

Abstract
Neurodegeneration is a hallmark of many neurological diseases, including Alzheimer's, Parkinson's and the polyglutamine diseases, which are all caused by misfolded proteins that accumulate in neuronal cells of the brain. Although apoptosis is believed to contribute to neurodegeneration in these cases, genetic mouse models of these diseases often fail to replicate apoptosis and overt neurodegeneration in the brain. Using nuclear transfer, we generated transgenic Huntington's disease (HD) pigs that express N-terminal (208 amino acids) mutant huntingtin with an expanded polyglutamine tract (105Q). Postnatal death, dyskinesia and chorea-like movement were observed in some transgenic pigs that express mutant huntingtin. Importantly, the transgenic HD pigs, unlike mice expressing the same transgene, displayed typical apoptotic neurons with DNA fragmentation in their brains. Also, expression of mutant huntingtin resulted in more neurons with activated caspase-3 in transgenic pig brains than that in transgenic mouse brains. Our findings suggest that species differences determine neuropathology and underscore the importance of large mammalian animals for modeling neurological disorders.
INTRODUCTION
Apoptosis, or programmed cell death, is a highly organized and orchestrated form of cell death that is common in a variety of biological processes and pathological conditions. Cellular apoptosis is best characterized by morphological nuclear changes that are distinct from those elicited by necrotic death. In the central nervous system, aberrant apoptosis has been implicated in the pathogenesis of a wide range of neurological disorders, including neurodegenerative diseases caused by protein misfolding (1–3). These neurodegenerative diseases consist of Alzheimer's, Parkinson's, amyotrophic lateral sclerosis (ALS) and polyglutamine (polyQ) diseases, which are characterized by selective neuronal loss with the accumulation of toxic and misfolded proteins (4,5).
Well-known examples of protein misfolding-mediated neurodegenerative disorders are the inherited polyglutamine diseases, which include Huntington's disease (HD) and eight other disorders that are all caused by an expanded polyglutamine repeat in the associated disease proteins (6). In HD, the disease protein huntingtin carries an expanded polyQ tract (>36 glutamines) in its N-terminal region. Various cellular and animal models of HD indicate that N-terminal fragments of htt with an expanded repeat are pathogenic, as they are prone to misfolding and aggregation and are also more toxic than full-length mutant htt in terms of causing cytotoxicity or severe neurological phenotypes in HD mouse models (7,8). Genetic mutations in other neurological disorders, such as Alzheimer's and Parkinson's diseases, have also been linked to disease protein misfolding and neurotoxicity (4). Molecular mechanistic studies reveal that misfolded proteins can affect mitochondrial function or alter intracellular signaling to induce apoptosis (1–3).
Identification of the genetic mutations responsible for misfolded protein-mediated neurodegenerative disorders has allowed the establishment of transgenic mouse models for these diseases. These mouse models have been widely used to uncover the pathogenesis of neurodegenerative disorders and to develop treatments for them. However, most of these mouse models show no apoptosis or overt neurodegeneration in their brains. Species differences as well as the expression levels and protein context of transgenes probably determine the nature of neurological phenotypes and neurodegeneration. For example, a transgenic monkey model of HD, which expresses exon 1 htt (1–67 amino acids) with an expanded polyQ repeat, shows more severe neurological symptoms than HD mice expressing exon 1 or larger N-terminal mutant htt (9) and displays prominent axonal pathology not seen in HD mouse models (10). On the other hand, transgenic HD sheep expressing full-length mutant htt with a 73Q tract live normally and show only a decrease in the expression of medium spiny neuron marker DARPP-32 (11). Thus, as with HD mouse models, the expression of N-terminal mutant htt may be necessary for large animals to display robust neurological phenotypes and pathological changes. However, whether mutant htt can cause apoptosis in large animals remains unknown.
Recently, pigs have been used to model genetic human diseases, because they are more similar to humans than mice in anatomy, neurobiology, life span and genetics (12–14). Pigs have a long life span (12–15 years), are easily bred and reach puberty at 5–6 months, and they offer major advantages for biomedical research over other large animals, such as primates, for ethical and economic reasons. In the present study, we used nuclear transfer to generate transgenic Tibetan miniature pigs that express N-terminal mutant htt (1–208 amino acids) with 105Q. Mutant htt causes the postnatal death of transgenic pigs and apoptosis in their brains, unlike transgenic mice expressing the same transgene. Our findings stress the importance of large animals for modeling neurodegenerative diseases and for the development of effective therapeutics.
RESULTS
Generation of transgenic HD pigs
Studies of various HD mouse models demonstrate that N-terminal htt fragments with an expanded polyQ tract are pathogenic and cause more severe neurological symptoms than full-length mutant htt (3144 amino acids) (7). Since transgenic miniature pigs generated by pronuclear injection of a 3.3 kb cDNA encoding ∼1100 amino acids of human htt with a 75 polyQ tract were reported to have no phenotypes (15), we chose to express the first 208 amino acids of human htt with 105Q (N208-105Q) in Tibetan miniature pigs, because expression of this fragment in astrocytes even below the endogenous level can cause age-dependent neurological symptoms in transgenic mice (16).
N208-105Q was linked with an enhanced cyan fluorescent protein (ECFP) via F2A, a small viral peptide that can be self-cleaved (17) in mammalian cells to separate N208-105Q and ECFP. The transgenes were expressed under the control of the cytomegalovirus enhancer and chicken beta-actin (CAG) promoter (Fig. 1A), which can lead to the ubiquitous expression of transgenes in all tissues. After transfection of this htt construct into early-passage primary porcine fetal fibroblast cells by electroporation, live cells expressing the transgenes were selected and verified by polymerase chain reaction (PCR; Fig. 1B). We selected five cloned cell lines that express either N208-105Q or N208-160Q (Table 1). The cloned cells show the expression of both transfected htt and ECFP (Fig. 1C and Supplementary Material, Fig. S1). These cells were used to generate transgenic HD pigs via nuclear transfer as described previously (18,19). A total of 4213 reconstructed embryos were transferred into 26 gilts that exhibited a natural estrus. Six early pregnancies were established, and four of them went to term with five live births (Table 1). However, all five of these piglets came from 2355 embryos that were reconstructed with cloned N208-105Q cells and transferred into 15 piglets. In contrast, 1858 embryos that were reconstructed with cloned N208-160Q cells and transferred into 11 piglets failed to develop to term, suggesting that N208-160Q with a larger polyQ repeat is more toxic and prevents the early development of transgenic pigs. PCR analysis of DNA samples from the ears of the five live piglets revealed that they were positive for the N208-105Q transgene (Fig. 1D). The live piglets appeared normal at birth (Fig. 1E and Supplementary Material, Fig. S2). Fibroblasts isolated from the ears of transgenic piglets were cultured and showed the expression of ECFP (Fig. 1F). Southern blotting analysis of the genomic DNAs from fibroblast cell lines of the transgenic pigs indicated the existence of multi-copy transgene loci, with the insertion of estimated three to four sites (Fig. 2).
