

Abstract
High-risk human papillomavirus (HPV) infection of the cervical epithelium is causally linked with the generation of cervical cancer. HPV does not activate Langerhans cells (LC), the antigen presenting cell (APC) at the site of infection, leading to immune evasion. The HPV protein responsible for inducing this immune escape has not been determined. We demonstrate that LC exposed to the minor capsid protein L2 in HPV16L1L2 virus-like particles (VLP) do not phenotypically or functionally mature. However, HPV16L1 VLP significantly induce activation of LC. Our data suggest that the L2 protein plays a specific role in the induction of this immune escape of HPV16 through the manipulation of LC. This novel function is the first immune modulating action attributed to the L2 protein and adds significantly to our understanding of the mechanism of HPV immune escape.
This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
Keywords: Human, Dendritic Cells, Viral, Cell Activation
Introduction
Cervical cancer is the second most common cancer among women worldwide (1) and is causally linked with high-risk human papillomavirus (HPV)4 infection (2). The majority of women will acquire a genital HPV infection at some point in their lifetime (3), and although most women will clear the infection, the average time for clearance is close to a year (4). About 15% of women with a high-risk HPV infection cannot induce an effective immune response against the virus (5). These observations indicate that HPV is escaping immune detection and clearance.
The life cycle of HPV is dependent on the differentiation of cells in the epithelium and thus it is difficult to produce large quantities of HPV virions in vitro, therefore HPV virus-like particles (VLP)4 have been developed. When the major capsid protein L1 is expressed, it can self-assemble into a L1 VLP with a 72-pentamer icosahedral structure (6). If both L1 and the minor capsid protein L2 are simultaneously expressed, the proteins assemble into L1L2 VLP that contain up to 72 L2 proteins per VLP (7, 8). Because HPV virions are composed of both L1 and L2 proteins, HPV L1L2 VLP are morphologically equivalent to HPV virions while VLP comprised of L1 alone are not.
While the HPV minor capsid protein L2 is not required for VLP formation, it has been shown to possess a variety of critical functions. The carboxy-terminus of L2 binds directly to L1 primarily through hydrophobic interactions. This region of L2 is proline-rich allowing for sharp bending of the protein that may facilitate L2 to loop through the central cavity of the L1 pentamer (9). It has also been demonstrated that L2 interacts with the viral genome and is integral in the encapsidation of viral DNA (10). Collectively, these interactions imply a significant role for L2 in the formation of the virion. Furthermore, L2 facilitates HPV infection through an interaction between between the N-terminus region of the L2 protein and an unknown cell surface receptor (11, 12). Further functions of L2 include binding of the virion to the cytoskeleton, transport within the cytoplasm (13), and facilitation of endosomal escape of the viral genome after infection (14).
Langerhans cells (LC)4 are APC located in the epithelium of the skin and mucosa (15). Due to the site of HPV infection, LC are responsible for initiating an immune response against HPV. It has been found in various studies that HPV L1 VLP and HPV L1L2 VLP can bind to and activate human dendritic cells (DC) (16-18), providing evidence that the structural surface components of HPV can induce the maturation of APC. However, we have previously demonstrated that human LC exposed to HPV16L1L2 VLP are not activated, implicating an HPV immune escape mechanism that targets LC (19). We have shown that this HPV16 immune escape mechanism is due to the deregulation of the PI3K-Akt pathway in LC (20). Thus, even though DC and LC are both potent APC, they respond differently to HPV.
In apparent contradiction to our studies, LC exposed to HPV L1 VLP were shown to generate cytolytic T cells in vitro (21). HPV L1 VLP were also shown to be taken up by LC through either a clathrin mediated (22) or caveolae-dependent mechanism (21), while we demonstrated that HPV16L1L2 VLP were taken up by LC through a clathrin-, caveolae-, actin-independent pathway (23). These contrasting studies highlight differences in the interaction between LC and HPV L1 VLP versus HPV L1L2 VLP, and point to the potentially important presence of L2. Therefore, we sought to elucidate if the minor capsid protein L2 is responsible for the induction of immune escape of HPV16.
Materials and Methods
Abs
The Abs against conformational HPV16L1 epitopes (H16.V5, and H16.E70) or linear HPV16L1 epitopes (Camvir-1, H16.D9, and H16.H5) were gifts from Neil Christensen (Penn State, Hershey, PA), except Camvir-1 (BD Biosciences). Polyclonal serum (DK44214) for HPV16L2 was a gift from John Schiller (NIH, Bethesda, MD). Additionally, the following Abs were used in this study: anti-CD1a-PE, CD80-FITC, CD86-FITC, HLA-DR, DQ, DP-FITC, isotype controls, biotinylated anti-rabbit IgG, streptavidin-PE, and streptavidin-HRP (BD Biosciences); anti-CD207 (langerin) (Immunotech); anti-E-cadherin (Millipore); anti-phosphorylated (p)-PI3K (Tyr 508), PI3K, p-Akt (Ser 473), Akt (Santa Cruz Biotechnology); anti-GAPDH (Chemicon); goat anti-mouse-FITC and goat anti-rabbit-HRP (Biosource); goat-anti-mouse-IR Dye 800 (Rockland), goat-anti-rabbit-Alexa Fluor 680 (Molecular Probes); and anti-IFN-γ and biotinylated anti-IFN-γ (Mabtech).
