

Abstract
Increasing evidence suggests that phosphorylation may play an important role in the oligomerization, fibrillogenesis, Lewy body (LB) formation, and neurotoxicity of α-synuclein (α-syn) in Parkinson disease. Herein we demonstrate that α-syn is phosphorylated at S87 in vivo and within LBs. The levels of S87-P are increased in brains of transgenic (TG) models of synucleinopathies and human brains from Alzheimer disease (AD), LB disease (LBD), and multiple system atrophy (MSA) patients. Using antibodies against phosphorylated α-syn (S129-P and S87-P), a significant amount of immunoreactivity was detected in the membrane in the LBD, MSA, and AD cases but not in normal controls. In brain homogenates from diseased human brains and TG animals, the majority of S87-P α-syn was detected in the membrane fractions. A battery of biophysical methods were used to dissect the effect of S87 phosphorylation on the structure, aggregation, and membrane-binding properties of monomeric α-syn. These studies demonstrated that phosphorylation at S87 expands the structure of α-syn, increases its conformational flexibility, and blocks its fibrillization in vitro. Furthermore, phosphorylation at S87, but not S129, results in significant reduction of α-syn binding to membranes. Together, our findings provide novel mechanistic insight into the role of phosphorylation at S87 and S129 in the pathogenesis of synucleinopathies and potential roles of phosphorylation in α-syn normal biology.
Introduction
Increasing evidence suggests that phosphorylation may play an important role in the fibrillogenesis, Lewy body (LB) formation, and neurotoxicity of α-synuclein (α-syn) in vivo (Fujiwara et al., 2002; Anderson et al., 2006). Immunohistochemical and biochemical studies revealed that the majority of α-syn within inclusions isolated from patients with Parkinson disease (PD) and other synucleinopathies (Kahle et al., 2000; Okochi et al., 2000; Fujiwara et al., 2002; Takahashi et al., 2003a; Anderson et al., 2006) is phosphorylated at S129 (S129-P). Proteinaceous inclusions formed in cellular and animal models overexpressing wild-type (WT) or mutant α-syn (Lo Bianco et al., 2002; Takahashi et al., 2003a; Yamada et al., 2004; Chen and Feany, 2005) can also be stained with an antibody against S129-P. Although the kinases and phosphatases responsible for regulating α-syn phosphorylation at S129 in vivo are still not known, a series of in vitro and cell culture-based studies have identified a number of kinases, which phosphorylate α-syn at S129 and/or S87, including CKI (S87 and S129) and CKII (S129) (Okochi et al., 2000) and the G protein-coupled receptor kinases (GRKs 1, 2, 5, and 6, S129) (Pronin et al., 2000), LRRK2 (S129) (Qing et al., 2009), and Polo-Like Kinases (S129) (Inglis et al., 2009; Mbefo et al., 2010).
A close comparison of the amino acid sequences of all synucleins from humans and other species shows that the majority of potential α-syn phosphorylation sites, including Y125, S129, Y133, and Y136, are highly conserved in all species (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Considering all the α-syn phosphorylation sites identified in vivo [S129 (Fujiwara et al., 2002; Chen and Feany, 2005)] and in vitro [S87 (Okochi et al., 2000; Kim et al., 2006), S129 (Okochi et al., 2000; Pronin et al., 2000; Fujiwara et al., 2002; Takahashi et al., 2003a; Chen and Feany, 2005; Kim et al., 2006), Y125, Y133, and Y136 (Ellis et al., 2001; Nakamura et al., 2001; Ahn et al., 2002; Negro et al., 2002; Takahashi et al., 2003b)], it is striking that only S87 lies in the hydrophobic non-amyloid component (NAC) region of α-syn, which is essential for α-syn aggregation and fibrillogenesis (El-Agnaf et al., 1998). S87 is one of the few residues and phosphorylation sites that distinguish the human α-syn sequence from that of mouse and rat α-syn (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). It is noteworthy that the sequences of mouse and rat α-syn contains the PD-associated mutation A53T, but do not develop PD pathology and symptoms. Therefore, we hypothesized that S87 might be an important contributor to PD-like pathology and symptoms induced in human, but not mouse and rat α-syn, where this residue is absent.
These observations prompted us to investigate the physiological relevance of S87 phosphorylation in vivo and its consequences on the structural and aggregation properties of α-syn in vitro. Toward these goals, we generated an antibody against phosphor-S87 and used it to investigate the presence of S87-P in LBs and compare the levels of S87-P α-syn in the brains of transgenic (TG) models of α-synucleinopathies as well as in human Alzheimer disease (AD), Lewy body disease (LBD), and multiple system atrophy (MSA) brains. To examine further the role of S87 phosphorylation in modulating the structural, aggregation, and membrane-binding properties of α-syn, we carried out a thorough comparative characterization of the phosphomimic S87E and the in vitro phosphorylated S87-P α-syn, as well as the S87A and WT α-syn using an array of biochemical and biophysical methods. Our findings and their implications for the understanding of the normal biology of α-syn and its role in the pathogenesis of PD are discussed.
Materials and Methods
Validation of the anti-S87P antibody in vivo
Plasmid construction and production of recombinant AAV2/6 viral vectors.
A single-nucleotide mutation coding for S87D was introduced into the cDNA of human α-syn already containing the A30P pathogenic mutation using the QuickChange Site-Directed Mutagenesis kit (Stratagene). Beforehand, the A30P α-syn cDNA had been cloned into the AAV-CMV-MCS backbone (Stratagene). The S129A mutation had been generated previously (Azeredo da Silveira et al., 2009). For the S87D variant, the mutagenesis primers were: sense 5′-GGAGGGAGCAGGGGACATTGCAGCAGCCAC-3′ and antisense 5′-GTGGCTGCTGCAATGTCCCCTGCTCCCTCC-3′. Production and titration of the recombinant pseudotyped AAV2/6 vectors (serotype 2 genome/serotype 6 capsid) were performed as previously described by Azeredo da Silveira et al. (2009).
Stereotaxic injections.
Male Wistar rats (Charles River Laboratories), weighing 180–200 g at the time of surgery were maintained under 12 h light/dark cycle, with ad libitum access to food and water, in accordance with the Swiss legislation and the European Community council directive (86/609/EEC) for the care and use of laboratory animals. Stereotaxic injections were performed under xylazine/ketamine anesthesia. Animals were placed in the stereotaxic frame (David Kopf Instruments) and received a unilateral intranigral injection of 2 μl of viral suspension, corresponding to a viral load of 2.5 × 107 transducing units. Injections were performed in the right brain hemisphere at a speed of 0.2 μl/min controlled by an automatic pump (CMA Microdialysis), using a 10 μl Hamilton syringe connected to a 34-gauge blunt-tip needle. The needle was left in place for an additional 5 min before being slowly withdrawn. Stereotaxic coordinates for virus injections above the substantia nigra pars compacta were as follows: anteroposterior (AP): −5.2 mm, lateral (L): −2.0 mm; dorsoventral (DV): −7.8 from the skull surface; with the incisor bar at 0.0 mm, according to the rat stereotaxic atlas by Paxinos and Watson (1986). To examine the in vivo specificity of our antibody against S87-P, animals injected with AAV2/6-α-syn A30P S129A viruses were sacrificed 13 weeks postinjection and transcardially perfused with 4% paraformaldehyde (PFA, Fluka-Sigma). Brains were removed, post-fixed overnight in PFA 4%, and then transferred into 30% sucrose. Coronal sections (25 μm thick) were cut at −20°C with a microtome (SM2400; Leica) and slices were stored at −20°C in antifreeze medium.
Samples and tissue processing
Analysis of α-syn phosphorylation was performed with postmortem human temporal cortex samples and with the brains of α-syn TG mice. The human samples were obtained from the Alzheimer Disease Research Center at the University of San Diego. A total of 21 cases were included, of which six were nondemented controls, six were diagnosed as AD, six as LBD, and three as MSA. In all cases the brains were processed within 8 h after death. Brains were divided sagittally, the right hemibrain was serially sectioned and preserved at −70°C, and the left hemibrain was fixed in formaldehyde. For neuropathological diagnosis, paraffin sections from cortical and subcortical brain regions were stained with H&E, and thiofavine-S, and immunostained with antibodies against ubiquitin and α-syn.
Three different α-syn TG mouse lines were used for this study: PDGF α-syn WT (Rockenstein et al., 2002), mThy1 α-syn WT (van der Putten et al., 2000), and MBP α-syn WT (Shults et al., 2005). Control experiments were performed with age-matched non-TG littermates. The PDGF α-syn WT TG mice display abundant α-syn accumulation in the neocortex and limbic system and mimics some aspects of diffuse LBD, the mThy1-syn WT shows more abundant accumulation of α-syn in subcortical regions with motor defects and mimics some aspects of PD. The MBP α-syn TG line accumulates syn in oligodendrocytes in cortical and subcortical regions and mimics some aspects of MSA. In accordance with National Institutes of Health (NIH) guidelines for the humane treatment of animals, mice were killed (at 6 months of age) by deep anesthesia with chloral hydrate. Brains were removed and divided sagittally. One hemibrain was postfixed in phosphate-buffered 4% paraformaldehyde, pH 7.4, at 4°C for 48 h and sectioned at 40 μm with a Vibratome 2000 (Leica), while the other hemibrain was snap frozen and stored at −70°C for Western blot analysis.
Immunocytochemistry
S87-P in transgenic mice.
To investigate the distribution of the phosphorylated α-syn epitopes brains from non-TG and TG mice, serially-sectioned, free-floating, blind-coded vibratome sections were incubated overnight at 4°C with either the mouse monoclonal antibody against α-syn (syn-1, 1:500, Transduction Laboratories), rabbit polyclonal anti-human α-syn-specific antibody (Millipore Bioscience Research Reagents, 1:500), mouse monoclonal against S129-P α-syn and the rabbit polyclonal against S87-P α-syn, as described previously (Masliah et al., 2000). Incubation with the primary antibody was followed by anti-mouse IgG or goat anti-rabbit IgG (1:100, Vector) followed by avidin D-HRP and DAB. Sections were analyzed with an Olympus bright field video microscope. For each case, three sections were analyzed and the results were averaged and expressed as mean pixel intensity.
Analysis of injected rat brains.
For Immunofluorescence, slices were washed overnight in PBS and incubated 30 min in H2O2 for 30 min at room temperature, then blocked in a solution of 5% bovine serum albumin (BSA, Sigma), 10% normal goat serum (NGS, Vector Laboratories), and 0.1% Triton X-100 (AppliChem) in PBS, during 2 h at room temperature. Slices were then incubated overnight at 4°C with the primary anti-tyrosine hydroxylase (1:500; AB152; Millipore) and anti-Human α-syn (1:500; LB509, Zymed Laboratories) antibodies in blocking buffer and subsequently incubated for 2 h at room temperature with secondary antibodies conjugated to Alexa Fluor-488 or Alexa Fluor-568 (1:1000; Invitrogen AG). Slices were finally washed and mounted on glass slides using DABCO mounting solution (Sigma-Aldrich). For the enzymatic revelation, slices were washed overnight in PBS and incubated in phenylhydrazine (Sigma-Aldrich) for 1 h at 37°C and blocked in a solution of 5% BSA, 10% NGS, and 0.1% Triton X-100 in PBS, during 2 h at room temperature. Slices were incubated overnight at 4°C with the primary antibody anti-P Ser 87 (1:100) and then incubated for 2 h with the biotinylated secondary antibody (1:200; Vector Laboratories) followed by incubation in avidin–biotin–peroxidase complex (Vector Laboratories) for 30 min at room temperature. Sections were finally revealed with DAB and mounted on glass slides with Merckoglas mounting medium (Merck).
