

Abstract
We investigated the potential of human hair follicle cells for multilineage differentiation and as a source of functional smooth muscle cells (SMCs). We report that human hair follicle stem cells (HFCs) isolated from individual follicles expressed surface markers that are characteristic of mesenchymal stem cells such as CD44, CD49b, CD73, CD90, and CD105 but lacked hematopoietic markers CD45 and CD34. In addition, HFCs differentiated toward adipocytes, chondrocytes, osteoblasts, or SMCs in the appropriate induction medium. Treatment with basic fibroblast growth factor increased proliferation and prevented myogenic differentiation, suggesting that basic fibroblast growth factor can be used to expand the population of undifferentiated HFCs to the large numbers needed for therapeutic applications. SMCs were isolated from HFCs using tissue-specific promoters and flow cytometry sorting. Cylindrical vascular constructs engineered with HF-SMCs showed remarkable contractility in response to receptor and nonreceptor agonists such KCl, endothelin-1, and the thromboxane mimetic, U46619, as well as superior mechanical properties compared to their counterparts with human vascular SMCs. Our results suggest that HF is a rich source of mesenchymal stem cells with great potential for myogenic differentiation providing functional SMCs for tissue regeneration and cell therapies.
Introduction
The hair follicle, one of the skin appendages, is a mini organ that forms early in embryonic development as a result of epithelial–mesenchymal cell interactions. The hair follicle is one of the few organs in the body with the ability to undergo cycles of degeneration and regeneration throughout life. During catagen, cells in the lower part of the follicle undergo apoptosis and the tissue retracts to the level of the bulge. A resting phase, telogen, may ensue during which the follicles remain dormant. During catagen and telogen, follicles prepare their stem cells for the next growth phase, anagen, when induction signals from the dermal papilla promote proliferation and downward migration of bulge stem cells to regenerate the inner and outer root sheath, matrix, and hair shaft.1
The hair follicle is a very rich source of multipotent adult stem cells and as such it may be an easily accessible alternative source of autologous smooth muscle cells (SMCs). Label-retaining assays localized stem cells in the bulge area of the hair follicle several years ago,2,3 and these results were recently verified by transgenic mice designed to express enhanced green fluorescent protein in the same region.4,5 In a pioneering study Lako et al. first demonstrated that cells from the dermal papilla or dermal sheath of hair follicles could reconstitute multiple lineages of the hematopoietic system in lethally irradiated mice, suggesting the presence of multipotent stem cells.6 Others showed that dermal papilla or dermal sheath cells from the rat follicles were similar to bone marrow mesenchymal stem cells (BM-MSCs) in terms of their ability to differentiate toward the adipogenic, osteogenic, and chondrogenic lineages.7,8 In a recent study, we employed the smooth muscle alpha-actin promoter (P-αSMA) to isolate smooth muscle progenitor cells from ovine hair follicles.9 We showed that smooth muscle progenitor cells from ovine hair follicles were highly clonogenic and expressed smooth-muscle-specific markers at the RNA and protein level. These results demonstrated that the hair follicle is a rich source of highly functional smooth muscle progenitors that can be used for vascular tissue regeneration.
In this communication, we present evidence that adult human hair follicles contain multipotent stem cells, similar to previous findings with rat and mouse follicle cells. Human hair follicle stem cells (HFCs) express surface markers characteristic of BM-MSCs such as CD90, CD105, CD44, and CD73, but they lack hematopoietic (CD34 and CD45) and endothelial (CD144) markers. Interestingly, in the presence of an appropriate induction medium, HFCs exhibited tri-lineage differentiation potential toward fat, bone, and cartilage, indicating the presence of MSCs in human hair follicles. Using tissue-specific promoters, we isolated highly contractile HF-SMCs from the total population of HFCs. Treatment with basic fibroblast growth factor (bFGF) increased proliferation but decreased the P-αSMA activity, expression of smooth-muscle-specific markers, and contractility of HFCs and to a much lesser extent HF-SMCs. Conversely, removal of bFGF promoted myogenic differentiation as shown by expression of SMC markers and increased contractility. Taken together, our results suggest that human hair follicles from adult donors contain a population of MSCs, thereby providing an easily accessible source of adult multipotent stem cells for regenerative medicine.
Materials and Methods
Isolation and cultivation of HFCs
Human HFCs were isolated as described previously.9 Briefly, hair-follicle-containing full-thickness skin biopsies from the scalp of two donors (41-year-old woman and 69-year-old man) were obtained from the Cooperative Human Tissue Network. The investigation conforms to the principles outlined in the Declaration of Helsinki.
After extensive washes with phosphate-buffered saline (PBS) (EMD Biosciences) containing antibiotic–antimycotic (Gibco), the tissues were trimmed to remove underlying fat tissue, cut into 2 × 4 mm pieces, and subsequently digested with 0.5% collagenase type I (Invitrogen) at 37°C with occasional agitation. After 4 h of digestion, single hair follicles were released from the full-thickness skin using fine forceps, filtered through 40 μm cell strainer (BD Biosciences), and washed extensively with PBS. Then, hair follicles were transferred each in a well of a 96-well plate (BD Biosciences) and cultured in 100 μL of Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco) to allow for cell migration onto the tissue culture plastic. Cells that originated from the bulge region were visually identified as epidermal keratinocytes, whereas cells migrating from the dermal sheath or papilla had the morphological appearance of mesenchymal cells. The wells populated with cells originating from the dermal sheath or papilla were selected, pooled, and expanded.
Cloning of tissue-specific promoters
P-αSMA or myosin heavy chain promoter (P-MHC) was polymerase chain reaction amplified from human genomic DNA using the following primers:
P-αSMA: forward primer, ACAACAATCGATAACAGCTGGTCATGGCTGTA; reverse primer, TGTTGTACCGGTGCATGAACCCAGCCAAATCC
P-MHC: forward primer, ACAACAATCGATAGCCTGTGCGGAGCAGCTCA; reverse primer, ACAACAGCTAGCGATGGAGAGCTCGGATCTGA
The underlined sequences represent the restriction sites ClaI (ATCGAT), AgeI (ACCGGT), or NheI (GCTAGC). The promoter sequences for P-αSMA or P-MHC include 1438 or 2452 bp upstream and 29 or 53 bp downstream of the transcriptional start site and were ligated into a promoterless lentiviral vector upstream of ZsGreen (P-αSMA) or DsRed (P-MHC) between ClaI and AgeI for P-αSMA, and ClaI and NheI for P-MHC restriction sites, respectively.
