

Abstract
We found that development of obesity was coupled with the emergence of a progressively worsening brain reward deficit. Similar changes in reward homeostasis induced by cocaine or heroin is considered a critical trigger in the transition from casual to compulsive drug-taking. Accordingly, we detected compulsive-like feeding behavior in obese but not lean rats, measured as palatable food consumption that was resistant to disruption by an aversive conditioned stimulus. Striatal dopamine D2 receptors (D2R) were downregulated in obese rats, similar to previous reports in human drug addicts. Moreover, lentivirus-mediated knockdown of striatal D2R rapidly accelerated the development of addiction-like reward deficits and the onset of compulsive-like food seeking in rats with extended access to palatable high-fat food. These data demonstrate that overconsumption of palatable food triggers addiction-like neuroadaptive responses in brain reward circuitries and drives the development of compulsive eating. Common hedonic mechanisms may therefore underlie obesity and drug addiction.
Feeding is influenced by pleasure and reward, and obtaining food reward can powerfully motivate consumption 1, 2. Nevertheless, hedonic mechanisms contributing to obesity remain poorly understood. In hyperphagic human patients with congenital leptin deficiency, activity in the dorsal and ventral striatum, core components of brain reward circuitries, is markedly increased in response to images of food 3, and leptin replacement therapy attenuates both striatal activity and self-reported “liking” of food 3. This suggests that the striatum plays an important role in hedonic aspects of feeding behavior. It was shown recently that activation of the striatum is blunted in obese individuals when compared with lean controls in response to highly palatable food 4. Moreover, hypofunctionality of the dorsal striatum and long-term weight gain is most pronounced in individuals with the TaqIA allele of the D2R gene locus that results in decreased striatal D2R expression, shown previously to be a predisposing factor to substance dependence disorders 4, 5. Based on these and similar observations it has been proposed that deficits in reward processing may be an important risk factor for the development of obesity, and that obese individuals may compulsively consume palatable food to compensate for reward hyposensitivity 6. Importantly, it is presently unclear if deficits in reward processing are constitutive and precede obesity, or if excessive consumption of palatable food is sufficient to drive reward dysfunction and thereby contribute to diet-induced obesity.
A defining characteristic of overweight and obese individuals is the fact that they will continue to overeat despite the well-known negative health and social consequences. Indeed, many overweight individuals express a desire to limit their food consumption, yet struggle to control their intake and repeatedly consume past energy requirements 7, 8. Development of feeding behavior that is insensitive to negative outcome is analogous to the compulsive drug-taking behavior seen in human drug addicts that is similarly impervious to negative consequences 9. Here, we investigated the effects of extended access to a palatable high-fat diet on the sensitivity of brain reward systems in rats. We also examined the link between diet-induced hedonic dysregulation and the emergence of compulsive food seeking. Finally, the role for striatal D2Rs in these addiction-like behavioral responses was investigated.
Results
Addiction-like reward deficits in obese rats
To test the effects of restricted or extended access to a palatable high-fat diet, male Wistar rats (300–350 g) were prepared with a bipolar stimulating electrode in the lateral hypothalamus and trained for 10–14 days in a discrete-trial current-threshold brain stimulation reward (BSR) procedure until stable reward thresholds were established 4. In the BSR procedure, rats respond vigorously to obtain rewarding electrical self-stimulation via the indwelling stimulating electrode, with the minimal stimulation intensity that maintains self-stimulation behavior termed the reward threshold 10. Because reward thresholds remain stable and unaltered over prolonged time periods under baseline conditions, this procedure provides a sensitive measure of the responsiveness of brain reward systems. After establishment of stable BSR thresholds (defined as < 10% variation in thresholds across three consecutive sessions), rats were allocated to three groups that exhibited no differences in mean body weights or reward thresholds between groups. The three groups were given differential access to a “cafeteria style” diet consisting of palatable energy-dense food readily available for human consumption (see Methods). Rats had 0 h (chow-only rats; n = 9), 1 h (restricted access rats; n = 11), or 18–23 h (extended access rats; n = 11) access to the diet per day for 40 consecutive days. Cafeteria diets are well known to result in diet-induced obesity in rats 11. Importantly, all rats also had ad libitum access to standard laboratory chow, with reward thresholds, weight gain, and caloric intake recorded throughout.
Weight gain increased strikingly in rats with extended access to the cafeteria diet compared to the chow-only or restricted access groups (Fig. 1a). Weight gain also tended to be increased in the restricted access rats compared to chow-only rats, but this effect did not reach statistical significance. The development of obesity in extended access rats was closely associated with a worsening deficit in brain reward function, reflected in progressively elevated BSR thresholds (Fig. 1b). Because no differences in response latencies were observed between the three groups of rats (Supplementary Fig. 1), performance variables cannot account for this observation. Similar deficits in brain reward function have been reported in rats with extended but not restricted access to intravenous cocaine or heroin self-administration 12–14. Thus, extended access to palatable high-fat food can induce addiction-like deficits in brain reward function, considered an important source of motivation that may drive overeating and contribute to the development of obesity 1, 6.
Figure 1. Weight gain and reward dysfunction in rats with extended access to a cafeteria diet.
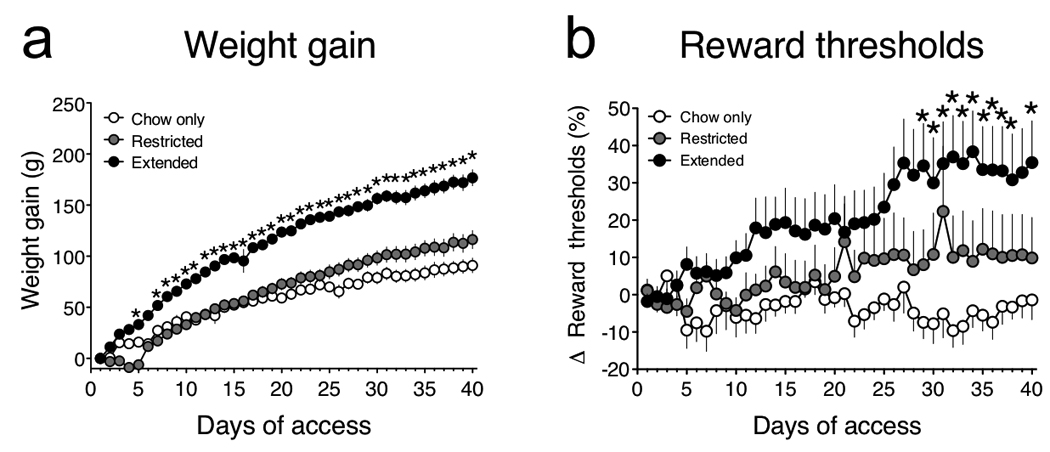
(a) Mean (± SEM) weight gain in chow-only, restricted access and extended access rats (Access × Day interaction: F39,702 = 7.9, P < 0.0001; *P < 0.05 compared with chow-only group, post-hoc test). (b) Mean (± SEM) percentage change from baseline reward thresholds (Access × Time interaction: F78,1092 = 1.7, P < 0.0005; *P < 0.05 compared with chow-only group, post-hoc test).
When we examined feeding behavior in detail we found that total daily caloric intake was similar between chow-only and restricted access rats (Fig. 2a, d). In contrast, total caloric intake in extended access rats was almost twice that of the restricted access and chow-only rats (Fig. 2a, d). Although the restricted access and chow-only rats maintained approximately the same daily caloric intake (Fig. 2a, d), restricted access rats obtained only <33% of their daily calories from chow (Fig. 2b, d), suggesting that they developed binge-like feeding behavior and consumed ~66% of their daily caloric intake during their 1 h access session to the cafeteria diet (Fig. 2d); see also Ref. 15. Extended access rats obtained only a small fraction (~5%) of their total caloric intake from chow (Fig. 2b), and consumed the cafeteria diet almost exclusively (Fig. 2d). The shift in dietary preference in the restricted and extended access groups was also reflected in a marked increase in fat intake compared with chow-only rats (Fig. 2c; see Supplementary Fig. 2). Consistent with previous reports 16, there was a tendency for consumption of the cafeteria diet to decrease over time in the extended access rats. This may reflect the development of tolerance to the palatability of the food items provided as part of the cafeteria diet over time. Nevertheless, the preference for the cafeteria diet versus standard chow remained consistently high throughout the experiment in these rats (Supplementary Fig. 3). These data demonstrate that extended but not restricted access to a palatable high-fat diet induces addiction-like reward deficits, overeating and loss of homeostatic energy balance. In contrast, restricted access to palatable food gives rise to binge-like patterns of consumption, but does not disrupt homeostatic levels of energy balance nor brain reward function. However, it is possible that exposure to the cafeteria diet for greater than 40 consecutive days in restricted access rats may induce significant weight gain and disruption of brain reward function.