LC and DC Generation
Human PBL from healthy donors were obtained by leukapheresis (19). LC and DC were generated from human PBL as previously described (24). HPV serology of all donors was negative. All studies were approved by USC’s IRB and informed consent was obtained from donors.
Virus-Like Particles
HPV16L1 VLP and HPV16L1L2 VLP were produced as previously described (19). Western blot analyses confirmed the presence of L1 and L2 while an ELISA and transmission electron microscopy confirmed the presence of intact particles. An E-toxate kit (Sigma-Aldrich) was used to semi-quantitate endotoxin. The endotoxin level in the preparations was less than 0.06 endotoxin units/ml and this level does not activate LC (19). Baculovirus DNA used in VLP production procedure does not activate LC (19).
Activation Assay
LC or DC were either left untreated, treated with 10 μg LPS (Sigma-Aldrich), 10 μg HPV16L1 VLP/106 cells or 10 μg HPV16L1L2 VLP/106 cells. For titration experiments, LC were either left untreated, treated with 10 μg LPS, 10 μg HPV16L1 VLP/106 cells, 10 μg HPV16L1L2 VLP/106 cells, 6.6 μg HPV16L1 VLP/106 cells and 3.3 μg HPV16L1L2 VLP/106 cells (2:1), 5 μg HPV16L1 VLP/106 cells and 5 μg HPV16L1L2 VLP/106 cells (1:1), 3.3 μg HPV16L1 VLP/106 cells and 6.6 μg HPV16L1L2 VLP/106 cells (1:2), or with 5 μg HPV16L1 VLP/106 cells and 5 μg heated (10 min, 95°C) HPV16L1L2 VLP/106 cells (1:1Δ). To validate results, three different VLP preparations were used over the course of our experiments. The cells were then incubated for 1h at 37°C, mixed occasionally, and placed at 37°C for 48 h in 20 ml complete media containing 1000 U/ml rGM-CSF. Supernatants were collected and cells were harvested, washed, stained for surface markers or isotype controls, and analyzed by flow cytometry. Supernatants were analyzed at the USC Beckman Immune Monitoring Center using the Bio-Plex Suspension Array System (Bio-Rad).
Migration Assay
Chemokine directed migration of LC was carried out using 24-well Transwell plates with 5μm-pore-size polycarbonate filters (Corning Costar). Briefly, media was added to the lower chamber containing either 250 ng/ml human r6Ckine/CCL21 (R&D Systems) or complete media alone to control for spontaneous migration. We added 2 × 105 LC, untreated or treated as indicated in the activation assay to the upper chamber and incubated for 3.5 h at 37°C. The cells that migrated to the lower chamber were counted using a hemacytometer.
In Vitro Immunization Assay
In vitro immunization assays were performed as previously described (19) with the same treatments as describe in the activation assay. After 28 days, effector CD8+ T cells were pooled and tested for IFN-γ production in an ELISPOT assay against a L1 peptide (aa 323-331, ICWGNQLFV) (25) as a measurement of HPV16L1 specific CD8+ T cell responses as described (19). Spots were counted using the KS ELISPOT analysis system (Carl Zeiss).
Western Blot
LC were treated as described in the activation assay at 37 °C for 15 min. Cellular extracts were prepared using the Mammalian Protein Extraction Reagent (Pierce). Normalized aliquots of cell lysates were electrophoresed on 10% NuPage Novex Bis-Tris gels (Invitrogen) and transferred to nitrocellulose membranes. Immunoblotting was performed using p-PI3K, PI3K, p-Akt, Akt or GAPDH Abs. Visualization was performed using the Odyssey Infrared Imaging System (LI-COR Bioscience).
HPV16 VLP Uptake Assay
HPV16L1L2 VLP and HPV16L1 VLP were labeled with carboxyfluorescein diacetate, succinimidyl ester (CFDA-SE) using the Vybrant CFDA-SE cell tracer kit (Invitrogen) as directed by the manufacturer’s instructions. CFDA-SE labeled HPV16 VLP were dialyzed against 4 L of cold PBS/0.5 M NaCl to remove all the excess free dye. As a control, CFDA-SE was added to PBS and dialyzed as described above. LC were harvested, washed with PBS, and aliquoted at a concentration of 1 × 106 cells/400 μl PBS into 1.5 ml amber tubes. Next, CFDA-SE/PBS control, CFDA-SE labeled HPV16L1L2 VLP or HPV16L1 VLP (1 μg VLP/1 × 106 cells) were incubated with the LC at 37°C. After 15 min, LC were harvested and fixed in 2% paraformaldehyde. Finally, HPV16 VLP uptake by LC was assessed via flow cytometry.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism (GraphPad Software Inc., San Diego, CA).