Flow cytometry and immunostaining
HFCs were harvested, resuspended in PBS, and fixed with 4% paraformaldehyde (room temperature [RT], 10 min). After three washes in PBS, cells were permeabilized in 0.1% Triton-X-100 (RT; 10 min; Fisher Scientific), washed once in PBS, and blocked with 2% bovine serum albumin (BSA)/PBS (106 cells/100 μL; RT, 30 min). HFCs were then incubated with mouse anti-human alpha-actin antibody (1:100 dilution, RT, 30 min; Serotec) in blocking solution (0.01% Triton-X-100 in 1% BSA/PBS). For immunostaining for surface markers the cells were blocked with 1% BSA/PBS (106 cells/100 μL; RT, 30 min) without fixation or permeabilization. The following antibodies were used: CD34, CD44, CD45, CD73, CD90, CD105, CD144, and Flk-1 (1:100 dilution in blocking solution, RT, 30 min; BD Biosciences). After three washes in PBS the cells were incubated with Alexa fluor 488–conjugated goat anti-mouse antibody (1:100 dilution, RT, 30 min in dark; Invitrogen) or Alexa Fluor® 647-R-phycoerythrin–conjugated goat anti-mouse (1:100 dilution, RT, 30 min; Invitrogen), washed once with PBS, and processed for flow cytometry. HFCs stained only with secondary antibody served as negative control. Immunostaining and gel compaction assays were performed as described previously.9,10
Adipogenic and osteogenic differentiation of HFCs
The differentiation of HFCs into adipocytes, chondrocytes, and osteoblasts was performed as described elsewhere.11 Briefly, HFCs (passage 6) were seeded at a density of 2.5 × 104/well in 12-well plates (n = 4 wells per condition in each experiment) and cultured in DMEM containing 10% FBS overnight. The next day, the culture medium was changed to the appropriate differentiation medium as follows. The adipogenic medium included DMEM with 1% FBS, 0.5 mM of isobutyl-methylxanthine (Sigma), 1 μM of dexamethasone (Sigma), 10 μM of insulin (Sigma), and 200 μM of Indomethacin (MP Biomedicals). The osteogenic medium comprised DMEM with 1% FBS, 0.1 μM dexamethasone, 50 μM ascorbate-2-phosphate (Sigma), and 10 mM β-glycerolphosphate (Alfa Aesar). The cells were cultured for 2 weeks and the medium was changed every 3 days. Oil Red O or alkaline phosphatase staining was performed as follows.
Oil Red O staining
After 2 weeks of culture in the adipogenic medium, the cells were washed once with PBS, fixed in 4% paraformaldehyde/1% calcium/PBS at room temperature for 60 min, washed three times with PBS and once with 70% ethanol, and incubated with 2% (wt/vol) Oil Red O (Alfa Aesar) at room temperature for 5 min. After washing once with 70% ethanol and twice with distilled water, the cells were imaged using a Zeiss Axio Observer inverted microscope and photographed with an ORCA-ER CCD camera (Hamamatsu).
Alkaline phosphatase enzymatic activity
After the cell layers were fixed in 4% formaldehyde for 1 min, they were washed twice with PBS, and alkaline phosphatase activity was detected using a kit (Chemicon) per manufacturer's instructions.
von Kossa staining
After differentiation in the osteogenic medium for 4 weeks, the cells were fixed in 4% paraformaldehyde (RT; 60 min), rinsed with distilled water, and overlaid with 1% silver nitrate solution in dark (RT; 30 min). After several washes with distilled water, cells were exposed to UV light for 60 min and washed three times, and un-reacted silver nitrate was removed by incubation with 5% sodium thiosulfate (RT; 5 min) and imaged with a Zeiss Axio Observer inverted microscope.
Chondrogenic differentiation of HFCs
For chondrogenic differentiation, cell spheres of HFCs were generated by adding 20 μL of 8 × 106 cell/mL HFCs drop wise in non-tissue-culture-treated 24-well plates. After 4 h of incubation at 37°C and 10% CO2, the medium was changed to the chondrogenic differentiation medium: DMEM with 1% FBS, 6.25 μg/mL insulin, 10 ng/mL transforming growth factor-beta 1 (TGF-β1; US Biological), and 50 nM of ascorbate-2-phosphate. The culture medium was replenished every 3 days for 2 weeks. At that time, cell spheres of HFCs were fixed in 10% buffered formalin phosphate (Fisher Scientific) and embedded in paraffin.
Alcian blue staining
Tissue sections (5 μm thick) were deparaffinized and rehydrated via a series of graded ethanol washes, immersed three times in 3% acetic acid (EMD Biosciences), and incubated in 1% Alcian blue (Sigma) in 3% acetic Acid (pH 2.5) for 30 min. The slides were then washed with running water for 10 min, rinsed in distilled water, counterstained in Nuclear Red Solution (Diagnostic Biosystems) for 5 min, and washed with running water. After dehydration by successive immersions in 95% alcohol followed by absolute alcohol and CitriSolv xylene substitute (Fisher Scientific), the slides were mounted with Permount, sealed with coverslips, and imaged as described above.
Cylindrical smooth muscle tissue equivalents
Tissue equivalents containing HF-SMCs or human vascular (V)-SMCs were prepared as described previously.10,12,13 Briefly, human HF-SMCs or V-SMCs were suspended in thrombin and mixed with fibrinogen at a ratio of 1:4, yielding fibrin gels with a final concentration of fibrinogen and thrombin at 2.5 mg/mL and 2.5 U/mL, respectively. For each tissue, 1 mL of fibrin containing 1 × 106 cells was polymerized around a 6.0 mm mandrel of poly(di-methyl siloxane) placed in the middle of each well. After 1 h the gels were detached from the walls and incubated in 2 mL of DMEM containing 10% FBS supplemented with 2 μg/mL insulin, 2 ng/mL TGF-β1, 300 μM ascorbic acid phosphate, and 2 mg/mL ɛ-amino-n-caproic acid. Thereafter, the cell culture medium was replenished every 3 days.
Contractility and mechanical properties of tissue equivalents
After 2 weeks in culture, the tissue constructs were photographed, wall thickness was measured using ImageJ, and the length of each construct was measured with a digital micrometer. Then, the tissues were released from the mandrel and mounted on two stainless hooks in an isolated tissue bath and incubated in Krebs–Ringer solution. The tissues were continuously bubbled with 94% O2, 6% CO2 to obtain a pH of 7.4, a 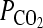 of 38 mmHg, and a
of 38 mmHg, and a 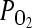 > 500 mm Hg at 37°C. Each construct was mounted on stainless steel hooks through the lumen: one was fixed and the other one was connected to a force transducer. Tissues were equilibrated at a basal tension of 1.0 g and constant length for 30–60 min. After equilibration, endothelin-1 (20 nM), the thromboxane A2 mimetic U46619 (1 μM), or KCl (118 mM) was added to the tissue bath, and isometric contraction was recorded using a PowerLab data acquisition unit and analyzed with Chart5 software (ADInstruments). For measuring mechanical properties, tissue equivalents were mounted on the force transducer and stretched incrementally until they broke, yielding the ultimate tensile strength (UTS) and break length of each tissue. The initial tissue length corresponds to the length under a passive tension of 1.0 g. Contractility and UTS were normalized by the tissue area applying force and expressed in kPa.