Figure 1.

Generation of cloned HD transgenic pigs. (A) DNA construct for expression of transgenic N-terminal mutant htt (N208-105Q or N208-160Q). CAG: the cytomegalovirus enhancer and chicken beta-actin promoter. (B) PCR analysis of the CAG repeats (105 and 160) in transgenic htt in pig fibroblast cell clones. (C) Expression of transgenic htt (N208-105Q) and ECFP in cloned Tibetan miniature pig fibroblast cells. (D) PCR reveals the presence of transgenic htt in tail tissues of all cloned transgenic pigs. (E) A live Tibetan miniature pig (7-1-3) transgenic for N208-105Q htt at 10 days after birth. (F) Expression of transgenic ECFP in cultured cells isolated from transgenic HD pigs (7-9 and 7-1-3).
Table 1.
Generation of HD transgenic pigs
| Cell line | CAG repeats | Recipient | Pregnant (%) | Live birth | Alive pig |
|---|---|---|---|---|---|
| 6–15 | 105 | 5 | 1 (20) | 1 | 0 |
| 7–1 | 105 | 5 | 3 (60) | 3 | 1 |
| 7–9 | 105 | 5 | 2 (40) | 1 | 0 |
| 7–101 | 160 | 9 | 0 (0) | 0 | 0 |
| 5–37 | 160 | 2 | 0 (0) | 0 | 0 |
Figure 2.
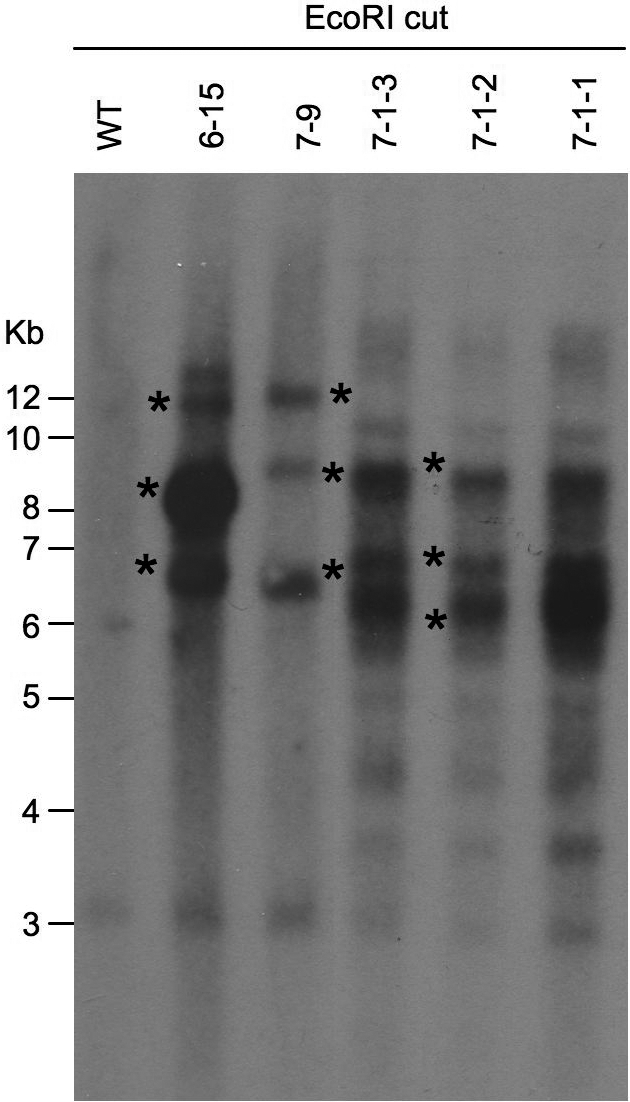
Genomic Southern blotting of cloned cells. Genomic DNAs were isolated from wild type and transgenic fibroblast cells from transgenic HD pigs (6-15, 7-9, 7-1-1, 7-1-2 and 7-1-3). The DNAs were digested with EcoRI and probed with the ECFP probe. Asterisks indicate strong bands that represent the integration sites of the transgene. Weak bands may be those that were not completely digested, degraded products or cross-reactive products.
Neurological phenotypes of transgenic HD pigs
Although newborn transgenic pigs appeared normal at birth, the 7-1-1, 7-1-2 and 7-9 piglets were weak or unable to gain weight and died at 53, 44 and 19 h after birth, respectively (Table 2). Such early death is similar to the postnatal death of transgenic monkeys that express exon 1 (the first 67 amino acids) mutant htt with 83–88Q (9). Another transgenic pig (6–15) lived for 25 days and then died prematurely (Table 2). Only the 7-1-3 pig is still alive (4 months old now) and has shown no obvious symptoms. Although the cause for the postnatal death of transgenic HD piglets remains to be determined, transgenic HD pigs (71-1, 7-1-2 and 7-9) do have sucking difficulty, which could contribute to their failure to gain weight and early death. In addition, the 7-1-1 piglet showed striking involuntary movement (hyperkinesia) before death (Supplementary Material, Fig. S3). Such a phenotype is similar to the chorea seen in HD patients and transgenic HD monkeys (9) and has not been found in more than 50 pigs transgenic for other genes in our previous experiments or reported in other transgenic pigs via nuclear transfer (12–14).
Table 2.
Summary of HD transgenic pigs
| Pig | Sex | CAG repeats | Early death time | Body weight at birth (g) | Body weight at death (g) | Htt protein expressiona | Sucking difficultyb | Chorea-like symptom | Respiratory difficultyb |
|---|---|---|---|---|---|---|---|---|---|
| 6–15 | F | 105 | 25 days | 330 | 800 | ++ | − | − | + |
| 7–1–1 | M | 105 | 53 h | 320 | 315 | +++ | ++ | +++ | +++ |
| 7–1–2 | M | 105 | 44 h | 390 | 410 | +++ | ++ | − | + |
| 7–1–3 | M | 105 | Alive | 670 | N/a | + | − | − | + |
| 7–9 | M | 105 | 19 h | 410 | ND | +++ | +++ | − | + |
F, female; M, male; N/a, not applicable; ND, not determined.
aHtt protein expression was examined using western blotting of cultured cells from transgenic pigs.
bSucking and respiratory difficulty was observed after birth.