Double labeling and confocal microscopy.
To determine the colocalization between α-syn-immunolabeled neurons and CK1, 40-μm-thick vibratome sections from α-syn TG mice were immunolabeled with the rabbit polyclonal antibodies against α-syn (Millipore Bioscience Research Reagents, affinity purified polyclonal, 1:500) (Masliah and Rockenstein, 2000) and CK1δ (C-18; Santa Cruz Biotechnology, 1:100). The CK1-positive cells were detected with the Tyramide Signal Amplification-Direct (Red) system (1:100, NEN Life Sciences), whereas α-syn immunoreactive was detected with the goat anti-rabbit fluorescein isothiocyanate antibody (Vector, 1:75). This system allows the simultaneous detection of signals from antibodies from the same species. All sections were processed under the same standardized conditions. The immunolabeled blind-coded sections were serially imaged with the laser-scanning confocal microscope (MRC1024, Bio-Rad). For each mouse, a total of three sections were analyzed and for each section, four fields in the frontal cortex and hippocampus were examined.
Detection of S87-P in Lewy bodies.
Lewy body enrichment was conducted as previously described (Gai et al., 2000). Fresh frozen brain tissues were from three diffuse Lewy body disease cases [sex/age at death/postmortem interval (h): m/80/6, m/74/24, f/81/12]. Gray matter was dissected temporal cortex comprising the superior, middle, inferior, and fusiform gyri from a 1-cm-thick coronal slice at the level of caudal amygdaloid, and homogenized in four volumes of homogenization buffer (HB; 0.32 m sucrose, 50 mm Tris-HCl at pH 7.4, 5 mm EDTA, leupeptin 1 mg/ml, pepstatin 1 mg/ml, phenylmethylsulfonyl fluoride 17.4 mg/ml), and homogenized in a Dounce homogenizer (Wheaton) by 10 strokes of pestle A and 10 strokes of pestle B. The homogenate was filtered through glass wool and centrifuged for 10 min at 1000 × g. The pellet was adjusted to 6 ml with HB and Percoll to 14% (v/v) of Percoll concentration. The suspension was overlaid on 2.4 ml of 35% Percoll (v/v in HB) and centrifuged for 30 min at 35,000 × g. The material between the sample-35% Percoll interface was collected and washed once in TBS (50 mm Tris–HCl buffered saline pH 7.4 and protease inhibitors) by centrifugation for 10 min at 4000 × g. The above procedures were conducted either on ice or at 4°C. The pellet was resuspended in 10 volumes of TBS and smeared onto gelatin-coated slides and air-dried at room temperature.
The smears were fixed for 10 min with 2% paraformaldehyde with 0.2% picric acid in PBS, followed by 3 × 5 min washes with TBS-azide (20 mm Tris-HCl, 0.15M NaCl, 0.1% sodium azide, pH 7.4), blocked for 60 min with 20% normal horse serum (NHS) in TBS-azide, incubated overnight with primary antibody pairs constituted in 1% NHS TBS-azide. Primary antibodies were affinity-purified sheep anti-α-syn (Gai et al., 1999) used at 5 μg/ml, affinity purified rabbit anti-S87-P α-syn, and mouse monoclonal11A5 anti-pS129 α-syn (Anderson et al., 2006). Omitting primary antibodies were used as negative controls. Following 3 × 5 min washes with TBS-azide, the smears were incubated for 1 h with Alexa 594 or Alexa 488 conjugated donkey anti-sheep, donkey anti-rabbit, or donkey anti-mouse (Invitrogen). The smears were washed 3 × 5 min washes with TBS-azide, and coverslipped with 20 μl of Vectashield mounting medium, sealed with nail polish, and examined using a Leica TCS SP5 Spectral Confocal Microscope.
Cloning, expression, and purification of α-syn variants
The S129E, S129A, S87A, and S87E α-syn mutants were generated using site-directed mutagenesis employing complementary internal mutagenic primers and two-step PCR. All cDNA sequences were confirmed by sequencing. All proteins used in these studies were expressed as previously described (Kessler et al., 2003), except those used for nuclear magnetic resonance (NMR) studies (see below). Cell lysis (Microfluidics Corporation M110-EHI) was followed by ammonium sulfate precipitation [30% (w/v) solution], anion exchange chromatography (GE Healthcare Q Sepharose) in 10 mm Tris-HCl/1 mm EDTA/pH 8.0, and cation exchange chromatography (GE Healthcare S Sepharose) in 10 mm sodium acetate/ 1 mm EDTA/pH 4.0. The final homogeneity of each preparation was determined by densitometric analysis of Coomassie-stained SDS-PAGE gel scans (NIH Image 1.61/ppc program). Purified preparations were lyophilized from ammonium bicarbonate buffer (100 mm; buffer exchange achieved by GE Healthcare G25 chromatography) and stored at −20°C until use. For the NMR studies, expression and purification of unlabeled and 15N-labeled α-syn were performed as described (Eliezer et al., 2001; Hoyer et al., 2002). To enable attachment of a spin label, a single Cys was introduced into α-syn at position 18 (A18C). The nitroxide spin label chosen for reaction with the Cys-containing mutant was MTSL (1-oxy-2, 2, 5, 5-tetramethyl-D-pyrroline-3-methyl)-methanethiosulfonate (Toronto Research Chemicals). MTSL has been shown previously to efficiently react with α-syn Cys mutants and the reaction was carried out as described previously (Bertoncini et al., 2005).
In vitro phosphorylation and dephosphorylation assays
WT or mutant α-syn was phosphorylated by CK1delta (specific activity 2,000,000 U/mg; NEB) at concentrations of 1.446 mg/ml (unless otherwise stated), in the presence of 1.09 mm ATP (Sigma), 1× reaction solution supplied with the enzyme and 1200U of CK1/145 μg of α-syn. The phosphorylation reaction was incubated at 30°C for the stated time points, and the reaction was stopped with EDTA disodium salt (5 mm final concentration; Axon Lab). The progress of the reaction was monitored by RP-HPLC and mass spectrometry (Wilkins et al., 1999). α-Syn dephosphorylation was performed with calf intestinal alkaline phosphatase (CIAP) (Promega). Briefly, α-syn previously phosphorylated at both S87 and S129 by CK1 was incubated 1 h at 37°C with CIAP at the concentration of 200 U/μl (2000U/μg of protein) in 1× CIAP buffer. The reaction was stopped by adding the loading buffer, and the progress of the phosphorylation reaction was monitored by Western blot using antibodies against S87-P and S129-P.
Fibrillization studies
To probe the effect of CK1-mediated phosphorylation on the aggregation of α-syn, 100 μm WT and mutant α-syn were first phosphorylated and then subjected to fibrillization conditions, at 37°C with continuous shaking for the indicated time points. The unphosphorylated controls were treated under the same conditions as the phosphorylated samples, but CK1 was not added to them. The extent of fibril formation was monitored by ThT fluorescence assay and by determining the amount of soluble monomeric and oligomeric α-syn in solution at indicated time point during the aggregation process (Paleologou et al., 2008).
Gel electrophoresis (SDS-PAGE) and immunoblotting
α-Syn samples were diluted in loading buffer and separated on 12% SDS 1 mm gel. Gels were stained with Simply Blue Safe stain (Invitrogen) or silver stained (Invitrogen) according to manufacturer's instructions. For Western blots, membranes were probed with the primary antibody [mouse monoclonal anti-α-syn (121–125) (211) at a dilution of 1:500 (Santa Cruz Biotechnology) or mouse anti-α-syn (15–123) at a dilution of 1:1000 (BD Transduction), or mouse monoclonal anti-S129-P α-syn at a dilution of 1:5000 (Wako), or rabbit polyclonal anti-S87-P α -syn at a dilution of 1:100] at room temperature (RT) for 1–2 h. After incubation with the secondary antibody (i.e., goat anti-mouse ALEXA Fluor 680) protected from the light at RT, for 1 h and washing with PBST (4×), the immunoblots were scanned in a Li-COR scanner at a wavelength of 700 nm. The Western blot analysis for brain homogenates was carried out as follows. Brain homogenates that include the frontal cortex and the striatum were separated by ultracentrifugation as previously described into cytosolic and membrane fractions (Masliah and Rockenstein, 2000). Samples from both cytosolic and membrane fractions were separated on 12% or 4–12% SDS-PAGE gels (NuPAGE, Invitrogen) and transferred onto 0.22 μm nitrocellulose membranes (Schleicher & Schuell) using 1× 3-[cyclohexylamino]-1-propaneosulfonic acid (CAPS) transfer buffer containing 20% methanol. Membranes were blocked with 3% milk in PBS containing 0.1% Tween-20 (Sigma) (PBS-T), followed by incubation in primary antibody (1:1000) in PBS-T overnight at 4°C. The primary antibodies used were as follows: rabbit polyclonal against total α-syn (Millipore Bioscience Research Reagents); mouse monoclonal against S129-P α-syn (courtesy of Dr. T. Iwatsubo, University of Tokyo, Tokyo, Japan), rabbit polyclonal against S87-P α-syn and mouse monoclonal against α-actin (Millipore Bioscience Research Reagents). Membranes were further incubated with goat anti-mouse or anti-rabbit IgG secondary antibodies conjugated to horseradish peroxidase (1:5000, American Qualex) and visualized by enhanced chemiluminescence (ECL, NEN Life Sciences) and exposed to film. For determinations of levels of immunoreactivity, ECL-treated membranes were analyzed in the VersaDoc imaging system (Bio-Rad) using the Quantity One software (Bio-Rad).
Preparation of large unilamellar vesicles and small unilamellar vesicles andα-syn-liposome complexes
1-Palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (sodium salt, POPG; Avanti Polar Lipids) was purchased in chloroform which was removed by evaporation and lyophilization. The residual phospholipid was hydrated with 50 mm HEPES (Fluka), 150 mm NaCl (Fluka), pH 7.4 solution, giving rise to a phospholipid suspension of 10 mg/ml. To increase the efficiency of large unilamellar vesicles formation, 10 cycles of freezing in dry ice and thawing at 37°C water bath were carried out. Small unilamellar vesicles were prepared by extrusion through a 100 nm polycarbonate membrane (Avestin) according to manufacturer's instructions. The small unilamellar vesicles were stored at 4°C and used within 3–5 d. The appropriate amount and volume of α-syn in PBS or sodium phosphate, pH 7.5, was mixed with the appropriate volume of POPG to generate a mass ratio of α -syn:POPG of 1:20. The α-syn-liposome complex was incubated for 2 h at RT before circular dichroism (CD) spectroscopy.
Circular dichroism
The average secondary structure of monomeric α-syn in the presence and absence of POPG (Avanti Polar Lipids) vesicles was determined by circular dichroism (CD) spectroscopy using a Jasco 810 Spectrometer. The Far UV-CD spectra (190–250 nm, integration time of 2 s for 0.2 nm) were collected at RT in a 1 mm path length quartz cuvette containing 0.1 mg/mL of α-syn in PBS or sodium phosphate buffer and vesicles were prepared as described by Paleologou et al. (2008).