Figure 2. Patterns of consumption in rats with extended access to a cafeteria diet.
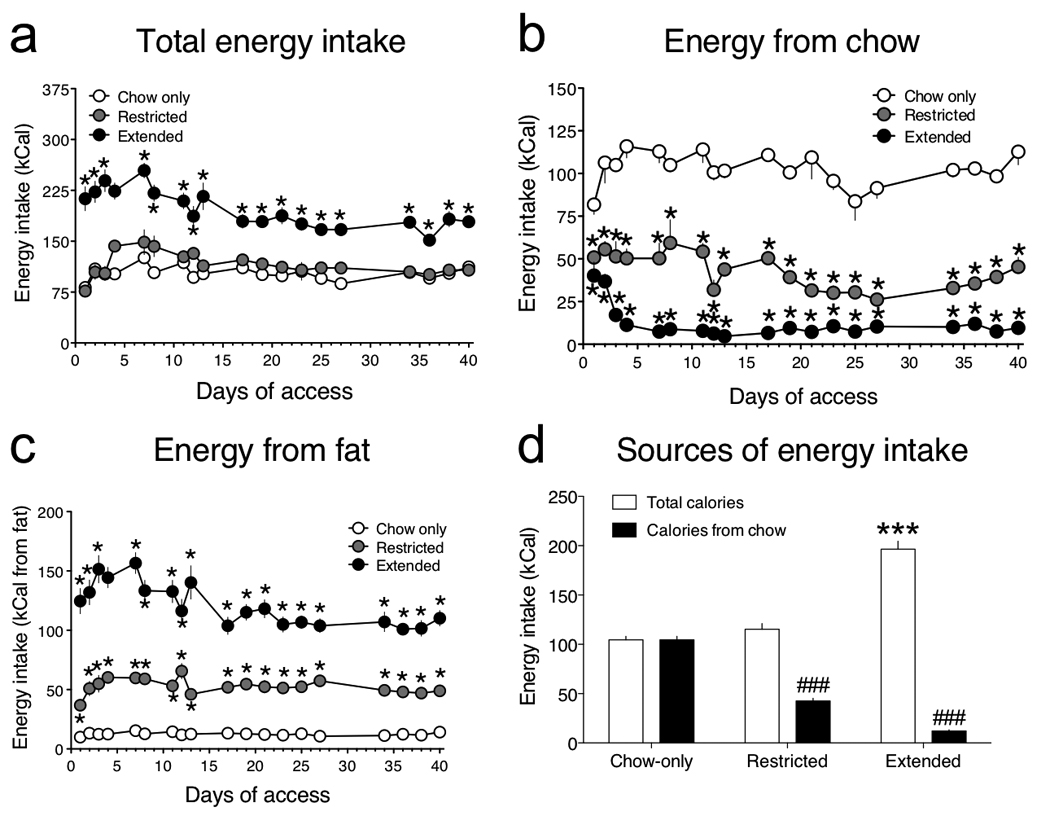
(a) Mean (± SEM) daily caloric intake in chow-only, restricted access and extended access rats (Access: F1,324 = 100.6, P < 0.0001; Time: F18,324 = 7.8, P < 0.0001; Access × Time interaction: F18,324 = 4.6, P < 0.0001; *P < 0.05 compared with chow-only group, post-hoc test). (b) Mean daily caloric intake (± SEM) from chow (Access: F2,504 = 349.1, P < 0.0001; Time: F18,504 = 5.9, P < 0.0001; Access × Time interaction: F36,504 = 3.52, P < 0.0001; *P < 0.05 compared with chow-only group, post-hoc test). (c) Mean daily caloric intake (± SEM) from fat (Access: F2,486 = 118.7, P < 0.0001; Time: F18,486 = 8.8, P < 0.0001; Access × Time interaction: F36,486 = 6.2, P < 0.0001; *P < 0.05 compared with chow-only group, post-hoc test). (d) Comparison of mean (± SEM) total caloric intake, and calories consumed exclusively from chow, during the entire 40-day period of access (Access: F2,54 = 25.0, P < 0.0001; Calorie source: F2,54 = 1235.2, P < 0.0001; Access × Calorie source interaction: F2,54 = 485.7, P < 0.0001; ***P < 0.001 compared with total calories in chow-only group, ###P < 0.001 compared with total calories in the same group of rats, post-hoc test).
After 40 days rats were no longer permitted access to the palatable diet, but continued to have ad libitum access to standard laboratory chow. Reward thresholds and chow consumption were assessed daily during this enforced “abstinence” period. We found that elevations in reward thresholds were long lasting and persisted for at least 2 weeks in the extended access rats when they no longer had access to the palatable diet (Fig. 3a). This contrasts with the relatively transient (~48 h) deficits in reward function reported in rats undergoing abstinence from self-administered cocaine 13. There was also a marked decrease in caloric intake (Fig. 3b) and a gradual decrease in body weight (Fig. 3c) in extended access rats, and to a lesser extent in restricted access rats, during this abstinence period when only standard chow was available, effects consistent with previous reports 11, 15. After 14 days of abstinence rats were euthanized and electrode placements were determined via cresyl violet staining (Fig. 3d).
Figure 3. Persistent reward dysfunction and hypophagia during abstinence in rats with extended access to a cafeteria diet.
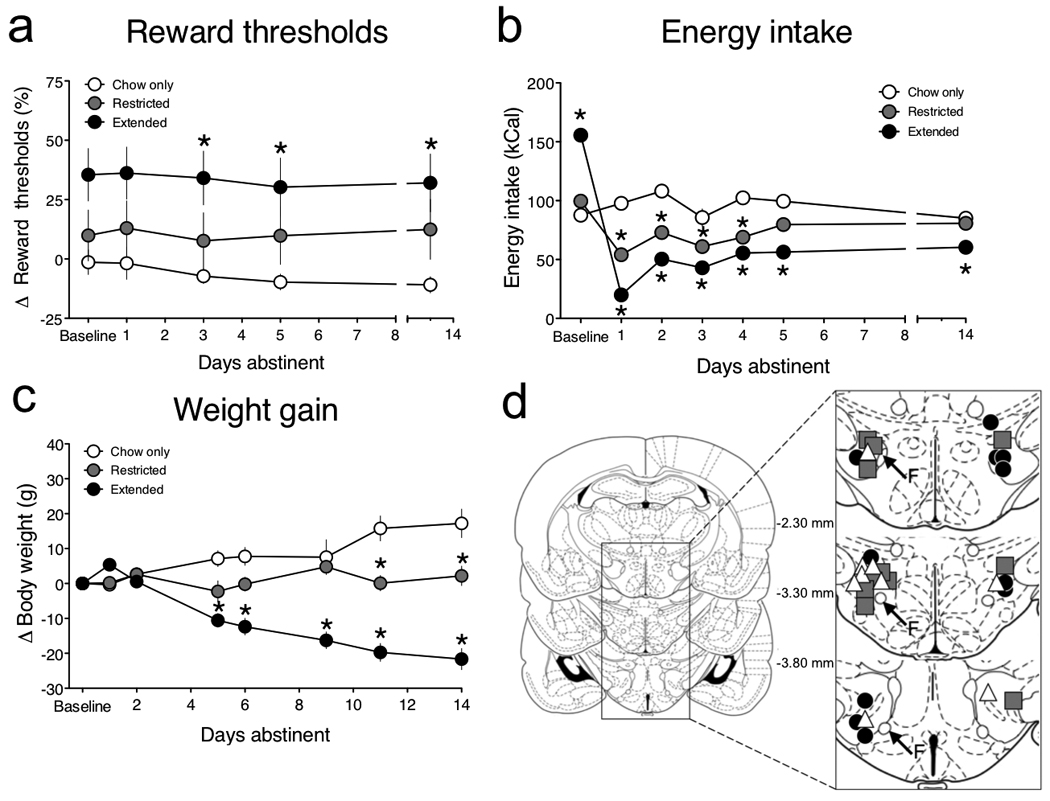
(a) Mean percentage change from baseline reward thresholds (± SEM) during abstinence from a palatable high-fat diet (Access: F2,112 = 3.7, P < 0.05; Time: F4,112 = 2.3, P > 0.05; *P < 0.05 compared with chow-only group, post-hoc test). (b) Mean caloric intake (± SEM) on the last day of access to the high-fat diet (Baseline) and during the 14 days of abstinence when only standard chow was available (Access: F2,168 = 41.7, P < 0.0001; Time: F6,168 = 65.6, P < 0.0001; Access × Time interaction: F12,168 = 38.3, P < 0.0001; *P < 0.05 compared with chow-only group, post-hoc test). (c) Change in mean body weight (± SEM) compared with body weight on the last day of access to the high-fat diet (Baseline) and during the 14 days of abstinence when only standard chow was available (Access: F1,126 = 37.2, P < 0.0001; Time: F7,126 = 3.1, P < 0.01; Access × Time interaction: F7,126 = 40.9, P < 0.0001; *P < 0.05 compared with chow-only group, post-hoc test). (d) Histological reconstruction of the location of BSR stimulating electrodes in the lateral hypothalamus of chow-only (△), restricted access ( ) and extended access (●) rats.