Expression of transgenic mutant htt in pigs and mice
The transgenic HD piglets were cloned from cell colonies that may have had varying copy numbers of transgenes at different integration sites. Also, a number of variables, including cell cycle stages, passages and treatment of donor cells, as well as different reprogramming states of the donor nuclei in oocytes after nuclear transfer, can influence the expression levels of mutant htt in transgenic piglets and the corresponding phenotypes. We thus performed western blot analysis of cultured fibroblasts isolated from the ears of transgenic piglets. Anti-htt (mME48) staining clearly shows the uncleaved transgenic protein and cleaved N208-105Q (arrow in Fig. 3A), which were only present in transgenic cells, but not in cells from a wild-type pig. We also performed western blotting using 1C2, an antibody against the expanded polyQ domain that is more sensitive to soluble mutant htt than mEM48. 1C2 western blotting revealed that cells from the 7-1-3 pig, which is still alive, expressed the lowest level of N208-105Q and its degraded product compared with cells from other piglets. Cells from the 6-15 pig expressed the second lowest level of N208-105Q and its degraded product (Fig. 3A), consistent with the fact that the 6-15 pig lived longer (25 days) than the other HD pigs. Thus, the expression levels of mutant htt may contribute to the life span of transgenic HD pigs.
Figure 3.

Expression of mutant htt in transgenic HD pigs and mice. (A) Western blots of cultured fibroblast cells isolated from HD transgenic or wild-type (WT) pigs. The blots were probed with antibodies to htt (mEM48) and polyQ domain (1C2). Arrowhead indicates the uncleaved transgenic N208-105Q-F2A-ECFP protein. Arrow indicates the cleaved N208-105Q protein (105Q). (B) Western blotting of the brain cortical tissues from transgenic pigs (7-9, 7-1-2 and 7-1-3) and wild-type (WT) pig. Sample (Cell) from cultured fibroblast cells of a 7-1-2 pig was also included. Arrowhead indicates the uncleaved N208-105Q-F2A-ECFP. Arrow indicates the N208-105Q protein. The blots were probed with mEM48 for htt and 1C2 for the expanded polyQ tract. (C) Western blots of the brain cortical tissues from F1 transgenic HD mice (2 months old) of lines 11 and 23. Some mice (35, 55, 56) express the N208-105Q protein (arrow) and the uncleaved N208-105Q-F2A-ECFP protein (arrowhead), which are not seen in other littermates.
We also performed western blot analysis of brain cortical tissues from transgenic pigs (Fig. 3B). We found the uncleaved (arrowhead) and cleaved N208-105Q htt (arrow) in the brain tissues of all dead transgenic pigs (Fig. 3B), which were recognized by anti-htt (mEM48) and 1C2 antibodies, but were not present in the brain tissue of the wild-type pig. Similarly, western blotting also showed the expression of transgenic htt in the peripheral tissues of the 6-15 HD pig (Supplementary Material, Fig. S4). We also generated transgenic mice expressing the same N208-105Q construct via pronuclear injection and obtained two lines (lines 11 and 23) of the N208-105Q mice. These transgenic HD mice were normal at birth, though their founders died at 5–6 months. Western blot analysis of F1 mice (2 months old) of these two lines with anti-htt verified the expression of N208-105Q in their brain tissues (Fig. 3C). In the HD mouse brains, we also observed the uncleaved transgenic protein (arrowhead) and N208-105Q (arrow), as well as its degraded product (Fig. 3C).
Apoptotic cells in the brains of transgenic HD pigs
1C2 immunocytochemistry confirmed the expression of N208-105Q in the transgenic pig brains. Interestingly, we found typical apoptotic neurons with nuclear DNA fragmentation in the brains of transgenic HD pigs (Fig. 4A). However, examination of the brains of the N208-105Q transgenic mice revealed no apoptotic neurons, although their brain neurons also expressed mutant htt (Fig. 4A). This negative phenomenon also occurs in the brains of Hdh140CAG knock-in mice expressing full-length mutant htt (Fig. 4B). To compare the relative expression levels of mutant htt in the brains of different HD animal models, we performed EM48 immunostaining, as this assay is more sensitive to aggregated htt than 1C2 immunostaining. The striatum of Hdh140CAG knock-in mouse showed more abundant expression of mutant htt than those of N208-105Q transgenic pig and mouse brains (Fig. 4C). Thus, species and protein context, rather than expression levels of mutant htt, are likely to contribute to apoptosis seen in the HD pig brain.
Figure 4.

Apoptotic neurons in the brains of transgenic HD pigs. (A) 1C2 immunocytochemistry of the brain regions (cortex and striatum) of wild-type (WT) mouse, N208-105Q transgenic mouse or the 7-9 HD pig. Arrows indicate apoptotic cells in the transgenic pig brain. (B) 1C2 immunocytochemistry of the brain cortex and striatum of HdhCAG140 knock-in mice. (C) Anti-htt (EM48) immunostaining of the brain striatal sections of the HD pig (7-9), 3-month-old N208-105Q transgenic mouse and 12-month-old Hdh140CAG knock-in mouse. Arrows indicate EM48-positive cells in the HD transgenic pig and mouse brain sections. Scale bars: (A) 10 µm; (B) 5 µm; (C) 20 µm.
Immunocytochemistry with an anti-htt antibody (EM48) verified the presence of mutant htt in the neuropil and cytoplasm of neurons in the brains of transgenic HD pigs, but not in the wild-type pig (Fig. 5A). Some mEM48-positive cells in the HD pig brains also showed fragmented DNA in the nuclei (arrow in Fig. 5A). When the brain sections were stained with a low concentration of EM48 to reveal nuclear morphology more clearly, we saw apoptotic cells with DNA fragmentation in the brains of all transgenic HD pigs examined (Fig. 5B). Hematoxylin staining of the nuclear morphology of peripheral tissues, including the liver, heart, muscle and kidney, revealed no apoptotic cells (Supplementary Material, Fig. S5). Furthermore, electron microscopy (EM) provided evidence for the condensation of chromatin and for fragmented nuclei in the neuronal cells of transgenic HD pigs (Fig. 6), but not in the wild-type pig brain (Supplementary Material, Fig. S6). Although glial nuclear DNA condensation and fragmentation were also seen, they were not as obvious as neuronal nuclear fragmentation (Fig. 6), suggesting that neuronal degeneration is more severe than glial degeneration in the HD pig brain.
Figure 5.

mEM48 immunostaining of the brain striatum of wild-type and HD pig brains. (A) mEM48 immunocytochemistry revealed the presence of mutant htt in the neurons of HD transgenic pig (7-1-2), but not in the wild-type pig. (B) mEM48 immunocytochemistry also revealed apoptotic neurons (arrows) in transgenic HD pigs (7-1-1, 7-1-2, 7-9 and 6-15). Scale bars: 10 µm.
Figure 6.