Transmission electron microscopy
For electron microscopy (EM) studies, WT or mutant α-syn samples were deposited on Formvar-coated 200 mesh copper grids (Electron Microscopy Sciences) at a concentration of 25 μm. Grids were washed with two drops of water and stained with two drops of freshly prepared 0.75% (w/v) uranyl acetate (Electron Microscopy Sciences). Specimens were inspected on a Philip CMX 10 electron microscope, operated at 100 kV. Digitized photographs were recorded with a slow scan CCD camera (Gatan, Model 679).
Nuclear magnetic resonance
NMR samples of free α-syn contained ∼ 0.1 mm 15N-labeled WT or mutant α-syn in 90% H2O/10% D2O, 50 mm phosphate buffer at pH 7.4, 100 mm NaCl. NMR experiments were acquired on Bruker Avance 600 and 700 MHz NMR spectrometers. The temperature was set to 15°C. NMR data were processed and analyzed using NMRPipe (Delaglio et al., 1995) and Sparky 3 (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco, San Francisco, CA) and NMRView (Johnson and Blevins, 1994). Spectra were referenced indirectly to DSS and ammonia using the known chemical shift of water (Wishart et al., 1995). Tentative assignments for the spectra of mutant and phosphorylated proteins were obtained by transferring each previously assigned cross peak in the 1H-15N heteronuclear single quantum coherence (HSQC) spectrum of the WT protein to the nearest unassigned cross peak in each new spectrum. Subsequently, they were verified by three-dimensional HNHA and three-dimensional NOESY-HSQC spectra (Bax and Grzesiek, 1993). 3J(HN,Hα) scalar couplings were measured using intensity modulated HSQC experiments (Permi et al., 2000). 15N R1ρ spin relaxation rates were measured with a pulse sequence based on Farrow et al. (Farrow et al., 1994) on a Bruker Avance 600 MHz spectrometer. Relaxation delays were set to 8, 48, 64, 128, and 184 ms. The spin lock frequency was 2.5 kHz. Relaxation times were calculated by fitting an exponential function to the decaying signal integrals. Steady-state heteronuclear 15N[1H]-NOE values are reported as the ratio of peak heights in paired spectra collected with and without an initial period (4 s) of proton saturation during the 5-s recycle delay. Pulse field gradient NMR experiments were acquired on unlabeled WT and mutant α-syn (concentration of 200 μm) dissolved in 99.9% D2O, 50 mm phosphate buffer pH 7.4 and containing dioxane (concentration ∼20 mm) as an internal radius standard and viscosity probe (Wilkins et al., 1999). Twenty one-dimensional 1H spectra were collected as a function of gradient amplitude employing the PG-SLED sequence (Jones et al., 1997; Wilkins et al., 1999). Each experiment was repeated at least two times. The gradient strength was increased from 1.69 to 33.72 Gauss/cm, in a linear manner. Signals corresponding to the aliphatic region of the 1H spectra (3.3–0.5 ppm) were integrated and diffusion data (signal intensity versus gradient strength) were fitted to Gaussian functions using XWINNMR (Bruker Instruments). Stokes radii of α-syn were calculated from the apparent diffusion coefficients of α-syn and dioxane, and the known Stokes radius of dioxane (Jones et al., 1997). Errors in stokes radii estimated from repeat measurements are about 0.5 Å.
For NMR studies of micelle-bound α-syn, lyophilized protein was dissolved in sample buffer (100 mm NaCl, 10 mm Na2HPO4, pH 7.4 in 90%/10% H2O/D2O) with 40 mm deuterated SDS (Cambridge Isotope). Spectra were recorded on either Varian INOVA 600 MHz (Weill Cornell, New York, NY) or a Bruker Avance 800 MHz (New York Structural Biology Center, New York, NY) spectrometers at sample temperature of 40°C. Two-dimensional and three-dimensional double and triple resonance data, including HSQC, HNCACB, CBCACONH, HNCO, HNCACO, and HSQC-NOESY-HSQC, were collected using standard pulse sequences. A mixing time of 150 ms was used for the NOESY experiment. Typical spectral widths were 10, 20, 8, and 53 PPM in the proton, nitrogen, CO, and C dimensions. Data were processed using NMRPipe (Delaglio et al., 1995) and analyzed using NMRView (Johnson and Blevins, 1994).
Results
Characterization of α-syn phosphorylation in TG models of synucleinopathies and human brain from AD, LBD, and MSA patients
To determine the physiological and pathological relevance of S87 phosphorylation, we generated a rabbit polyclonal antibody, anti-S87-P, against a synthetic peptide that corresponds to amino acid residues 81–93 of human α-syn, with S87 being phosphorylated. Figure 1A demonstrate that our anti-S87-P antibody specifically detects serine 87 phosphorylated forms (S87-P) of α-syn, and reveals the absence of immunoreactivity against either unphosphorylated, S129A, or S129-P α-syn. To rule out the possibility that the lack of immunoreactivity of S87A is due to a loss of the epitope rather than lack of phosphorylation, the specificity of the antibody was verified by probing its immunoreactivity toward diphosphorylated α-syn (S87-P/S129-P) after phosphatase (CIAP) treatment. Phosphatase treatment resulted in the loss of immunoreactivity toward both anti-S87-P and anti-S129-P antibodies (Fig. 1B), consistent with the loss of the phosphate group on both residues, thus providing further verification of the specificity of both antibodies.
Figure 1.

Specificity of anti-S87-P antibody. A, WT and mutant α-syn phosphorylated for 24 h were separated on a 12%SDS gel and probed with anti-α-syn (211, 1:500), anti-S129-P (1:5000), or anti-S87-P (1:100) α-syn antibodies. B, S87-P and S129-P antibodies failed to detect phosphorylated forms of α-syn treated with calf intestinal alkaline phosphatase. C–N, Virus mediated-overexpression of human α-syn A30P S129A (C–H) and human α-syn A30P S87D S129A (I–N) in the rat substantia nigra. The staining for human α-syn (LB509) colocalized with tyrosine hydroxylase (TH) staining (C, D, I, J) in the injected side (E, K), but was absent in the noninjected side (F, L). The staining for S87-P was restricted to the side injected with the virus coding for human α-syn A30P S129A (G), whereas no staining could be detected on the side injected with the mutant α-syn form S87D (M). Noninjected sides also appeared negative (H, N). Scale bar, 500 μm.
To probe the specificity of our S87-P antibody and determine whether phosphorylation at this residue occurs in vivo, we characterized the level of S87-P in slices from rat brains injected on one side with an AAV2/6 viral vector overexpressing the S129A variant of α-syn A30P (Fig. 1C–H) (n = 3). As a control, we also examined tissues from rat brains injected a mutant form of the same protein, in which phosphorylation at S87 was blocked by a serine to aspartate substitution at position 87 (S87D/S129A-A30P) (Fig. 1I–N) (n = 3). In both cases, the uninjected side served as an additional control to assess the level of background signal due to nonspecific interactions and antibody cross-reactivity with other proteins. We detected a strong staining against human A30P α-syn in the injected substantia nigra (Fig. 1E,K) compared to the noninjected side (Fig. 1F,L). However, anti-S87-P staining was exclusively detected in the substantia nigra overexpressing human S129A α-syn (Fig. 1G) and no labeling was detectable, neither in the noninjected side (Fig. 1H,N) nor in the substantia nigra overexpressing the S87D/S129A mutant (Fig. 1M). These findings demonstrate conclusively that phosphorylation at serine 87 does occur in vivo, at least under conditions of increased α-syn expression [corresponding to an approximate doubling of the total α-syn level in nigral tissue extracts (Azeredo da Silveira et al., 2009)] and validate the specificity of our antibody on brain tissue. However, it is not possible to determine the overexpression levels in individual neurons.
α-Syn in LBs is phosphorylated at S87-P and S87-P levels are increased in human brains from AD, LBD, and MSA patients
Having established the specificity of the antibody, anti-S87-P, together with anti-α-syn and anti-S129-P antibodies, was tested in human brain (frontal cortex) homogenates from control, AD, LBD, and MSA cases. Immunoblot analysis of cortical samples, from control or diseased human brains, showed a native α-syn band at 14 kDa in the cytosolic fractions (Fig. 2A), which was significantly increased in the AD, LBD, and MSA cases relative to the control (Fig. 2A,G). In the membrane fractions increased α-syn levels are detected in the LBD and MSA cases, with higher molecular weigh species being present in the LBD cases only (Fig. 2B,G). Anti-S87-P antibody detected a weak band at 14 kDa in the control cases. This band became more pronounced in the diseased brains notably in the LBD cases (Fig. 2D,H), where it was accompanied by a weaker band at around 17 kDa. The antibody against S129-P detected a band at 14 kDa as well as higher MW bands (Fig. 2E,F,I). With both antibodies against phosphorylated α-syn most of the immunoreactivity was detected in the membrane fractions in the LBD and MSA cases but not in the controls and AD cases (Fig. 2C–F). Several α-syn immunoreactive bands at molecular weights of 38 kDa and higher were detected in the control samples from LBD cases. These bands were more abundant in the membrane fractions and showed clear immunoreactivity toward the anti-S129-P, but at best weak reactivity to the anti-S87-P antibody.
Figure 2.

Comparison of the levels of phosphorylated α-syn immunoreactivity by immunoblot in human brains from control, AD, LBD, and MSA cases. Samples from the frontal cortex were divided into cytosolic and particulate fractions and analyzed by Western blot with antibodies against total and phosphorylated α-syn. A, B, Cytosolic and particulate fractions probed with an antibody against total α-syn. Native α-syn is identified at 14 kDa. Compared to controls, in LBD cases there was increased levels of native and aggregated α-syn above 28 kDa preferentially in the membrane fraction. C, D, S87-P α-syn was identified as a single band at 14 kDa in the membrane fraction. The intensity of this band was greater in LBD, MSA, and AD cases compared to controls. E, F, The native S129-P α-syn was identified as a single band at 14 kDa and oligomerized α-syn was identified as multiple bands ranging from 28 kDa to 98 kDa. S129-P α-syn was only identified in the membrane fraction and was most abundant in LBD cases. G, H, I, Quantitative analysis of brain homogenate from 21 AD, LBD, MSA, and control cases including the representative samples shown in A–F. Statistical analyses are performed using a one-way ANOVA followed by post hoc Dunnet's test. *p < 0.05 compared with control values.
To further investigate the pathological relevance of S87 phosphorylation, we assessed S87-P levels in Lewy body samples from three diffuse Lewy body (DLB) cases. Lewy bodies were intensely labeled by the sheep anti-α-syn antibody. Therefore, the signal gain was set low (50% of S87-P α-syn signal) to show characteristic homogenous and concentric patterns, with little background or neuropile labeling (Fig. 3, left column). The S87-P α-syn immunoreactivity was less intense but readily detected in all Lewy bodies (>500 Lewy bodies were analyzed by confocal microscopy). The S87-P α-syn labeling appeared smooth within Lewy bodies (Fig. 3B,E). In addition, granular S87-P α-syn labelings of 0.2–0.5 μm size were also detected, more obvious in Lewy body periphery, neural processes, and neuropil (Fig. 3A,D). No such labeling was seen when anti-S87-P α-syn antibody was omitted. These granular labelings may be complexes or cellular particles containing more concentrated S87-P α-syn. Figure 3, J and K, illustrates the relative intensities for α-syn (red line), S87-P α-syn (green), and DNA (blue) labelings across a Lewy body. However, it must be noted that fluorescence intensity depends on many factors, including the dye used, the affinity of each antibody for its epitope, the accessibility of the epitope, etc.
