

Abstract
Aims/hypothesis
The single nucleotide polymorphism (SNP) rs9939609 in the fat mass and obesity associated gene (FTO) and the rs7566605 SNP located 10 kb upstream of the insulin-induced gene 2 gene (INSIG2) have been proposed as risk factors for common obesity.
Methods
We tested for genotype–treatment interactions on changes in obesity-related traits in the Diabetes Prevention Program (DPP). The DPP is a randomised controlled trial of 3,548 high-risk individuals from 27 participating centres throughout the USA who were originally randomised to receive metformin, troglitazone, intensive lifestyle modification or placebo to prevent the development of type 2 diabetes. Measures of adiposity from computed tomography were available in a subsample (n=908). This report focuses on the baseline and 1 year results.
Results
The minor A allele at FTO rs9939609 was positively associated with baseline BMI (p=0.003), but not with baseline adiposity or the change at 1 year in any anthropometric trait. For the INSIG2 rs7566605 genotype, the minor C allele was associated with more subcutaneous adiposity (second and third lumbar vertebrae [L2/3]) at baseline (p=0.04). During follow-up, CC homozygotes lost more weight than G allele carriers (p=0.009). In an additive model, we observed nominally significant gene–lifestyle interactions on weight change (p=0.02) and subcutaneous (L2/3 [p=0.01] and L4/5 [p=0.03]) and visceral (L2/3 [p=0.02]) adipose areas. No statistical evidence of association with physical activity energy expenditure or energy intake was observed for either genotype.
Conclusions/interpretation
Within the DPP study population, common variants in FTO and INSIG2 are nominally associated with quantitative measures of obesity, directly and possibly by interacting with metformin or lifestyle intervention.
Keywords: Adiposity, Diabetes Prevention Program, FTO, Gene–environment interaction, Genetic, INSIG2, Lifestyle, Metformin, Obesity, Randomised controlled trial, Troglitazone
Introduction
The most convincing findings to date for an obesity-related genotype have been for variants at the fat mass and obesity associated (FTO) gene. Frayling and coworkers reported a strong and robust association between a single nucleotide polymorphism (SNP) located within the first FTO intron (rs9939609) and BMI [1], which concurred with the results of two independent studies published almost simultaneously [2, 3]. In the initial studies of Chinese and Oceanic populations, no evidence of association between the previously reported FTO genotypes and obesity was observed [4, 5]. However, more recently, reports have emerged of associations between rs9939609 and obesity-related traits in people of Asian descent [6–9]. Elsewhere, Andreasen and coworkers reported evidence that rs9939609 interacts with physical activity, where the risk of obesity conveyed by the minor allele was completely offset in physically active individuals [10].
Herbert et al. undertook the initial genome-wide association study for obesity in the Framingham Heart Study [11]. In that study, the SNP rs7566605, located 10 kb upstream of the insulin-induced gene 2 (INSIG2), was the only one found to be associated with obesity at a genome-wide level of statistical significance; a recessive effect of the SNP rs7566605 was reproduced in nine independent cohorts comprising 9,187 participants (p=0.008). Individuals homozygous for the high-risk, minor C allele incurred a 1.22-fold increased risk of being obese compared with individuals without the putative risk allele. Although several subsequent confirmatory analyses were published [11], others found no evidence of association [12–16]. In 6,599 British whites [16], for example, a non-significant p value of 0.09 was obtained for a trend in a direction opposite to that reported by Herbert et al. [11], with C allele homozygotes appearing to be leaner than individuals carrying the G allele. In a recent meta-analysis of 16,969 individuals, the summary effects of rs7566605 genotype on BMI and obesity (as a dichotomous trait) were calculated [17]. The authors reported that, although the allelic effects across studies were heterogeneous, CC homozygotes had a marginally higher BMI than G allele carriers, an effect that was nominally statistically significant (p=0.046) [17].
In the present report from the Diabetes Prevention Program (DPP), we tested the hypotheses that the SNPs rs9939609 and rs7566605 are associated with obesity at baseline and modify the effects of the DPP interventions on changes in weight, adipose distribution and energy balance behaviours at 1 year. Specifically, we hypothesised that the effects of the metformin and lifestyle interventions on weight loss are dependent on the FTO and INSIG2 genes, and that variants at these loci, which have previously been associated with obesity traits, would modify the effects of the interventions.
Methods
The DPP
The DPP is a multicentre randomised clinical trial in which the effects of metformin, troglitazone or an intensive lifestyle intervention on the incidence of type 2 diabetes were assessed, as described in detail previously [18, 19]. Briefly, non-diabetic overweight persons (n=3,234) with raised fasting and post-challenge glucose levels were randomised to receive placebo, metformin (850 mg twice daily) or a programme of intensive lifestyle modification (aimed at ~7% weight loss and ~150 min of physical activity per week); a fourth arm of individuals (n=585) assigned to receive troglitazone (400 mg daily) was terminated 2 years after the trial commenced because of hepatotoxicity [20]. The principal endpoint in the DPP was the development of diabetes by confirmed OGTT. Other phenotypes, such as changes in weight, waist circumference, abdominal adipose tissue distribution, lipids, insulin and glucose, were also collected. The primary focus of this report is the obesity-related traits; the secondary focus is diabetes incidence. Written, informed consent was obtained from each participant, and each of the 27 DPP centres obtained approval from their respective institutional review boards prior to initiation of the study protocol.
Participants
Consent for genetic analyses was obtained for 93.3% (n=3,356/3,597) of the participants who were originally assigned to a treatment arm in the DPP and for whom data at baseline and 1 year were available. Of the participants studied here, 56.1% were white, 20.4% were African American, 16.7% were Hispanic, 4.4% were Asian/Pacific Islander and 2.5% were American Indian by self-report. Concordant with the entire DPP, the participants' mean age at enrolment was 51 years and mean BMI was 34.1 kg/m2.