Electron microscopy of the striatum of wild type (WT) transgenic HD pig brain (6–15) at low (A) and high (B–D) magnifications. (A) Degenerated neuron (arrow) shows nuclear chromatin condensation and is larger than a glial cell (double arrows). (B–C) Apoptotic cells (arrows) in the striatum of the HD pig brain show typical chromatin condensation and fragmentation. (D) The nuclei of glial cell (double arrows) also shows slight chromatin condensation or fragmentation. Scale bars: (A) 5 µm; (B) 2 µm; (C) 1 µm; (D) 2 µm.
Although fragmented nuclei provide strong evidence for apoptosis in the brains of transgenic HD pigs, they occur at the end stage of cell degeneration and are not frequently seen by light microscopy. We therefore performed immunohistochemistry with an antibody to the activated form of caspase-3; increased staining of activated caspase-3 results from caspase activation and reflects early apoptotic events. We observed an increased number of caspase-3-positive neurons in transgenic pig brains (Fig. 7). Mouse brains expressing the same mutant htt (N208-105Q), but not those expressing full-length mutant htt in HdhCAG140 knock-in mice, also showed caspase-3-positive neurons (Fig. 7A), supporting the earlier findings that expression of N-terminal mutant htt can activate caspases in cellular and mouse models of HD (20–24). Many caspase-3-positive cells are larger than glial cells that contain smaller and more intense hematoxylin-stained nuclei (Fig. 7B), indicating that more neuronal cells undergo apoptosis. Increased numbers of caspase-3-positive cells were seen in the brain striatum of different HD transgenic pigs (Fig. 8A). Furthermore, using a low concentration of the anti-caspase-3, we were able to verify that caspase-3-positive cells do contain fragmented nuclei in the brains of transgenic HD pigs (Fig. 8B). Importantly, we did not find nuclear fragmentation in the brains of transgenic N208-105Q mice, despite the presence of caspase-3-positive cells, suggesting that species differences may cause neurons to respond differently to caspase activation. We next quantified the relative numbers of caspase-3-positive neurons in N208-105Q transgenic mouse and pig brains. By counting the percentage of caspase-3-positive cells out of total cells per field (20 times magnification), we found more caspase-3-positive cells in the pig striatum than in the pig cortex (Fig. 8C). Compared with the N208-105Q transgenic mouse brains, transgenic HD pig brains also show more caspase-3-positive neurons in the striatum and cortex (Fig. 8C). This difference also points to the impact of species differences on apoptosis and neuropathology.
Figure 7.

Immunohistochemistry with an antibody to the activated form of caspase-3 of the mouse brain striatal tissues. (A) The brain striatum of wild-type (WT), HdhCAG140 knock-in (140Q KI), N208-105Q mice and N208-105Q transgenic pig (7–9) were immunostaininged with anti-caspase-3. Arrows indicate caspase-3-positive cells. (B) High power images showing caspase-3-positive neurons, which are distinct from negative cells and small glial nuclei. Scale bars: (A) 20 µm; (B) 10 µm.
Figure 8.

Increased numbers of activated caspase-3-positive neurons in the brains of transgenic HD pig brains. (A) Increased numbers of caspase-3-positive cells (arrows) in the brain striatal tissues of transgenic HD pigs (7-9, 7-1-2 and 6-15) but not in wild-type pig (control). (B) Active caspase-3-positive neurons (arrows) in transgenic HD pig (7-1-1 and 7-9) brains show the DNA fragmentation feature of apoptosis. (C) Quantification of the relative number of caspase-3-positive cells in the transgenic HD pig and mouse brains. The images of HD mice are presented in Figures 4 and 7. The data were collected by examining the wild-type and HD (7-1-2, 7-9 and 6-15) pig brains and are presented as mean ± SE. **P<0.01 compared with control. Scale bars: 5 µm.
DISCUSSION
Although we know that biological differences between humans and mice may account for the failure of some mouse models to replicate pathology in humans, whether larger transgenic animal models can mimic important neurodegenerative features caused by misfolded proteins, such as apoptosis, has not been rigorously tested. By comparing transgenic pigs and mice expressing the same transgenic mutant htt, our findings provide evidence that mutant htt can indeed cause apoptosis in the pig brain. Our studies underscore the importance of establishing neurodegenerative disease models by using large animals to investigate the pathogenesis of these diseases and to develop therapeutics for them.
Pigs are much closer to humans than mice in anatomy, complexity, physiological function and genetics. For example, a genetic pig model of cystic fibrosis replicates abnormalities seen in cystic fibrosis patients that do not occur in mouse models (13). However, successful generation of transgenic pigs relies on nuclear transfer, a cloning strategy that has a low rate (<1–2%) for transferred pig embryos to develop to term (18). Nevertheless, we have obtained five piglets despite this low rate. These piglets, however, survived for different periods of time and did not show the same phenotypes, which is probably contributed by different expression levels of mutant htt in these pigs. Although the causes of early death and chorea-like symptom in transgenic HD pigs remain to be determined, we found that apoptotic neurons are presented in the brains of all four transgenic pigs examined. The presence of apoptosis in the brains of the transgenic HD pigs is linked to the expression of mutant htt in the brain. First, all transgenic HD pig brains examined show apoptotic cells, but there were none in the control wild-type pig. Second, caspase-3 staining clearly indicates that the fragmented nuclei are associated with increased caspase-3 staining. Third, by comparing our pigs with HD mouse models that also show increased caspase-3 staining, we observed that the only casapse-3-positive pig cells display apoptotic features. Also, examination of the peripheral tissues of the HD pigs revealed no apoptotic cells, suggesting specificity in neuronal apoptosis. Finally, we observed more apoptotic cells in the striatum than in the cortex. The preferential neuronal loss in the HD striatum may be associated with pathogenic cell–cell interactions by mutant htt (16,25) and the effect of mutant htt on those proteins that have specific function or are enriched in the striatum, such as the small G protein Rhes, which has the properties of a SUMO-ES ligase (26).
Recent studies have also established other large transgenic animal HD models. The HD transgenic monkey models expressing exon 1 htt (N-terminal 67 amino acids with an 83–88 or 147 glutamine repeat) show no apoptosis (9,10). It is possible that exon 1 htt lacks the protein context necessary for caspase activation in the monkey brain. Protein context-related neuropathology has been found in various HD mouse models, expressing different forms of mutant htt (23). In a transgenic pig (15) expressing a larger mutant htt fragment (1100 amino acids) and in transgenic HD sheep (11) expressing full-length (3144 amino acids) mutant htt, no apoptosis, early animal death or neurological phenotype was reported. It is possible that neurodegeneration in large animals occurs only when sufficient degraded N-terminal fragments have accumulated in old animals. Thus, expressing N-terminal mutant htt fragments can facilitate disease progression, resulting in the early postnatal death of transgenic HD pigs and apoptosis in their brains.