> 500 mm Hg at 37°C. Each construct was mounted on stainless steel hooks through the lumen: one was fixed and the other one was connected to a force transducer. Tissues were equilibrated at a basal tension of 1.0 g and constant length for 30–60 min. After equilibration, endothelin-1 (20 nM), the thromboxane A2 mimetic U46619 (1 μM), or KCl (118 mM) was added to the tissue bath, and isometric contraction was recorded using a PowerLab data acquisition unit and analyzed with Chart5 software (ADInstruments). For measuring mechanical properties, tissue equivalents were mounted on the force transducer and stretched incrementally until they broke, yielding the ultimate tensile strength (UTS) and break length of each tissue. The initial tissue length corresponds to the length under a passive tension of 1.0 g. Contractility and UTS were normalized by the tissue area applying force and expressed in kPa.
Statistical analysis
Data were expressed as mean ± standard deviation, and statistical significance (defined as p < 0.05) was determined using Student's t-test.
Results
Isolation of cells from individual hair follicles
After collagenase digestion, hair follicles were released from the skin and washed extensively, and individual follicles were placed each in a well of a 24-well plate to allow for cell migration onto the tissue culture plastic. Mesenchymal cells migrating from individual follicles were expanded and pooled from several follicles of a 41-year-old woman or a 69-year-old man donor and cultured in the presence or absence of bFGF as indicated. Some wells were populated with epidermal keratinocytes from the bulge region of the hair follicles and were excluded from this study. As expected from previous studies with MSCs from other sources, in the presence of bFGF (1 ng/mL) HFCs appeared smaller and spindle-shaped (Fig. 1A) and proliferated faster, yielding 12-fold increase in cell number after 3 weeks in culture (Fig. 1B). In the bFGF-containing medium, HFCs could be routinely subcultured for at least 12–13 passages without exhausting their proliferative potential.
FIG. 1.
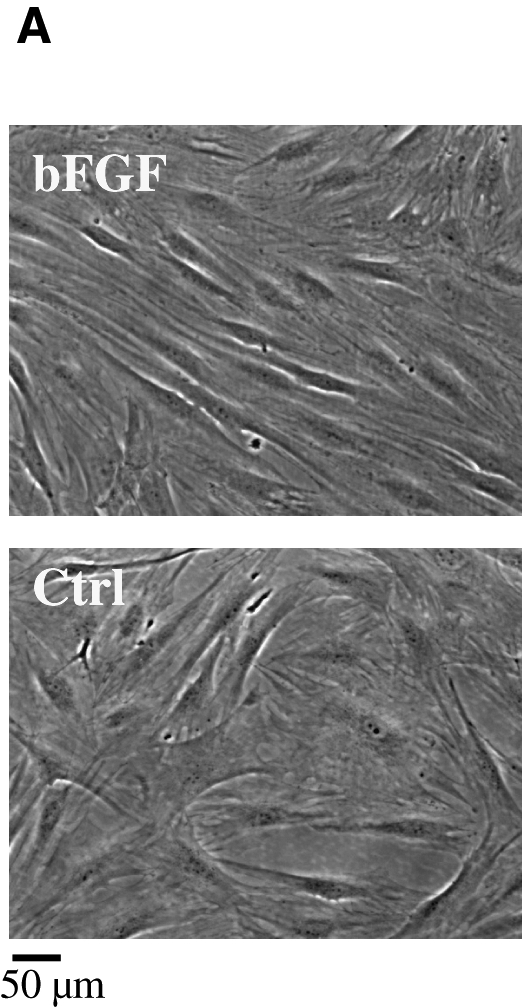

HFC morphology and proliferation potential. HFCs were plated in six-well plates at 104 cells/well and cultured in DMEM with 10% FBS alone (Ctrl) or with 2 ng/mL bFGF. (A) Photomicrograph of HFCs in the presence or absence of bFGF. (B) Cells were cultured in the presence or absence of bFGF and at the indicated times trypsinized, counted, and re-plated at the same density. The cumulative cell number was plotted as mean ± SD of three independent experiments (n = 3). HFCs, hair follicle stem cells; bFGF, basic fibroblast growth factor; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; SD, standard deviation.
HFCs displayed surface markers and multilineage differentiation potential similar to BM-MSCs
The mesenchymal morphology prompted us to examine whether HFCs expressed surface markers that characterize BM-MSCs. As shown in Figure 2 HFCs displayed nearly the same molecular profile as BM-MSCs. Specifically, HFCs expressed high levels of CD44, CD73, CD90, and CD105, but lacked CD34, CD45, or CD144. This result suggested that human HFCs might display the multilineage differentiation potential that characterizes BM-MSCs.
FIG. 2.

HFC surface marker profile. Flow cytometry with antibodies for mesenchymal stem-cell-specific markers as indicated. Representative flow cytometry plots from three independent experiments are shown. Color images available online at www.liebertonline.com/ten.
To address this hypothesis we examined whether HFCs could differentiate into fat, bone, or cartilage cells under appropriate differentiation conditions.11 Indeed, we found that under conditions that promote adipogenesis, HFCs expressed adipocyte-specific transcription factors aP2 and peroxisome proliferator-activated receptor (PPAR)-γ (Fig. 3A) and accumulated triglycerides that were observed by staining with Oil Red O (Fig. 3B–D). Under conditions that are known to promote chondrogenic differentiation (cell pellets cultured in chondrogenic differentiation medium), HFCs expressed the genes for collagen II and Sox9 (Fig. 3E) and immunostaining confirmed collagen II expression at the protein level (Fig. 3F, G). In the presence of the osteogenic medium, HFCs expressed osteogenic markers such as alkaline phosphatase, Runx2, and osteocalcin (Fig. 4A). Immunostaining demonstrated alkaline phosphatase activity in differentiated cells (Fig. 4B) but not in control cells (Fig. 4C), and von Kossa staining revealed calcium deposits (Fig. 4D, E). These results demonstrate that HFCs are similar to BM-MSCs in terms of the surface marker profile and multilineage differentiation potential.