Figure 3.

Analysis of the colocalization of S87-P and α-syn in LBs isolated from fresh brain of DLB cases (A–K). Immunostaining anti-S87-P in conjunction with sheep anti-α-syn antibody showed a colocalization of the two forms of α-syn in a mature (A–C) and a less mature cortical LB (D–F). G–I correspond to control staining in which anti-S87-P antibody has been omitted, no labeling for S87-P was detected in the Lewy body. J and K illustrate a fluorescence intensity analysis for α-syn (red line), S87-P (green), and DNA (blue) labelings across a Lewy body, indicating a good colocalization between total α-syn and pS87 α-syn reactivities. Scale bars: (in B, E, H) A–J, 10 μm. S87-P is enriched in SDS/Urea soluble fractions of synucleinopathies (L–O). Samples from the temporal cortex were separated by high-speed centrifugation at 18,000 × g for 30 min, and pellet containing vesicles and particulate material were future solubilized in SDS/Urea. Increased S87-P α-syn reactivity was detected in SDS/Urea soluble fraction M compared to buffer soluble fraction L. In contrast, total α-syn reactivity was more prominent in buffer soluble fraction N than in SDS/Urea soluble fraction O.
The localization of S87-P α-syn in Lewy bodies suggests it is an integrated part of the inclusions. We further examined whether S87-P α-syn is enriched in buffer insoluble fractions in brains of synucleinopathies and Alzheimer's disease. The buffer insoluble fractions contained numerous Lewy bodies and glial inclusions and were further solubilized in 5%SDS/Urea. As shown in Figure 3L–O, S87-P α-syn reactivity was more prominent in 5%SDS/Urea fraction, compared to total α-syn reactivity. Figure 3M demonstrates that the level of S87-P in these fractions is higher in diseased brains relative to control cases.
The levels of S87-P are increased in brains of TG models of synucleinopathies
To further assess whether S87 phosphorylation is a pathological event, we assessed its levels in brain homogenates from TG mouse experimental models of PD/LBD and MSA (Fig. 4). As expected the highest levels of α-syn expression were observed in the mThy1 α-syn TG mice followed by the PDGF α-syn and the MBP-α-syn TG both in the cytosolic (Fig. 4A,C,E) and membrane fractions (Fig. 4B,D,F). Bands reflecting monomers (at 14 kDa) and the oligomers (at 38–62 kDa) were detected. Consistent with the finding in the human brain homogenates, antibodies against S87-P detected a band at 14 kDa, which were significantly more abundant in the membrane fraction of the mThy1 α-syn TG mice (Fig. 4D). With antibody against S129-P, a strong 14 kDa band was detected in the cytosolic fraction of both the mThy1 α-syn and MBP α-syn TG mice (Fig. 4E). S129-P α-syn was detected at 14 and 42 kDa in the membrane fractions of the α-syn TG mice (Fig. 4F). Consistent with the Western blots, immunocytochemical analysis with the antibody against total α-syn showed immunoreactivity in the neuropil corresponding to nerve terminals in the non-TG mice (Fig. 5A). In the PDGF and mThy1-α-syn TG mice abundant accumulation of α-syn was detected in the neuronal cell bodies, whereas in MBP α-syn TG mice immunoreactivity was associated with oligodendrocytes (Fig. 5B–D). With S129-P antibody abundant α-syn immunoreactivity was detected in the three lines of α-syn TG mice (Fig. 5E–H). With the S87-P antibody abundant immunoreactivity was observed in the neuronal cell bodies of the mThy1-α-syn TG (but not in the PDGF-α-syn) mice and in the inclusions in the oligodendrocytes in the MBP-α-syn TG mice (Fig. 5K,L).
Figure 4.

Comparison of the levels of phosphorylated α-syn immunoreactivity by immunoblot in brains of α-syn models of LBD. Samples from the neocortex were divided into cytosolic and particulate fractions and analyzed by Western blot with antibodies against total and phosphorylated α-syn. The PDGF-α-syn express moderate levels of α-syn in the neocortex and hippocampus, the thy1-α-syn expresses high levels of α-syn in cortex and subcortex, the MBP-α-syn accumulates syn in oligos and is a model for aspects of MSA. A, B, Cytosolic and particulate fractions probed with an antibody against total α-syn. Native α-syn is identified at 14 kDa. In the TG mice there is an increase in native α-syn compared to non-TG. Compared to non-TG, in thy1-α-syn cases there was increased accumulation of α-syn aggregates above 28 kDa preferentially in the membrane fraction. C and D, S87-P α-syn was identified as a single band at 14 kDa. This band was exclusive to the membrane fraction and detected only in the TG mice cases. E, F, The native S129-P α-syn was identified as a single band at 14 kDa and oligomerized α-syn was identified as multiple bands ranging from 28 kDa to 98 kDa. The aggregated S129-P α-syn identified in the α-syn TG mice was more abundant in the membrane fraction. The native form was more abundant in the cytosolic fraction.
Figure 5.

Patterns of phosphorylated α-syn immunoreactivity in brains of α-syn models of LBD. Vibratome sections were immunostained with antibodies against total and phosphorylated α-syn. A–D, The PDGF-α-syn express moderate levels of α-syn in the neocortex and hippocampus, the thy1-α-syn expresses high levels of α-syn in cortex and subcortex, and the MBP-α-syn accumulates syn in oligos and is a model for aspects of MSA. Compared to non-TG in α-syn TG mice there is abundant accumulation of total α-syn and the most abundant is in Thy1-α-syn mice. In MBP-α-syn accumulation is in oligos. E–H, S129-P α-syn accumulates in neurons in PDFG-α-syn and Thy1-α-syn neurons and in MBP-α-syn TG mice in oligos (Abeliovich et al., 2000). I–L, S87-P α-syn was found in some neurons in Thy1-α-syn and in MBP-α-syn cases but not in PDGF-α-syn.
S87-P colocalization with CK1
Among the various kinases reported to phosphorylate α-syn in vitro, CK1 and the dual specificity tyrosine regulated kinase 1A (Dyrk1A) are the only two that phosphorylate α-syn at S87. To determine the colocalization between S87-P immunolabeled-neurons and CK1, 40-μm-thick vibratome sections from the temporal cortex of α-syn TG mice and LBD/PD patients were immunolabeled with the antibodies against S87-P and CK1. Figure 6 demonstrates a great degree of colocalization of S87-P and CK1 in neuronal inclusions in Tg mice (Fig. 6A) and in LB-like structures in LBD/PD diseased brains (Fig. 6B), consistent with in vitro observations and suggesting that CK1 may by directly involved in modulating α-syn phosphorylation in vivo. These findings are consistent with previous cell culture studies implicating CK1 in the phosphorylation of α-syn at S129 and S87 (Okochi et al., 2000).
Figure 6.
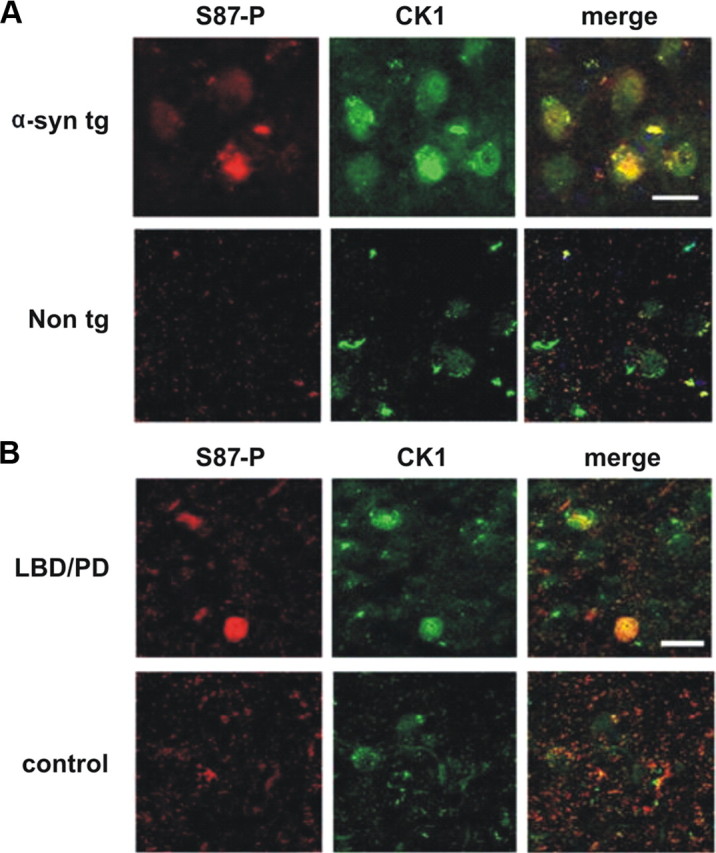
Colocalization of S87-P and CK1 within inclusions in the thy1-α-syn TG mice and LBD/PD diseased brains. Vibratome sections (40 μm thick) from α-syn TG mice and LDB/PD cases were stained against S87-P and CK1. A, B, A representative immunostaining from the temporal cortex demonstrates a great degree of colocalization of S87-P and CK1 in thy1-α-syn TG mice inclusions (A) and LBD/PD LB-like structure (B), compared to the controls.
Serine→glutamate substitution or phosphorylation at S87 inhibits the fibrillization of WT and mutant (S129A and S129E) α-syn
Having verified the pathological relevance of S87 phosphorylation, we sought to understand the role of S87 phosphorylation in modulating the structure and aggregation properties of α-syn. Toward this goal, we compared the structural, oligomerization, fibrillization, and membrane-binding properties of monomeric WT α-syn to those of the phosphorylation mimics (S87E) as well as the purified in vitro S87-phosphorylated form of α-syn using NMR, CD, SEC, SDS-PAGE, ThT, and transmission EM (TEM).
Phosphorylation at S87 is sufficient to block α-syn fibrillization
Recent studies from our laboratory demonstrated that CK1-mediated phosphorylation blocks α-syn fibrillization (Paleologou et al., 2008). In vitro CK1 phosphorylation of WT α-syn followed by tryptic digestion and mapping of phosphorylation sites revealed that CK1 phosphorylates α-syn at multiple sites (S87, T92, S129), with S87 and S129 being the major phosphorylation sites (supplemental Figs. 2, 3, available at www.jneurosci.org as supplemental material). To determine the relative contributions of phosphorylation at S87 to the CK1-induced inhibition of α-syn fibril formation, we examined the effect of CK1-mediated phosphorylation on the fibrillization of the S129A and S129E, both of which cannot be phosphorylated at S129. Prephosphorylation of both variants with CK1 results in significant retardation of α-syn fibrillization and a reduction in amyloid fibril formation relative to the unphosphorylated forms of both proteins (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). S129A, S129E, and the WT α-syn formed significant amounts of amyloid fibrils, whereas prephosphorylated forms of S129E (S129E/S87P) and WT (S129P/S87P) showed predominantly soluble oligomeric species. S129A rapidly forms short protofibrillar/fibrillar structures, consistent with its marked increased propensity to fibrillize (Paleologou et al., 2008). However, the amount of fibrils formed by prephosphorylated S129A is significantly less than that observed for unphosphorylated S129A.