Results
LC acquire a mature phenotype when exposed to HPV16L1 VLP but not when exposed to HPV16L1L2 VLP
We sought to determine if L2 is responsible for initiating immune escape of HPV16 in LC. First, to verify the purity of the LC used in this study, we assessed by flow cytometry the presence of surface markers commonly used to identify LC: langerin, CD1a, and E-cadherin. Our results show that LC generated from human monocytes are a pure population and phenotypically equivalent to LC found in the epidermis (Fig. 1a). Our LC derived from human monocytes contain Birbeck granules, as we have also previously shown (19). Although it is possible to isolate human LC from epidermal sheets, the isolation process induces the activation of LC (26) and therefore cannot be used. Thus, human monocyte-derived LC are the most appropriate model to critically examine the interaction between HPV and human LC.
Figure 1. Expression of surface markers on LC and DC.

A. Purity of human monocyte-derived LC was confirmed by their expression of langerin, CD1a, and E-cadherin by flow cytometry. B. HPV16L1 VLP induce the upregulation of CD86 on LC, however CD86 is not increased on LC exposed to HPV16L1L2 VLP. Both types of HPV16 VLP induce the upregulation of CD86 on DC. LC and DC were treated as indicated in the activation assay and analyzed by flow cytometry. Grey lines represent isotype matched controls. One representative experiment of eleven is shown. C. Fold change in expression of MHC class II, CD80, and CD86 on HPV VLP exposed LC relative to untreated LC are depicted. The mean of eleven separate experiments ± SEM is presented (*P<.05 **P<.01 determined by a two-tailed, paired t-test, as compared to LC exposed to HPV16L1 VLP).
To determine the effects of L2 on the phenotypic maturation of LC, we assessed the expression of cell surface activation markers on LC after exposure to either HPV16L1 VLP or HPV16L1L2 VLP. After exposure to HPV16L1 VLP, LC upregulated CD86 (Fig. 1b, Fig. 1c), CD80, and MHC class II molecules (Fig. 1c) in comparison to untreated LC, while LC exposed to HPV16L1L2 VLP had only a minor upregulation of these markers (Fig. 1b, Fig. 1c). As a control, DC were exposed to HPV16L1 VLP or HPV16L1L2 VLP and were found to be phenotypically activated by both VLP types (Fig. 1b).
Differential expression of cytokines and chemokines by LC exposed to HPV16L1 VLP or HPV16L1L2 VLP
In addition, we analyzed the types of cytokines and chemokines that are secreted by LC upon exposure to HPV16L1 VLP or HPV16L1L2 VLP. LC exposed to HPV16L1 VLP highly secreted pro-inflammatory cytokines and chemokines indicative of a Th1 cell-mediated immune response, specifically TNF-α, IL-12p70, IL-6, IL-8, interferon-inducible protein 10 (IP-10), MCP-1, MIP-1β, and RANTES (Fig. 2a). The secretion of these cytokines and chemokines was similar to that of the positive control, LPS stimulated LC. LC incubated with HPV16L1L2 VLP secreted comparable levels of pro-inflammatory cytokines and chemokines produced by untreated LC (Fig. 2a). As a control, DC were exposed to HPV16L1 VLP or HPV16L1L2 VLP and were found to be functionally activated by both VLP types (Fig. 2b).
Figure 2. Differential secretion of Th1-associated cytokines and chemokines by DC and LC exposed to HPV16L1 VLP or HPV16L1L2 VLP.