FIG. 3.

HFCs demonstrated adipogenic and chondrogenic differentiation potential. HFCs were cultured in the adipogenic (A–D) or chondrogenic (E–G) differentiation medium for 2 weeks. (A) reverse-transcription (RT)-PCR for adipogenic markers aP2 and PPARγ. Oil Red O staining of HFCs that were treated with the adipogenic (B) or control (C) medium. (D) Higher magnification of (B). (E) RT-PCR for chondrogenic markers Sox9 and Col II. Type II collagen immunostaining of HFC pellets cultured with chondrogenic (F) or control (G) medium. Cells stained with secondary antibody only served as negative control. All samples were counterstained with 4′,6-diamidino-2-phenylindole (DAPI): to observe cell nuclei. Representative images are shown from one out of three independent experiments. PCR, polymerase chain reaction; GAPDH, glyceraldehyde 3-phosphate dehydrogenase. Color images available online at www.liebertonline.com/ten.
FIG. 4.

HFCs demonstrated osteogenic differentiation potential. HFCs were cultured in osteogenic differentiation medium for 2 weeks. (A) RT-PCR for osteogenic markers alkaline phosphatase (ALP), Runx2, and osteocalcin (OC). Alkaline phosphates activity of HFCs that were treated with the osteogenic (B) or control (C) medium. von Kossa staining of HFCs that were cultured in the osteogenic (D) or control (E) medium for 4 weeks. Representative images are shown from one out of three independent experiments. Color images available online at www.liebertonline.com/ten.
We also examined whether HFCs were able to differentiate into SMCs. To this end, the cells were cultured in the myogenic differentiation medium containing 2 ng/mL of TGF-β1, a cytokine that has been shown to promote differentiation of embryonic stem cells14 or BM-MSCs15–17 into mature contractile SMCs. Under these conditions, RT-polymerase chain reaction showed increased expression of SMC markers that have been associated with contractility such as αSMA and smoothelin (Fig. 5A). Analysis at the protein level using flow cytometry demonstrated that in the presence of the myogenic medium the percentage of αSMA-expressing cells increased from 9.3% ± 1.6% to 67.5% ± 2.0% and the mean fluorescence intensity increased from 55.5 ± 2.3 to 205.8 ± 11.9 fluorescence intensity units (p < 0.05, n = 3). In agreement, immunofluorescence microscopy confirmed increased expression of αSMA and calponin, and revealed that both proteins were organized in a network of well-defined fibers (Fig. 5B). Note that to enable observation of control cells the exposure time was much longer (723 ms) compared with cells cultured in the myogenic medium (160 ms).
FIG. 5.


HFCs demonstrated myogenic differentiation potential and force generation ability. HFCs were cultured in the myogenic differentiation medium for 1 week. (A) RT-PCR for αSMA, and smoothelin. (B) Immunostaining for αSMA and calponin. Cells stained with Alexa 488 secondary antibody only served as negative control. All samples were counterstained with DAPI to observe the nuclei. Representative images are shown from one out of three independent experiments. (C) HFCs were cultured in the presence of bFGF (2 ng/mL) for 5 days until they reached 85%–90% confluence. At that time the cells were trypsinized and embedded in fibrin that was allowed to polymerize in 24-well plates to form disks. One hour after polymerization, the gels were detached from the walls and allowed to compact in the presence of FBS alone or supplemented with bFGF (2 ng/mL) or TGF-β1 (2 ng/mL). At the indicated times, gels were photographed and their area was measured using ImageJ software. The ratio of gel area at the indicated times over the initial area was plotted as percentage of initial hydrogel area over time. (D) HFCs were cultured in DMEM with 10% FBS alone or supplemented with bFGF (2 ng/mL) or TGF-β1 (2 ng/mL) for 5 days. At that time, the cells were embedded in fibrin hydrogels that were incubated with the medium of the same composition. The percentage of initial hydrogel area was plotted over time. All values represent the mean ± SD of triplicate samples in a representative experiment (n = 3). Asterisks denote p < 0.05 between the TGF-β1- and FBS-treated hydrogels at the same time point. bFGF-treated samples were significantly different (p < 0.05) than either the TGF-β1- or FBS-treated samples at all times. P-αSMA, smooth muscle alpha-actin promoter; TGF-β1, transforming growth factor-beta 1. Color images available online at www.liebertonline.com/ten.
Fibrin gel contraction by HFCs: Effects of bFGF and TGF-β1
The defining property of SMCs is their ability to generate force, which has been previously correlated with expression of αSMA.18,19 Since the mRNA and protein levels of αSMA were low in the bFGF-containing growth medium and high in the TGF-β1 containing differentiation medium, we examined the effect of bFGF or TGF-β1 on the ability of HFCs to generate force. To this end, HFCs were cultured in the presence of bFGF (2 ng/mL) for 7 days before they were embedded (106 cells/mL) in fibrin hydrogels and overlaid with the medium containing 10% FBS alone or supplemented with bFGF (2 ng/mL) or TGF-β1 (2 ng/mL). An hour after gelation the hydrogels were released from the walls and gel compaction was monitored by measuring the area of each gel at the indicated times. As shown in Figure 5C, after a 48 h delay, HFCs contracted fibrin hydrogels significantly to ∼20% of their initial surface area within ∼7 days (169 h) when incubated with FBS or TGF-β1. The time to compact to 50% of the original area was approximately 60 h (t50% = 60 h). Treatment with bFGF abolished fibrin contraction, suggesting that bFGF adversely affected the ability of HFCs to develop a contractile phenotype.
We also examined whether pretreatment of HFCs with bFGF or TGF-β1 influenced HFC contractility. To this end, HFCs were cultured in the presence of bFGF, TGF-β1, or FBS alone for one week and then embedded in fibrin hydrogels and incubated under the same conditions for the indicated times. Compaction of fibrin hydrogels prepared with cells that were pretreated with TGF-β1 occurred much faster (t50%∼15 h) and without lag-phase reaching less than 10% of their original area within 48 h (Fig. 5D). Removal of bFGF during pretreatment (FBS alone) had similar effect, suggesting that bFGF prevented differentiation of HFCs toward a contractile SMC phenotype.