To further prove that inhibition of α-syn fibrillogenesis is due to phosphorylation at S87, we prepared and purified the S87-P phosphorylated forms of S129A and S129E α-syn and compared their aggregation properties to those of the corresponding unphosphorylated forms of α-syn. The phosphorylated (S87-P) and unphosphorylated α-syn forms of each protein were separated by RP-HPLC and their purities were verified by MALDI-TOF mass spectrometry. We consistently observed that the mono (S87-P) and diphosphorylated (S87-P/ T92-P) forms of S129A and S129E species did not form fibrils even after 48 h, whereas the unphosphorylated forms of these proteins exhibited extensive fibril formation after 24 h of incubation (Fig. 7A). These observations were confirmed by SEC and TEM studies, which revealed the absence of any fibrillar aggregates and presence of predominantly monomers in the S129A/S87-P and S129E/S87-P samples (Fig. 7B,C). Interestingly, monophosphorylated S129E/S87-P appears to form significantly less oligomeric/protofibrillar-like aggregates than S129A/S87-P, suggesting that the presence of both phosphate and Glu substitutions at S87 and S129, respectively results in a greater inhibition of α-syn fibrillization.
Figure 7.

Phosphorylation at S87 inhibits S129A α-syn aggregation propensity. A, ThT fluorescence was monitored at indicated time points from samples of 70 μm solutions of WT, S129A, S129A/S87-P, and S129A/S87-P, T92-P S87E α-syn incubated at 37°C. B, The samples were collected at the indicated time points and centrifuged at 18,000 × g for 30 min at 4°C before their supernatants were separated on a 12% SDS gel or applied to an analytical Superedex 200 PC 3.2/30 column. The analysis was carried out under isocratic conditions (10 mm Tris, pH 7.4) and the signal was monitored at 280 nm. C, Negatively stained TEM images of S129A, S129A/S87-P, and S129A/S87-P, T92-P S87E α-syn after 96 h of incubation at 37°C under agitating conditions (Scales bar, 200 nm). D–F, Assessing and comparing the aggregation propensity of WT, S87A, and S87E α-syn. D, ThT fluorescence was monitored at indicated time points from samples of 60 μm solutions of WT, S87A, and S87E α-syn incubated at 37°C. E, The samples were collected at the indicated time points and centrifuged at 18,000 × g for 30 min at 4°C before their supernatants were separated on a 12% SDS gel (F) or applied to an analytical Superedex 200 PC 3.2/30 column. The analysis was carried out under isocratic conditions (10 mm Tris, pH 7.4) and the signal was monitored at 280 nm. F, Negatively stained TEM images of WT, S87A, and S87E α-syn after 0 h and 72 h of incubation at 37°C under agitating conditions (Scales bar 200 nm). Coomassie-stained gel of the samples after 0 h and 72 h of aggregation.
In the fibrillar state, only S129 undergoes phosphorylation by CK1 in vitro
According to the solid state NMR data α-syn residue 87 is in a region that is part of the rigid structure of the fibrils and within one of the β-strands participating in the formation of the fibril structure (Kloepper et al., 2007; Heise et al., 2008; Vilar et al., 2008), suggesting that it might not be subject to phosphorylation after fibril formation has occurred. To determine whether S87 is accessible and can undergo phosphorylation in the fibrillar state, we generated monomer-free fibrillar samples of WT α-syn and subjected them to in vitro phosphorylation with CK1. Phosphorylation was assessed by Western blotting using antibodies against α-syn, S87-P, and S129-P. We observed that only S129 undergoes phosphorylation by CK1, suggesting that S87, within α-syn fibrils, is not accessible or exists in a conformation that is not recognized by CK1 (supplemental Fig. 5, available at www.jneurosci.org as supplemental material).
The phosphomimic S87E aggregates slower than WT and S87A α-Syn
Selective phosphorylation and/or overexpression of S87-P in vivo is currently not possible, as CKI phosphorylates at both S87 and S129 and DYRK1A-mediated phosphorylation at S87 is less efficient. Therefore, future efforts to assess the role of S87 phosphorylation in modulating the normal biology and aggregation of α-syn in vivo are likely to rely on the use of the phosphomimics S87E and S87A. Recent studies from our group (Paleologou et al., 2008) and others (Waxman and Giasson, 2008; McFarland et al., 2009) demonstrate that substitution of S129 by glutamate or aspartate does not reproduce the effect of phosphorylation at this site on α-syn structure and aggregation properties in vitro. Having established that S87-P is sufficient to inhibit α-syn oligomerization and fibrillogenesis, the 87 mutants S87A and S87E were generated to assess whether the phosphorylation mimic S87E is likely to mimic S87 phosphorylation in vivo. Comparison of the fibrillization of WT, S87A, and S87E α-syn showed that S87A aggregated at a similar rate and to similar levels as WT α-syn, whereas for S87E the observed aggregation rate was slower than those of the WT and S87A proteins (Fig. 7D). TEM images further confirmed these findings, as both WT and S87A formed dense networks of fibrils as opposed to S87E, which formed a few short fibrils after 72 h of aggregation (Fig. 7F). In accordance with these findings, the consumption of monomeric protein over time monitored by SEC and SDS-PAGE analysis also revealed a significant decrease in monomeric WT and S87A (Fig. 7B,E), whereas the levels of the monomeric S87E α-syn remained virtually unchanged over the 72 h of aggregation, providing further evidence that S87E does not aggregate.
α-Syn is disordered independent of phosphorylation at S87
Serine 87 lies in the hydrophobic NAC region of α-syn, suggesting that the introduction of a negative charge through the substitution of serine by glutamate at this position is unfavorable and may alter the secondary structure and hydrodynamic properties of monomeric α-syn. Figure 8A demonstrates that WT, S87A, and S87E α-syn exhibit virtually identical random coil and α-helical CD spectra in solution and upon binding to synthetic membranes, respectively. Given that α-syn exists predominantly in a random coil conformation, subtle changes in the secondary structure of the protein are unlikely to be discernible by CD studies. Therefore, to further elucidate the consequences of phosphorylation on the structure and dynamics of monomeric α-syn, high-resolution NMR studies were also conducted. For all proteins, the resonances in 1H-15N HSQC spectra were sharp and showed only a limited dispersion of chemical shifts, reflecting a high degree of backbone mobility (Fig. 8B). After phosphorylation of WT α-syn by CK1, the resonances of S87 and S129 were strongly attenuated at the position seen in the proton-nitrogen correlation (HSQC) of the unphosphorylated protein, but new signals appeared in the region, in which resonances of phosphorylated amino acids are usually found (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). For S129D α-syn, phosphorylation at S129 was blocked so that only the resonance of S87 was attenuated at its original position and appeared at its phosphorylated position (Fig. 8B). Other chemical shift changes induced by phosphorylation were generally small. In addition, steady-state heteronuclear 15N[1H]-NOEs (Fig. 8C), 15N R1ρ relaxation rates and 3J(HN,Hα) couplings (supplemental Fig. 4, available at www.jneurosci.org as supplemental material) were very similar for unphosphorylated and phosphorylated α-syn, indicating that phosphorylation at S87 has no apparent effect on the secondary structure of α-syn.
Figure 8.

α-syn is disordered independent of phosphorylation at S87. A, Circular dichroism spectroscopy reveals that WT (blue), S87A (red), and S87E (green) α-syn adopt random coil structure. α-Syn at 10 μm. B, Comparison of two-dimensional 1H-15N HSQC spectra of nonphosphorylated S129D α-syn (blue) and phosphorylated S129D α-syn (red). Resonance assignments are indicated with residue numbers. A dashed rectangle marks glutamine and asparagine side chain resonances. C, Heteronuclear 15N [1H]-NOEs in nonphosphorylated WT α-syn (black), phosphorylated WT α-syn (gray), and phosphorylated S129D α-syn (red). D, Hydrodynamic radii of various α-syn mutants in phosphate buffer at 15°C with and without 8 m urea. In phosphorylated S129A/D α-syn, only S87 was phosphorylated.
Pulse field gradient NMR experiments allow accurate determination of the diffusion coefficient of a molecule. From the diffusion coefficient, a hydrodynamic radius Rh can be calculated that provides an estimation of the overall dimensions of a biomolecule (Farrow et al., 1994). For WT and S129D α-syn, we determined hydrodynamic radii of 28.2 Å and 28.1 Å, respectively (Fig. 8D). The phosphorylation mimic S87E did not change the hydrodynamic radius of α-syn. On the other hand, phosphorylation of S129A and S129D α-syn at S87 increased Rh by 2.6 and 1.9 Å (Fig. 8D). Addition of 8 m urea to phosphorylated S87A α-syn further increased the Rh value to 36.2 Å. If α-syn were a true random coil, a hydrodynamic radius of 36.9 Å would be expected (Kohn et al., 2004). Interestingly, phosphorylation of S129 appears to have a more dramatic effect on the hydrodynamic radius of monomeric α-syn as it results in an increase of Rh by ∼5.0 Å. This result is consistent with our previous studies demonstrating disruption of the intramolecular interactions between the C- and N-terminal regions of α-syn upon phosphorylation at S129 and suggest that phosphorylation at S87 may only partially disrupt this interaction (Paleologou et al., 2008).
Phosphorylation at S87 results in changes in protein conformation upon membrane binding
In the presence of SDS micelles, the lipid-binding domain of α-syn adopts a conformation consisting of two helical segments, which have been previously characterized using NMR (Eliezer et al., 2001; Bussell and Eliezer, 2003; Chandra et al., 2003; Ulmer and Bax, 2005). A comparison of proton-nitrogen correlation spectra of micelle-bound phosphorylated WT (S87-P and S129-P) α-syn and its unphosphorylated counterpart shows that the resonances of S87 and S129 of the phosphorylated protein are dramatically shifted. In addition to local changes, many resonances of amino acids in the vicinity of S87-P were also shifted (79, 81, 83, 84, 85, 86, 88, 89, 91, 92, 93, 94, 95, 96) (Fig. 9A). Most of these amino acids are part of the second helix that α-syn adopts upon binding to micelles. Relatively less extensive shifts were observed in the resonances of amino acids close to S129, consistent with our previous studies of the effects of phosphorylation at this site (Paleologou et al., 2008). Similar changes in the vicinity of S87-P were made when comparing the HSQC spectra of S129A/S87-P (Fig. 9B) and S129E/S87-P (Fig. 9C) and their unphosphorylated counterparts, strongly suggesting that changes occur in the environment of the second helix are a result of S87 phosphorylation.
Figure 9.
