 mmol) in CH3CN (2 mL) was added to a solution of 3-((trimethylsilyl)methyl)but-3-en-1-amine (60 mg, 0.36 mmol) in CH3CN (1 mL) with 4 Å molecular sieves (5 beads). The reaction was heated under reflux for 4 h under nitrogen. The reaction was cooled, and stirring was continued for 2 h at room temperature. The 4 Å molecular sieves were removed, the reaction was cooled to −40 °C, and CF3CO2H (3 mL, 40 mmol) was added. The reaction was allowed to warm to room temperature over 3 h and then stirred at room temperature for 21 h. The reaction was concentrated under reduced pressure, and the residue was dissolved in CH2Cl2 (5 mL). The reaction was cooled to 0 °C, NaCN aq. (1.9 mL of a 2 M soln.) was added, the ice bath was removed, and the reaction was stirred at room temperature for 4 h. The reaction was cooled to 0 °C, saturated K2CO3 (5 mL) was added, and the mixture was extracted with Et2O (3 × 5 mL). The combined organic layers were washed with brine (10 mL), dried (K2CO3), filtered and concentrated under reduced pressure. The crude material was purified by silica gel chromatography eluting with a 10–20% gradient of ether/pentane to yield 66 mg (68%) of 52a, 52b, 53a, and 53b as a (2.8:1:1.7:1.7) mixture of isomers. The major isomer 52a was crystallized from ether/pentanes. For 52a: 1H NMR (400 MHz) δ 4.79 (app dd, J = 3.5, 1.6 Hz, 1 H), 4.66 (app dd, J = 3.9, 2.0 Hz, 1 H), 3.77-3.75 (m, 1 H), 3.10 (td, J = 11.9, 3.9 Hz, 1 H), 2.88 (dd, J = 13.1, 3.2 Hz, 1 H), 2.54 (ddd, J = 11.5, 6.3, 1.8 Hz, 1 H) 2.38-2.20 (m, 2 H) 2.12-1.71 (m, 6 H) 1.60 (m, 1 H) 2.37-2.22 (m, 2 H), 1.69-1.50 (m, 5 H), 1.50-1.37 (m, 2 H); 13C NMR (100 MHz) 143.5, 120.7, 109.9, 58.1, 50.1, 48.6, 43.5, 41.9, 34.1, 29.2, 28.2, 22.7, 22.2, 20.5; IR (neat) 2932, 2863, 2360, 1652, 1456, 888 cm−1; mass spectrum (CI) m/z 231.1861 [C15H23N2 (M+1) requires 231.1858], 243, 231 (base), 230.
4.13 2-Methylenedodecahydropyrido[1,2-j]quinoline (54, 55)
Solid NaBH4 (27 mg, 0.71 mmol) was added to a solution of 53a–d (32.4 mg, 0.14 mmol) in MeOH (5 mL) at 0 °C. The reaction was warmed to room temperature and then heated at 50 °C (oil bath) for 3 h. The reaction mixture was cooled to room temperature, HCl (1 M, 2 mL) was added, and the mixture was washed with Et2O (3 × 4 mL). NaOH aq. (2.5 M, 3 mL) was added, and the mixture was extracted with Et2O (3 × 3 mL). The combined ethereal extracts were concentrated under reduced pressure to yield 17 mg (60%) of an inseparable mixture (1.1:1.0) of 54 and 55. 1H NMR of the mixture (400 MHz, CDCl3) δ 4.77-4.61 (comp, 4 H), 4.08 (dd, J = 12.1, 5.2 Hz, 1 H), 3.38-3.21 (m, 1 H), 2.87-2.83 (m, 1 H), 2.70-2.38 (comp, 6 H), 2.28-1.16 (comp, 29 H); 13C NMR (100 MHz) 145.0, 143.4, 120.3, 109.6, 108.8, 58.7, 57.0, 49.7, 49.2, 47.0, 45.3, 44.4, 43.2, 41.8, 34.4, 34.1, 31.2, 28.5, 28.0, 27.9, 27.6, 25.9, 25.8, 25.7, 25.6, 22.0, 21.9, 20.5; IR (neat) 2928, 2862, 2810, 1650, 1454, 1352, 1120 cm−1; mass spectrum (CI) m/z 206.19146 [C14H24N (M+1) requires 206.1909], 204, 206 (base).
Supplementary Material
Scheme 8.

Acknowledgments
We thank the National Institutes of Health (GM 25439), the Robert A. Welch Foundation, Pfizer, Inc., Merck Research Laboratories, and Boehringer Ingelheim Pharmaceuticals for their generous support of this research. We are also grateful to Dr. Richard Pederson (Materia, Inc.) for catalyst support. We additionally thank Dr. Vincent Lynch (The University of Texas) for X-ray crystallography data and Dr. Andrew S. Judd (Abbott Laboratories) for helpful discussions.