Quantification of energy intake, energy expenditure and body composition
Physical activity (leisure time and occupational) was assessed at baseline and 1 year using the validated Modifiable Activity Questionnaire (MAQ) [21–23]. The MAQ asks questions about activities common within the DPP cohort, and the responses were subsequently converted into metabolic equivalent (MET) h/week, which we use in this report as a surrogate marker of physical activity energy expenditure (PAEE).
Diet information was collected by face-to-face interviews at baseline and 1 year later using the semi-quantitative DPP food frequency questionnaire [24, 25].
Body composition measurements included height, weight, waist circumference and, in a subgroup, abdominal computed tomography (CT) quantification of subcutaneous and visceral fat areas. The CT instruments used included the GE High Speed Advantage (GE Healthcare, Wauwatosa, WI, USA) (five centres), the Picker PQ 5000 (TransAmerican Imaging, North Lindon, UT, USA) (five centres), the Siemens and Siemens Somatom Plus (Siemens Health Care, Malvern, PA, USA) (two centres), the GE 9800 (GE Healthcare) (three centres), and the GE Highlite (GE Healthcare) (two centres). Two 10 mm thick axial images were obtained at the second and third lumbar vertebrae (L2/3) and L4/5 disc spaces, and were used to define the different adipose compartments and areas by trained staff at a central reading facility (University of Colorado, Denver, CO, USA), as described previously [26].
Genotyping
Genotyping was performed by allele-specific primer extension of multiplex amplified products and detection with matrix-assisted laser desorption/ionisation time-of-flight mass spectroscopy on a Sequenom iPLEX platform (Sequenom, San Diego, CA, USA). The genotyping success rates for the SNPs rs9939609 and rs7566605 were 97.3% and 99.6%, respectively. The allele frequencies at both SNPs in each of the five ethnic groups were in Hardy–Weinberg equilibrium (p>0.2).
Statistical power
We estimated the power of the present study at baseline to detect previously reported effect sizes for BMI using Quanta software (version 1.2.3) [27]. For the FTO rs9939609 genotype, assuming an additive mode of inheritance (TT vs TA vs AA), our study was powered at 85% (p<0.05) to detect the previously reported effect of 0.36 kg/m2 per allele [1]. For the INSIG2 rs7566605 genotype, assuming a recessive mode of inheritance (GG+GC vs CC), our study was powered at 70% (p<0.05) to detect the previously reported effect of 0.69 kg/m2 [17]. We also calculated the power available within the DPP cohort to detect the interaction effect sizes reported here (Electronic supplementary material [ESM] Table 1).
Table 1.
Baseline characteristics of the entire DPP cohort stratified by FTO rs9939609 genotype
| Baseline characteristic | TT (n=1,235) | TA (n= 1,623) | AA (n=593) | p value |
|---|---|---|---|---|
| Men | 420 (36.5) | 528 (45.9) | 202 (17.6) | 0.65a |
| Age (years) | 50.4±10.5 | 51.0±10.4 | 51.1±11.3 | 0.25 |
| Weight (kg) | 93.2±20.0 | 94.3±19.4 | 98.1 ±21.4 | <0.001 |
| BMI (kg/m2) | 33.7±6.6 | 33.9±6.5 | 35.1±7.2 | <0.001 |
| Waist circumference (cm) | 104±14.3 | 105±14.1 | 107±15.5 | <0.001 |
| LTPA (MET h/week) | 15.9±20.5 | 16.4±29.2 | 15.2±19.9 | 0.62b |
| Energy intake (kJ) | 9,010 ±4,61 8 | 8,918±4,320 | 8,700±3,852 | 0.3 7b |
| Treatment | ||||
| Placebo | 357 (37.0) | 434 (44.9) | 175 (18.1) | 0.08a |
| Metformin | 354 (36.7) | 447 (46.3) | 164 (17.0) | |
| Lifestyle | 337 (34.6) | 456 (46.9) | 180 (18.5) | |
| Troglitazone | 187 (34.2) | 286 (52.3) | 74 (13.5) | |
| Self-reported ethnicity | ||||
| European American | 621 (31.8) | 952 (48.8) | 378 (19.4) | <0.001c |
| African American | 193 (27.9) | 345 (49.9) | 154 (22.3) | |
| Hispanic | 279 (48.4) | 246 (42.6) | 52 (9.0) | |
| Asian/Pacific Islander | 86 (57.7) | 58 (38.9) | 5 (3.4) | |
| American Indian | 56 (68.3) | 22 (26.8) | 4 (4.9) |
Data are n (%) or mean±SD
The p values are from F tests
χ2 tests
the Wilcoxon test
Fisher's exact test
Genotype percentages represent the proportion of each genotypic category within each treatment arm or ethnic group
MET, metabolic equivalent
Statistical analysis
As a general principle of the DPP genetics project, statistical analyses are planned a priori and the decision to undertake specific tests and to report the results from these tests is made before embarking upon statistical analyses. The primary endpoints were changes in obesity-related traits (i.e. weight, waist circumference or fat mass) and changes in energy balance behaviours (i.e. leisure time physical activity [LTPA] or energy intake) at 1 year after randomisation. A secondary endpoint was diabetes incidence at study end, which was assessed using Cox proportional hazards models. LTPA and energy intake were logarithmically transformed owing to skewed distributions. All other traits were normally distributed and so no transformations were applied. All residuals from the general linear models were normally distributed. All genotype-by-treatment group interaction models were tested separately by intervention arm; we used analysis of covariance with genotype, intervention and genotype by intervention interactions as the independent variables predicting the change in weight, waist circumference or adiposity. Interactions with troglitazone were also assessed, although owing to insufficient numbers, these were confined to analyses in which weight change was the outcome variable of interest. To determine whether it was appropriate to combine data from different ethnic groups, we began by testing interaction terms for genotype by ethnicity. If these tests were statistically significant (p<0.05), we progressed by undertaking adjusted ethnicity-stratified analyses. In all other cases, data were pooled and results are adjusted for age, sex, ethnicity and the baseline value for the respective obesity trait. When no significant genotype by treatment group interaction was evident, the statistical models were adjusted for treatment group. Differences between means were tested using pairwise contrasts. Additive genetic models were analysed for both SNPs. For the INSIG2 rs7566605 SNP, we also analysed recessive genetic models, since both additive and recessive models have been reported in the literature. For general linear models using genotype as a class variable, nominal two-sided p values are reported, adjusted for multiple comparisons (unless otherwise stated, three genotypic groups within each trait: AA vs Aa, AA vs aa, Aa vs aa) using the Holm procedure [28]. Experiment-wide adjustments for multiple hypothesis testing was not undertaken. This is because previous studies have reported associations with obesity traits at the loci of interest [1–3, 11, 17]. As a consequence, the prior probability for association is higher than when testing de novo hypotheses.