Isolation of contractile SMCs using tissue-specific promoters
These results clearly indicated that HFCs could differentiate into contractile SMCs and suggested that obtaining a relatively pure population of these cells could facilitate their use in tissue engineering and cell therapies. To this end, we employed smooth-muscle-specific promoters to identify and purify SMCs from HFCs and examined their contractile function. Since different promoters were previously shown to derive different SMC populations from embryonic stem cells,20 we employed two promoters, namely, αSMA and MHC, representing an early and a late marker gene of SMC differentiation. To this end, we generated two lentiviral vectors encoding either for ZsGreen (a bright variant of enhanced green fluorescent protein) under the αSMA promoter (P-αSMA) or DsRed under the MHC promoter (P-MHC) (Fig. 6A). As bFGF decreased αSMA expression, HFCs were cultured in the absence of bFGF for 5–7 days before lentivirus gene transfer and subsequent sorting of green or red cells, respectively.
FIG. 6.


Derivation of SMCs from HFCs using tissue-specific promoters. (A) Schematics of lentiviral vectors encoding for ZsGreen under the control of αSMA promoter (P-αSMA) or DsRed under the P-MHC. (B) HFCs were transduced with either lentivirus and P-αSMA (ZsGreen+) or P-MHC (DsRed+) cells were sorted by flow cytometry. Fluorescence images of P-αSMA (ZsGreen+) or P-MHC (DsRed+) that were cultured in the presence or absence of bFGF (2 ng/mL). (C) Sorted cells were plated in six-well plates (104 cells/well) and cultured in the presence or absence of bFGF (2 ng/mL). On day 7, the cells were trypsinized and counted, and the cell number was plotted as mean ± SD of triplicate samples in a representative experiment (n = 3). SMCs, smooth muscle cells; P-MHC, myosin heavy chain promoter. Color images available online at www.liebertonline.com/ten.
P-MHC cells (DsRed + , ∼5% of the total population) appeared large and exhibited slow proliferation, consistent with a mature SMC phenotype. On the other hand, P-αSMA cells (ZsGreen + , ∼17% of the total population) were smaller and elongated, and proliferated faster, especially in the presence of bFGF (Fig. 6B, C). Due to greater potential for expansion, the rest of the experiments were conducted with P-αSMA cells.
bFGF decreases αSMA expression and contractility of P-αSMA
Since bFGF prevented myogenic differentiation of HFCs, we examined whether bFGF had an effect on αSMA expression. As shown in Figure 7A the percentage of HF-SMCs carrying active P-αSMA (i.e., ZsGreen+ cells) decreased modestly from ∼99% to ∼91%, but the mean fluorescence intensity decreased by ∼40% after bFGF treatment (Fig. 7B). In agreement, flow cytometry using an antibody against αSMA showed that the percentage of αSMA-positive cells decreased somewhat from 92% to 85% (Fig. 7C) and the mean fluorescence intensity decreased by ∼50% (Fig. 7D). In contrast, TGF-β1 did not affect the level of αSMA expression compared to P-αSMA cells cultured in 10% FBS (data not shown). These results suggested that bFGF decreased but did not eliminate αSMA expression in P-αSMA cells.
FIG. 7.

HFC-derived P-αSMA cells decrease αSMA expression in response to bFGF. P-αSMA cells were seeded at 103 cells/cm2 and cultured in the presence or absence of bFGF (2 ng/mL). On day 7, the percentage of ZsGreen+ cells (A) and mean green fluorescence intensity (B) of ZsGreen+ cells were measured by flow cytometry. All values are the mean ± SD of triplicate samples in a representative experiment (n = 3). (C, D) P-αSMA cells were fixed, permeabilized, and stained mouse anti-αSMA followed by incubation with Alex Fluor 647-R-phycoerythin goat anti-mouse secondary antibody. The percentage of cells and the level of αSMA expression were measured by flow cytometry. (C) Percentage of αSMA+ cells and (D) mean green fluorescence intensity of αSMA+ cells. All values are the mean ± SD of triplicate samples in a representative experiment (n = 3). (E) P-αSMA cells were cultured in the presence of bFGF (2 ng/mL) for 5 days before embedding in fibrin gels that were allowed to compact in DMEM with 10% FBS alone or supplemented with bFGF (2 ng/mL) or TGF-β1 (2 ng/mL). The percentage of initial hydrogel area was plotted over time. All values represent the mean ± SD of triplicate samples in a representative experiment (n = 3). Symbols (*) and (#) denote significant difference (p < 0.05) between bFGF- and FBS- or bFGF- and TGF-β1-treated hydrogels, respectively, at the indicated time point.
In agreement with αSMA expression, bFGF delayed but did not abolish fibrin hydrogel compaction. Specifically, within the first 6 h the hydrogel volume decreased to ∼44% of the original volume for control, ∼50% for TGF-β1-treated samples, and ∼70% for bFGF-treated samples (p < 0.05 between bFGF and the other two conditions), respectively (Fig. 7E). On the other hand, the final extent of contraction was not significantly different between control (FBS alone), TGF-β1-, or bFGF-treated hydrogels. P-αSMA cells contracted fibrin hydrogels much faster than the original population of HFCs (t50% = 6 h for HF-SMCs compared to t50% = 58 h for HFCs in FBS alone) and even HFCs that were pretreated with TGF-β1 (t50% = 18 h), suggesting that αSMA cells comprised a more homogeneous population of SMCs than the original HFC population.
P-αSMA cells displayed significant receptor and nonreceptor mediated vascular reactivity
The defining property of mature V-SMCs is their ability to contract and generate force in response to vasoactive agonists. To examine whether P-αSMA cells exhibited functional properties of mature V-SMCs, we measured the isometric tension generated by cylindrical rings of fibrin-based tissue constructs prepared with P-αSMA cells or V-SMCs. To this end, cylindrical tissue equivalents were cultured around 6-mm-diameter mandrels in the presence of TGF-β1 (2ng/mL) and ascorbic acid (50 μg/mL) as we reported previously10,12,13 for 2 weeks. At that time, P-αSMA cells or V-SMC tissues compacted significantly reaching wall thickness of 0.22 ± 0.06 mm (n = 8) and 0.17 ± 0.04 mm (n = 4), respectively. In addition, P-αSMA cells distributed uniformly within the hydrogel (hematoxylin and eosin) and stained positive for αSMA and calponin (Fig. 8A–C).
FIG. 8.

Vascular constructs with P-αSMA cells expressed human vascular SMC markers. P-αSMA cells were embedded in fibrin hydrogels and cultured around 6-mm mandrels for 2 weeks to form rings. (A) Hematoxylin and eosin (H&E) staining showed uniform distribution of P-αSMA cells within fibrin hydrogels (scale bar = 100 μm). Immunostaining for (B) αSMA and (C) calponin (scale bar = 20 μm). Color images available online at www.liebertonline.com/ten.