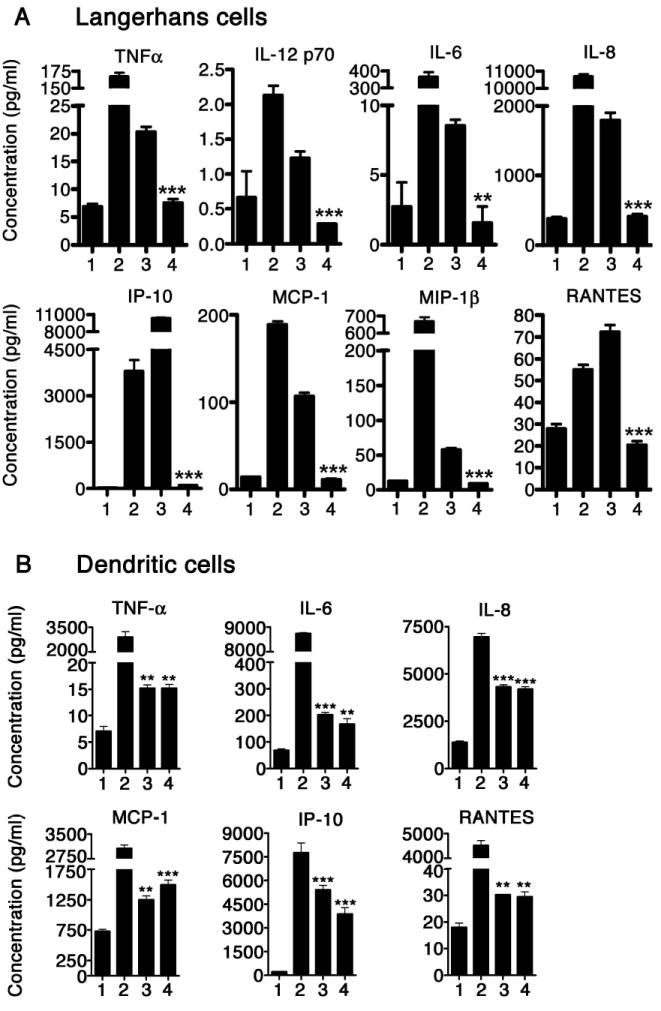
A. LC exposed to HPV16L1 VLP secrete Th1-associated cytokines and chemokines while LC exposed to HPV16L1L2 VLP do not. Supernatants collected from untreated LC (1), LPS treated LC (2), HPV16L1 VLP-exposed LC (3), and HPV16L1L2 VLP-exposed LC (4) were analyzed in triplicate for the presence of cytokines and chemokines. These data are expressed as the mean concentration +/- SEM (**, p<0.01; ***, p<0.001 determined by a two-tailed, unpaired t test, as compared with LC exposed to HPV16L1 VLP). B. DC incubated with either HPV16L1 VLP or HPV16L1L2 VLP secrete Th1-associated cytokines and chemokines. Supernatants collected from untreated DC (1), LPS treated DC (2), HPV16L1 VLP-exposed DC (3), and HPV16L1L2 VLP-exposed DC (4) were analyzed in triplicate for the presence of cytokines and chemokines. Levels of cytokines and chemokines were quantified using a human cytokine LINCOplex assay. These data are expressed as the mean concentration ± SEM (**P<.01 ***P<.001 determined by a two-tailed, unpaired t-test, as compared to untreated DC). The experiment was repeated three times and yielded similar results.
LC increase migration when exposed to HPV16L1 VLP but not HPV16L1L2 VLP
In order to initiate an adaptive immune response, mature APC migrate to the lymph node via the expression of CCR7, which binds to CCL21 (27). To assess the migratory capacity of LC incubated with either HPV16L1 VLP or HPV16L1L2 VLP, we performed a transwell migration assay using CCL21. Exposure to HPV16L1 VLP induced statistically significant increased LC migration towards CCL21 compared to that of untreated LC and LC exposed to HPV16L1L2 VLP (Fig. 3).
Figure 3. HPV16L1 VLP induce LC migration.

LC exposed to HPV16L1 VLP migrate towards CCL21, however LC exposed to HPV16L1L2 VLP do not migrate towards CCL21. LC were treated as indicated in the activation assay, used in a migration assay and analyzed in triplicate. (*P<.05 determined by a two-tailed, unpaired t-test, as compared to LC exposed to HPV16L1 VLP). The mean number of migrating cells ± SEM is presented. The experiment was repeated four times and yielded similar results.
LC exposed to HPV16L1L2 VLP fail to induce an HPV-specific CD8+ T cell response in contrast to the strong response induced by LC exposed to HPV16L1 VLP
During viral infections, APC take-up viral particles and subsequently process and present viral peptides on MHC class I molecules to CD8+ T cells through a process known as cross presentation. Thus, we investigated whether LC exposed to HPV16L1 VLP or HPV16L1L2 VLP would lead to differential induction of HPV16-specific CD8+ T cell responses by performing in vitro immunization assays. LC exposed to HPV16L1L2 VLP failed to induce an HPV16L1-specific CD8+ T cell response. In contrast, LC exposed to HPV16L1 VLP induced a robust HPV16L1-specific CD8+ T cell response (Fig. 4). These results are of major impact because they demonstrate that the presence of L2 in the VLP silences the ability of LC to activate effector T cells thereby crippling the HPV specific immune response.
Figure 4. LC exposed to HPV16 VLP induce differential activation of HPV16-specific CD8+ T cells.

LC exposed to HPV16L1 VLP induce an HPV16L1-specific CD8+ T cells response yet LC exposed to HPV16L1L2 VLP do not. LC were treated as indicated in the activation assay and used in an in vitro immunization assay. Responder cells were analyzed for IFN-γ production in an ELISPOT assay against a L1 peptide. The number of spots in each well was counted and averaged over five wells, and background values (no peptide stimulation in the ELISPOT) were subtracted. These data are expressed as the mean of three separate experiments ± SEM (*P<.05 determined by a two-tailed, paired t-test, as compared to LC exposed to HPV16L1 VLP).