Results
Participant characteristics
Participant characteristics for the entire DPP cohort, stratified by the rs9939609 and rs7566605 genotypes, are reported in Tables 1 and 2, respectively. Tables 3 and 4 report the characteristics of the subgroup of DPP participants for whom CT scan data were available, stratified by the rs9939609 and rs7566605 genotypes, respectively. Table 5 reports Pearson correlation coefficients for each of the measures of adiposity in the CT scan subgroup. Allele frequencies at both loci differed significantly by ethnicity.
Table 2.
Baseline characteristics of the entire DPP cohort stratified by INSIG2 rs7566605 genotype
| Baseline characteristic | GG (n= 1,722) | GC (n= 1,469) | CC (n=342) | p value |
|---|---|---|---|---|
| Men | 546 (46.7) | 502 (42.9) | 121 (10.4) | 0.21a |
| Age (years) | 50.8±10.6 | 50.6±10.4 | 51.3±10.9 | 0.54 |
| Weight (kg) | 94.3±20.1 | 94.5±19.7 | 95.1 ±21.0 | 0.8 |
| BMI (kg/m2) | 34.0±6.8 | 33.9±6.5 | 34.2±6.7 | 0.76 |
| Waist circumference (cm) | 105±14.6 | 105±14.3 | 106±14.7 | 0.59 |
| LTPA (MET h/week) | 16.3±28.8 | 15.7±19.9 | 15.3±20.3 | 0.70b |
| Energy intake (kj) | 8,981 ±4,463 | 8,901 ±4,325 | 8,579±3,710 | 0.30b |
| Treatment | ||||
| Placebo | 500 (50.1) | 406 (40.6) | 93 (9.3) | 0.89a |
| Metformin | 470 (47.7) | 414 (42.0) | 101 (10.3) | |
| Lifestyle | 478 (47.8) | 421 (42.1) | 100 (10.0) | |
| Troglitazone | 274 (49.8) | 228 (41.5) | 48 (8.7) | |
| Self-reported ethnicity | ||||
| European American | 895 (44.9) | 875 (43.9) | 224 (11.2) | <0.001c |
| African American | 421 (59.0) | 254 (35.6) | 38 (5.3) | |
| Hispanic | 293 (49.6) | 245 (41.5) | 53 (9.0) | |
| Asian/Pacific Islander | 60 (40.0) | 67 (44.7) | 23 (15.3) | |
| American Indian | 53 (62.4) | 28 (32.9) | 4 (4.7) |
Data are n (%) or mean±SD
The p values are from F tests
χ2 tests
the Wilcoxon test
Fisher's exact test
Genotype percentages represent the proportion of each genotypic category within each treatment arm or ethnic group
MET, metabolic equivalent
Table 3.
Baseline characteristics of the DPP CT scan subgroup (n=869) stratified by FTO rs9939609 genotype
| Baseline characteristic | TT (n=315) | TA (n=398) | AA (n=156) | p value |
|---|---|---|---|---|
| Men | 111 (37.6) | 127 (43.1) | 57 (19.3) | 0.49a |
| Age (years) | 51.3±10.4 | 51.9±10.9 | 52.3 ±10.5 | 0.59 |
| Weight (kg) | 91.8±17.9 | 91.9±17.4 | 95.7±18.2 | 0.05 |
| BMI (kg/m2) | 32.9±5.7 | 32.9±5.8 | 34.2±6.1 | 0.04 |
| Waist circumference (cm) | 103±13.2 | 104±13.0 | 105±13.6 | 0.14 |
| SAT L2/L3 (cm2) | 303±126 | 310±124 | 316±138 | 0.54 |
| SAT L4/L5 (cm2) | 438±153 | 442±148 | 451±163 | 0.69 |
| VAT L2/L3 (cm2) | 198±86.2 | 195±85.1 | 208±90.7 | 0.29 |
| VAT L4/L5 (cm2) | 158±63.2 | 161 ±66.4 | 164±63.6 | 0.56 0.72b |
| LTPA (MET h/week) | 17.4±24.8 | 18.0±40.9 | 15.5 ±22.0 | |
| Energy intake (kj) | 8,889±4,534 | 8,796±3,881 | 7,796±2,826 | 0.01b |
| Treatment | ||||
| Placebo | 109 (38.7) | 126 (44.7) | 47 (16.7) | 0.75a |
| Metformin | 106 (36.1) | 131 (44.6) | 57 (19.4) | |
| Lifestyle | 100 (34.1) | 141 (48.1) | 52 (17.7) | |
| Self-reported ethnicity | ||||
| European American | 166 (33.1) | 238 (47.4) | 98 (19.5) | <0.001c |
| African American | 57 (31.5) | 86 (47.5) | 38 (21.0) | |
| Hispanic | 67 (45.3) | 62 (41.9) | 19 (12.8) | |
| Asian/Pacific Islander | 25 (65.8) | 12 (31.6) | 1 (2.6) |
Data are n (%) or mean±SD
The p values are from F tests
χ tests
the Wilcoxon test
Fisher's exact test
Genotype percentages represent the proportion of each genotypic category within each treatment arm or ethnic group
MET, metabolic equivalent
Table 4.