Cellular apoptosis is a complex process that is triggered by extrinsic and intrinsic signals. The extrinsic pathway involves the activation of a death receptor upon binding of its ligand, recruitment of specific proteins at the ‘death domain’ and downstream signaling through a cascade of protein–protein interactions. The intrinsic pathway involves the mitochondria and the release of pro-apoptotic factors into the cytosol, with subsequent activation of executioner caspases. It is well accepted that mitochondria are key players in the early induction and regulation of apoptotic cell death by releasing cytochrome C (27). Toxic insults, such as oxidative stress, DNA damage, and misfolded proteins, can damage mitochondria or initiate mitochondrial apoptotic signaling to trigger caspase activation cascades (28,29). It has been documented that mutant htt directly binds mitochondria to affect the respiratory function, membrane permeability or trafficking of mitochondria (30,31). Mutant htt also indirectly affects the biogenesis of mitochondrial proteins via gene transcription dysregulation (32,33). In addition, aggregated proteins recruit caspase-8 to activate caspase cascades (21). In support of this idea, a dominant-negative mutant of caspase-1 is found to delay disease progression in transgenic HD mice (20).
Although apoptosis is seen in the brains of patients with HD (34–37), as well as Alzheimer's, Parkinson's, and other neurodegenerative diseases (1–3,38), most transgenic mouse models of neurodegenerative disorders lack apoptotic neurons. In the brains of symptomatic HD mice that express N-terminal mutant htt and show an increased caspase activation (23,39), only dark degenerated neurons without typical apoptotic features are seen (23,40). Although ultrastructural evidence for apoptosis in the HD mouse brains is still lacking and no apoptosis has been found in YAC and BAC transgenic mice that express full-length mutant htt, an early study showed that transgenic mice expressing full-length mutant htt under the CMV promoter displayed apoptotic-like cells death in their brains (41). It is possible that protein context and polyQ repeat length in htt, the levels of transgenic htt and the ways to generate transgenic animals (i.e. pronuclear microinjection versus nuclear transfer) can influence the neuropathology in transgenic animals. However, our comparison of transgenic HD pigs and mice, which express the same mutant htt, clearly indicates the species differences in the generation of apoptosis. The pig brain is more similar to the human brain than the rodent in development, gross anatomy and biochemical and physiological function (42). Neuronal circuitry, anatomy and physiological functions specific to the pig brain could make pig neurons more vulnerable to misfolded proteins. The longer gestation period for pigs than mice (typically 113–115 versus 21 days) may allow the toxic form of mutant proteins, such as truncated N-terminal mutant htt, to cause more severe neurodegeneration in the pig brains. Although these possibilities need to be investigated, our findings clearly indicate that species differences play an important role in the nature of the neuropathology caused by misfolded proteins. Considering these differences, transgenic models using higher mammalian species or large animals to model important neurodegenerative diseases would provide deeper insight into the pathogenesis of these disorders. Furthermore, because pigs and humans are more physiologically similar than rodents and other typical laboratory animals, transgenic pig models of diseases can also help identify effective therapeutics for HD and other neurodegenerative diseases.
MATERIALS AND METHODS
Antibodies
Mouse monoclonal antibody (mEM48) against the N-terminal region (amino acids 1–256) of human htt was described in our previous study (43). The mouse anti-polyQ antibody (1C2) was purchased from Millipore (Temecula, CA, USA). A rabbit antibody to the activated form of caspase-3 was from Cell Signaling. The mouse anti-gamma-tubulin antibody was purchased from Sigma-Aldrich (St Louis, MO, USA) and used at 1:50 000 dilution. Secondary antibodies were peroxidase-conjugated donkey anti-mouse or donkey anti-rabbit IgG (H + L) from Jackson ImmunoResearch (West Grove, PA, USA).
Cell culture, transfection and selection
Primary fibroblasts were isolated from 35-day-old Tibet miniature swine fetuses, and digested by collagenase-DNase in cell culture medium containing 0.032% Collagenase (Sigma, C5138) and 2500 IU/ml DNase for 4–6 h at 37°C. The cells were plated on 10 cm diameter culture dish in the cell culture medium. Adherent cells were frozen the following day.
For transfection of N-terminal mutant htt (the first 208 amino acids including a normal 23Q repeat) with a total of 105Q in the repeat, A pgk-neo expression cassette as a selection marker was used to express a fusion protein of N208-105Q-F2A-ECFP under the control of the cytomegalovirus enhancer and chicken beta-actin (CAG) promoter. Primary fetal fibroblasts were thawed and cultured for 2 days to subconfluence. About 1 × 107 fibroblasts were electroporated using an electroporator (GenePulser Xcell, Bio-rad) at 230 V, 500 µF in 0.8 ml of Dulbecco's phosphate-buffered saline (D-PBS) containing pCAG-HTT-2A-ECFP (100 µg/ml) pre-linerized with Sfi I restriction endonuclease. Transfected cells were divided into 10 culture dishes (10 cm diameter) and cultured for 2 days in DMEM containing 15% fetal calf serum and G418 (1 mg/ml). After 7–10 days of culture with G418 (1 mg/ml), cell clones with ECFP expression were picked up and verified via immunofluorscence microscopy for expressing transfected htt. Htt-positive cells were thawed and cultured to subconfluence before nuclear transfer.
Nuclear transfer and generation of transgenic HD pigs and mice
We used similar methods described in our previous studies (18,19,44). Pig ovaries collected from local slaughter house were transported to the laboratory in 0.9% saline at 35–39°C. Cumulus oocyte complexes (COCs) were aspirated from 2 to 6 mm diameter antral follicles with an 18 gauge needle fixed to a 10 ml disposable syringe. The COCs were washed three times in maturation medium, which is a tissue culture medium 199 (Gibco, 31100–035) supplemented with 0.1% (w/v) polyvinyl alcohol (Sigma, P8136), 3.05 mm d-glucose, 0.91 mm sodium pyruvate (Sigma P-4562), 0.57 mm cysteine (Sigma C-7352), 0.5 µg/ml of luteinizing hormone (Sigma, L-5269), 0.5 µg/ml of follicle-stimulating hormone (Sigma, F-2293), 10 ng/ml of epidermal growth factor (Sigma, E-4127), 10% (v:v) of porcine follicular fluid, 75 µg/ml of penicillin G and 50 µg/ml of streptomycin. Fifty to 60 COCs were transferred to 500 µl of the same medium that had been covered with mineral oil in a four-well multidish (Nunc, Roskilde, Denmark) and pre-equilibrated at 39°C in an atmosphere of 5% CO2 in air overnight. After 42–44 h of culture, oocytes were freed from cumulus cells by vigorous vortexing for 4 min in TL–HEPES supplemented with 0.1% polyvinyl alcohol (Sigma P8136) and 0.1% hyaluronidase (Sigma, H3506). Cumulus-free (denuded) oocytes were enucleated by aspirating the first polar body and adjacent cytoplasm with a glass pipette (diameter, 30 µm) in manipulation medium supplemented with cytochalasin B (7.5 µg/ml). A single donor cell with ECFP expression was selected under an epifluorscent microscope and injected into the perivitelline space of the oocyte to contact the oocyte membrane.