Footnotes
Supporting Information Available: Experimental procedures for the preparation of 13, 28–30, 58 and 61. Copies of 1H NMR spectra for all new compounds, as well as 1H and 13C NMR spectra of epilupinine (1), tashiromine (2), and epimyrtine (3) and a CIF file for 52a.
References
- i.For representative syntheses of epilupinine, see: Comins DL, Brown JD. Tetrahedron Lett. 1986;27:2219–2222.Morley C, Knight DW, Share AC. J Chem Soc Perkin Trans. 1994;1:2903–2907.Pandey G, Reddy GD, Chakrabarti D. J Chem Soc, Perkin Trans. 1996;1:219–224.Naidu BN, West FG. Tetrahedron. 1997;53:16565–16574.Mangeney P, Hamon L, Raussou S, Urbain N, Alexakis A. Tetrahedron. 1998;54:10349–10362.Ma S, Ni B. Chem Eur J. 2004;10:3286–3300. doi: 10.1002/chem.200305581.Ahari MH, Perez A, Menant C, Vasse JL, Szymoniak JA. Org Lett. 2008;10:2473–2476. doi: 10.1021/ol800722a.
- ii.For representative syntheses of tashiromine, see: Kim SH, Kim SI, Lai S, Cha JK. J Org Chem. 1999;64:6771–6775. doi: 10.1021/jo9907383.Olivier D, Bellec C, Fargeau-Bellassoued MC, Lhommet G. Heterocycles. 2001;55:1689–1701.Bates RW, Boonsombat J. J Chem Soc Perkin Trans. 2001;1:654–656.Dieter RK, Watson R. Tetrahedron Lett. 2002;43:7725–7728.McElhinney AD, Marsden SP. SynLett. 2005;16:2528–2530.Belanger G, Larouche-Gauthier R, Menard F, Nantel M, Barabe F. J Org Chem. 2006;71:704–712. doi: 10.1021/jo052141v.Pohmakotr M, Prateeptongkum S, Chooprayoon S, Tuchinda P, Reutrakul V. Tetrahedron. 2008;64:2339–2347.
- iii.For representative syntheses of epimyrtine, see: Slosse P, Hootele C. Tetrahedron. 1981;37:4287–4292.Gardette D, Gelas-Mialhe Y, Gramain JC, Perrin B, Remuson R. Tetrahedron: Asymmetry. 1998;9:1823–1828.Davis FA, Zhang Y, Anilkumar G. J Org Chem. 2003;68:8061–8064. doi: 10.1021/jo030208d.
- iv.For a recent review see: Clive DL, Yu M, Wang J, Yeh VS, Kang S. Chem Rev. 2005;105:4483–4514. doi: 10.1021/cr0406209.For representative syntheses of halichlorine, see: Trauner D, Schwartz JB, Danishefsky SJ. Angew Chem Int Ed. 1999;38:3542–3545. doi: 10.1002/(sici)1521-3773(19991203)38:23<3542::aid-anie3542>3.0.co;2-i.Christie HS, Heathcock CH. Proc Natl Acad Sci USA. 2004;101:12079–12084. doi: 10.1073/pnas.0403887101.Matsumura Y, Aoyagi S, Kibayashi C. Org Lett. 2004;6:965–968. doi: 10.1021/ol0301431.Zhang HL, Zhao G, Ding Y, Wu B. J Org Chem. 2005;70:4954–4961. doi: 10.1021/jo047882v.Andrade R, Martin SF. Org Lett. 2005;7:5733–5735. doi: 10.1021/ol0525009.
- v.For a recent review see: Weinreb SM. Chem Rev. 2006;106:2531–2549. doi: 10.1021/cr050069v.For representative syntheses of cylindricines, see: Snider BB, Liu T. J Org Chem. 1997;62:5630–5633.Molander GA, Roen M. J Org Chem. 1999;64:5183–5187. doi: 10.1021/jo990363l.Liu JF, Heathcock CH. J Org Chem. 1999;64:8263–8266. doi: 10.1021/jo991020q.Trost BM, Rudd MT. Org Lett. 2003;5:4599–4602. doi: 10.1021/ol035752n.Arai T, Abe H, Ayogai S, Kibayashi C. Tetrahedron Lett. 2004;45:5921–5924.Canesi S, Bouchu D, Ciufolini MA. Angew Chem, Int Ed. 2004;43:4336–4338. doi: 10.1002/anie.200460178.Liu J, Hsung RP, Peters SD. Org Lett. 2004;6:3989–3992. doi: 10.1021/ol048353g.Mihara H, Shibuguchi T, Kuramochi A, Ohshima T, Shibasaki M. Heterocycles. 2007;72:421–438.Flick AC, Caballero MJ, Padwa A. Org Lett. 2008;10:1871–1874. doi: 10.1021/ol8006056.