LC activate PI3K but down-regulate Akt after exposure to HPV16L1L2 VLP but not after exposure to HPV16L1 VLP
Furthermore, we examined if L2 plays a role in immune escape of HPV16 through deregulation of the PI3K pathway in LC, a mechanism implicated in our earlier studies (18). LC exposed to HPV16L1 VLP did not induce the activation, i.e. phosphorylation of PI3K, while exposure to HPV16L1L2 VLP highly induced the activation of PI3K in LC compared to that detected in untreated LC (Fig. 5). We also demonstrate that HPV16L1L2 VLP down-regulated Akt activation, as shown by a decrease in the phosphorylation of Akt when compared to untreated LC, while HPV16L1 VLP maintained baseline levels of phosphorylated Akt (Fig. 5). Previously, we have demonstrated that blocking PI3K activation during LC exposure to HPV16L1L2 VLP allowed for LC maturation and the induction of an HPV16-specific CD8+ T cell response, indicating that PI3K activation by HPV16L1L2 VLP is an active immune evasion mechanism (20). This earlier study, combined with our current data, suggest that the L2 protein’s mechanism of action is the deregulation of the PI3K-Akt pathway, which leads to the suppression of LC maturation and therefore immune evasion.
Figure 5. HPV16L1L2 VLP induce an immune suppressive signal transduction cascade in LC.

LC were left untreated, treated with LPS, incubated with HPV16L1 VLP or incubated with HPV16L1L2 VLP for 15min. Cellular lysates were isolated and subjected to western blot analysis. HPV16L1L2 VLP induce the activation of PI3K but down-regulation of p-Akt in LC while LC exposed to HPV16L1 VLP do not upregulate PI3K activity and maintain a baseline level of p-Akt. One representative experiment of three is shown.
Differential internalization of HPV16L1 VLP and HPV16L1L2 VLP in LC
The differences in LC activation and signaling led us to investigate whether there is a difference in internalization of HPV16L1 VLP versus HPV16L1L2 VLP. We decided to assess uptake at 15 min because we have previously demonstrated that LC readily internalize VLP by 15 min (19) and this is the time point in which a difference in PI3K and Akt signaling is observed (20). LC were exposed to CFDA-SE labeled-HPV16 VLP, fixed with 2% paraformaldehyde following incubation and internalization was assessed by flow cytometry. CFDA-SE attaches to proteins, via amines, and once internalized it generates a fluorescent signal after cleavage by intracellular esterases that is detectable by flow cytometry. Therefore, HPV16 VLP that have been internalized by LC will fluoresce, while HPV16 VLP bound to the cell surface will not be detected. Previously, we have shown that CFDA-SE labeling does not interfere with the initial binding interaction between VLP and APC (19). We found that LC internalize over twice as much HPV16L1L2 VLP compared to HPV16L1 VLP (Fig. 6), suggesting that there exists a specific L2 receptor and HPV16L1L2 VLP internalization pathway.
Figure 6. LC internalize HPV16L1L2 VLP twice as much as HPV16L1 VLP.

LC were incubated with either CFDA-SE/PBS control, CFDA-SE labeled-HPV16L1 VLP or CFDA-SE labeled-HPV16L1L2 VLP for 15 min. Internalization was assessed by flow cytometry. The percent uptake is noted in the upper right quadrant. One representative experiment of three is shown.
Differential activation of LC exposed to ratios of HPV16L1 VLP to HPV16L1L2 VLP
To determine if the suppressing effects of HPV16L1L2 VLP are dominant over the activating effects of HPV16L1 VLP, we exposed LC to different ratios of HPV16L1 VLP to HPV16L1L2 VLP and assessed the activation of LC. We demonstrate that HPV16L1 VLP phenotypically (Fig. 7a) and functionally (Fig. 7b) activated LC in a dose dependent manner in the presence of HPV16L1L2 VLP. As a control, we disrupted the conformational structure of HPV16L1L2 VLP by boiling them for 10 minutes. We exposed LC to a 1:1 ratio of HPV16L1 VLP to disrupted HPV16L1L2 VLP and determined the activation status of LC. LC were similarly activated when exposed to either a 1:1 ratio of VLP or a 1:1 ratio of VLP with heated HPV16L1L2 VLP, suggesting that the conformational structure of L2 does not inhibit the activation of LC by HPV16L1 VLP. These data support our previous studies, which demonstrated that HPV16L1L2 VLP-exposed LC can be subsequently activated by many activating signals including TLR agonists and CD40L (19, 23, 24). We conclude that L2 is dominant within a HPV L1L2 VLP, however it cannot inhibit the maturation of LC by an independent activation signal.
Figure 7. The suppressive effect of HPV16L1L2 VLP is not dominant over potent activating signals.