The isometric tension was measured in response to non-receptor- and receptor-mediated vasoagonists, such as KCl (118 mM), endothelin-1 (20 nM), or the thromboxane mimetic U46619 (10−6 M), and representative plots of force versus time are shown in Figure 9A. As shown in Figure 9B vascular constructs prepared with P-αSMA cells (n = 4) exhibited contractility of similar magnitude as tissues prepared with V-SMCs (n = 4), suggesting that P-αSMA cells had developed active pathways of receptor- and non-receptor-mediated vasoconstriction.
FIG. 9.

Vascular constructs with P-αSMA cells demonstrated remarkable vasoreactivity. P-αSMA cells were embedded in fibrin hydrogels and cultured around 6-mm mandrels for 2 weeks to form rings. Vascular reactivity was measured using an isolated tissue bath system. (A) Representative graphs of isometric contraction over time in response to the indicated agonists. (B) Vascular reactivity (N/g dry tissue weight) in response to KCl (118 mM), endothelin (ET)-1 (20 nM), or U46619 (10−6 M). All values represent the mean ± SD of triplicate samples in a representative experiment (n = 3).
P-αSMA-cell-based tissue equivalents exhibited significantly better mechanical properties than those prepared with V-SMCs as evidenced by the higher UTS (P-αSMA cells: 490.54 ± 41.73 kPa, n = 8; V-SMCs: 160.20 ± 143.74 kPa, n = 4; p < 0.03) and Young's modulus (slope of the linear part of the stress–strain curve; P-αSMA cells: 401.77 ± 15.95 kPa, n = 8; V-SMCs: 157.35 ± 11.43 kPa, n = 4; p < 0.03). Taken together, these results clearly showed that P-αSMA cells generated force when placed in a three-dimensional matrix and contracted in response to vascular agonists. On the basis of these findings P-αSMA cells were termed hair follicle derived SMCs (HF-SMCs).
Discussion
The bulge area of the hair follicle has been identified as a rich source of epidermal stem cells several years ago,2,3 and recent studies using transgenic mice verified these observations4,5 and suggested that HF may contain pluripotent stem cells. The present results showed that HFCs express many of the surface markers that characterize MSCs such as CD44, CD73, CD90, and CD105. Even more important, HFCs displayed multipotency as they differentiated toward fat, bone, cartilage, and SMCs, suggesting that these cells may be very similar to MSCs derived from the BM, adipose, or other tissues. Two previous studies showed that rat hair follicle cells exhibited tri-lineage differentiation potential,7,8 but this is the first report to show that human HFCs exhibit similar properties. Although we recently showed that ovine hair follicle cells contained functional and contractile SMCs,9 they failed to differentiate toward osteogenic, adipogenic, or chondrogenic lineages (our data not shown), suggesting that interspecies differences may be significant.
Our results are consistent with several recent studies demonstrating the multipotency of rat or human skin dermal fibroblasts.21–25 Comparison between fibroblasts from interfollicular skin and hair follicles prompted some investigators to suggest that the niche of skin multipotent stem cells resides in the hair follicle.7,26,27 Indeed, two recent studies confirmed the multipotent nature of rat hair follicle cells and showed similar differentiation potential to BM-MSCs.7,8 Our results extent these findings to the human system and confirm that our isolation protocol renders cells from human hair follicles with the potential to differentiate toward osteogenic, adipogenic, chondrogenic, and myogenic lineage. Therefore, the hair follicle may be a readily accessible source of autologous human MSCs (hMSCs) that can be used for tissue engineering and regenerative medicine.
The HFCs that were used in the present study originated from two adult donors (over 40 years old), suggesting that these cells maintained their multipotency regardless of donor age, further supporting their potential for tissue regeneration. In the presence of the embryonic stem cell medium containing knockout serum and bFGF, human hair-follicle-derived cells formed neurospheres and remained undifferentiated for several passages while maintaining the ability to differentiate toward melanocytic, myogenic, and neuronal lineages.28 It is likely that our method of cell isolation by organ culture and outgrowth selected for mesenchymal cells were different from those forming neurospheres. It would be interesting to examine the effect of the embryonic stem cell medium and gelatin in preventing differentiation and maintaining the pluripotency of HFCs.
Using tissue-specific promoters, namely, P-αSMA and P-MHC, and fluorescence-activated cell sorting, we isolated a population of SMCs from HFCs. HFCs isolated using P-MHC appeared flat, large cells with limited proliferation potential, possibly due to terminal differentiation. In contrast, use of P-αSMA yielded a proliferative population of HFCs in agreement with previous results with ovine hair follicle and BM-SMCs.9,10 Using biochemical and functional assays, we showed that P-αSMA cells expressed several markers of the SMC lineage, including αSMA, calponin, and MHC. What is more, P-αSMA cells exhibited contractile phenotype as evidenced by hydrogel compaction and strong contractility in response to vasoactive agonists acting through receptor- or non-receptor-mediated pathways. These results suggest that HF-SMCs may be a great source of functional SMCs for tissue engineering of blood vessels as well as other SMC-containing tissues, including bladder, urethra, and intestine.
Recent studies correlated the magnitude of cell traction forces and hydrogel contractility with the level of αSMA expression in corneal stromal cells, subcutaneous fibroblasts, or lung fibroblasts.18,19 In addition, application of bFGF-containing collagen sponges in full-thickness wounds reduced αSMA expression and wound contraction in a rat model.29,30 In agreement, we showed that treatment of HFCs with bFGF decreased αSMA fibers significantly and diminished the ability of these cells to contract fibrin. Interestingly, the effect of bFGF on P-αSMA was less dramatic. Although bFGF decreased the level of αSMA per cell, the majority of cells remained αSMA-positive and continued to contract fibrin hydrogels though to a lesser extent than control cells. Since P-αSMA cells were sorted from the total population of HFCs based on the highest level of P-αSMA expression, the αSMA reduction induced by bFGF might not have been enough to impair their contractile ability. This result also suggests that bFGF is very effective in preventing differentiation of HFCs toward the myogenic lineage, but it may be less effective in affecting high αSMA-expressing cells.
Several studies showed that bFGF increased proliferation and prevented differentiation of BM-MSCs (hMSCs)31,32 and embryonic stem cells.33 For example, bFGF has been shown to promote proliferation but prevent osteogenic34 or adipogenic35 differentiation of hMSCs. Recent studies started to reveal that bFGF may promote proliferation and maintain pluripotency through distinct pathways. The effect of bFGF on hMSC proliferation was mediated via Erk2 but not Erk1 mitogen-activated kinase.35 On the other hand, reduced myogenic differentiation by bFGF was attributed to decreased TGF-β2 expression,36 but the mechanism remains elusive. It will be interesting to determine how bFGF prevents expression of genes that induce differentiation of HFCs to the myogenic lineage.