- vi.For leading reviews of quinolizidine and indolizidine alkaloids, see: Daly JW, Spande TF, Garraffo HM. J Nat Prod. 2005;68:1556–1575. doi: 10.1021/np0580560.Michael JP. Nat Prod Rep. 2007;24:191–222. doi: 10.1039/b509525p.
- vii.(a) Williamson SA, Gist RP, Smith KM, Martin SF. J Org Chem. 1983;47:5170–5180. [Google Scholar]; (b) Grzejszczak S, Wiliamson SA, Rueger H, Martin SF. J Am Chem Soc. 1987;109:6124–6134. [Google Scholar]; (c) Martin SF, Yang CP, Laswell WL, Rueger H. Tetrahedron Lett. 1988;29:6685–6688. [Google Scholar]; (d) Martin SF, Campbell CL. J Org Chem. 1988;53:3184–3190. [Google Scholar]; (e) Martin SF, Liao Y, Chen HJ, Paetzel M, Ramser MN. Tetrahedron Lett. 1994;35:6005–6008. [Google Scholar]; (f) Martin SF, Bur SK. Tetrahedron. 1999;55:8905–8914. [Google Scholar]; (g) Deiters A, Martin SF. Org Lett. 2002;4:3243–3245. doi: 10.1021/ol026470a. [DOI] [PubMed] [Google Scholar]; (h) Reichelt A, Bur SK, Martin SF. Tetrahedron. 2002;58:6323–6328. [Google Scholar]; (i) Deiters A, Chen K, Eary CT, Martin SF. J Am Chem Soc. 2003;125:4541–4550. doi: 10.1021/ja0296024. [DOI] [PubMed] [Google Scholar]; (j) Dieters A, Pettersson M, Martin SF. J Org Chem. 2006;71:6547–6561. doi: 10.1021/jo061032t. [DOI] [PubMed] [Google Scholar]
- viii.For reviews of N-alkyl iminium ions, see: Blumenkopf TA, Overman LE. Chem Rev. 1986;86:857–874.Kleinman EF, Volkmann RA. Comp Org Syn; Trost, B, M, Ed. 1991;2:975–1006.Overman LE. Aldrichimica Acta. 1995;28:107–120.Royer J, Bonin M, Micouin L. Chem Rev. 2004;104:2311–2352. doi: 10.1021/cr020083x.For reviews of N-acyl iminium ions, see: Speckamp WN, Moolenaar MJ. Tetrahedron. 2000;56:3817–3856.Maryanoff BE, Zhang HC, Cohen JH, Turchi IJ, Maryanoff CA. Chem Rev. 2004;104:1431–1628. doi: 10.1021/cr0306182.For a review of vinylogous Mannich reactions, see: Martin SF. Acc Chem Res. 2002;35:895–904. doi: 10.1021/ar950230w.
- ix.For some reviews, see: Ho TL. Tandem Organic Reactions. 1992Tietze LF. Chem Rev. 1996;96:115–136. doi: 10.1021/cr950027e.Padwa A. Pure Appl Chem. 2003;75:47–62.Tietze LF, Rackelmann N. Pure Appl Chem. 2004;76:1967–1983.
- x.For some leading references, see: Nicolaou KC, Edmonds David J, Bulger Paul G. Angew Chem Int Ed. 2006;45:7134–7186. doi: 10.1002/anie.200601872.Chapman CJ, Frost CG. Synthesis. 2007:1–21.Enders D, Grondal C, Huettl MRM. Angew Chem Int Ed. 2007;46:1570–1581. doi: 10.1002/anie.200603129.
- xi.For a preliminary account of some of these results, see: Amorde SM, Judd AS, Martin SF. Org Lett. 2005:2031–2033. doi: 10.1021/ol050544b.
- xii.For leading examples of related cyclizations, see: Cellier M, Gelas-Mialhe Y, Husson HP, Perrin B, Remuson R. Tetrahedron: Asymmetry. 2000;11:3913–3919.Agami C, Comesse S, Kadouri-Puchot C. J Org Chem. 2002;67:2424–2428. doi: 10.1021/jo010780+.