A. LC were treated as indicated in the activation assay and analyzed by flow cytometry for the expression of CD86. Grey lines represent isotype matched controls. One representative experiment of three is shown. B. Supernatants were collected from each of the following treatments: untreated LC (1), LC treated with LPS (2), LC exposed to HPV16L1 VLP (3), LC exposed to HPV16L1L2 VLP (4), LC exposed to 2:1 ratio of HPV16L1 VLP to HPV16L1L2 VLP (5), LC exposed to 1:1 ratio of HPV16L1 VLP to HPV16L1L2 VLP (6), LC exposed to 1:2 ratio of HPV16L1 VLP to HPV16L1L2 VLP (7), and LC exposed to 1:1Δ ratio of HPV16L1 VLP to heated HPV16L1L2 VLP (8). Supernantants were analyzed in triplicate for the presence of cytokines and chemokines. Levels were quantified using a human cytokine LINCOplex assay. These data are expressed as the mean concentration ± SD. The experiment was repeated three times and yielded similar results.
Discussion
HPV has evolved to evade human immune detection in multiple ways in order to establish an infection and maintain a persistent lifecycle within a hostile, anti-viral environment (28). Persistence of an HPV infection is the greatest risk factor in the development of cervical cancer (29). By comparing the effects of HPV16L1 VLP and HPV16L1L2 VLP on LC, we investigated the role of the minor capsid protein L2 in the induction of immune escape. Herein, we demonstrate that HPV16L1 VLP induce LC maturation as shown through the upregulation of surface markers, the increased production of pro-inflammatory cytokines and chemokines, the increased migration of LC, and the induction of an HPV16-specific CD8+ T cell response. In contrast, HPV16L1L2 VLP do not induce LC maturation but instead suppress the generation of an effective HPV-specific immune response via the deregulation of the PI3K-Akt pathway in LC. Collectively, our results strongly suggest a novel role for L2 in the initiation of HPV16 immune escape through LC.
Previously, it was demonstrated that LC exposed to HPV6bL1 VLP are activated as assessed by the generation of effector CD8+ T cells (21). This activation of LC by the HPV6bL1 VLP is likely due to the lack of the L2 protein. Notably, HPV6b is a low-risk genotype, while HPV16 used in this study is a high-risk genotype. It remains to be addressed whether L2 mediates immune evasion across all genotypes or whether it is a genotype specific response. L2’s role may be analogous among varied genotypes due to its highly conserved sequence across distantly related human and animal papillomavirus types (11, 30).
Research regarding the uptake of HPV by human LC has been at times contradictory. We previously demonstrated that the mode of uptake of HPV16L1L2 VLP by LC is clathrin-, caveolae-, and actin-independent (23). In a study by Yan et al., LC were shown to internalize HPV6bL1 VLP through a caveolae-dependent pathway (21). Meanwhile, Bousarghin et al. demonstrated that HPV16L1 VLP entered LC through a clathrin-dependent pathway (22). Although these studies came to different conclusions, they are likely due to the use of HPV L1 VLP versus HPV L1L2 VLP. When viewed in the contexts of our current results which demonstrate the LC internalize HPV16L1L2 VLP twice as much as HPV16L1 VLP, these studies indicate the possible presence of a specific L2 receptor and uptake mechanism.
This concept of a specific L2 receptor is supported by several studies examining L2 mediated infectivity (11, 12). Kawana et al., demonstrated that preincubation of COS-1 cells with the HPV16 L2 peptide aa 108-120 decreased infectivity of HPV16 pseudovirions (11). Additionally, Yang et al., suggested that HPV16L1L2 VLP binding to the cell surface of HeLa cells causes aa 13-31 of the L2 protein to be displayed on the virion surface, interact with a secondary receptor and facilitate infection (12). The existence of an L2-specific receptor becomes highly conceivable when these studies are viewed in the context of our results.
Due to the close interaction of the L1 and L2 proteins within the HPV capsid, we cannot rule out the possibility that L1 and L2 together are mediating the immune evasion. Upon binding of HPV to the cell surface, there may be a conformational change in the capsid that exposes regions of L1 and/or L2 that lead to the suppression of LC activation. Nonetheless, our results clearly demonstrate that the presence of L2 in the capsid not only leads to increased uptake but also is necessary for the induction of HPV immune evasion through the suppression of LC.
Additionally, it has been demonstrated that APC displaying peptides in the absence of both co-stimulation and pro-inflammatory cytokines have the ability to both anergize T cells (31) and generate regulatory T cells (Tregs) (32). By silencing maturation but continuing to present peptides, LC exposed to HPV16L1L2 VLP are likely to become tolerizing APC that possess the ability to induce anergic HPV16-specific T cells and/or Tregs. This L2 mediated immune escape mechanism allows the virus to remain infectious by selectively eliminating beneficial T cells and actively suppressing HPV16-specific immune responses.
Our data indicate that the L2 protein dominates the interaction between HPV16L1L2 VLP and LC, likely by preferentially binding to a specific L2 receptor and initiating a L2 mediated signaling cascade, which leads to immune evasion. Our results further suggest that, in the absence of L2, HPV16L1 VLP enter LC through a secondary activating pathway. However, in a natural infection, HPV16L1 virions do not exist and therefore only the effects of HPV16L1L2 virions on LC are physiologically relevant. Thus, this study guides the field towards the use of HPV16L1L2 VLP when examining the interaction between HPV16 and host cells. These results are of major impact because they identify L2 as a critical protein, which drives immune evasion of HPV16 through the interaction with human LC.