Recent studies demonstrated that introduction of four transcription factors was sufficient to induce de-differentiation of human adult stem cells to embryonic-like stem cells, which retain high proliferation potential and pluripotency.37–40 Although most studies employed human skin fibroblasts, the process was 100 times more efficient for epidermal keratinocytes,41 which are highly clonogenic and well adapted to regenerate the epidermis repeatedly throughout life. Since mesenchymal-like stem cells from HF exhibit multilineage differentiation potential, it would be interesting to investigate whether they might be more readily reprogrammable into an embryonic like state. The ease of accessibility makes the hair follicle an ideal source of adult stem cells or iPS cells for tissue engineering and regenerative medicine.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01 HL086582) and New York State Stem Cell Fund (NYSTEM, Contract no. C024316). The authors would also like to thank Vivek Bajpai and Panagiotis Mistriotis for their help with flow cytometry experiments.
Disclosure Statement
No competing financial interests exist.
References
- 1.Alonso L. Fuchs E. The hair cycle. J Cell Sci. 2006;119:391. doi: 10.1242/jcs.02793. [DOI] [PubMed] [Google Scholar]
- 2.Cotsarelis G. Sun T.T. Lavker R.M. Label-retaining cells reside in the bulge area of pilosebaceous unit: implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell. 1990;61:1329. doi: 10.1016/0092-8674(90)90696-c. [DOI] [PubMed] [Google Scholar]
- 3.Morris R.J. Potten C.S. Highly persistent label-retaining cells in the hair follicles of mice and their fate following induction of anagen. J Invest Dermatol. 1999;112:470. doi: 10.1046/j.1523-1747.1999.00537.x. [DOI] [PubMed] [Google Scholar]
- 4.Tumbar T. Guasch G. Greco V. Blanpain C. Lowry W.E. Rendl M. Fuchs E. Defining the epithelial stem cell niche in skin. Science. 2004;303:359. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morris R.J. Liu Y. Marles L. Yang Z. Trempus C. Li S. Lin J.S. Sawicki J.A. Cotsarelis G. Capturing and profiling adult hair follicle stem cells. Nat Biotechnol. 2004;22:411. doi: 10.1038/nbt950. [DOI] [PubMed] [Google Scholar]
- 6.Lako M. Armstrong L. Cairns P.M. Harris S. Hole N. Jahoda C.A. Hair follicle dermal cells repopulate the mouse haematopoietic system. J Cell Sci. 2002;115:3967. doi: 10.1242/jcs.00060. [DOI] [PubMed] [Google Scholar]
- 7.Jahoda C.A. Whitehouse J. Reynolds A.J. Hole N. Hair follicle dermal cells differentiate into adipogenic and osteogenic lineages. Exp Dermatol. 2003;12:849. doi: 10.1111/j.0906-6705.2003.00161.x. [DOI] [PubMed] [Google Scholar]
- 8.Hoogduijn M.J. Gorjup E. Genever P.G. Comparative characterization of hair follicle dermal stem cells and bone marrow mesenchymal stem cells. Stem Cells Dev. 2006;15:49. doi: 10.1089/scd.2006.15.49. [DOI] [PubMed] [Google Scholar]
- 9.Liu J.Y. Peng H.F. Andreadis S.T. Contractile smooth muscle cells derived from hair-follicle stem cells. Cardiovasc Res. 2008;79:24. doi: 10.1093/cvr/cvn059. [DOI] [PubMed] [Google Scholar]
- 10.Liu J.Y. Swartz D.D. Peng H.F. Gugino S.F. Russell J.A. Andreadis S.T. Functional tissue-engineered blood vessels from bone marrow progenitor cells. Cardiovasc Res. 2007;75:618. doi: 10.1016/j.cardiores.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 11.Zuk P.A. Zhu M. Mizuno H. Huang J. Futrell J.W. Katz A.J. Benhaim P. Lorenz H.P. Hedrick M.H. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng. 2001;7:211. doi: 10.1089/107632701300062859. [DOI] [PubMed] [Google Scholar]
- 12.Yao L. Swartz D.D. Gugino S.F. Russell J.A. Andreadis S.T. Fibrin-based tissue-engineered blood vessels: differential effects of biomaterial and culture parameters on mechanical strength and vascular reactivity. Tissue Eng. 2005;11:991. doi: 10.1089/ten.2005.11.991. [DOI] [PubMed] [Google Scholar]
- 13.Swartz D.D. Russell J.A. Andreadis S.T. Engineering of fibrin-based functional and implantable small-diameter blood vessels. Am J Physiol Heart Circ Physiol. 2005;288:H1451. doi: 10.1152/ajpheart.00479.2004. [DOI] [PubMed] [Google Scholar]
- 14.Sinha S. Hoofnagle M.H. Kingston P.A. McCanna M.E. Owens G.K. Transforming growth factor-beta1 signaling contributes to development of smooth muscle cells from embryonic stem cells. Am J Physiol Cell Physiol. 2004;287:C1560. doi: 10.1152/ajpcell.00221.2004. [DOI] [PubMed] [Google Scholar]
- 15.Kinner B. Zaleskas J.M. Spector M. Regulation of smooth muscle actin expression and contraction in adult human mesenchymal stem cells. Exp Cell Res. 2002;278:72. doi: 10.1006/excr.2002.5561. [DOI] [PubMed] [Google Scholar]
- 16.Wang D. Park J.S. Chu J.S. Krakowski A. Luo K. Chen D.J. Li S. Proteomic profiling of bone marrow mesenchymal stem cells upon transforming growth factor beta1 stimulation. J Biol Chem. 2004;279:43725. doi: 10.1074/jbc.M407368200. [DOI] [PubMed] [Google Scholar]
- 17.Ross J.J. Hong Z. Willenbring B. Zeng L. Isenberg B. Lee E.H. Reyes M. Keirstead S.A. Weir E.K. Tranquillo R.T. Verfaillie C.M. Cytokine-induced differentiation of multipotent adult progenitor cells into functional smooth muscle cells. J Clin Invest. 2006;116:3139. doi: 10.1172/JCI28184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hinz B. Celetta G. Tomasek J.J. Gabbiani G. Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. 2001;12:2730. doi: 10.1091/mbc.12.9.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J. Li H. SundarRaj N. Wang J.H. Alpha-smooth muscle actin expression enhances cell traction force. Cell Motil Cytoskeleton. 2007;64:248. doi: 10.1002/cm.20178. [DOI] [PubMed] [Google Scholar]