) and extended access (●) rats.
Reduced striatal D2R levels in obese rats: Role in brain reward deficits
We next tested the hypothesis that overconsumption of a palatable cafeteria diet may reduce striatal D2R density, contributing to the development of addiction-like reward hyposensitivity. A new cohort of chow-only, restricted and extended access rats were permitted access to the cafeteria diet until there was a statistically significant increase in body weight in the extended rats compared to the chow-only group (P < 0.05; Fig. 4a). Striatal expression of the heavily glycosylated (~70 kDa) membrane-bound form of the D2R was downregulated in extended access rats compared to the restricted access or chow-only rats (Fig. 4b). In fact, when animals in each access condition were sub-divided into two groups based on a median split of body weights (light or heavy), a clear inverse relationship between body weight and striatal D2R expression levels was observed (Fig. 4a, c). No statistically significant decreases in expression of the unglycosylated immature (~39 kDa) and intermediate glycosylated cytoplasmic (~51 kDa) forms of the D2R were detected (Supplementary Fig. 4) 17, suggesting that striatal D2R expression levels in extended access rats are likely regulated through post-transcriptional mechanisms.
Figure 4. Weight gain is inversely related to striatal D2R levels.
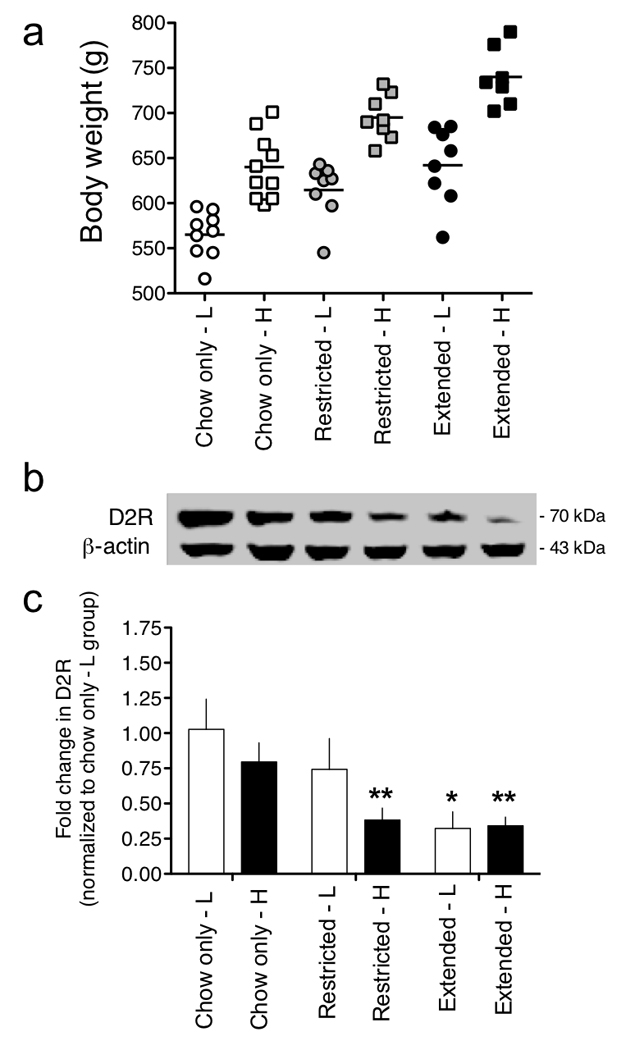
(a) Chow-only, restricted access and extended access rats were sub-divided into two groups per access condition based on a median split of body weights; light (L) or heavy (H). (b) The entire striatal complex was collected from all rats and D2R levels in each group measured by Western blotting. The membrane-associated post-synaptic D2R band was resolved at 70 kDa, and the protein-loading control is displayed below (β-actin, 43 kDa). Full-length immunoblots are displayed in Supplementary Figure 12. (c) Relative amounts of D2R in the striatum of chow-only, restricted and extended access rats were quantified by densitometry (F2,6=5.2, P < 0.05, main effect of Access; *P < 0.05 compared with Chow-only-L group).
Next, to directly test the functional relevance of diet-induced reductions in striatal D2R levels on brain reward function we designed and validated a lentivirus vector to deliver a short-hairpin interfering RNA (shRNA) to knockdown D2R (Lenti-D2Rsh) (Fig. 5; Supplementary Fig. 5). Reward thresholds began elevating in Lenti-D2Rsh rats almost immediately upon being permitted extended access to the cafeteria diet, whereas reward thresholds remained stable and unaltered in extended access rats treated with an empty lentivirus vector (Lenti-Control) over the relatively short period of access to the cafeteria diet (14-days; Fig. 6a). Response latencies were unaltered in both groups of rats, demonstrating that this effect was not secondary to deficits in task performance (Supplementary Fig. 6). Reward thresholds were also unaltered in Lenti-D2Rsh and Lenti-Control rats with chow-only access over the same period (Fig. 6b). Thresholds remained persistently elevated during an additional 15-days of abstinence when all rats had access only to standard chow (Supplementary Fig. 7). Knockdown of striatal D2R therefore increases vulnerability to diet-induced reward hypofunctionality but does not alter the baseline activity of brain reward systems.
Figure 5. Lentivirus-mediated knockdown of striatal D2R expression.
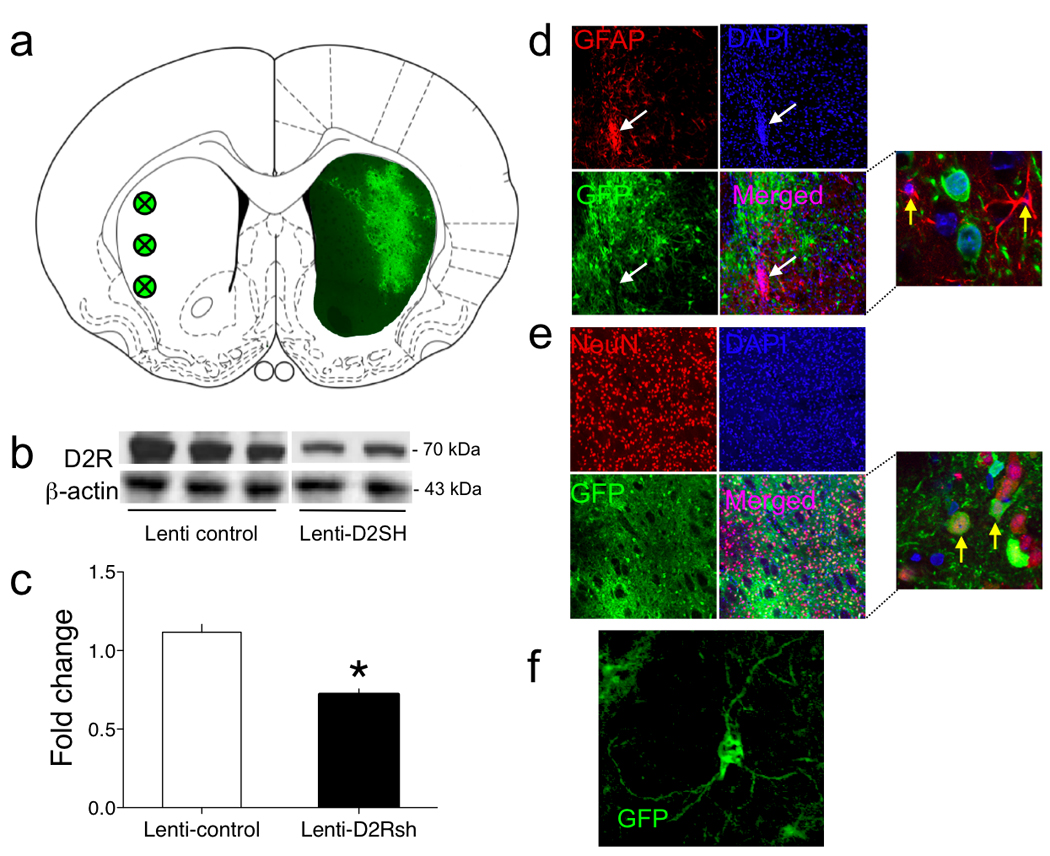
(a) Graphical representation of the striatal areas in which Lenti-D2Rsh was overexpressed. Green circles in the left striatal hemisphere represent the locations at which viral infusions were targeted. Green staining in the right striatal hemisphere is a representative immunochemistry staining for GFP from the brain of a Lenti-D2Rsh rat. (b) Representative immunoblot of the decreased D2R expression in the striatum of Lenti-D2Rsh rats. Full-length immunoblots are displayed in Supplementary Figure 13. (c) Relative amounts of D2R in the striatum of Lenti-Control and Lenti-D2Rsh rats were quantified by densitometry (*P < 0.05 compared with the Lenti-Control group, post-hoc test). (d) Infection of glial cells in the striatum by the Lenti-D2Rsh vector was not detected. Green staining is GFP from virus; red is the astrocyte marker glial fibrillary acidic protein [GFAP]; cell nuclei are highlighted by DAPI staining in blue. The white arrows indicate a highly localized area of gliosis found only at the site of virus injection in the striatum and not in the surrounding tissues into which the virus has diffused. Even in this area none of the astrocytes were GFP-positive. The yellow arrows in the magnified image highlight the typical GFP-negative astrocytes that were detected. (e) High levels of neuronal infection in the striatum by the Lenti-D2Rsh vector were detected. Green staining is GFP from virus; red is the neuronal nuclear marker NeuN; cell nuclei are highlighted by DAPI staining in blue. The yellow arrows in the magnified image highlight GFP-positive and NeuN-positive neurons in the striatum. (f) A higher magnification image of a virally infected (GFP-positive) neuron in the striatum of Lenti-D2Rsh rats that displays the typical morphological features of medium spiny neurons.