Baseline characteristics of the DPP CT scan subgroup (n=908) stratified by INSIG2 rs7566605 genotype
| Baseline characteristic | GG (n=438) | GC (n=375) | CC (n=95) | p value |
|---|---|---|---|---|
| Men | 139 (45.6) | 131 (43.0) | 35 (19.3) | 0.49a |
| Age (years) | 51.9±10.8 | 51.3±10.3 | 53.0±11.1 | 0.35 |
| Weight (kg) | 92.6±18.2 | 91.8±17.4 | 94.7±17.5 | 0.37 |
| BMI (kg/m2) | 33.3±6.1 | 32.8±5.6 | 33.7±5.6 | 0.31 |
| Waist circumference (cm) | 104±13.5 | 103±12.6 | 106±13.8 | 0.15 |
| SAT L2/L3 (cm2) | 313±130 | 297±125 | 324± 119 | 0.09 |
| SAT L4/L5 (cm2) | 448±156 | 431±150 | 457±141 | 0.16 |
| VAT L2/L3 (cm2) | 197±85.8 | 198±86.0 | 203 ±90.1 | 0.81 |
| VAT L4/L5 (cm2) | 159±64.4 | 160±66.5 | 160±62.8 | 0.98 |
| LTPA (MET h/week) | 16.4±37.3 | 17.4±26.2 | 19.2±28.1 | 0.72b |
| Energy intake (kj) | 8,692±4,053 | 8,725±4,120 | 8,068±2,981 | 0.33b |
| Treatment | ||||
| Placebo | 143 (47.8) | 130 (43.5) | 26 (16.7) | 0.47a |
| Metformin | 153 (50.2) | 122 (40.0) | 30 (19.4) | |
| Lifestyle | 142 (46.7) | 123 (40.5) | 39 (17.7) | |
| Self-reported ethnicity | ||||
| European American | 237 (45.1) | 226 (43.0) | 62 (11.8) | 0.02c |
| African American | 113 (58.5) | 66 (34.2) | 14 (7.3) | |
| Hispanic | 76 (50.0) | 62 (40.8) | 14 (9.2) | |
| Asian/Pacific Islander | 12 (31.6) | 21 (55.3) | 5 (13.2) |
Data are n (%) or mean±SD
The p values are from F tests
χ2 tests
the Wilcoxon test
Fisher's exact test
Genotype percentages represent the proportion of each genotypic category within each treatment arm or ethnic group
MET, metabolic equivalent
Table 5.
Pairwise correlations (r) between measures of obesity in the DPP CT scan subgroup (n=908)
| Measure of obesity | SAT L4/5 (cm2) | VAT L2/3 (cm2) | VAT L4/5 (cm2) | Waist circumference (cm) | Weight (kg) | BMI (kg/m2) |
|---|---|---|---|---|---|---|
| SAT L2/3 (cm2) | 0.89b | 0.13b | 0.22b | 0.65b | 0.64b | 0.80b |
| SAT L4/5 (cm2) | 0.07a | 0.16b | 0.54b | 0.58b | 0.79b | |
| VAT L2/3 (cm2) | 0.79b | 0.68b | 0.58b | 0.39b | ||
| VAT L4/5 (cm2) | 0.63b | 0.49b | 0.41b | |||
| Waist circumference (cm) | 0.88b | 0.76b | ||||
| Weight (kg) | 0.82b |
Data are Pearson correlation coefficients
p<0.05
p<0.0001
Results for the FTO SNP (rs9939609)
Baseline associations of rs9939609
In baseline analyses adjusted for age, sex and ethnicity, and consistent with prior reports [1–3], the minor A allele at rs9939609 was associated with heavier weight (p=0.03) and higher BMI (p=0.003). The association with waist circumference was not statistically significant (p=0.06) (see Table 1 for unadjusted results).
As indicated in Table 3, the allele frequencies for the subgroup in which abdominal adiposity had been assessed (n=869) were similar to those in the cohort as a whole. The genotypexethnicity interaction term was statistically significant for subcutaneous adipose tissue at L2/3 (p=0.044). However, none of the tests of association between the rs9939609 genotype and this slice site was statistically significant when performed within ethnic group. No genotype associations were observed for area of subcutaneous or visceral adiposity at either slice site (L2/3 or L4/5, see Table 3) or for PAEE or energy intake.
FTO rs9939609 genotype–treatment interactions
We tested whether the rs9939609 genotype modifies the effects of treatment on the change in obesity-related traits at 1 year. In these models, it appeared that the minor allele was associated with a greater increase in subcutaneous adipose tissue (L2/3) in the placebo group but not in the metformin and lifestyle groups (see Fig. 1) (p=0.06 for interaction with metformin, p=0.05 for interaction with lifestyle).
Fig. 1.