Acknowledgments
We thank Dr. Barbara Gitlitz for screening donors.
Footnotes
This study was supported by NIH grant RO1 CA 74397 to WMK, NIH training grant T32 AI07078 to LMF, and NIH training grant T32 GM0607587 to ABR.
Abbreviations used: HPV, human papillomavirus; VLP, virus-like particles; LC, Langerhans cells
The authors declare no conflict of interest.
References
- 1.Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118:3030–3044. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 2.Walboomers JM, Jacobs V, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;182:12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 3.Syrjänen K, Hakama M, Saarikoski S, Väyrynen M, Yliskoski M, Syrjänen S, Kataja V, Castrén O. Prevalence, incidence, and estimated life-time risk of cervical human papillomavirus infections in a nonselected Finnish female population. Sex Trans Dis. 1990;17:15–19. [PubMed] [Google Scholar]
- 4.Woodman CB, C S, Winter H, Bailey A, Ellis J, Prior P, Yates M, Rollason TP, Young LS. Natural history of cervical human papillomavirus infection in young women: a longitudinal cohort study. Lancet. 2001;9:1831–1836. doi: 10.1016/S0140-6736(00)04956-4. [DOI] [PubMed] [Google Scholar]
- 5.Stanley MA, Pett MR, Coleman N. HPV: from infection to cancer. Biochem Soc Trans. 2007;35:1456–1460. doi: 10.1042/BST0351456. [DOI] [PubMed] [Google Scholar]
- 6.Kirnbauer R, Booy F, Cheng N, Lowy DR, Schiller JT. Papillomavirus L1 major capsid protein self-assemble into virus-like particles that are highly immunogenic. Proc Natl Acad Sci U S A. 1992;89:12180–12184. doi: 10.1073/pnas.89.24.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kirnbauer R, Taub J, Greenstone H, Roden R, Durst M, Gissmann L, Lowy DR, Schiller JT. Efficient self-assembly of human papillomavirus type 16 L1 and L1-L2 into virus-like particles. J Virol. 1993;67:6929–6939. doi: 10.1128/jvi.67.12.6929-6936.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buck CB, Cheng N, Thompson CD, Lowy DR, Steven AC, Schiller JT, Trus BL. Arrangement of L2 within the papillomavirus capsid. J Virol. 2008;82:5190–5197. doi: 10.1128/JVI.02726-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finnen RL, Erickson KD, Chen XS, Garcea RL. Interactions between papillomavirus L1 and L2 capsid proteins. J Virol. 2003;77:4818–4826. doi: 10.1128/JVI.77.8.4818-4826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao KN, Sun XY, Frazer IH, Zhou J. DNA packaging by L1 and L2 capsid proteins of bovine papillomavirus type 1. Virology. 1998;243:482–491. doi: 10.1006/viro.1998.9091. [DOI] [PubMed] [Google Scholar]
- 11.Kawana Y, Kawana K, Yoshikawa H, Taketani Y, Yoshiike K, Kanda T. Human papillomavirus type 16 minor capsid protein L2 N-terminal region containing a common neutralization epitope binds to the cell surface and enters the cytoplasm. J Virol. 2001;75:2331–2336. doi: 10.1128/JVI.75.5.2331-2336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang R, Day PM, Yutzy WH, IV, Lin K, Hung C, Roden RBS. Cell surface-binding motifs of L2 that facilitate papillomavirus infection. J Virol. 2003;77:3531–3541. doi: 10.1128/JVI.77.6.3531-3541.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang R, Yutzy WH, IV, Viscidi RP, Roden RBS. Interaction of L2 with β-actin directs intracellular transport of papillomavirus and infection. J Biol Chem. 2003;278:12546–12553. doi: 10.1074/jbc.M208691200. [DOI] [PubMed] [Google Scholar]
- 14.Kämper N, D PM, Nowak T, Selinka HC, Florin L, Bolscher J, Hilbig L, Schiller JT, Sapp M. A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes. J Virol. 2006;80:759–768. doi: 10.1128/JVI.80.2.759-768.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 16.Lenz P, Day PM, Pang YS, Frye SA, Jensen PN, Lowy DR, Schiller JT. Papillomavirus-like particles induce acute activation of dendritic cells. J Immunol. 2001;166:5346–5355. doi: 10.4049/jimmunol.166.9.5346. [DOI] [PubMed] [Google Scholar]
- 17.Rudolf M, Fausch SC, Da Silva DM, Kast WM. Human dendritic cells are activated by chimeric human papillomavirus type-16 virus-like particles and induce epitope-specific human T cell responses in vitro. J Immunol. 2001;166:5917–5924. doi: 10.4049/jimmunol.166.10.5917. [DOI] [PubMed] [Google Scholar]