- xiii.(a) Ziegler FE, Spitner EB. J Am Chem Soc. 1973;95:7146–7149. doi: 10.1021/ja00802a040. [DOI] [PubMed] [Google Scholar]; (b) Deslongchamps P. Organic Chemistry Series, Vol 1: Stereoelectronic Effects in Organic Chemistry. 1983;211:221. [Google Scholar]; (c) Maruoka K, Miyazaki T, Ando M, Matsumura Y, Sakane S, Hattori K, Yamamoto H. J Am Chem Soc. 1983;105:2831–2843. [Google Scholar]; (d) Stevens RV. Acc Chem Res. 1984;17:289–296. [Google Scholar]
- xiv.Wasserman HH, Vu CB, Cook JD. Tetrahedron. 1992;48:2101–2112. [Google Scholar]
- xv.Schreiber SL, Claus RE, Reagan J. Tetrahedron Lett. 1982;23:3867–3870. [Google Scholar]
- xvi.Mooiweer HH, Hiemstra H, Fortgens HP, Speckamp WN. Tetrahedron Lett. 1987;28:3285–3288. [Google Scholar]
- xvii.Bertrand S, Hoffmann N, Pete JP. Tetrahedron Lett. 1999;40:3173–3174. [Google Scholar]
- xviii.(a) Enders D. Chem Soc Rev. 2000;29:359–373. [Google Scholar]; (b) Enders D, Thiebes C. Synlet. 2000;12:1745–1748. [Google Scholar]; (c) Agami C, Couty F, Evano G. Org Lett. 2000;2:2085–2088. doi: 10.1021/ol0059908. [DOI] [PubMed] [Google Scholar]; (d) Meyer N, Opatz T. Synlett. 2003;10:1427–1430. [Google Scholar]; (e) Wolckenhaur SA, Rychnovsky SD. Org Lett. 2004;6:2745–2748. doi: 10.1021/ol049039p. [DOI] [PubMed] [Google Scholar]; (f) Werner F, Blank N, Opatz T. Eur J Org Chem. 2007;23:3911–3915. [Google Scholar]; (g) Liermann JC, Opatz T. J Org Chem. 2008;73:4526–4531. doi: 10.1021/jo800467e. [DOI] [PubMed] [Google Scholar]
- xix.(a) Bruylants P. Bull Soc Chem de Belg. 1924;33:467–478. [Google Scholar]; (b) Bernardi L, Bonini BF, Capito E, Dessole G, Fochi M, Comes-Franchini M, Ricci M. Synlet. 2003;12:1778–1782. [Google Scholar]; (c) Reimann E, Ettmayer A. Monatsch Chem. 2004;135:1289–1295. [Google Scholar]; (d) Beaufort-Droal V, Pereira E, Thery V, Aitken DJ. Tetrahedron. 2006;62:11948–11954. [Google Scholar]
- xx.The products 30 (α-CN) and 30 (β-CN) were readily separable by flash chromatography. The ratio of the two compounds appears to be thermodynamic as 30 (β-CN) equilibrated to a mixture (87:13) 30 (α-CN) and 30 (β-CN) upon standing in CD3CN or upon heating in the presence of basic alumina.
- xxi.Husson HP, Roulland E, Cecchin F. J Org Chem. 2005;70:4474–4477. doi: 10.1021/jo050258d. [DOI] [PubMed] [Google Scholar]
- xxii.In this context it is significant that a nOe interaction was observed between the hydrogen atom at C(9a) and the hydrogen atoms on the pendant butenyl group at C(10) in45.
- xxiii.Monfray J, Gelas-Mialhe Y, Gramain JC, Remuson R. Tetrahedron Lett. 2003;44:5785–5787. [Google Scholar]
- xxiv.(a) Davis FA, Zhang Y, Anilkumar G. J Org Chem. 2003;68:8061–8064. doi: 10.1021/jo030208d. [DOI] [PubMed] [Google Scholar]; (b) Gardette D, Gelas-Mialhe Y, Gramain JC, Perrin B, Remuson R. Tetrahedron: Asymmetry. 1998;9:1823–1828. [Google Scholar]; (c) Slosse P, Hootele C. Tetrahedron. 1981;37:4287–94. [Google Scholar]
- xxv.Enders D, Shilvock JP. Chem Soc Rev. 2000;29:359–373. [Google Scholar]
- xxvi.Rubiralta M, Diez A, Miguel D, Remuson R, Gelas-Mialhe Y. Synth Commun. 1992;22:359–367. [Google Scholar]
- xxvii.Polniaszek RP, Belmont SE. J Org Chem. 1990;55:4688–93. [Google Scholar]
- xxviii.(a) Heathcock C. J Org Chem. 2001;66:7751–7756. doi: 10.1021/jo0106391. [DOI] [PubMed] [Google Scholar]; (b) Overman LE. J Org Chem. 1997;62:6379–6387. [Google Scholar]
- xxix.For a related route to similar dienyl silanes, see: Barrett E. Org Lett. 2003;7:671–672.Yaun H. J Am Chem Soc. 2001;123:2964–2969.
- xxx.For an intermolecular example see: Larsen SD, Grieco PA, Fobare WF. J Am Chem Soc. 1986;108:3512–3515.
- xxxi.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518. [Google Scholar]
- xxxii.Still WC, Kahn M, Mitra A. J Org Chem. 1978;43:2923. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.