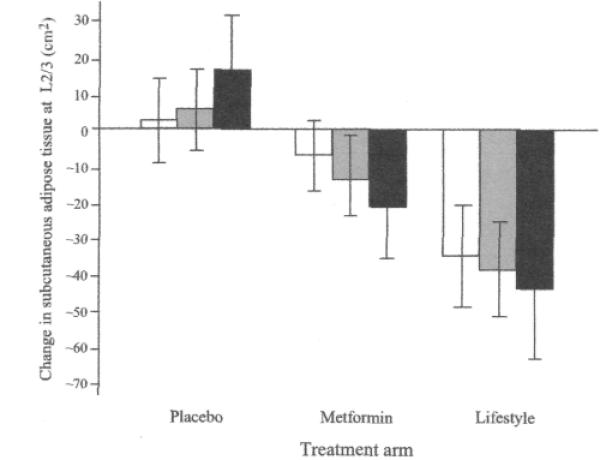
Interaction between the FTO rs9939609 genotype and metformin (p=0.06) or lifestyle intervention (p=0.05) on change in subcutaneous adipose tissue area at L2/3 during the first year after randomisation in the DPP (n=869). Black bars, major allele homozygotes (TT); grey bars, heterozygotes (TA); white bars, minor allele homozygotes (AA). The p values for main effects within treatment arms are: placebo, 0.18; metformin, 0.18; lifestyle, 0.75. Data are means and 95% CIs adjusted for age, sex and ethnicity
Results for the INSIG2 SNP (rs7566605)
Baseline associations for rs 7566605
In Table 2, the unadjusted means for weight, waist circumference and BMI are shown stratified by rs7566605 genotype for the entire DPP cohort. In Table 4, these obesity indices and mean values for subcutaneous and visceral adipose areas are shown for the subgroup of individuals for whom CT assessments of adipose area were available (n=908). As indicated in Table 4, the allele frequencies for those with abdominal fat area assessed using CT (n=908) were similar to those in the whole DPP cohort.
Following adjustment for age, sex and ethnicity, there was no evidence of association between the rs75 66605 genotypes and baseline weight, waist circumference or BMI. However, the minor CC genotype was nominally associated with more subcutaneous adipose tissue at L2/3 compared with the other genotypes (mean difference 26.0 cm2, 95% CI 1.2–50.9 cm2, p=0.04). No significant genotype associations with visceral adiposity, PAEE or energy intake were observed.
INSIG2 rs 7566605 genotype–treatment interactions
As shown in Fig. 2, in an additive model, the test of interaction between the rs7566605 genotype and lifestyle intervention (vs placebo) was nominally statistically significant for change in weight (p=0.02); no statistical association between genotype and weight loss was observed in the placebo group, but in the lifestyle intervention group, CC homozygotes had lost, on average, 1.7 kg (95% CI −3.3 to −0.2 kg, p=O.Q2) more weight at 1 year after randomisation than G allele carriers.
Fig. 2.
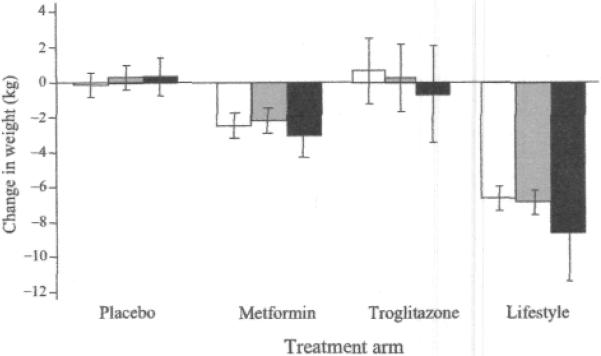
Interaction between the rs7566605 genotype located upstream of the INSIG2 gene and lifestyle intervention on change in weight (p=0.02) during the first year of the DPP (n=3,556). Black bars, major allele homozygotes (GG); grey bars, heterozygotes (GC); white bars, minor allele homozygotes (CC). The p values for main effects within treatment arms are: placebo, 0.50; metformin 0.17; troglitazone, 0.51; lifestyle, 0.06. Data are means and 95% CIs adjusted for age, sex and ethnicity
We observed nominally significant gene-lifestyle interactions on subcutaneous adipose area at L2/3 (p=0.01) (Fig. 3a) and L4/5 (p=0.03) (Fig. 3b), and for visceral adipose area at L2/3 (p=0.02) (Fig. 3c); in the placebo group, subcutaneous and visceral adipose areas increased irrespective of genotype, whereas with lifestyle intervention, a reduction in subcutaneous and visceral adipose areas tended to occur, which was greatest for C allele homozygotes.
Fig. 3.

a–d Interaction between the rs7566605 genotype located upstream of the INSIG2 gene and treatment on changes in subcutaneous adiposity during the first year of the Diabetes Prevention Program (n=725). Black bars are major allele homozygotes (GG), grey bars are heterozygotes (GC), and white bars are minor allele homozygotes (CC). Nominally significant genotype–lifestyle interactions were observed under an additive genetic model on change in subcutaneous adipose tissue area at L2/3 (a) (p=0.01) and L4/5 (b) p=0,03), and visceral adipose area at L2/3 (c) (p=0.02). The interaction on visceral adipose tissue at L4/5 (d) was not statistically significant. The p values for main effects within treatment arms by figure panel (a–d) are: placebo, 0.70, 0.82, 0.76, 0.97; metformin, 0.59, 0.049, 0.06, 0.95; lifestyle, 0.11, 0.14, 0.20, 0.24. Data are means and 95% CIs adjusted for age, sex and ethnicity
FTO (rs9939609) and INSIG2 (rs7566605) genotypes and the incidence of diabetes
In adjusted Cox proportional hazards models, we observed no evidence of association with incident diabetes in additive models for either rs9939609 (HR 1.05, 95% CI 0.94–1.18) or rs7566605 (HR 1.0, 95% CI 0.76–1.31) genotypes.