Figure 6. Knockdown of striatal D2R increases vulnerability to reward dysfunction in rats with extended access to a cafeteria diet.

(a) Mean (± SEM) percentage change from baseline reward thresholds in Lenti-Control and Lenti-D2Rsh rats that had extended access to the cafeteria diet for 14 consecutive days (Virus: F1,156 = 5.9, P < 0.05; Time: F13,156 = 2.2, P < 0.05; Virus × Time interaction: F13,156 = 2.2, P < 0.05; #P < 0.05, interaction effect). (b) Mean (± SEM) percentage change from baseline reward thresholds in Lenti-Control and Lenti-D2Rsh rats that had chow-only access (c) Mean (± SEM) caloric intake during 14 days of chow-only or extended access animals (Access: F2,28 = 135.6, ***P < 0.0001). (d) Mean (± SEM) weight gain during 14 days of chow-only or extended access animals (Access: F2,28 = 96.4, P < 0.0001; ***P < 0.001, main effect of access).
We found that caloric intake (Fig. 6c) and weight gain (Fig. 6d) was similar between the Lenti-D2Rsh and corresponding Lenti-Control groups under chow-only or extended access conditions (total intake of chow and palatable food are shown in Supplementary Figs. 8 and 9). Thus, striatal D2R knockdown does not alter preference for the cafeteria diet nor total caloric intake when the palatable food is freely available for consumption.
Compulsive eating in obese rats: Role for striatal D2R
We next tested the hypothesis that compulsive-like eating may emerge in rats with extended access to the cafeteria diet and that deficits in striatal D2R signaling may contribute to this effect. A new cohort of chow-only, restricted access and extended access rats were permitted access to the cafeteria diet for ~40 days until statistically significant weight increases were observed in the extended rats (P < 0.05 compared with chow-only rats; data not shown). All three groups of rats were then permitted only 30 min access per day to the cafeteria diet for 5–7 days in an operant chamber until stable intake was achieved (defined as < 10 % variation in daily intake). Half the animals in each access condition then were exposed to a light (conditioned stimulus; CS) paired with delivery of footshocks (punished group), whereas the remaining rats in each group were exposed to the CS in the absence of footshock (unpunished group). On the test day, the effects of CS exposure alone on palatable food consumption were examined (see Methods). We found that mean caloric intake during the 30 min baseline sessions was higher in the chow-only and restricted access rats compared with extended access rats (Fig. 7 a, b). This suggests that the chow-only and restricted access rats binged on the palatable food during the intermittent 30 min access sessions, reflected in the fact that these rats consumed ~40–50% of their daily caloric intake, typically ~100 kCal, during the 30 min access sessions (Fig. 7a, b). In contrast, extended access rats appear resistant to developing this binge-like feeding behavior, perhaps because their prior history of almost unlimited access to the palatable food for >40 consecutive days established patterns of eating that were relatively inflexible to change. On the test day, no statistically significant effects of CS replay on food consumption were detected in the unpunished animals from the chow-only, restricted or extended access groups when compared with levels of intake seen during the baseline period (Fig. 7a). The CS alone therefore has no motivational salience. In punished rats the shock-paired CS significantly decreased palatable food intake in the chow-only and restricted access rats. However, the CS had no effect on palatable food intake in the extended access rats, demonstrating that their consumption was insensitive to aversive environmental cues predicting adversity. Baseline levels of energy intake in the extended access rats were lower than those of the other groups. However, because levels of chow intake during similar time-periods are far lower (see Fig. 7d), it is unlikely that this represents a “floor effect” that confounds our findings. Taken together, our data suggest that compulsive-like eating behavior can emerge in extended access rats in a manner analogous to the compulsive cocaine-taking seen in rats with a history of extended access to the drug 18.
Figure 7. Compulsive-like responding for palatable food.
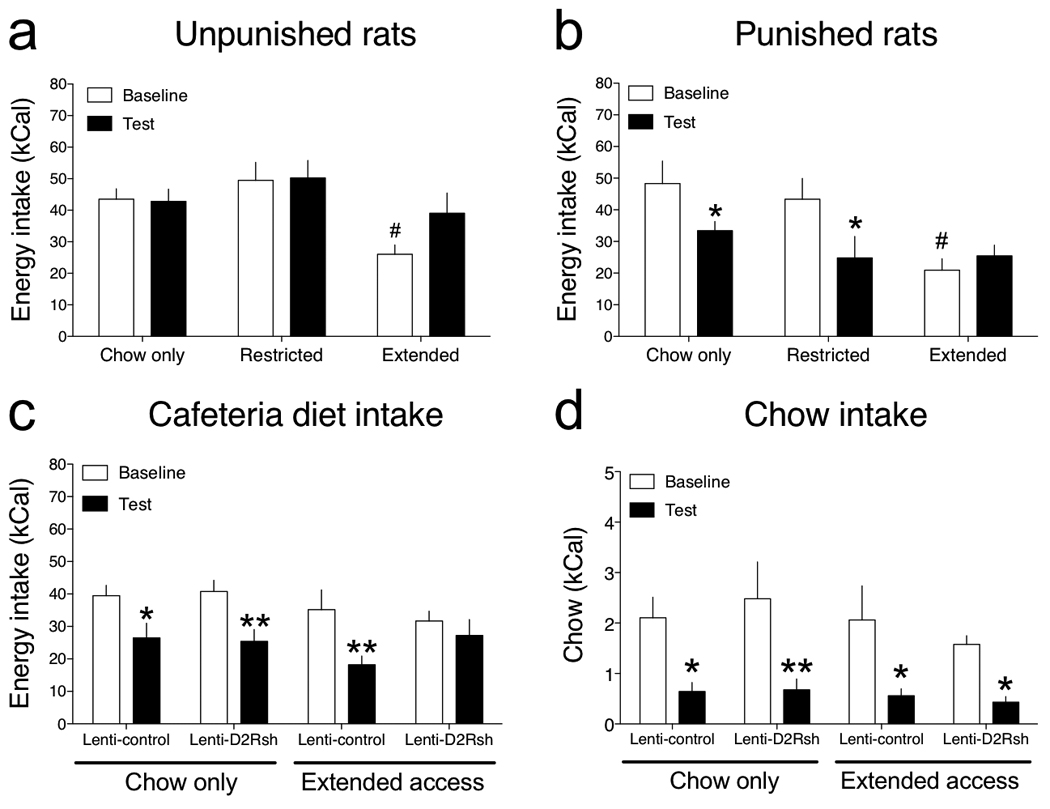
(a) Mean (± SEM) palatable diet consumption (kCal; ± SEM) in unpunished rats during the 30 min baseline sessions and on the test day when rats were exposed to a neutral conditioned stimulus that was not previously paired with noxious foot shock (Access: F2,20 = 5.2, P < 0.05; #P < 0.05 compared with chow-only rats). (b) Mean (± SEM) palatable diet consumption (kCal; ± SEM) in punished rats during the 30 min baseline sessions and on the test day when rats were exposed to a conditioned stimulus that was previously paired with noxious foot shock (Access: F2,21 = 3.9, P < 0.05; Cue: F1,21 = 8.6, P < 0.01; Access × Cue interaction: F2,21 = 4.7, P < 0.05; *P < 0.05 compared with intake during the baseline session, #P < 0.05 compared with chow-only rats). (c) Mean (± SEM) palatable diet consumption during the 30 min baseline sessions and on the test day in Lenti-Control and Lenti-D2Rsh rats that previously had chow-only or extended access to a cafeteria diet (Cue: F1,26 = 29.7, P < 0.0001; *P < 0.05, **P < 0.01 compared with intake during the baseline sessions, post-hoc test). (d) Mean (± SEM) chow consumption during the 30 min baseline sessions and on the test day in Lenti-Control and Lenti-D2Rsh rats that previously had chow-only or extended access to a cafeteria diet (Cue: F1,26 = 44.9, P < 0.0001; *P < 0.05, **P < 0.01 compared with intake during the baseline sessions, post-hoc test).