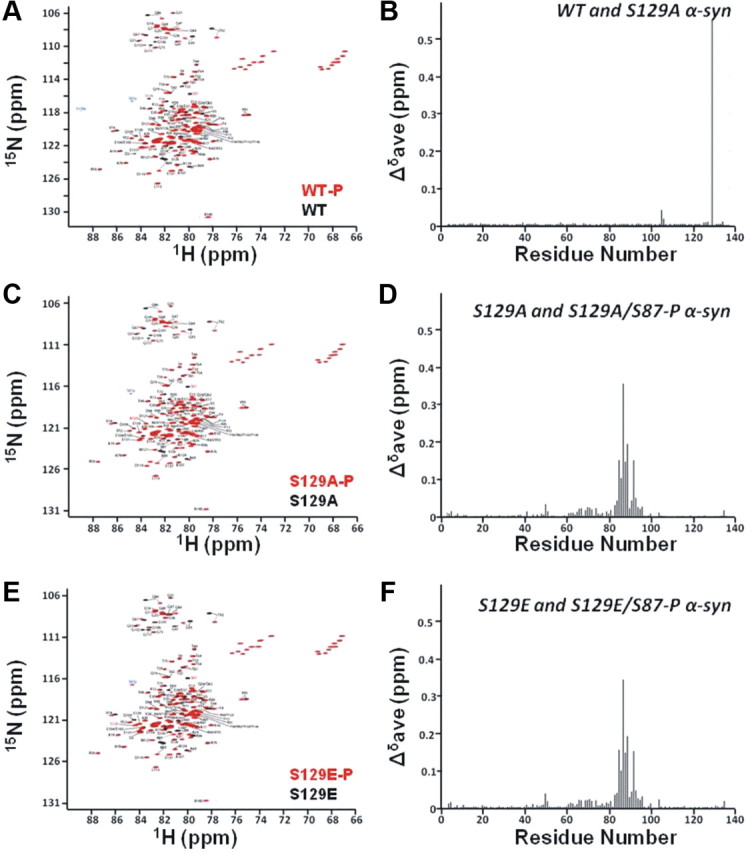
Phosphorylation at S87 results in changes in protein conformation upon membrane binding. A–C, Overlaid contour plots of 1H-15N HSQC spectra of monophosphorylated WT (black) and its unphosphorylated control (red; A), monophosphorylated S129A (black) and its unphosphorylated control (red; B), and monophosphorylated S129E (black) and its unphosphorylated control (C). Amino acid residues that shift with respect to unphosphorylated proteins are labeled. D–F, Mean weighted 1H-15N chemical shift differences [calculated as [(Δδ1H) + (Δδ15N)2/25]1/2/2] between WT and S129A α-syn in the SDS-bound state (D), unphosphorylated S129A and S129A/S87-P in the SDS-bound state (E), and unphosphorylated S129E and S129E/S87-P in the SDS-bound state (F).
To quantify the chemical shift changes between the phosphorylated and unphosphorylated micelle-bound proteins, the resonances of each form of S129A α-syn were directly assigned using triple resonance methods. Graphs of the resulting weighted average of the amide proton and nitrogen chemical shift changes between the phosphorylated and unphosphorylated proteins are shown (Fig. 9E) for S129A /S129A-S87-P. A similar graph was generated for S129E/S129E-S87-P (Fig. 9F) by transferring the S129A assignments. The data demonstrate clearly that relatively large chemical shift changes take place at sites close to S87 in both phosphorylated proteins, with more minor changes spanning the region between residues 60 and 80. Chemical shift changes observed upon comparing micelle-bound unphosphorylated S129A or S129E with WT α-syn were restricted to the immediate sites of the mutation and were essentially identical to those previously observed for the free protein (Paleologou et al., 2008), indicating that the mutations themselves do not cause long-range effects.
To better understand the effects of S87 phosphorylation on the micelle-bound structure of α-syn, we analyzed both sequential HN-HN NOE and Cα chemical shift data. The NOE data (Fig. 10A) reveal a decrease in signal intensity, when compared with those previously obtained for the WT protein, in the second half of the second micelle-bound helix, coinciding with the location of the phosphorylated S87 residue and suggesting a possible decrease in the stability of the helical structure in this region. Accordingly, secondary Cα chemical shift data (Fig. 10B) also show smaller positive amplitudes in this region when compared with previous observations for the unphosphorylated WT protein. Interestingly, these structural effects are similar to those observed for the protein β-syn, a close homolog of α-syn, which is however missing 11 residues from the NAC region (Sung and Eliezer, 2006, 2007).
Figure 10.

The effects of S87 phosphorylation on the micelle-bound structure of α-syn. A, B, Sequential amide proton NOEs (A) and α carbon secondary shifts (B) for SDS micelle bound S129A/S87-P α-syn. NOEs represent the average of the forward and backward crosspeak intensities between resonances originating from any two consecutive residues. Black bars show data previously reported for the unphosphorylated WT protein (Bussell and Eliezer, 2003), whereas blue bars represent the S87-P form of the protein. Green bars represent regions of resonance overlap that preclude reliable detection of the presence or absence of an NOE signal. Secondary shifts were calculated using NMRView (Delaglio et al., 1995) based on random coil shifts determined for small peptides in 1 M urea at pH 5.0 (Wishart et al., 1995). C, Circular dichroism spectroscopy reveals that WT (yellow line), S129A (red line), S129A/S87-P (green line), and S129A/S87-P, T92-P (blue line) α-syn adopt random coil structure. α-Syn at 10 μm. D, Circular dichroism spectroscopy of WT (yellow line), S129A (red line), and S129A/S87-P (green line), S129A/S87-P, T92-P (blue line) α-syn in the presence of POPG vesicles. α-syn: POPG mass ratio is 1:20.
To further probe the effect of phosphorylation on α-syn-membrane interactions, we compared the CD spectra of mono- (S129A/S87-P) and diphosphorylated S129A (S129A/S87-P,T92-P) to that of S129A and WT α-syn in solution and upon binding to POPG vesicles. Although the effect of phosphorylation at S87 and T92 at the monomer is not discernible by Far-UV/CD (Fig. 10C), both mono- (S129A/S87-P) and diphosphorylated (S129A/S87-P,T92-P) exhibit reduced α-helical propensity in the presence of synthetic vesicles with diphosphorylation having the most dramatic affect (Fig. 10D).
Discussion
α-Syn S87 phosphorylation occurs in vivo and is increased in synucleinopathies
Phosphorylation is an important reversible post-translational modification that regulates the structural and functional properties of proteins in health and disease. The role of phosphorylation in modulating the aggregation and fibrillogenesis of tau and α-syn is currently a subject of intense investigation and pathways involved are being pursued as viable targets to prevent or slow down the progression of AD and PD (Glicksman et al., 2007; Wen et al., 2008; Rezai-Zadeh et al., 2008). While hyperphosphorylation of tau at different sites has been well characterized and the role of tau phosphorylation in AD pathology was investigated in several mouse models of AD, a thorough mapping and investigation of all phosphorylation sites within α-syn and of the kinases involved remains to be accomplished. Several kinases phosphorylate α-syn at S129 (Okochi et al., 2000; Pronin et al., 2000; Fujiwara et al., 2002; Takahashi et al., 2003a; Chen and Feany, 2005; Kim et al., 2006; Qing et al., 2009; Inglis et al., 2009; Mbefo et al., 2010) and S87 (Okochi et al., 2000; Pronin et al., 2000), as well as Y125 (Ellis et al., 2001; Nakamura et al., 2001; Hasegawa et al., 2002; Takahashi et al., 2003a), Y133 (Ellis et al., 2001; Negro et al., 2002), and Y136 (Ellis et al., 2001; Negro et al., 2002) in vitro. However, only phosphorylation at S129 has been shown to correlate with the pathology of PD and related α-synucleinopathies. Here we demonstrate that α-syn is phosphorylated at S87 in vivo and the level of S87-P is increased in brains of TG models of synucleinopathies and human brains from AD, LBD, and MSA patients. To determine whether α-syn in LBs is also phosphorylated at S87, we performed immunofluorescence stainings of LBs isolated from fresh human brains using our anti-S87-P antibody. S87-P immunoreactivity was consistently detected in homogeneous presumably earlier stages of formation as well as mature LBs, suggesting S87-P α-syn occurs throughout the life span of LB development.
Neither S87 nor S129 phosphorylation are found in LB, but they are not responsible for LB formation
Previous studies from our group and others demonstrated that phosphorylation at S129 blocks, rather than inhibit α-syn fibrillization. Herein, we show that phosphorylation at S87 also inhibits α-syn fibrillization in vitro. Together, these findings suggest that phosphorylation at these residues should inhibit, rather than promote LB formation in vivo. Our findings that S129, but not S87, undergoes phosphorylation by CK1 in the fibrillar state is consistent with the solid state NMR results and favor the hypothesis that phosphorylation at S129 is a late event that occurs after α-syn fibrillization and/or during the development and maturation of LBs. The increased level of S129-P is consistent with its being further away from the core of the fibrils and in a region that is highly disordered and thus more accessible to phosphorylation by kinases. The colocalization of several kinases (Arawaka et al., 2006; Ryu et al., 2008) and α-syn within LBs combined with recent studies from our group (Mbefo et al., 2010) and by Waxman and Giasson (2008) demonstrating that aggregated α-syn species are better substrate for CK1, CK2, and the PLK 1–3 suggest that S129 phosphorylation could easily occur subsequent to LB formation. When combined with our own observations that S129 phosphorylation actually inhibits α-syn fibrillization [also confirmed by a recent study by Waxman and Giasson (2008)], these observations provide strong support for the hypothesis that phosphorylation at S129 is in fact not a prerequisite for LB formation and more likely occurs after α-syn fibrillization (Fig. 10B).
Recent studies by Fournier et al. (2009), using our anti-S87-P antibody, demonstrated that S87-P was only observed in the TBS soluble fractions, whereas S129-P was observed in both soluble and SDS fractions from healthy and symptomatic mice overexpressing α-syn or α-syn and Parkin. These results combined with the presence of S87 in a β-strand conformation within the core of fibrils are consistent with our results showing the inhibition of α-syn fibrillization upon phosphorylation of this site and the lack of phosphorylation in the preformed fibrillar state, but do not explain how this residue becomes phosphorylated within LBs. These observations could be explained if α-syn within LBs exists in different aggregated and/or fibrillar states, some of which may possess different core-structure than α-syn fibrils formed in vitro. From the Lewy body data we have gathered, it appears that s87P has been incorporated in Lewy bodies at early (with homogeneous morphology) as well as later (concentric morphology) stages of maturation. Alternately, the Lewy bodies we typically observe may perhaps reflect later events of the disease process and are likely generated from small α-syn aggregates originating in sites where α-syn turnover is most active, for instance at synapses. Small α-syn aggregates may be retrogradely transported through axons, sometimes leading to the formation of Lewy neurites. We did not detect S87-P in Lewy neurites, suggesting that S87 phosphorylation is indeed a later event occurring after the formation of early aggregates. Thus, S87 phosphorylation may occur subsequent to early α-syn aggregation, but before the formation of the mature amyloid fibril form, in which S87 is no longer accessible to the responsible kinases. Further studies will be required to determine the aggregation states of S87-P immunoreactive material within LBs and to determine the kinetics of, and interplay between, S87 and S129 phosphorylation during the evolution of LBs.
S → E mutation at S87 or S129 mimic only some aspects of phosphorylation
Recent studies by two independent research groups demonstrated that AAV-mediated overexpression of the S129A in the substantia nigra of rats results in both increased aggregation and loss of dopaminergic neurons relative to overexpression of WT and S129D α-syn (Gorbatyuk et al., 2008; Azeredo da Silveira et al., 2009). Thus, it appears that the increased in vivo aggregation caused by the S129A mutation (which also promotes α-syn aggregation in vitro) may lead to toxicity that is unrelated to its effect in blocking phosphorylation at S129. To properly probe the role of phosphorylation in vivo it is essential that mutants designed to abolish phosphorylation should exhibit aggregation properties similar to that of the WT protein. Similarly, phosphorylation mimicking mutations should reproduce the structural and functional consequences of α-syn phosphorylation. Neither the S129A nor S129D/E mutants fulfill these criteria as demonstrated by both the in vivo studies cited above and our own recent in vitro studies. In contrast, as we show here, the S87A and S87E mutants come closer to fulfilling these criteria. Figure 7 demonstrates that S87A exhibits similar fibrillization properties as the WT protein, whereas S87E does not form fibrils, suggesting S → E substitution at S87 reproduces the effect of phosphorylation on α-syn aggregation. Together, these findings suggest that overexpression of S87E and S87A mutants should facilitate the elucidation of the role of phosphorylating this residue on modulating α-syn oligomerization and fibrillogenesis in vivo.