Discussion
We tested whether genotypes at the previously associated SNPs rs9939609 in FTO and rs7566605 upstream of INSIG2 modify the effects of treatment with metformin, troglitazone or intensive lifestyle modification on obesity-related traits in individuals at high risk of type 2 diabetes. We observed strong baseline associations with anthropometric measures of obesity for FTO rs9939609 and nominally significant differences in baseline levels of subcutaneous adipose tissue for 1NSIG2 rs7566605. For INSIG2 rs7566605, we observed nominal evidence of statistical interactions between genotype and treatment groups on change in abdominal adipose area during the first year of follow-up; the common G allele was associated with a lesser reduction in subcutaneous and visceral adipose areas following treatment with lifestyle intervention, whereas change in adipose area in those assigned to the placebo group was similar across genotypes.
The mechanisms that underpin the associations between the FTO and INSIG2 variants and obesity remain undefined. Weight gain can result from defects in molecular pathways influencing behavioural drive (e.g. centrally mediated energy intake, dietary nutrient preference [29, 30] and the tendency to be physically active [31]) and from cellular mechanisms that lead to variations in dietary-induced thermogenesis or basal energy expenditure [32]. The amino acid sequence of the protein encoded by FTO is highly conserved among vertebrates and is present in non-vertebrates [33]. In vitro murine studies show that Fto is highly expressed throughout the hypothalamus, particularly in the arcuate nucleus [33, 34], the feeding centre of the brain. Thus, it seems probable that FTO has played an important role in the regulation of food intake throughout the evolution of several diverse species. The protein product of INSIG2 is believed to be primarily involved in the control of cholesterol and fatty acid metabolism via regulation of sterol regulatory element binding transcription factors (SREBFs) [35]. SREBFs regulate the proteolysis and transport of specific membrane proteins involved in lipid synthesis and metabolism [36]. The mechanisms through which INSIG2 influences obesity are yet to be established, although some have speculated that the gene may regulate rates of adipogenesis via SREBF1 (also known as SREBP1) [37].
For both the FTO and INSIG2 SNPs studied here, we were unable to detect statistically significant associations with any of the available behavioural measures of energy balance. This may be because our measures of energy balance were derived using questionnaires, and their imprecision limits the ability to detect genetic associations with these traits. Consequently, it is possible that an allelic association with these phenotypes exists but was undetectable in this study owing to insufficient statistical power. Alternatively, the genetic effects may influence basal energy metabolism or dietary thermogenesis, for which measurements are lacking in the DPP, and play no role in feeding or physical activity per se.
Two recent reports emanating from observational epidemiological cohorts suggest that physical activity may interact with FTO variants to attenuate the risk of obesity [10, 38]. We found tentative support for an interaction between the FTO variant and lifestyle intervention on change in adipose area. However, because our lifestyle intervention combined physical activity, dietary modification and weight loss, our study does not provide direct confirmation of those previous studies. In a report of a lifestyle intervention study of 293 obese German children [39], the body mass (expressed as age- and sex-standardised BM units) of CC homozygotes at INSIG2 rs7566605 decreased to a lesser extent following lifestyle intervention than that of G allele carriers; this observation is in the opposite direction to our current result in adult DPP participants. There are various plausible explanations for the disparate findings which include the possibility that the findings of one or both studies are false-positives. An alternative explanation is that interactions with other unobserved factors influence the direction of genetic effects.
DPP participants were specifically recruited because they were at high risk of type 2 diabetes. It is possible that the specific nature of this cohort may hinder the detection of genetic effects and inhibit the generalisability of findings from the DPP to cohorts at lower risk. Nonetheless, we were able to detect the effect of the previously associated FTO variant on baseline levels of obesity, illustrating that the data for the DPP population can be used to study obesity genetics. A further possible limitation of our study is that the ethnic diversity of the DPP cohort could have reduced the ability to detect genetic associations if the markers we have focused on are tagging polymorphisms in some but not all of the DPP ethnic groups. In this scenario the effect estimates and corresponding probability statistics could be diluted. However, we found no statistical evidence to suggest that this was the case in the present analyses. For example, the effects for the models that were significant in the overall cohort are consistent in direction when stratified by the three main ethnic groups in this study (European Americans, African Americans and Hispanics). The association between the FTO SNP and baseline BMI was somewhat stronger in magnitude in European Americans than in Hispanics or African Americans, whereas the association between the INSIG2 SNP and change in weight over the first year after randomisation was marginally stronger in Hispanics than in European Americans or African Americans. However, none of these associations differed statistically between ethnic group.
In conclusion, we were able to confirm the previous reports that the minor allele at the FTO rs9939609 locus is associated with measures of obesity. We also found modest evidence of association and interaction with the lifestyle intervention for the INSIG2 rs7566605 genotype. This latter observation may help explain why some of the attempts to replicate the association between INSIG2 and obesity, initially reported in the Framingham Heart Study, in cohorts that may have different lifestyle behaviours have yielded variable results. It is important to stress, however, that our observations are from a setting of multiple hypothesis testing, and most results only reach a nominal level of statistical significance. Thus, one should remain cognisant of this limitation when interpreting our findings of gene–treatment interactions at the FTO and INSIG2 loci.