Finally, we examined the effects of the punishment-paired CS on food intake in the Lenti-Control and Lenti-D2sh rats that previously had chow-only or extended access to the cafeteria diet (animals from Fig. 6). We found that baseline levels of palatable food intake during the 30-min baseline sessions were similarly high (~40 kCal) in all four groups of rats (Fig. 7c). In addition, total daily chow consumption (in the home cage) was similar between all four groups of rats during the conditioning sessions and on the test day (Supplementary Fig. 10). The 14 days of prior access to the cafeteria diet was therefore not sufficient to block binge-like eating behavior in a manner similar to that seen in rats that had <40 days extended access to the cafeteria diet (Fig. 7a, b). The aversive CS disrupted palatable food intake in Lenti-Control and Lenti-D2Rsh rats that previously had chow-only access (Fig. 7c). Similarly, the aversive CS disrupted palatable food intake in the Lenti-Control rats that previously had 14 days extended access to the cafeteria diet. In contrast, the aversive CS had no impact on palatable food consumption in the Lenti-D2Rsh rats that previously had 14 days extended access to the cafeteria diet (Fig. 7c). BSR thresholds remained significantly elevated in these rats when recorded 48 h after the test session, whereas thresholds remained stable and unaltered in the other three groups of rats (Supplementary Fig. 11). To verify that resistance to CS-induced suppression of palatable food intake in the Lenti-D2Rsh extended access rats was not secondary to impairments in classical conditioning processes, we tested the effects of the aversive CS on consumption of less palatable standard chow in all four groups of rats. In contrast to the binge-like consumption of palatable food, we found that all four groups of rats consumed low levels of chow (~2 kCal) during the 30 min baseline sessions (Fig. 7d), and that chow intake was disrupted in all four groups by a similar magnitude upon exposure to the aversive CS (Fig. 7d). These data demonstrate that knockdown of striatal D2R dramatically accelerates the emergence of compulsive-like eating of palatable food, but only in rats with a prior history of extended access. Moreover, because compulsive eating was detected only in Lenti-D2Rsh rats that had elevated BSR thresholds, diet-induced reward hypofunctionality may be a necessary antecedent to the emergence of compulsive food seeking.
Discussion
Ease of access to palatable high-fat food is considered a major environmental risk factor for obesity 19. We found that extended access to a highly palatable cafeteria-style diet results in overeating and weight gain coupled with progressively elevating BSR thresholds in rats. This effect on BSR thresholds can be explained by gradually decreasing responsiveness of brain reward circuitries, an interpretation consistent with the fact that food restriction and weight loss can increase 20, whereas acute overfeeding can transiently decrease 21, responding for BSR stimulation in rats. This finding represents an important extension of the pioneering work of Hoebel and colleagues, who have shown that acute overfeeding of rats through intra-gastric feeding tube 21, and gastric distention or intravenous glucagon infusion that mimic postprandial satiety 22–24, decreases responding for rewarding lateral hypothalamic BSR and increases aversion-like responses to the stimulation 25. Moreover, Hoebel and colleagues have also shown that repeatedly force-feeding rats through intra-gastric tube until their weight had increased by ~200 g similarly decreased rates of responding for BSR stimulation, an effect that persisted until body weight had normalized; described in Ref.23. Similar to these findings in rats, responding for lateral hypothalamic BSR was inhibited by prior feeding of cats to satiation 26, demonstrating that interactions between brain reward function and metabolic state is highly-conserved and likely to also occur in humans. Ease of access and consequent overeating of cafeteria-style diets in humans is considered a major environmental factor contributing to the current obesity epidemic in Western societies 19. Our data show that reward hypofunctionality arises in rats that volitionally overeat a palatable cafeteria diet similar to that consumed by humans and that this effect becomes progressively worse as their weight gain increases. Notably, all rats with reward threshold elevations ≥ 20% had BSR electrodes located within ~500 µm of the fornix dorsolaterally. Sensitivity of reward-related neurons in this area is increased in food restriction in a manner sensitive to the fat-derived hormone leptin, and this brain region is considered an important substrate for food reward 27. Brain circuitries regulating food hedonics are therefore inhibited by post-ingestive signals that predict satiety, consistent with recent human imaging studies demonstrating that gastric distention 28 and the gut-derived postprandial factor peptide YY3-36 (PYY) 29 modulate the activity of regions of the brain involved in reward processing. In addition, reward systems are also inhibited by excessive weight gain. Interesting in this regard are the recent reports that circulating leptin, a key regulator of energy balance, may penetrate into brain tissues and inhibit the activity of reward circuitries 3, 27, 30, 31.
Reward deficits in overweight rats may reflect counteradaptive decreases in the baseline sensitivity of brain reward circuitries to oppose their overstimulation by palatable food. Such diet-induced reward hypofunctionality may have a pathophysiological role in the development of obesity by increasing the motivation to consume high-reward “obesogenic” diets to avoid or alleviate this state of negative reward 6. This may account for the hypophagia we observed in extended access rats and to a lesser degree in restricted access rats when the palatable food was withdrawn and only the less palatable chow was available. Such a scenario is also consistent with data from human brain imaging studies in which blunted activation of the striatum in response to highly palatable food, particularly in individuals with genetic polymorphisms thought to decrease striatal D2R expression, is associated with long-term weight gain 4. It has been unclear if such reward hyposensitivity in obese individuals is manifested before the development of obesity and related solely to genetic factors (“reward deficiency syndrome”) or if overeating can cause disruption in reward processing. Our data demonstrate that extended access to palatable high-energy food and subsequent overeating blunts reward sensitivity and may therefore represent an important hedonic mechanism that promotes the development of obesity. Similar reward dysfunction to that reported here in obese rats is also detected in rats with a history of extended but not restricted access to intravenous cocaine or heroin self-administration 12–14. Moreover this drug-induced reward dysfunciton is hypothesized to trigger the transition from casual to compulsive drug seeking to alleviate this persistent state of diminished reward 12, 32. Thus, our data suggest that obesity and drug addiction may share common underlying hedonic mechanisms.
Downregulation of striatal D2R expression is a notable neuroadaptive response to overconsumption of palatable food. Indeed, reductions in striatal D2R density are seen in overweight individuals 4, 33, and rodents 34, 35. Conversely, patients suffering from anorexia nervosa have elevated striatal D2R levels 36, and weight loss in obese patients following bariatric (gastric bypass) surgery is associated with increased striatal D2R density 28. The gene polymorphism referred to as the TaqIA A1 allele results in decreased striatal D2R density, and individuals harboring this allele are over-represented in obese populations 4. The TaqIA A1 allele also increases vulnerability to alcohol, opioid and psychomotor stimulant addiction 37. Reductions in striatal D2R density occurring either through constitutive genetic factors or as a consequence of overeating may therefore play a role in the neurobiological mechanisms of obesity. We found that striatal levels of the 70 kDa D2R isoform, thought to reflect the membrane-associated post-synaptic D2R, were inversely related to body weight in chow-only, restricted access and extended access rats (Fig. 4). Knockdown of striatal D2R expression, most prominently in dorsolateral regions of the striatum (see Fig. 5), resulted in BSR thresholds elevating almost immediately upon exposure to the cafeteria diet. Decreases in striatal D2R levels therefore rapidly accelerates the emergence of reward hypofunctionality in rats with extended access to highly palatable food, a finding consistent with human brain imaging data in which deficits in striatal D2R density are thought to contribute to reward hypofunctionality in obese individuals 4. There are three interesting features of the Lenti-D2Rsh rats that are also important to highlight. First, although striatal D2R knockdown combined with extended access to the palatable diet resulted in elevating BSR thresholds, there were no differences in caloric intake or weight gain in these rats compared to control rats. This may reflect that fact that rats only had 14 days access to the cafeteria diet and longer periods of access may have resulted in higher weight gain over time similar to the increased susceptibility to weight gain seen in humans with deficits in striatal D2R signaling 4. However, a major advantage of limiting access to the cafeteria diet to only 14 days is that the knockdown rats with extended access were the only group to demonstrate elevated BSR thresholds, and this permitted us to assess the potential role for reward hypofunctionality in the development of compulsive eating behavior (see below). Second, BSR thresholds remained stable and unaltered in the knockdown rats that had chow-only access. This suggests that reduced striatal D2R expression alone is not sufficient to induce reward hyposensitivity, but instead seems to interact with overconsumption of palatable food to accelerate the emergence of this state of negative reward. Other adaptive responses in brain reward circuitries may therefore trigger reward hyposensitivity in rats with extended access to the cafeteria diet. With this in mind, it is interesting to note that the D2R agonist bromocriptine reduces circulating levels of leptin 38, and leptin inhibits feeding behavior at least in part through inhibition of striatal regions controlling hedonic responses to food 3, 30, 31. Thus, it is an interesting possibility that downregulation of striatal D2R in response to increasing body weight may serve to increase leptin signaling and thereby boost the inhibitory effects of this adipokine on brain reward systems. Finally, it is important to note that we targeted our lentivirus vectors toward dorsolateral regions of the striatum. This was primarily for technical reasons, as lateral placement of cannulae for virus delivery into the striatum enabled us also to accommodate the indwelling hypothalamic stimulating electrode for BSR threshold determination. Thus, it is possible that targeting of D2Rs for knockdown in other areas of the striatum, particularly the dorsomedial and ventral areas (nucleus accumbens core and shell), may have elevated BSR thresholds even in the absence of the palatable diet.