Injected oocyte-fibroblast pairs were placed between two 0.2 mm diameter platinum electrodes 1 mm apart in fusion and activation medium with a position vertical to the electrodes. Fusion/activation was induced with two successive DC pulses of 1.2 kV/cm for 30 µs on an electro fusion instrument (CF-150B, BLS, Hungary). Embryos were kept in micromanipulation medium without CB for another 30 min before the fusion rate was evaluated. Reconstructed embryos (50–100) were transferred to a 500 µl of culture medium covered with mineral oil in a four-well multidish, and the dishes were held in 5% CO2 in air at 39°C until embryo transfer. Most embryos were cultured for 20 h and surgically transferred into the oviduct of a surrogate the day after observed estrus.
A total of 4213 (2355 from the 7-1, 7-9 and 6-15 cell lines, 1858 from the 7-101 and 5-37 cell lines) reconstructed embryos were generated. These reconstructed embryos were cultured for 20 h in vitro and then surgically transferred into the oviduct of 26 Tibetan miniature gilts exhibiting a natural estrus. Of these gilts, 15 gilts were used for transferring embryos derived from the 7-1, 7-9 and 6-15 cell lines and 11 gilts for transferring embryos derived from the 7-101 and 5-37 cell lines. Gestation was monitored weekly via ultrasound from 24 days after transfer. Six early pregnancies were established, and all were from the 7-1, 7-9 and 6-15 cells. Four of these pregnancies went to term, with five liveborn piglets. Ear skin tissues were obtained from the newborn piglets mechanically, treated with 70% ethanol for 5 min and washed three times with D-PBS. The tissues were minced, and cells were isolated for culture in the same way as for the fetal fibroblast cell culture.
For generation of transgenic HD mice, microinjection of the N208-105Q-F2A-ECFP linearized vector into the pronucleus of fertilized oocytes from FVB mice was performed by the Emory University transgenic mouse core facility. Genomic DNA was isolated from mouse or pig tails, and the PCR genotyping method was employed for screening transgenic mice and pigs. Primers with sequences flanking the CAG repeat were used for PCR. Sequences of the primers are: forward primer (5′-ATGAAGGCCTTCGAGTCCCTCAAGTCCTTC-3′) and reverse primer (5′-AAACTCACGGTCGGTGCAGCGGCTCCTCAG-3′). Positive founders and their corresponding lines carry the expected length (105Q) of the polyQ repeat in transgenic htt. HdhCAG140 knock-in mice were provided by Dr Michael Levine at the University of California, Los Angeles, and bred in the Department of Animal Resources of Emory University.
Western blotting and immunocytochemistry
For western blots, cultured cells or brain tissues were homogenized in RIPA buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 1 mm EDTA, pH 8.0, 1 mm EGTA, pH 8.0, 0.1% SDS, 0.5% deoxycholate and 1% Triton X-100) with 1× Protease inhibitor from Sigma (P8340) and 100 mg/ml of phenylmethylsulfonyl fluoride. The cell or tissue lysates were diluted in 1× SDS sample buffer (62.6 mm Tris–HCl, pH 6.8, 2% SDS, 10% glycerol and 0.01% bromophenol blue) and sonicated for 10 s after incubation at 100°C for 5 min. The total lysates were resolved in a 10% or 4–12% Tris–glycine gel (Invitrogen) and blotted to a nitrocellulose membrane. The western blots were developed using the ECL-Plus Kit (GE Health Care/Amersham).
For immunohistochemistry labeling, mice were anesthetized and then perfused intracardially with 0.9% NaCl, for 30 s followed by 4% paraformaldehyde in 0.1 m phosphate buffer at pH 7.2. The postmortem pig brains were removed and cut into 5 mm slices and soaked in 4% paraformildehyde for 72 h with two changes of fresh 4% paraformildehyde. Brains were cryoprotected in 30% sucrose at 4°C. Fixed mouse or pig brains were sectioned at 40 µm using a cryostate (Leica CM 1850). Free-floating sections were preblocked in 4% normal goat serum in PBS, 0.1% Triton X-100 and then incubated with mEM48 antibody (42) or 1C2 (1:2000, Millipore) at 4°C for 48 h. The immunoreactive product was visualized with the avidin–biotin complex kit (Vector ABC Elite; Vector Laboratories). For hematoxylin staining, brain sections were stained with modified Lillie-Mayer hematoxylin for 1–3 min and blue hematoxylin in 0.3% sodium borate for 15 s. After washes with distilled water, brain sections were placed in 95% alcohol for 30 s. Light micrographs were taken using a Zeiss microscope (Axiovert 200 MOT) equipped with a digital camera (Orca-100; Hamamatsu) and the image acquisition software Openlab (Improvision). A 20× (LD-Achroplan 20′/0.4 numerical aperture NA) or 63× lens (63′/0.75 numerical aperture) was used for light microscopy.
Quantification of caspase-3-positive cells was performed using the same method described previously (23). Each micrograph at a magnification of ×20 was examined. The percentage of caspase-3-positive cells of total cells in hematoxylin-stained brain sections was obtained. For each pig or mouse brain region, three to four randomly selected images were captured. More than three brain sections of each animal were examined. The data were obtained from the brains of HD pigs (7-1-2, 7-9 and 6–15) and wild-type pig as well as N208-105Q transgenic and control mice.
Electron microscopy
For EM examination of the pig brain morphology, postmortem pig brains that were removed within 2–3 h after death were fixed for 72 h in 4% paraformaldehyde in 1× PBS. Cortex and striatum vibratome sections (50 µm) were further fixed in 4% paraformaldehyde/2.5% glutaradehyde/0.1 m PB for 24 h. The sections were then fixed in 1% buffered osmium tetroxide, dehydrated with ethanol and embedded in Eponate 12 resin (Ted pella, Inc., Redding, CA, USA). Ultrathin sections were cut on a Leica UC6rt ultramicrotomm (Leica Microsystems, Bannockburn, IL, USA) at 70–80 nm, placed on Formvar-coated grids and counter-stained with 4% aqueous uranyl acetate and 2% lead citrate. Sections were examined using a Hitachi H-7500 transmission electron microscope (Hitachi High Technologies of America, Inc., Pleasanton, CA, USA) equipped with a Gatan BioScan CCD camera.