Acknowledgements
The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the National Institutes of Health (NIH) provided funding to the clinical centres and the coordinating centre for the design and conduct of the study and the collection, management, analysis and interpretation of the data. The Southwestern American Indian Centres were supported directly by the NIDDK and the Indian Health Service. The General Clinical Research Center Program of the National Center for Research Resources supported data collection at many of the clinical centres. Funding for data collection and participant support was also provided by the Office of Research on Minority Health, the National Institute of Child Health and Human Development, the National Institute on Aging, the Centers for Disease Control and Prevention, Office of Research on Women's Health, and the American Diabetes Association. Bristol-Myers Squibb and Parke-Davis provided medication. This research was also supported, in part, by the intramural research programme of the NIDDK. LifeScan, Health O Meter, Hoechst Marion Roussel, Merck-Medco Managed Care, Merck, Nike Sports Marketing, Slim Fast Foods and Quaker Oats donated materials, equipment or medicines for concomitant conditions. McKesson BioServices, Matthews Media Group, and the Henry M. Jackson Foundation provided support services under subcontract with the coordinating centre. The opinions expressed are those of the investigators and do not necessarily reflect the views of the Indian Health Service or other funding agencies. A complete list of centres, investigators and staff can be found in the ESM. The investigators gratefully acknowledge the commitment and dedication of all participants in the DPP, without whom this work would not have been possible. This work was funded by grant no. R01 DK072041-02 to D. Altshuler and J. C. Florez (P. W. Franks and W. C. Knowler are co-investigators). P. W. Franks was supported by grants from Västerbotten's Health Authority (ALF strategic appointment 2006–2009), Novo Nordisk (370579201), the Swedish Heart-Lung Foundation (20070633) and the Swedish Diabetes Association (DIA2006-013). J. C. Florez is supported by NIH Research Career Award K23 DK65978-04. S. E. Kahn is supported in part by the Department of Veterans Affairs.
Funding: The study was funded by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), the National Institute on Aging (NIA), the National Institute of Child Health and Human Development (NICHD), the National Center on Minority Health and Health Disparities (NCMHD) and the Office of Research on Women's Health (ORWH).
Abbreviations
- CT
computed tomography
- DPP
Diabetes Prevention Program
- L
lumbar vertebra
- LTPA
leisure time physical activity
- PAEE
physical activity energy expenditure
- SNP
single nucleotide polymorphism
- SREBF
sterol regulatory element binding transcription factor
Footnotes
Trial registration: ClinicalTrials.gov NCT00004992
Electronic supplementary material The online version of this article (doi:10.1007/s00125-008-1158-x) contains supplementary material, including a list of members of the Diabetes Prevention Program Research Group, which is available to authorised users.
Duality of interest J. C. Florez has received consulting honoraria from Merck and from Publicis Healthcare Communications Group, a global advertising agency engaged by Amylin Pharmaceuticals. No other potential conflict of interest is declared by any author.
References
- 1.Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dina C, Meyre D, Gallina S, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39:724–726. doi: 10.1038/ng2048. [DOI] [PubMed] [Google Scholar]
- 3.Scott LJ, Mohlke KL, Bonnycastle LL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li H, Wu Y, Loos RJ, et al. Variants in the fat mass- and obesity-associated (FTO) gene are not associated with obesity in a Chinese Han population. Diabetes. 2008;57:264–268. doi: 10.2337/db07-1130. [DOI] [PubMed] [Google Scholar]
- 5.Ohashi J, Naka I, Kimura R, et al. FTO polymorphisms in oceanic populations. J Hum Genet. 2007;52:1031–1035. doi: 10.1007/s10038-007-0198-2. [DOI] [PubMed] [Google Scholar]
- 6.Al-Attar SA, Pollex RL, Ban MR, et al. Association between the FTO rs9939609 polymorphism and the metabolic syndrome in a non-Caucasian multi-ethnic sample. Cardiovasc Diabetol. 2008;7:5. doi: 10.1186/1475-2840-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chu X, Erdman R, Susek M, et al. Association of morbid obesity with FTO and INSIG2 allelic variants. Arch Surg. 2008;143:235–240. doi: 10.1001/archsurg.2007.77. [DOI] [PubMed] [Google Scholar]
- 8.Hotta K, Nakata Y, Matsuo T, et al. Variations in the FTO gene are associated with severe obesity in the Japanese. J Hum Genet. 2008;53:546–553. doi: 10.1007/s10038-008-0283-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marvelle AF, Lange LA, Qin L, Adair LS, Mohlke KL. Association of FTO with obesity-related traits in the Cebu Longitudinal Health and Nutrition Survey (CLHNS) cohort. Diabetes. 2008;57:1987–1991. doi: 10.2337/db07-1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andreasen CH, Stender-Petersen KL, Mogensen MS, et al. Low physical activity accentuates the effect of the FTO rs9939609 polymorphism on body fat accumulation. Diabetes. 2008;57:95–101. doi: 10.2337/db07-0910. [DOI] [PubMed] [Google Scholar]
- 11.Herbert A, Gerry NP, McQueen MB, et al. A common genetic variant is associated with adult and childhood obesity. Science. 2006;312:279–283. doi: 10.1126/science.1124779. [DOI] [PubMed] [Google Scholar]
- 12.Dina C, Meyre D, Samson C, et al. A common genetic variant is associated with adult and childhood obesity. Science. 2007;315:187. doi: 10.1126/science.1129402. [DOI] [PubMed] [Google Scholar]
- 13.Kumar J, Sunkishala RR, Karthikeyan G, Sengupta S. The common genetic variant upstream of INSIG2 gene is not associated with obesity in Indian population. Clin Genet. 2007;71:415–418. doi: 10.1111/j.1399-0004.2007.00795.x. [DOI] [PubMed] [Google Scholar]