As noted above, we targeted our lentivirus injections toward the dorsolateral striatum. This brain region has been heavily implicated in stimulus-response (S-R) habit-type learning, reflected in the development of consummatory behavior that is insensitive to devaluation by prior feeding to satiation or being paired with noxious stimuli 39. By targeting predominately the dorsolateral striatum we may have impacted populations of D2Rs that regulate vulnerability to develop compulsive-like consummatory behaviors. In keeping with a role for striatal D2Rs in compulsive behaviors, the human TaqIA allele of the D2R gene locus that results in deficits in striatal D2R density 5, blunted striatal activation in response to palatable food 4, and increased vulnerability to obesity 4, is also associated with deficits in learning to avoid actions with negative consequences 40. Loss of inhibitory control over behavior that can have a negative outcome is a characteristic feature of both obesity and drug addiction, in which consummatory behaviors persist despite negative social, health or financial consequences. Cocaine-taking behavior in rats with a history of extensive drug intake can become inflexible and resistant to disruption by an aversive conditioned stimulus that predicts negative outcome (i.e., delivery of footshock) 18. Similarly, mice that previously had access to a palatable high-fat diet will spend more time in an aversive environment (brightly lit) to obtain the palatable food than mice that had no prior experience of the diet 41. We found that palatable food consumption in extended access rats is similarly insensitive to an aversive CS. Consistent with a role for striatal D2Rs in this effect, compulsive-like eating was detected in the striatal D2R knockdown rats that previously had 14 days extended access to the cafeteria diet but not in the control groups of rats. From a neurocircuitry perspective, extended access to palatable food may trigger plasticity in corticostriatal pathways rendering animals more vulnerable to developing compulsive-like behaviors, with deficits in striatal D2R signaling enhancing this process. Indeed, deficits in striatal D2R density in obese individuals is correlated with reduced metabolism in prefrontal and orbitofrontal cortical areas 42, brain regions that exert inhibitory control over behavior 43. Intriguingly, compulsive-like consumption of palatable food was detected only in the knockdown rats that previously had extended access to the cafeteria diet, but not in control rats that had extended access to the cafeteria diet for the same time period, nor in knockdown rats that had chow-only access. The major difference between the knockdown rats with prior extended access compared to the other groups was the fact that they had persistently elevated BSR thresholds. This may reflect that reward hypofunctionality and the emergence of compulsive-like eating arise from the same underlying neurobiological mechanisms and hence are temporally coincident yet independent phenomena. Alternatively, it is interesting possibility that diet-induced reward hypofunctionality may serve as a substrate for negative reinforcement that facilitates the development of compulsive-like eating 14, 32. Whatever the underlying mechanisms, our findings demonstrate that addiction-like compulsive responding for palatable food can emerge in obese rats, and suggest that deficits in striatal D2R signaling increases vulnerability to develop this behavior.
In summary, we have shown that over-stimulation of brain reward systems through excessive consumption of palatable energy-dense food induces a profound state of reward hyposensitivity and the development of compulsive-like eating. These maladaptive behavioral responses in obese rats likely arise from diet-induced deficits in striatal D2R signaling. Overconsumption of drugs of abuse similarly decreases striatal D2R density, induces a profound state of reward hypofunctionality and triggers the emergence of compulsive-like drug-taking behaviors. Our findings therefore support the previous work of Hoebel and many others 4, 19, 41, 44–46 suggesting that obesity and drug addiction may arise from similar neuroadaptive responses in brain reward circuitries.
Methods
Animals
Male Wistar rats weighing 300–350 g at the start of the experiments were obtained from Charles River. Upon arrival animals were housed individually at constant temperature on a 12 h light/dark cycle (lights on at 2200 h). Animals were permitted ad libitum access to standard laboratory chow and water for the duration of the experiment. All procedures were approved by the Institutional Animal Care and Use Committee of Scripps Florida, and animals were treated in accordance with the guidelines set forth by the National Institutes of Health regarding the principles of animal care.
Surgical procedures
Rats prepared with BSR stimulating electrodes were first anaesthetized by inhalation of 1–3% isoflurane in oxygen and positioned in a stereotaxic frame (Kopf Instruments, Tujunga, CA). Bipolar BSR electrodes (11 mm in length) were implanted into the posterior lateral hypothalamus (AP: −0.5 mm from bregma; ML: ± 1.7 mm from midline; DV: 8.3 mm from dura; incisor bar was adjusted to 5 mm above the interaural line) according to the atlas of Pellegrino et al. 47. Animals receiving virus injections were also prepared with bilateral guide cannulae (23 gauge, 14 mm in length) positioned above the striatum (AP: 2.8 from bregma; ML: ± 3.1 from midline; DV: −2.4)47 and filled with 14 mm stylets. Four stainless steel skull screws and dental acrylic held the electrode and cannulae in place. The surgical wound was treated with topical antibiotic (once every 12 h) for five days after the surgery. Animals were allowed 7−10 days to recover from surgery, and were then trained in the BSR threshold procedure.
BSR procedure
Animals were trained to respond for BSR stimulation according to a discrete-trial current-threshold procedure similar to that described in detail elsewhere 10, 14. Briefly, BSR current levels were varied in alternating descending and ascending series in 5 µA steps. In each testing session, four alternating descending/ascending series were presented. The threshold for each series was defined as the midpoint between two consecutive current intensities for which animals responded in at least three of the five trials, and two consecutive current intensities for which animals did not respond in three or more of the five trials. The overall threshold of the session was defined as the mean of the thresholds for the four individual series. Each testing session was approximately 30 min in duration. Stable BSR thresholds were defined as ≤10% variation in thresholds over 5 consecutive days, usually established after 10–14 days of training. The response latency for each test session was defined as the mean response latency of all trials during which a positive response occurred.
Viral packaging and delivery
Short-hairpin RNA was delivered and constitutively expressed using the pRNAT-U6.2/Lenti vector system (GenScript, Piscataway, NJ). Viral particles were prepared according to the manufacturer’s protocol. Briefly, HEK 293FT cells were transfected with vector containing the shRNA insert (5’-GGA TCC CGC GCA GCA GTC GAG CTT TCT TCA AGA GAG AAA GCT CGA CTG CTG CGC TTT TTT CCA ACT CGA G-3’) or the empty vector, and ViraPower Packaging Mix (Invitrogen, Carolsbad, CA) for 72-h (media replaced after 24-h). Supernatant was then collected and concentrated by ultracentrifugation (25,000 RPM, 90 min, 4°C) and viral titer was determined fluorescence-activated cell sorting? (FACS) according to the manufacturer’s instructions. Virus was aliquoted and stored in light-protected boxes at −80°C until use.
Rats with stable BSR thresholds received bilateral virus injections at three sites in the striatum of each brain hemisphere (2 µl per injection, 1 µl/min, 1 min between injections; total of six injections per animal). Rats were allowed at least 2–3 days recovery from intra-striatal injections before BSR thresholds assessment was resumed. Daily BSR threshold assessment continued for 33 days after virus injections to ensure maximal striatal D2R knockdown was achieved prior to permitting rats access to the cafeteria diet. There were no differences in BSR thresholds between the Lenti-Control and Lenti-D2Rsh rats during these 33 days (data not shown).