- 18.de Witte L, Zoughlami Y, Aengeneyndt B, David G, van Kooyk Y, Gissmann L, Geijtenbeek TB. Binding of human papillomavirus L1 virus-like particles to dendritic cells is mediated through heparan sulfates and induces immune activation. Immunobiology. 2008;212:679–691. doi: 10.1016/j.imbio.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 19.Fausch SC, Da Silva DM, Rudolf MP, Kast WM. Human papillomavirus virus-like particles do not activate Langerhans cells: A possible immune escape mechanism used by human papillomaviruses. J Immunol. 2002;169:3242–3249. doi: 10.4049/jimmunol.169.6.3242. [DOI] [PubMed] [Google Scholar]
- 20.Fausch SC, Fahey LM, Da Silva DM, Kast WM. HPV can escape immune recognition through Langerhans cell PI3-Kinase activation. J Immunol. 2005;174:7172–7178. doi: 10.4049/jimmunol.174.11.7172. [DOI] [PubMed] [Google Scholar]
- 21.Yan M, Peng J, Jabbar IA, Liu X, Filgueira L, Frazer IH, Thomas R. Despite differences between dendritic cells and Langerhans cells in the mechanism of papillomavirus-like particle antigen uptake, both cells cross-prime T cells. Virology. 2004;324:297–310. doi: 10.1016/j.virol.2004.03.045. [DOI] [PubMed] [Google Scholar]
- 22.Bousarghin L, Hubert P, Franzen E, Jacobs N, Delvenne P. Human papillomavirus 16 virus-like particles use heparan sulfates to bind dendritic cells and colocalize with langerin in Langerhans cells. J Gen Virol. 2005;86:1297–1305. doi: 10.1099/vir.0.80559-0. [DOI] [PubMed] [Google Scholar]
- 23.Fausch SC, Da Silva DM, Kast WM. Differential uptake and cross-presentation of human papillomavirus virus-like particles by dendritic cells and Langerhans cells. Cancer Res. 2003;63:3478–3482. [PubMed] [Google Scholar]
- 24.Fahey LM, Raff AB, Da Silva DM, Kast WM. Reversal of human papillomavirus-specific T cell immune suppression through TLR agonist treatment of Langerhans cells exposed to human papillomavirus type 16. J Immunol. 2009;182:2919–2928. doi: 10.4049/jimmunol.0803645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaufmann AM, Nieland J, Schinz M, Nonn M, Gabelsberger J, Meissner H, Müller RT, Jochmus I, Gissmann L, Schneider A, Dürst M. HPV16L1E7 chimeric virus-like particles induce specific HLA-restricted T cells in humans after in vitro vaccination. Int J Cancer. 2001;92:285–293. doi: 10.1002/1097-0215(200102)9999:9999<::aid-ijc1181>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 26.Klechevsky E, Morita R, Liu M, Cao Y, Coquery S, Thompson-Snipes L, Briere F, Chaussabel D, Zurawski G, Palucka AK, Reiter Y, Banchereau J, Ueno H. Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity. 2008;29:497–510. doi: 10.1016/j.immuni.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saeki H, Moore AM, Brown MJ, Hwang ST. Secondary lymphoid tissue chemokine (SLC) and CC chemokine receptor 7 (CCR7) participate in the emigration pathway of mature dendritic cells from the skin to regional lymph nodes. J Immunol. 1999;162:2472–2475. [PubMed] [Google Scholar]
- 28.Kanodia S, Fahey LM, Kast WM. Mechanisms used by human papillomavirus to escape the host immune response. Curr Cancer Drug Targets. 2007;7:79–89. doi: 10.2174/156800907780006869. [DOI] [PubMed] [Google Scholar]
- 29.Schlecht NF, Kulaga S, Robitaille J, Ferreira S, Santos M, Miyamura RA, Duarte-Franco E, Rohan TE, Ferenczy A, Villa LL, Franco EL. Persistent human papillomavirus infection as a predictor of cervical intraepithelial neoplasia. JAMA. 2001;286:3106–3114. doi: 10.1001/jama.286.24.3106. [DOI] [PubMed] [Google Scholar]
- 30.Gambhira R, Karanam B, Jagu S, Roberts JN, Buck CB, Bossis I, Alphs H, Culp T, Christensen ND, Roden RB. A protective and broadly cross-neutralizing epitope of human papillomavirus L2. J Virol. 2007;81:13927–13931. doi: 10.1128/JVI.00936-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan P, Anasetti C, Hansen JA, Melrose J, Brunvand M, Bradshaw J, Ledbetter JA, Linsley PS. Induction of alloantigen-specific hyporesponsiveness in human T lymphocytes by blocking interactions of CD28 with its natural ligand B7/BB1. J Exp Med. 1993;177:165–173. doi: 10.1084/jem.177.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jonuleit H, Schmitt E, Schuler G, Knop J, Enk AH. Induction of interleuikin10-producing, non-proliferating CD4+ T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J Exp Med. 2000;192:1213–1222. doi: 10.1084/jem.192.9.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]