Nevertheless, the fact that phosphorylation at S87 (S87-P), but not the phosphomimic S87E, induced significant changes in the conformation of membrane bound α-syn that coincided with a slight reduction in the propensity of S87-P to form α-helical structure during binding to membranes, suggests that S87E may not recapitulate phosphorylation-dependent protein-membrane interactions. Our findings demonstrate that detailed and careful biophysical characterization of the phosphorylated proteins and their phosphomimics can assist in designing and interpreting in vivo studies aimed at elucidating the role of phosphorylation in health and disease.
Potential role of S87 and S129 in regulating phosphorylation dependent protein-protein interactions
Phosphorylation of proteins represents an important molecular switch for regulating protein-protein and protein-ligand interactions and thus protein function. The C-terminal region of α-syn has been implicated in the majority of α-syn interactions with proteins (Jensen et al., 1999; Giasson et al., 2003; Fernández et al., 2004) and metal ions (Paik et al., 1999; Brown, 2007), and phosphorylation in this region is likely to influence the affinity of α-syn for other proteins and thereby alter the biochemical and biological processes regulated by its interactions. Indeed, McFarland et al. recently showed differences in the set of proteins pulled down by S129- or Y125-phosphorylated forms of α-syn (McFarland et al., 2008).
S87 lies in the NAC region, which is crucial in mediating α-syn fibrillization, and our results accordingly demonstrate that S87 phosphorylation exerts a profound effect on α-syn aggregation. Interestingly, our results also show that S87 phosphorylation alters the conformation of membrane bound α-syn and decreases its affinity to lipid vesicles (Fig. 10), probably by destabilizing the helical conformation and decreasing the lipid-binding affinity of the protein around the phosphorylation site (Figs. 9, 10). S87 is also located in a region involved in the interactions of α-syn with at least some proteins, such as the enzyme PLD2 (Payton et al., 2004). Thus, phosphorylation at position S87 may offer further opportunities, beyond phosphorylation in the C-terminal tail region, for regulating synuclein protein-protein, as well as protein-membrane interactions. Further studies of the effects of S87 phosphorylation in vivo will be required to evaluate this hypothesis.
Conclusions
The results presented here indicate that phosphorylation of α-syn at S87 clearly occurs in vivo and is increased in synucleinopathies. However, the role of α-syn S87 phosphorylation in disease is unlikely to encompass directly increasing the fibrillization or deposition of the protein as phosphorylation at this residue blocks rather than promotes α-syn fibrillization. Whether phosphorylation enhances or protects against α-syn toxicity in vivo remains controversial. Previous studies aimed at addressing this question in the case of S129 phosphorylation are based on overexpression of the phosphomimics (S129E /D), which do not reproduce all aspects of phosphorylation and have yielded contradictory results (Chen and Feany 2005; Gorbatyuk et al., 2008; Azezredo da Silveira et al., 2009; McFarland et al., 2009). Our work here suggests that S87E should provide a more robust phosphomimic, at least as regards its effects on a-syn aggregation. To date, emphasis has been placed on trying to elucidate the role of phosphorylation in modulating α-syn aggregation and toxicity. However, α-syn phosphorylation at S87 may also influence the normal function of the protein, either by perturbing the membrane-bound conformation of the protein as suggested by our data, or by modulating protein-protein interactions, as has been shown previously for other phosphorylation sites. The identification of the natural kinases and phosphatases involved in regulating α-syn phosphorylation at S87 (and S129) should facilitate studies of its effects on both synuclein toxicity and function and is of crucial importance both to understanding the role of phosphorylation in the pathogenesis of PD and to the identification of novel targets to treat PD and related synucleinopathies.
Footnotes
This work was supported by the Swiss Federal Institute of Technology Lausanne (to H.A.L., K.E.B., A.W.S.), grants from the Swiss National Science Foundation (to H.A.L., E.P., A.S., 310000-110027 and to P.A. and B.L.S., 31003A and 120653), by the Max Planck Society (to M.Z., C.O.F.), a Deutsche Forschungsgemeinschaft Heisenberg scholarship (to Z.W. 71/2-1 and 3-1), and by National Institutes of Health (NIH)/National Institute on Aging Grant AG019391, NIH Grants AG5131 and AG18440 (to E.M.), the Irma T. Hirschl Foundation, the Michael J. Fox Foundation, and a gift from Herbert and Ann Siegel (to D.E.) and Marie Curie fellowship (to A.O.). D.E. is a member of the New York Structural Biology Center, supported by NIH Grant GM66354, which is a STAR center supported by the New York State Office of Science, Technology, and Academic Research, by NIH Grant P41 GM66354 and which received funds from NIH, the Keck Foundation, New York State, and the NYC Economic Development Corporation for the purchase of 900 MHz spectrometers. We thank Dr. Omar Elagnaf and Dr. Ruth Luthi-Carter for critical review of this manuscript.
References
- Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Ahn BH, Rhim H, Kim SY, Sung YM, Lee MY, Choi JY, Wolozin B, Chang JS, Lee YH, Kwon TK, Chung KC, Yoon SH, Hahn SJ, Kim MS, Jo YH, Min DS. alpha-Synuclein interacts with phospholipase D isozymes and inhibits pervanadate-induced phospholipase D activation in human embryonic kidney-293 cells. J Biol Chem. 2002;277:12334–12342. doi: 10.1074/jbc.M110414200. [DOI] [PubMed] [Google Scholar]
- Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281:29739–29752. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- Arawaka S, Wada M, Goto S, Karube H, Sakamoto M, Ren CH, Koyama S, Nagasawa H, Kimura H, Kawanami T, Kurita K, Tajima K, Daimon M, Baba M, Kido T, Saino S, Goto K, Asao H, Kitanaka C, Takashita E, et al. The role of G-protein-coupled receptor kinase 5 in pathogenesis of sporadic Parkinson's disease. J Neurosci. 2006;26:9227–9238. doi: 10.1523/JNEUROSCI.0341-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azeredo da Silveira S, Schneider BL, Cifuentes-Diaz C, Sage D, Abbas-Terki T, Iwatsubo T, Unser M, Aebischer P. Phosphorylation does not prompt, nor prevent, the formation of alpha-synuclein toxic species in a rat model of Parkinson's disease. Hum Mol Genet. 2009;1:872–887. doi: 10.1093/hmg/ddn417. [DOI] [PubMed] [Google Scholar]
- Bax A, Grzesiek S. Methodological advances in NMR. Acc Chem Res. 1993;26:131–138. [Google Scholar]
- Bertoncini CW, Jung YS, Fernandez CO, Hoyer W, Griesinger C, Jovin TM, Zweckstetter M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc Natl Acad Sci U S A. 2005;102:1430–1435. doi: 10.1073/pnas.0407146102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DR. Interactions between metals and alpha-synuclein–function or artefact? FEBS J. 2007;274:3766–3774. doi: 10.1111/j.1742-4658.2007.05917.x. [DOI] [PubMed] [Google Scholar]
- Bussell R, Jr, Eliezer D. A structural and functional role for 11-mer repeats in alpha-synuclein and other exchangeable lipid binding proteins. J Mol Biol. 2003;329:763–778. doi: 10.1016/s0022-2836(03)00520-5. [DOI] [PubMed] [Google Scholar]
- Chandra S, Chen X, Rizo J, Jahn R, Südhof TC. A broken alpha -helix in folded alpha -Synuclein. J Biol Chem. 2003;278:15313–15318. doi: 10.1074/jbc.M213128200. [DOI] [PubMed] [Google Scholar]
- Chen L, Feany MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- El-Agnaf OM, Bodles AM, Guthrie DJ, Harriott P, Irvine GB. The N-terminal region of non-A beta component of Alzheimer's disease amyloid is responsible for its tendency to assume beta-sheet and aggregate to form fibrils. Eur J Biochem. 1998;258:157–163. doi: 10.1046/j.1432-1327.1998.2580157.x. [DOI] [PubMed] [Google Scholar]
- Eliezer D, Kutluay E, Bussell R, Jr, Browne G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- Ellis CE, Schwartzberg PL, Grider TL, Fink DW, Nussbaum RL. alpha-synuclein is phosphorylated by members of the Src family of protein-tyrosine kinases. J Biol Chem. 2001;276:3879–3884. doi: 10.1074/jbc.M010316200. [DOI] [PubMed] [Google Scholar]
- Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- Fernández CO, Hoyer W, Zweckstetter M, Jares-Erijman EA, Subramaniam V, Griesinger C, Jovin TM. NMR of alpha-synuclein-polyamine complexes elucidates the mechanism and kinetics of induced aggregation. EMBO J. 2004;23:2039–2046. doi: 10.1038/sj.emboj.7600211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier M, Vitte J, Garrigue J, Langui D, Dullin JP, Saurini F, Hanoun N, Perez-Diaz F, Cornilleau F, Joubert C, Ardila-Osorio H, Traver S, Duchateau R, Goujet-Zalc C, Paleologou K, Lashuel HA, Haass C, Duyckaerts C, Cohen-Salmon C, Kahle PJ, Hamon M, Brice A, Corti O. Parkin deficiency delays motor decline and disease manifestation in a mouse model of synucleinopathy. PLoS One. 2009;4:e6629. doi: 10.1371/journal.pone.0006629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Gai WP, Power JH, Blumbergs PC, Culvenor JG, Jensen PH. Alpha-synuclein immunoisolation of glial inclusions from multiple system atrophy brain tissue reveals multiprotein components. J Neurochem. 1999;73:2093–2100. [PubMed] [Google Scholar]
- Gai WP, Yuan HX, Li XQ, Power JT, Blumbergs PC, Jensen PH. In situ and in vitro study of colocalization and segregation of alpha-synuclein, ubiquitin, and lipids in Lewy bodies. Exp Neurol. 2000;166:324–333. doi: 10.1006/exnr.2000.7527. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300:636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- Glicksman MA, Cuny GD, Liu M, Dobson B, Auerbach K, Stein RL, Kosik KS. New approaches to the discovery of cdk5 inhibitors. Current Alzheimer Research. 2007;4:547–559. doi: 10.2174/156720507783018181. [DOI] [PubMed] [Google Scholar]
- Gorbatyuk OS, Li S, Sullivan LF, Chen W, Kondrikova G, Manfredsson FP, Mandel RJ, Muzyczka N. The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc Natl Acad Sci U S A. 2008;105:763–768. doi: 10.1073/pnas.0711053105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Fujiwara H, Nonaka T, Wakabayashi K, Takahashi H, Lee VM, Trojanowski JQ, Mann D, Iwatsubo T. Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J Biol Chem. 2002;277:49071–49076. doi: 10.1074/jbc.M208046200. [DOI] [PubMed] [Google Scholar]