Immunoblotting
Rats were euthanized approximately 1 h after their regularly scheduled access to the cafeteria diet, and brains were rapidly removed. Brain sections of approximately 1–2 mm thickness were prepared using a coronal brain matrix (1 mm slice interval; Plastics One) stationed on an ice block, and tissue punches of dorsal striatum (Bregma: ~2.2 to −0.26) were taken. Striatal tissue punches were rapidly collected, snap frozen, and stored at −80°C until use. Individual samples were thawed on ice and equal amounts of striatal tissue were pooled based on a weight-dependent median split of access groups (7–10 animals per pool). Tissue was resuspended in 500 µl ice-cold RIPA buffer (Thermo Scientific, Rockland, IL) containing sodium orthovanadate, phosphatase cocktail inhibitors 1 and 2 (Sigma-Aldrich, St. Louis, MO), leupeptin, and pepstatin prior to homogenization. Tissue lysates were boiled for 10 min in sample buffer and loaded onto 4–20% or 10% tris-glycine SDS gels (Invitrogen, Carlsbad, CA). Protein was transferred to nitrocellulose membranes, blocked for 1 h at room temperature (5% non-fat dry milk and 0.2% Tween 20 in PBS, pH 7.4), and incubated in primary antibody overnight at 4°C. The following primary antibodies were diluted in block solution: D2R mouse monoclonal (Santa Cruz, 1:100) or β-Actin mouse monoclonal (Santa Cruz, 1:200). Chemiluminescent ECL reagent was added after incubation with HRP-conjugated secondary antibodies (Amersham; 1:2000). The mature membrane-associated form of D2DR (~70 kDa) 17, 48 was normalized to a protein-loading control (β-Actin; 43 kDa) and quantified by densitometry using NIH Image J software.
Immunochemical analysis
Rats were anesthetized and transcardially perfused with 4% paraformadehyde in PBS (pH 7.6). Brains were removed, post-fixed overnight, and stored in sucrose (30% solution in PBS, pH 7.4) for at least 72 h. Frozen tissue sections (30 µm thickness) were collected from a microtome and blocked (3% BSA, 5% normal goat serum, and 0.3% Triton X-100 in PBS) for 1 h at room temperature. The following primary antibodies were added to the block solution and incubated overnight at 4°C: chicken polyclonal to GFP (Abcam, 1:1000); rabbit monoclonal to GFAP (Millipore, 1:1000); mouse monoclonal to NeuN (Millipore, 1:1000). Sections were incubated with fluorescent-dye-conjugated secondary antibodies at room temperature: anti-chicken 488 nm (Jackson Immunoresearch, 1:1000), anti-rabbit 594 nm (Invitrogen, 1:1000), and anti-mouse 594 nm (Invitrogen, 1:1000). Sections were mounted with Vectashield mounting media containing DAPI (Vector Labs, Burlingame, CA) and cover slipped. Images were taken using an Olympus BX61 fluorescence microscope (2X objective) or an Olympus confocal microscope (10X, 100X objectives).
Feeding procedure
Animals were housed individually on paper bedding (alpha pads; Shepherd Specialty Papers, Lawrenceville, GA) to prevent food products from being soiled with loose bedding materials. The cafeteria diet consisted of bacon, sausage, cheesecake, pound cake, frosting, and chocolate, which were individually weighed before being made available to the rats. The cafeteria diet food items were delivered in small metal receptacles. All food items, including standard laboratory chow, were weighed again following completion of the feeding session. Caloric intake from the various macronutrients was calculated based on the nutritional information provided by the manufacturer.
Cue-induced suppression of feeding behavior
Feeding procedures took place in sound-attenuated operant chambers identical in dimensions to those used in the BSR experiments. Rats were placed into an operant chamber, and had access to the cafeteria diet or chow for 30 min. The food products were delivered in small metal receptacles. All food items were weighed before and after feeding sessions, which were carried out during the animals’ normal feeding period. Chow consumption was assessed by consumption of 45 mg chow pellets identical in composition to chow provided in the animals’ home cages. Rats were then permitted 30 min access per day to the cafeteria diet until stable intake was achieved (defined as <10 % variation in daily intake), requiring 5–7 days. Following stabilization of palatable food intake during this baseline period, rats in each access condition were allocated to two groups: punished (those receiving foot shock) and unpunished (remaining foot shock naïve). Animals were then subjected to four conditioning sessions on consecutive days in the same operant chamber in which they previously had access to the palatable food. During the 30 min conditioning sessions a cue light (conditioned stimulus; CS) was activated for 10 min, turned off for 10 min, and then turned back on for 10 min. Punished animals received foot shock only during presentation of the CS (0.5 mA for 1.0 sec; 10 stimulations with ~1 min intervals). The unpunished rats were presented with the CS in the same manner, but without the delivery of foot shock. On the test day, occurring on the day after the final conditioning session, animals in the punished groups received intermittent foot shock (5 stimulations in total) paired with activation of the CS for 5 min. The unpunished animals were again exposed to the CS in the absence of foot shock. After the 5 min punishment period, all animals were permitted access to the palatable food for a 30 min session with the CS activated intermittently (10 min CS on, 10 min CS off, 10 min CS on).
Statistical analyses
Baseline reward thresholds were defined as the average threshold value for the five days preceding access to the cafeteria diet for each subject. Reward thresholds were expressed as the percent change from the baseline threshold value. Percentage of baseline reward threshold values, weight gain, caloric consumption, and caloric consumption from fat data were analyzed by two-factor repeated-measures analysis of variance (ANOVA), with Access (chow-only, restricted or extended access), Calorie Source (standard chow or cafeteria diet), Virus (Lenti-Control or Lenti-D2Rsh) and Cue (paired or unpaired with punishment) as the between-subjects factor, and Time as the within-subjects factor. When appropriate, main effects in the ANOVAs were further analyzed by Bonferroni post-tests. All statistical analyses were performed using GraphPad Prism software.
Supplementary Material
Acknowledgements
This work was supported by a Bank of America Fellowship (PMJ), The Landenberger Foundation (PJK) and by a grant from the National Institutes of Health (DA025983; PJK). This is publication number 19563 from The Scripps Research Institute.
Footnotes
Author Contributions
P.M.J. conducted all experiments. P.M.J. and P.J.K. designed the experiments, analyzed the data and wrote the manuscript.
References
- 1.Saper CB, Chou TC, Elmquist JK. The need to feed: homeostatic and hedonic control of eating. Neuron. 2002;36:199–211. doi: 10.1016/s0896-6273(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 2.Zheng H, Berthoud HR. Eating for pleasure or calories. Curr Opin Pharmacol. 2007;7:607–612. doi: 10.1016/j.coph.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farooqi IS, et al. Leptin regulates striatal regions and human eating behavior. Science. 2007;317:1355. doi: 10.1126/science.1144599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stice E, Spoor S, Bohon C, Small DM. Relation between obesity and blunted striatal response to food is moderated by TaqIA A1 allele. Science. 2008;322:449–452. doi: 10.1126/science.1161550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noble EP. Addiction and its reward process through polymorphisms of the D2 dopamine receptor gene: a review. Eur Psychiatry. 2000;15:79–89. doi: 10.1016/s0924-9338(00)00208-x. [DOI] [PubMed] [Google Scholar]
- 6.Wang GJ, Volkow ND, Fowler JS. The role of dopamine in motivation for food in humans: implications for obesity. Expert Opin Ther Targets. 2002;6:601–609. doi: 10.1517/14728222.6.5.601. [DOI] [PubMed] [Google Scholar]
- 7.Booth ML, Wilkenfeld RL, Pagnini DL, Booth SL, King LA. Perceptions of adolescents on overweight and obesity: the weight of opinion study. J Paediatr Child Health. 2008;44:248–252. doi: 10.1111/j.1440-1754.2007.01267.x. [DOI] [PubMed] [Google Scholar]
- 8.Puhl RM, Moss-Racusin CA, Schwartz MB, Brownell KD. Weight stigmatization and bias reduction: perspectives of overweight and obese adults. Health Educ Res. 2008;23:347–358. doi: 10.1093/her/cym052. [DOI] [PubMed] [Google Scholar]