Genomic Southern blotting
Genomic DNAs were extracted from pig primary fibroblast cells or mouse tails. Five micrograms of DNA were incubated with EcoRI at 37°C overnight. The digested DNA were loaded on a 0.7% agarose gel and run overnight. The gel were stained with 0.05 mg/ml of EtBr for revealing DNA and then washed in 1× TAE buffer for 30 min to remove EtBr. The DNA gel was denatured and neutralized. DNA were blotted to a supported nitrocellulose (GE health Care life science) and hybridized at 65°C for 3 h with a DNA probe of ECFP fragment released from the transgenic vector. The DNA probe was labeled using the Amersham Rediprime™ II DNA Labeling System (GE health care, Life science) and 32P-dCTP (Perkin Elmer Life Science). The blots were hybridized in the Rapid-hyb buffer (GE health care, life science) according to manufacture's instruction, and were exposed to a blue-sensitive film for 3 h at −80°C.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by grants AG019206 and NS041669 (X.J.L.) and NS045016 and AG031153 (S.H.L.) from the National Institutes of Health, and grants from the National High Technology Research and Development Program of China (2006AA02A103), the National 973 Program of China (2009CB941001) and Natural Science Foundation of Guangdong Province, China (9251066302000001, 8451066302001811) to L.X.L.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jianmei Wang for technical assistance with immunohistochemistry analysis, Qinghong Wu for assistance with animal management and Cheryl T. Strauss for her critical reading of the manuscript.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Yuan J., Yankner B.A. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. doi:10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 2.Mattson M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. doi:10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 3.Friedlander R.M. Apoptosis and caspases in neurodegenerative diseases. N. Engl. J. Med. 2003;348:1365–1375. doi: 10.1056/NEJMra022366. doi:10.1056/NEJMra022366. [DOI] [PubMed] [Google Scholar]
- 4.Bossy-Wetzel E., Schwarzenbacher R., Lipton S.A. Molecular pathways to neurodegeneration. Nat. Med. 2004;10((suppl.)):S2–S9. doi: 10.1038/nm1067. [DOI] [PubMed] [Google Scholar]
- 5.Haass C., Selkoe D.J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. doi:10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 6.Orr H.T., Zoghbi H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. doi:10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 7.Heng M.Y., Detloff P.J., Albin R.L. Rodent genetic models of Huntington disease. Neurobiol. Dis. 2008;32:1–9. doi: 10.1016/j.nbd.2008.06.005. doi:10.1016/j.nbd.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 8.Li X., Li H., Li X.J. Intracellular degradation of misfolded proteins in polyglutamine neurodegenerative diseases. Brain Res. Rev. 2008;59:245–252. doi: 10.1016/j.brainresrev.2008.08.003. doi:10.1016/j.brainresrev.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang S.H., Cheng P.H., Banta H., Piotrowska-Nitsche K., Yang J.J., Cheng E.C., Snyder B., Larkin K., Liu J., Orkin J., et al. Towards a transgenic model of Huntington's disease in a non-human primate. Nature. 2008;453:921–924. doi: 10.1038/nature06975. doi:10.1038/nature06975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang C.E., Tydlacka S., Orr A.L., Yang S.H., Graham R.K., Hayden M.R., Li S., Chan A.W., Li X.J. Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington's disease. Hum. Mol. Genet. 2008;17:2738–2751. doi: 10.1093/hmg/ddn175. doi:10.1093/hmg/ddn175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacobsen J.C., Bawden C.S., Rudiger S.R., McLaughlan C.J., Reid S.J., Waldvogel H.J., MacDonald M.E., Gusella J.F., Walker S.K., Kelly J.M., et al. An ovine transgenic Huntington's disease model. Hum. Mol. Genet. 2010;19:1873–1882. doi: 10.1093/hmg/ddq063. doi:10.1093/hmg/ddq063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carter D.B., Lai L., Park K.W., Samuel M., Lattimer J.C., Jordan K.R., Estes D.M., Besch-Williford C., Prather R.S. Phenotyping of transgenic cloned piglets. Cloning Stem Cells. 2002;4:131–145. doi: 10.1089/153623002320253319. doi:10.1089/153623002320253319. [DOI] [PubMed] [Google Scholar]
- 13.Rogers C.S., Stoltz D.A., Meyerholz D.K., Ostedgaard L.S., Rokhlina T., Taft P.J., Rogan M.P., Pezzulo A.A., Karp P.H., Itani O.A., et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science. 2008;321:1837–1841. doi: 10.1126/science.1163600. doi:10.1126/science.1163600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aigner B., Renner S., Kessler B., Klymiuk N., Kurome M., Wunsch A., Wolf E. Transgenic pigs as models for translational biomedical research. J. Mol. Med. 2010;88:653–664. doi: 10.1007/s00109-010-0610-9. [DOI] [PubMed] [Google Scholar]
- 15.Uchida M., Shimatsu Y., Onoe K., Matsuyama N., Niki R., Ikeda J.E., Imai H. Production of transgenic miniature pigs by pronuclear microinjection. Transgenic Res. 2001;10:577–582. doi: 10.1023/a:1013059917280. doi:10.1023/A:1013059917280. [DOI] [PubMed] [Google Scholar]
- 16.Bradford J., Shin J.Y., Roberts M., Wang C.E., Li X.J., Li S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc. Natl Acad. Sci. USA. 2009;106:22480–22485. doi: 10.1073/pnas.0911503106. doi:10.1073/pnas.0911503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szymczak A.L., Workman C.J., Wang Y., Vignali K.M., Dilioglou S., Vanin E.F., Vignali D.A. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat. Biotechnol. 2004;22:589–594. doi: 10.1038/nbt957. doi:10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- 18.Lai L., Prather R.S. Production of cloned pigs by using somatic cells as donors. Cloning Stem Cells. 2003;5:233–241. doi: 10.1089/153623003772032754. doi:10.1089/153623003772032754. [DOI] [PubMed] [Google Scholar]
- 19.Lai L., Kang J.X., Li R., Wang J., Witt W.T., Yong H.Y., Hao Y., Wax D.M., Murphy C.N., Rieke A., et al. Generation of cloned transgenic pigs rich in omega-3 fatty acids. Nat. Biotechnol. 2006;24:435–436. doi: 10.1038/nbt1198. doi:10.1038/nbt1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ona V.O., Li M., Vonsattel J.P., Andrews L.J., Khan S.Q., Chung W.M., Frey A.S., Menon A.S., Li X.J., Stieg P.E., et al. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature. 1999;399:263–267. doi: 10.1038/20446. [DOI] [PubMed] [Google Scholar]