- 14.Rosskopf D, Bornhorst A, Rimmbach C, et al. A common genetic variant is associated with adult and childhood obesity. Science. 2007;315:187. doi: 10.1126/science.1130571. [DOI] [PubMed] [Google Scholar]
- 15.Skelly T, Pinheiro AP, Lange LA, Sullivan PF. Is rs7566605, a SNP near INSIG2, associated with body mass in a randomized clinical trial of antipsychotics in schizophrenia. Mol Psychiatry. 2007;12:321–322. doi: 10.1038/sj.mp.4001956. [DOI] [PubMed] [Google Scholar]
- 16.Loos RJ, Barroso I, O'Rahilly S, Wareham NJ. A common genetic variant is associated with adult and childhood obesity. Science. 2007;315:187. doi: 10.1126/science.1130012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lyon HN, Emilsson V, Hinney A, et al. The association of a SNP upstream of INSIG2 with body mass index is reproduced in several but not all cohorts. PLoS Genet. 2007;3:e61. doi: 10.1371/journal.pgen.0030061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The Diabetes Prevention Program Research Group Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The Diabetes Prevention Program Research Group The Diabetes Prevention Program: recruitment methods and results. Control Clin Trials. 2002;23:157–171. doi: 10.1016/s0197-2456(01)00184-2. [DOI] [PubMed] [Google Scholar]
- 20.Knowler WC, Hamman RF, Edelstein SL, et al. Prevention of type 2 diabetes with troglitazone in the Diabetes Prevention Program. Diabetes. 2005;54:1150–1156. doi: 10.2337/diabetes.54.4.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aaron DJ, Kriska AM, Dearwater SR, Cauley JA, Metz KF, LaPorte RE. Reproducibility and validity of an epidemiologic questionnaire to assess past year physical activity in adolescents. Am J Epidemiol. 1995;142:191–201. doi: 10.1093/oxfordjournals.aje.a117618. [DOI] [PubMed] [Google Scholar]
- 22.Kriska AM, Knowler WC, LaPorte RE, et al. Development of questionnaire to examine relationship of physical activity and diabetes in Pima Indians. Diabetes Care. 1990;13:401–411. doi: 10.2337/diacare.13.4.401. [DOI] [PubMed] [Google Scholar]
- 23.Schulz LO, Harper IT, Smith CJ, Kriska AM, Ravussin E. Energy intake and physical activity in Pima Indians: comparison with energy expenditure measured by doubly-labeled water. Obes Res. 1994;2:541–548. doi: 10.1002/j.1550-8528.1994.tb00103.x. [DOI] [PubMed] [Google Scholar]
- 24.Mayer-Davis EJ, Sparks KC, Hirst K, et al. Dietary intake in the diabetes prevention program cohort: baseline and 1-year post randomization. Ann Epidemiol. 2004;14:763–772. doi: 10.1016/j.annepidem.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 25.Mayer-Davis EJ, Vitolins MZ, Carmichael SL, et al. Validity and reproducibility of a food frequency interview in a multi-cultural epidemiology study. Ann Epidemiol. 1999;9:314–324. doi: 10.1016/s1047-2797(98)00070-2. [DOI] [PubMed] [Google Scholar]
- 26.Fujimoto WY, Jablonski KA, Bray GA, et al. Body size and shape changes and the risk of diabetes in the Diabetes Prevention Program. Diabetes. 2007;56:1680–1685. doi: 10.2337/db07-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gauderman W, Morrison J. [accessed 17 September 2008];Quanta 1.1: A computer program for power and sample size calculations for genetic-epidemiology studies. 2006 Available from http://hydra.usc.edu/gxe.
- 28.Holm S. A simple sequentially rejective Bonferroni test procedure. Scand J Stat. 1979;6:5–70. [Google Scholar]
- 29.Goldstein GL, Daun H, Tepper BJ. Influence of PROP taster status and maternal variables on energy intake and body weight of pre-adolescents. Physiol Behav. 2007;90:809–817. doi: 10.1016/j.physbeh.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 30.O'Connor TM, Yang SJ, Nicklas TA. Beverage intake among preschool children and its effect on weight status. Pediatrics. 2006;118:el010–el018. doi: 10.1542/peds.2005-2348. [DOI] [PubMed] [Google Scholar]
- 31.Joosen AM, Gielen M, Vlietinck R, Westerterp KR. Genetic analysis of physical activity in twins. Am J Clin Nutr. 2005;82:1253–1259. doi: 10.1093/ajcn/82.6.1253. [DOI] [PubMed] [Google Scholar]
- 32.Kovacs P, Ma L, Hanson RL, et al. Genetic variation in UCP2 (uncoupling protein-2) is associated with energy metabolism in Pima Indians. Diabetologia. 2005;48:2292–2295. doi: 10.1007/s00125-005-1934-9. [DOI] [PubMed] [Google Scholar]
- 33.Fredriksson R, Hägglund M, Olszewski PK, et al. The obesity gene, FTO, is of ancient origin, upregulated during food deprivation and expressed in neurons of feeding-related nuclei of the brain. Endocrinology. 2008;149:2062–2071. doi: 10.1210/en.2007-1457. [DOI] [PubMed] [Google Scholar]
- 34.Gerken T, Girard CA, Tung YC, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318:1469–1472. doi: 10.1126/science.1151710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yabe D, Brown MS, Goldstein JL. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci USA. 2002;99:12753–12758. doi: 10.1073/pnas.162488899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bengoechea-Alonso MT, Ericsson J. SREBP in signal transduction: cholesterol metabolism and beyond. Curr Opin Cell Biol. 2007;19:215–222. doi: 10.1016/j.ceb.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 37.Dahlman I, Arner P. Obesity and polymorphisms in genes regulating human adipose tissue. Int J Obes (Lond) 2007;31:1629–1641. doi: 10.1038/sj.ijo.0803657. [DOI] [PubMed] [Google Scholar]
- 38.Rampersaud E, Mitchell BD, Pollin TI, et al. Physical activity and the association of common FTO gene variants with body mass index and obesity. Arch Intern Med. 2008;168:1791–1797. doi: 10.1001/archinte.168.16.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reinehr T, Hinney A, Nguyen TT, Hebebrand J. Evidence of an influence of a polymorphism near the INSIG2 on weight loss during a lifestyle intervention in obese children and adolescents. Diabetes. 2008;57:623–626. doi: 10.2337/db07-0408. [DOI] [PubMed] [Google Scholar]