- 9.American Medical Association. Diagnostic and Statistical Manual of Mental Disorders. Fourth Edition (DSM-IV) 1994. [Google Scholar]
- 10.Markou A, Koob GF. Construct validity of a self-stimulation threshold paradigm: effects of reward and performance manipulations. Physiol Behav. 1992;51:111–119. doi: 10.1016/0031-9384(92)90211-j. [DOI] [PubMed] [Google Scholar]
- 11.Rolls BJ, Rowe EA, Turner RC. Persistent obesity in rats following a period of consumption of a mixed, high energy diet. J Physiol. 1980;298:415–427. doi: 10.1113/jphysiol.1980.sp013091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmed SH, Kenny PJ, Koob GF, Markou A. Neurobiological evidence for hedonic allostasis associated with escalating cocaine use. Nat Neurosci. 2002;5:625–626. doi: 10.1038/nn872. [DOI] [PubMed] [Google Scholar]
- 13.Markou A, Koob GF. Postcocaine anhedonia. An animal model of cocaine withdrawal. Neuropsychopharmacology. 1991;4:17–26. [PubMed] [Google Scholar]
- 14.Kenny PJ, Chen SA, Kitamura O, Markou A, Koob GF. Conditioned withdrawal drives heroin consumption and decreases reward sensitivity. J Neurosci. 2006;26:5894–5900. doi: 10.1523/JNEUROSCI.0740-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cottone P, Sabino V, Steardo L, Zorrilla EP. Opioid-dependent anticipatory negative contrast and binge-like eating in rats with limited access to highly preferred food. Neuropsychopharmacology. 2008;33:524–535. doi: 10.1038/sj.npp.1301430. [DOI] [PubMed] [Google Scholar]
- 16.Llado I, et al. Effects of cafeteria diet feeding on beta3-adrenoceptor expression and lipolytic activity in white adipose tissue of male and female rats. Int J Obes Relat Metab Disord. 2000;24:1396–1404. doi: 10.1038/sj.ijo.0801390. [DOI] [PubMed] [Google Scholar]
- 17.Fishburn CS, Elazar Z, Fuchs S. Differential glycosylation and intracellular trafficking for the long and short isoforms of the D2 dopamine receptor. J Biol Chem. 1995;270:29819–29824. doi: 10.1074/jbc.270.50.29819. [DOI] [PubMed] [Google Scholar]
- 18.Vanderschuren LJ, Everitt BJ. Drug seeking becomes compulsive after prolonged cocaine self-administration. Science. 2004;305:1017–1019. doi: 10.1126/science.1098975. [DOI] [PubMed] [Google Scholar]
- 19.Volkow ND, Wise RA. How can drug addiction help us understand obesity? Nat Neurosci. 2005;8:555–560. doi: 10.1038/nn1452. [DOI] [PubMed] [Google Scholar]
- 20.Blundell JE, Herberg LJ. Relative effects of nutritional deficit and deprivation period on rate of electrical self-stimulation of lateral hypothalamus. Nature. 1968;219:627–628. doi: 10.1038/219627a0. [DOI] [PubMed] [Google Scholar]
- 21.Hoebel BG, Teitelbaum P. Hypothalamic control of feeding and self-stimulation. Science. 1962;135:375–377. doi: 10.1126/science.135.3501.375. [DOI] [PubMed] [Google Scholar]
- 22.Mount G, Hoebel BG. Lateral hypothalamic self-stimulation: Self-determined threshold increased by food intake. Psychon Science. 1967;9:265–266. [Google Scholar]
- 23.Hoebel BG. Feeding and self-stimulation. Ann N Y Acad Sci. 1969;157:758–778. doi: 10.1111/j.1749-6632.1969.tb12919.x. [DOI] [PubMed] [Google Scholar]
- 24.Hoebel BG, Balagura S. Self-stimulation of the lateral hypothalamus modified by insulin and glucagon. Physiol Behav. 1967;2:337–340. [Google Scholar]
- 25.Hoebel BG, Thompson RD. Aversion to lateral hypothalamic stimulation caused by intragastric feeding or obesity. J Comp Physiol Psychol. 1969;68:536–543. doi: 10.1037/h0027651. [DOI] [PubMed] [Google Scholar]
- 26.Wilkinson HA, Peele TL. Modification of intracranial self-stimulation by hunger satiety. Am J Physiol. 1962;203:537–540. doi: 10.1152/ajplegacy.1962.203.3.537. [DOI] [PubMed] [Google Scholar]
- 27.Fulton S, Woodside B, Shizgal P. Modulation of brain reward circuitry by leptin. Science. 2000;287:125–128. doi: 10.1126/science.287.5450.125. [DOI] [PubMed] [Google Scholar]
- 28.Wang GJ, et al. Gastric distention activates satiety circuitry in the human brain. Neuroimage. 2008;39:1824–1831. doi: 10.1016/j.neuroimage.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 29.Batterham RL, et al. PYY modulation of cortical and hypothalamic brain areas predicts feeding behaviour in humans. Nature. 2007;450:106–109. doi: 10.1038/nature06212. [DOI] [PubMed] [Google Scholar]
- 30.Hommel JD, et al. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron. 2006;51:801–810. doi: 10.1016/j.neuron.2006.08.023. [DOI] [PubMed] [Google Scholar]
- 31.Fulton S, et al. Leptin regulation of the mesoaccumbens dopamine pathway. Neuron. 2006;51:811–822. doi: 10.1016/j.neuron.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 32.Kenny PJ. Brain reward systems and compulsive drug use. Trends Pharmacol Sci. 2007;28:135–141. doi: 10.1016/j.tips.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 33.Wang GJ, et al. Brain dopamine and obesity. Lancet. 2001;357:354–357. doi: 10.1016/s0140-6736(00)03643-6. [DOI] [PubMed] [Google Scholar]
- 34.Huang XF, et al. Dopamine transporter and D2 receptor binding densities in mice prone or resistant to chronic high fat diet-induced obesity. Behav Brain Res. 2006;175:415–419. doi: 10.1016/j.bbr.2006.08.034. [DOI] [PubMed] [Google Scholar]
- 35.Thanos PK, Michaelides M, Piyis YK, Wang GJ, Volkow ND. Food restriction markedly increases dopamine D2 receptor (D2R) in a rat model of obesity as assessed with in-vivo muPET imaging ([11C] raclopride) and in-vitro ([3H] spiperone) autoradiography. Synapse. 2008;62:50–61. doi: 10.1002/syn.20468. [DOI] [PubMed] [Google Scholar]
- 36.Frank GK, et al. Increased dopamine D2/D3 receptor binding after recovery from anorexia nervosa measured by positron emission tomography and [11c]raclopride. Biol Psychiatry. 2005;58:908–912. doi: 10.1016/j.biopsych.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 37.Neville MJ, Johnstone EC, Walton RT. Identification and characterization of ANKK1: a novel kinase gene closely linked to DRD2 on chromosome band 11q23.1. Hum Mutat. 2004;23:540–545. doi: 10.1002/humu.20039. [DOI] [PubMed] [Google Scholar]
- 38.Mastronardi CA, Yu WH, Srivastava VK, Dees WL, McCann SM. Lipopolysaccharide-induced leptin release is neurally controlled. Proc Natl Acad Sci U S A. 2001;98:14720–14725. doi: 10.1073/pnas.251543598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin HH, Knowlton BJ, Balleine BW. Inactivation of dorsolateral striatum enhances sensitivity to changes in the action-outcome contingency in instrumental conditioning. Behav Brain Res. 2006;166:189–196. doi: 10.1016/j.bbr.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 40.Klein TA, et al. Genetically determined differences in learning from errors. Science. 2007;318:1642–1645. doi: 10.1126/science.1145044. [DOI] [PubMed] [Google Scholar]
- 41.Teegarden SL, Bale TL. Decreases in dietary preference produce increased emotionality and risk for dietary relapse. Biol Psychiatry. 2007;61:1021–1029. doi: 10.1016/j.biopsych.2006.09.032. [DOI] [PubMed] [Google Scholar]
- 42.Volkow ND, et al. Low dopamine striatal D2 receptors are associated with prefrontal metabolism in obese subjects: possible contributing factors. Neuroimage. 2008;42:1537–1543. doi: 10.1016/j.neuroimage.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clarke HF, Dalley JW, Crofts HS, Robbins TW, Roberts AC. Cognitive inflexibility after prefrontal serotonin depletion. Science. 2004;304:878–880. doi: 10.1126/science.1094987. [DOI] [PubMed] [Google Scholar]
- 44.Avena NM, Rada P, Hoebel BG. Evidence for sugar addiction: behavioral and neurochemical effects of intermittent, excessive sugar intake. Neurosci Biobehav Rev. 2008;32:20–39. doi: 10.1016/j.neubiorev.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Volkow ND, O'Brien CP. Issues for DSM-V: should obesity be included as a brain disorder? Am J Psychiatry. 2007;164:708–710. doi: 10.1176/ajp.2007.164.5.708. [DOI] [PubMed] [Google Scholar]
- 46.Cottone P, et al. CRF system recruitment mediates dark side of compulsive eating. Proc Natl Acad Sci U S A. 2009;106:20016–20020. doi: 10.1073/pnas.0908789106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pellegrino LJ, Pellegrino AS, Cushman AJ. A Stereotaxic Atlas of the Rat Brain. New York: Plenum Press; 1979. [Google Scholar]
- 48.David C, Fishburn CS, Monsma FJ, Jr, Sibley DR, Fuchs S. Synthesis and processing of D2 dopamine receptors. Biochemistry. 1993;32:8179–8183. doi: 10.1021/bi00083a018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.