- 21.Sanchez I., Xu C.J., Juo P., Kakizaka A., Blenis J., Yuan J. Caspase-8 is required for cell death induced by expanded polyglutamine repeats. Neuron. 1999;22:623–633. doi: 10.1016/s0896-6273(00)80716-3. doi:10.1016/S0896-6273(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 22.Li S.H., Lam S., Cheng A.L., Li X.J. Intranuclear huntingtin increases the expression of caspase-1 and induces apoptosis. Hum. Mol. Genet. 2000;9:2859–2867. doi: 10.1093/hmg/9.19.2859. doi:10.1093/hmg/9.19.2859. [DOI] [PubMed] [Google Scholar]
- 23.Yu Z.X., Li S.H., Evans J., Pillarisetti A., Li H., Li X.J. Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease. J. Neurosci. 2003;23:2193–2202. doi: 10.1523/JNEUROSCI.23-06-02193.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hermel E., Gafni J., Propp S.S., Leavitt B.R., Wellington C.L., Young J.E., Hackam A.S., Logvinova A.V., Peel A.L., Chen S.F., et al. Specific caspase interactions and amplification are involved in selective neuronal vulnerability in Huntington's disease. Cell Death Differ. 2004;11:424–438. doi: 10.1038/sj.cdd.4401358. doi:10.1038/sj.cdd.4401358. [DOI] [PubMed] [Google Scholar]
- 25.Gu X., Li C., Wei W., Lo V., Gong S., Li S.H., Iwasato T., Itohara S., Li X.J., Mody I., et al. Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron. 2005;46:433–444. doi: 10.1016/j.neuron.2005.03.025. doi:10.1016/j.neuron.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 26.Subramaniam S., Sixt K.M., Barrow R., Snyder S.H. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science. 2009;324:1327–1330. doi: 10.1126/science.1172871. doi:10.1126/science.1172871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang X., Wang X. Cytochrome C-mediated apoptosis. Annu. Rev. Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. doi:10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 28.Beal M.F. Mitochondrial dysfunction in neurodegenerative diseases. Biochim. Biophys. Acta. 1998;1366:211–223. doi: 10.1016/s0005-2728(98)00114-5. [DOI] [PubMed] [Google Scholar]
- 29.Lin M.T., Beal M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. doi:10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 30.Bossy-Wetzel E., Petrilli A., Knott A.B. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 2008;31:609–616. doi: 10.1016/j.tins.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X.J., Orr A.L., Li S. Impaired mitochondrial trafficking in Huntington's disease. Biochim. Biophys. Acta. 2010;1802:62–65. doi: 10.1016/j.bbadis.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cui L., Jeong H., Borovecki F., Parkhurst C.N., Tanese N., Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. doi:10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 33.Weydt P., Pineda V.V., Torrence A.E., Libby R.T., Satterfield T.F., Lazarowski E.R., Gilbert M.L., Morton G.J., Bammler T.K., Strand A.D., et al. Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. doi:10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 34.Dragunow M., Faull R.L., Lawlor P., Beilharz E.J., Singleton K., Walker E.B., Mee E. In situ evidence for DNA fragmentation in Huntington's disease striatum and Alzheimer's disease temporal lobes. Neuroreport. 1995;6:1053–1057. doi: 10.1097/00001756-199505090-00026. doi:10.1097/00001756-199505090-00026. [DOI] [PubMed] [Google Scholar]
- 35.Portera-Cailliau C., Hedreen J.C., Price D.L., Koliatsos V.E. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J. Neurosci. 1995;15:3775–3787. doi: 10.1523/JNEUROSCI.15-05-03775.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vis J.C., Schipper E., de Boer-van Huizen R.T., Verbeek M.M., de Waal R.M., Wesseling P., ten Donkelaar H.J., Kremer B. Expression pattern of apoptosis-related markers in Huntington's disease. Acta. Neuropathol. 2005;109:321–328. doi: 10.1007/s00401-004-0957-5. doi:10.1007/s00401-004-0957-5. [DOI] [PubMed] [Google Scholar]
- 37.Thomas L.B., Gates D.J., Richfield E.K., O'Brien T.F., Schweitzer J.B., Steindler D.A. DNA end labeling (TUNEL) in Huntington's disease and other neuropathological conditions. Exp. Neurol. 1995;133:265–272. doi: 10.1006/exnr.1995.1029. doi:10.1006/exnr.1995.1029. [DOI] [PubMed] [Google Scholar]
- 38.Tatton W.G., Chalmers-Redman R., Brown D., Tatton N. Apoptosis in Parkinson's disease: signals for neuronal degradation. Ann. Neurol. 2003;53((Suppl. 3)):S61–S70. doi: 10.1002/ana.10489. Discussion S70–62. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y., Ona V.O., Li M., Drozda M., Dubois-Dauphin M., Przedborski S., Ferrante R.J., Friedlander R.M. Sequential activation of individual caspases, and of alterations in Bcl-2 proapoptotic signals in a mouse model of Huntington's disease. J. Neurochem. 2003;87:1184–1192. doi: 10.1046/j.1471-4159.2003.02105.x. doi:10.1046/j.1471-4159.2003.02105.x. [DOI] [PubMed] [Google Scholar]
- 40.Turmaine M., Raza A., Mahal A., Mangiarini L., Bates G.P., Davies S.W. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc. Natl Acad. Sci. USA. 2000;97:8093–8097. doi: 10.1073/pnas.110078997. doi:10.1073/pnas.110078997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reddy P.H., Williams M., Charles V., Garrett L., Pike-Buchanan L., Whetsell W.O., Jr, Miller G., Tagle D.A. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nature Genet. 1998;20:198–202. doi: 10.1038/2510. doi:10.1038/2510. [DOI] [PubMed] [Google Scholar]
- 42.Lind N.M., Moustgaard A., Jelsing J., Vajta G., Cumming P., Hansen A.K. The use of pigs in neuroscience: modeling brain disorders. Neurosci. Biobehav. Rev. 2007;31:728–751. doi: 10.1016/j.neubiorev.2007.02.003. doi:10.1016/j.neubiorev.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 43.Zhou H., Cao F., Wang Z., Yu Z.X., Nguyen H.P., Evans J., Li S.H., Li X.J. Huntingtin forms toxic NH2-terminal fragment complexes that are promoted by the age-dependent decrease in proteasome activity. J. Cell Biol. 2003;163:109–118. doi: 10.1083/jcb.200306038. doi:10.1083/jcb.200306038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lai L., Prather R.S. A method for producing cloned pigs by using somatic cells as donors. Methods Mol. Biol. 2004;254:149–164. doi: 10.1385/1-59259-741-6:149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.