- 20.Sinha S. Wamhoff B.R. Hoofnagle M.H. Thomas J. Neppl R.L. Deering T. Helmke B.P. Bowles D.K. Somlyo A.V. Owens G.K. Assessment of contractility of purified smooth muscle cells derived from embryonic stem cells. Stem Cells. 2006;24:1678. doi: 10.1634/stemcells.2006-0002. [DOI] [PubMed] [Google Scholar]
- 21.Toma J.G. Akhavan M. Fernandes K.J. Barnabe-Heider F. Sadikot A. Kaplan D.R. Miller F.D. Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat Cell Biol. 2001;3:778. doi: 10.1038/ncb0901-778. [DOI] [PubMed] [Google Scholar]
- 22.Fernandes K.J. McKenzie I.A. Mill P. Smith K.M. Akhavan M. Barnabe-Heider F. Biernaskie J. Junek A. Kobayashi N.R. Toma J.G. Kaplan D.R. Labosky P.A. Rafuse V. Hui C.C. Miller F.D. A dermal niche for multipotent adult skin-derived precursor cells. Nat Cell Biol. 2004;6:1082. doi: 10.1038/ncb1181. [DOI] [PubMed] [Google Scholar]
- 23.Toma J.G. McKenzie I.A. Bagli D. Miller F.D. Isolation and characterization of multipotent skin-derived precursors from human skin. Stem Cells. 2005;23:727. doi: 10.1634/stemcells.2004-0134. [DOI] [PubMed] [Google Scholar]
- 24.Chen F.G. Zhang W.J. Bi D. Liu W. Wei X. Chen F.F. Zhu L. Cui L. Cao Y. Clonal analysis of nestin(−) vimentin(+) multipotent fibroblasts isolated from human dermis. J Cell Sci. 2007;120:2875. doi: 10.1242/jcs.03478. [DOI] [PubMed] [Google Scholar]
- 25.Lorenz K. Sicker M. Schmelzer E. Rupf T. Salvetter J. Schulz-Siegmund M. Bader A. Multilineage differentiation potential of human dermal skin-derived fibroblasts. Exp Dermatol. 2008;17:925. doi: 10.1111/j.1600-0625.2008.00724.x. [DOI] [PubMed] [Google Scholar]
- 26.Jahoda C.A. Cell movement in the hair follicle dermis—more than a two-way street? J Invest Dermatol. 2003;121:ix. doi: 10.1111/j.1523-1747.2003.12585.x. [DOI] [PubMed] [Google Scholar]
- 27.Sellheyer K. Krahl D. Cutaneous mesenchymal stem cells: status of current knowledge, implications for dermatopathology. J Cutan Pathol. 2009 doi: 10.1111/j.1600-0560.2009.01477.x. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 28.Yu H. Fang D. Kumar S.M. Li L. Nguyen T.K. Acs G. Herlyn M. Xu X. Isolation of a novel population of multipotent adult stem cells from human hair follicles. Am J Pathol. 2006;168:1879. doi: 10.2353/ajpath.2006.051170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ono I. Tateshita T. Inoue M. Effects of a collagen matrix containing basic fibroblast growth factor on wound contraction. J Biomed Mater Res. 1999;48:621. doi: 10.1002/(sici)1097-4636(1999)48:5<621::aid-jbm5>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 30.Akasaka Y. Ono I. Tominaga A. Ishikawa Y. Ito K. Suzuki T. Imaizumi R. Ishiguro S. Jimbow K. Ishii T. Basic fibroblast growth factor in an artificial dermis promotes apoptosis and inhibits expression of alpha-smooth muscle actin, leading to reduction of wound contraction. Wound Repair Regen. 2007;15:378. doi: 10.1111/j.1524-475X.2007.00240.x. [DOI] [PubMed] [Google Scholar]
- 31.Tsutsumi S. Shimazu A. Miyazaki K. Pan H. Koike C. Yoshida E. Takagishi K. Kato Y. Retention of multilineage differentiation potential of mesenchymal cells during proliferation in response to FGF. Biochem Biophys Res Commun. 2001;288:413. doi: 10.1006/bbrc.2001.5777. [DOI] [PubMed] [Google Scholar]
- 32.Solchaga L.A. Penick K. Porter J.D. Goldberg V.M. Caplan A.I. Welter J.F. FGF-2 enhances the mitotic and chondrogenic potentials of human adult bone marrow-derived mesenchymal stem cells. J Cell Physiol. 2005;203:398. doi: 10.1002/jcp.20238. [DOI] [PubMed] [Google Scholar]
- 33.Vallier L. Alexander M. Pedersen R.A. Activin/Nodal and FGF pathways cooperate to maintain pluripotency of human embryonic stem cells. J Cell Sci. 2005;118:4495. doi: 10.1242/jcs.02553. [DOI] [PubMed] [Google Scholar]
- 34.Go M.J. Takenaka C. Ohgushi H. Forced expression of Sox2 or Nanog in human bone marrow derived mesenchymal stem cells maintains their expansion and differentiation capabilities. Exp Cell Res. 2008;314:1147. doi: 10.1016/j.yexcr.2007.11.021. [DOI] [PubMed] [Google Scholar]
- 35.Carcamo-Orive I. Tejados N. Delgado J. Gaztelumendi A. Otaegui D. Lang V. Trigueros C. ERK2 protein regulates the proliferation of human mesenchymal stem cells without affecting their mobilization and differentiation potential. Exp Cell Res. 2008;314:1777. doi: 10.1016/j.yexcr.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 36.Ito T. Sawada R. Fujiwara Y. Seyama Y. Tsuchiya T. FGF-2 suppresses cellular senescence of human mesenchymal stem cells by down-regulation of TGF-beta2. Biochem Biophys Res Commun. 2007;359:108. doi: 10.1016/j.bbrc.2007.05.067. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi K. Tanabe K. Ohnuki M. Narita M. Ichisaka T. Tomoda K. Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 38.Zhao R. Daley G.Q. From fibroblasts to iPS cells: induced pluripotency by defined factors. J Cell Biochem. 2008;105:949. doi: 10.1002/jcb.21871. [DOI] [PubMed] [Google Scholar]
- 39.Park I.H. Zhao R. West J.A. Yabuuchi A. Huo H. Ince T.A. Lerou P.H. Lensch M.W. Daley G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 40.Park I.H. Arora N. Huo H. Maherali N. Ahfeldt T. Shimamura A. Lensch M.W. Cowan C. Hochedlinger K. Daley G.Q. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aasen T. Raya A. Barrero M.J. Garreta E. Consiglio A. Gonzalez F. Vassena R. Bilic J. Pekarik V. Tiscornia G. Edel M. Boue S. Belmonte J.C. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26:1276. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]