- Heise H, Celej MS, Becker S, Riedel D, Pelah A, Kumar A, Jovin TM, Baldus M. Solid-state NMR reveals structural differences between fibrils of wild-type and disease-related A53T mutant alpha-synuclein. J Mol Biol. 2008;380:444–450. doi: 10.1016/j.jmb.2008.05.026. [DOI] [PubMed] [Google Scholar]
- Hoyer W, Antony T, Cherny D, Heim G, Jovin TM, Subramaniam V. Dependence of alpha-synuclein aggregate morphology on solution conditions. J Mol Biol. 2002;322:383–393. doi: 10.1016/s0022-2836(02)00775-1. [DOI] [PubMed] [Google Scholar]
- Inglis KJ, Chereau D, Brigham EF, Chiou SS, Schöbel S, Frigon NL, Yu M, Caccavello RJ, Nelson S, Motter R, Wright S, Chian D, Santiago P, Soriano F, Ramos C, Powell K, Goldstein JM, Babcock M, Yednock T, Bard F, et al. Polo-like kinase 2 (PLK2) phosphorylates alpha-synuclein at serine 129 in central nervous system. J Biol Chem. 2009;284:2598–2602. doi: 10.1074/jbc.C800206200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen PH, Hager H, Nielsen MS, Hojrup P, Gliemann J, Jakes R. alpha-synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J Biol Chem. 1999;274:25481–25489. doi: 10.1074/jbc.274.36.25481. [DOI] [PubMed] [Google Scholar]
- Johnson BA, Blevins RA. NMRView: a computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- Jones JA, Wilkins DK, Smith LJ, Dobson CM. Characterisation of protein unfolding by NMR diffusion measurements. J Biomol NMR. 1997;10:199–203. [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, van Der Putten H, Probst A, Kremmer E, Kretzschmar HA, Haass C. Subcellular localization of wild-type and Parkinson's disease-associated mutant alpha-synuclein in human and transgenic mouse brain. J Neurosci. 2000;20:6365–6373. doi: 10.1523/JNEUROSCI.20-17-06365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler JC, Rochet JC, Lansbury PT., Jr The N-terminal repeat domain of alpha-synuclein inhibits beta-sheet and amyloid fibril formation. Biochemistry. 2003;42:672–678. doi: 10.1021/bi020429y. [DOI] [PubMed] [Google Scholar]
- Kim EJ, Sung JY, Lee HJ, Rhim H, Hasegawa M, Iwatsubo T, Min do S, Kim J, Paik SR, Chung KC. Dyrk1A phosphorylates alpha-synuclein and enhances intracellular inclusion formation. J Biol Chem. 2006;281:33250–33257. doi: 10.1074/jbc.M606147200. [DOI] [PubMed] [Google Scholar]
- Kloepper KD, Zhou DH, Li Y, Winter KA, George JM, Rienstra CM. Temperature-dependent sensitivity enhancement of solid-state NMR spectra of alpha-synuclein fibrils. J Biomol NMR. 2007;39:197–211. doi: 10.1007/s10858-007-9189-z. [DOI] [PubMed] [Google Scholar]
- Kohn JE, Millett IS, Jacob J, Zagrovic B, Dillon TM, Cingel N, Dothager RS, Seifert S, Thiyagarajan P, Sosnick TR, Hasan MZ, Pande VS, Ruczinski I, Doniach S, Plaxco KW. Random-coil behavior and the dimensions of chemically unfolded proteins. Proc Natl Acad Sci U S A. 2004;101:12491–12496. doi: 10.1073/pnas.0403643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bianco C, Ridet JL, Schneider BL, Deglon N, Aebischer P. alpha-synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson's disease. Proc Natl Acad Sci U S A. 2002;99:10813–10818. doi: 10.1073/pnas.152339799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E. Genetically altered transgenic models of Alzheimer's disease. J Neural Transm Suppl. 2000;59:175–183. doi: 10.1007/978-3-7091-6781-6_20. [DOI] [PubMed] [Google Scholar]
- Mbefo MK, Paleologou KE, Boucharaba A, Oueslati A, Schell H, Fournier M, Olschewski D, Yin G, Zweckstetter M, Masliah E, Kahle PJ, Hirling H, Lashuel HA. Phosphorylation of synucleins by members of the polo-like kinase family. J Biol Chem. 2010;285:2807–2822. doi: 10.1074/jbc.M109.081950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland MA, Ellis CE, Markey SP, Nussbaum RL. Proteomics analysis identifies phosphorylation-dependent alpha-synuclein protein interactions. Mol Cell Proteomics. 2008;7:2123–2137. doi: 10.1074/mcp.M800116-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland NR, Fan Z, Xu K, Schwarzschild MA, Feany MB, Hyman BT, McLean PJ. Alpha-synuclein S129 phosphorylation mutants do not alter nigrostriatal toxicity in a rat model of Parkinson disease. J Neuropathol Exp Neurol. 2009;68:515–524. doi: 10.1097/NEN.0b013e3181a24b53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Yamashita H, Takahashi T, Nakamura S. Activated Fyn phosphorylates alpha-synuclein at tyrosine residue 125. Biochem Biophys Res Commun. 2001;280:1085–1092. doi: 10.1006/bbrc.2000.4253. [DOI] [PubMed] [Google Scholar]
- Negro A, Brunati AM, Donella-Deana A, Massimino ML, Pinna LA. Multiple phosphorylation of alpha-synuclein by protein tyrosine kinase Syk prevents eosin-induced aggregation. FASEB J. 2002;16:210–212. doi: 10.1096/fj.01-0517fje. [DOI] [PubMed] [Google Scholar]
- Okochi M, Walter J, Koyama A, Nakajo S, Baba M, Iwatsubo T, Meijer L, Kahle PJ, Haass C. Constitutive phosphorylation of the Parkinson's disease associated alpha-synuclein. J Biol Chem. 2000;275:390–397. doi: 10.1074/jbc.275.1.390. [DOI] [PubMed] [Google Scholar]
- Paik SR, Shin HJ, Lee JH, Chang CS, Kim J. Copper(II)-induced self-oligomerization of alpha-synuclein. Biochem J. 1999;340:821–828. [PMC free article] [PubMed] [Google Scholar]
- Paleologou KE, Schmid AW, Rospigliosi CC, Kim HY, Lamberto GR, Fredenburg RA, Lansbury PT, Jr, Fernandez CO, Eliezer D, Zweckstetter M, Lashuel HA. Phosphorylation at S129, but not the phosphomimics S129E/D inhibits the fibrilization of alpha-synuclein. J Biol Chem. 2008;238:16895–16905. doi: 10.1074/jbc.M800747200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic; 1986. [DOI] [PubMed] [Google Scholar]
- Payton JE, Perrin RJ, Woods WS, George JM. Structural determinants of PLD2 inhibition by alpha-synuclein. J Mol Biol. 2004;337:1001–1009. doi: 10.1016/j.jmb.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Permi P, Kilpeläinen I, Annila A, Heikkinen S. Intensity modulated HSQC and HMQC: Two simple methods to measure (3)J(HNH alpha) in proteins. J Biomol NMR. 2000;16:29–37. doi: 10.1023/a:1008343926502. [DOI] [PubMed] [Google Scholar]
- Pronin AN, Morris AJ, Surguchov A, Benovic JL. Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J Biol Chem. 2000;275:26515–26522. doi: 10.1074/jbc.M003542200. [DOI] [PubMed] [Google Scholar]
- Qing H, Wong W, McGeer EG, McGeer PL. Lrrk2 phosphorylates alpha-synuclein at serine 129: Parkinson disease implications. Biochem Biophys Res Commun. 2009;387:149–152. doi: 10.1016/j.bbrc.2009.06.142. [DOI] [PubMed] [Google Scholar]
- Rezai-Zadeh K, Douglas Shytle R, Bai Y, Tian J, Hou H, Mori T, Zeng J, Obregon D, Town T, Tan J. Flavonoid-mediated presenilin-1 phosphorylation reduces Alzheimer's disease beta-amyloid production. J of Cellular and Mol Med. 2009;13:574–588. doi: 10.1111/j.1582-4934.2008.00344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68:568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- Ryu MY, Kim DW, Arima K, Mouradian MM, Kim SU, Lee G. Localization of CKII beta subunits in Lewy bodies of Parkinson's disease. J Neurol Sci. 2008;266:9–12. doi: 10.1016/j.jns.2007.08.027. [DOI] [PubMed] [Google Scholar]
- Shults CW, Rockenstein E, Crews L, Adame A, Mante M, Larrea G, Hashimoto M, Song D, Iwatsubo T, Tsuboi K, Masliah E. Neurological and neurodegenerative alterations in a transgenic mouse model expressing human alpha-synuclein under oligodendrocyte promoter: implications for multiple system atrophy. J Neurosci. 2005;25:10689–10699. doi: 10.1523/JNEUROSCI.3527-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung YH, Eliezer D. Secondary structure and dynamics of micelle bound beta- and gamma-synuclein. Protein Sci. 2006;15:1162–1174. doi: 10.1110/ps.051803606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung YH, Eliezer D. Residual structure, backbone dynamics, and interactions within the synuclein family. J Mol Biol. 2007;372:689–707. doi: 10.1016/j.jmb.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Kanuka H, Fujiwara H, Koyama A, Hasegawa M, Miura M, Iwatsubo T. Phosphorylation of alpha-synuclein characteristic of synucleinopathy lesions is recapitulated in alpha-synuclein transgenic Drosophila. Neurosci Lett. 2003a;336:155–158. doi: 10.1016/s0304-3940(02)01258-2. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Yamashita H, Nagano Y, Nakamura T, Ohmori H, Avraham H, Avraham S, Yasuda M, Matsumoto M. Identification and characterization of a novel Pyk2/related adhesion focal tyrosine kinase-associated protein that inhibits alpha-synuclein phosphorylation. J Biol Chem. 2003b;278:42225–42233. doi: 10.1074/jbc.M213217200. [DOI] [PubMed] [Google Scholar]
- Ulmer TS, Bax A. Comparison of structure and dynamics of micelle-bound human alpha-synuclein and Parkinson disease variants. J Biol Chem. 2005;280:43179–43187. doi: 10.1074/jbc.M507624200. [DOI] [PubMed] [Google Scholar]
- van der Putten H, Wiederhold KH, Probst A, Barbieri S, Mistl C, Danner S, Kauffmann S, Hofele K, Spooren WP, Ruegg MA, Lin S, Caroni P, Sommer B, Tolnay M, Bilbe G. Neuropathology in mice expressing human alpha-synuclein. J Neurosci. 2000;20:6021–6029. doi: 10.1523/JNEUROSCI.20-16-06021.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilar M, Chou HT, Lührs T, Maji SK, Riek-Loher D, Verel R, Manning G, Stahlberg H, Riek R. The fold of alpha-synuclein fibrils. Proc Natl Acad Sci U S A. 2008;105:8637–8642. doi: 10.1073/pnas.0712179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Giasson BI. Specificity and regulation of casein kinase-mediated phosphorylation of alpha-synuclein. J Neuropathol Exp Neurol. 2008;67:402–416. doi: 10.1097/NEN.0b013e3186fc995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Planel E, Herman M, Figueroa HY, Wang L, Liu L, Lau LF, Yu WH, Duff KE. Interplay between cyclin-dependent kinase 5 and glycogen synthase kinase 3 beta mediated by neuregulin signaling leads to differential effects on tau phosphorylation and amyloid precursor protein processing. J Neurosci. 2008;28:2624–2632. doi: 10.1523/JNEUROSCI.5245-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry. 1999;38:16424–16431. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. H-1, C-3 and N-15 chemical-shift referencing in biomolecular NMR. J Biomol NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- Yamada M, Iwatsubo T, Mizuno Y, Mochizuki H. Overexpression of alpha-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of alpha-synuclein and activation of caspase-9: resemblance to pathogenetic changes in Parkinson's disease. J Neurochem. 2004;91:451–461. doi: 10.1111/j.1471-4159.2004.02728.x. [DOI] [PubMed] [Google Scholar]