

Abstract
Wildtype (WT) gastrointestinal stromal tumors (GISTs), lacking mutations in KIT or PDGFRA, represent 85% of GISTs in pediatric patients. Treatment options for pediatric WT GIST are limited. Recently, expression profiling of a limited number of pediatric and adult WT GISTs and more in depth study of a single pediatric WT GIST implicated the insulin like growth factor 1 receptor (IGF1R) as a potential therapeutic target in pediatric WT GIST. We performed immunoblotting, SNP and FISH studies to determine the extent of expression, biochemical activation and genomic amplification of IGF1R in a larger number of pediatric WT GISTs. Pediatric WT GISTs expressed IGF1R strongly, whereas typical adult KIT mutant GISTs did not. IGF1R gene amplification was not detected in pediatric WT GISTs, and some KIT-mutant GISTs had IGF1R gene deletion due to monosomy 15. Despite the absence of apparent genomic activation mechanisms accounting for overexpression, clinical study of IGF1R-directed therapies in pediatric WT GIST is warranted.
The insulin-like growth factor 1 receptor (IGF1R) and its ligands, insulin-like growth factors (IGF) 1 and 2 serve crucial physiologic roles in growth and development 1, 2. The IGF pathway also has important pathophysiologic roles in cancer: IGF1R is required for neoplastic transformation in many experimental systems 3-5; IGF1R is expressed strongly in a variety of neoplasms; and IGFs promote proliferation of neoplastic cells 6, 7. Monoclonal antibodies and small molecules targeting IGF1R are currently undergoing clinical evaluation. In early phase clinical trials of anti-IGF1R antibodies, adverse effects have been minimal and objective responses have been documented in advanced neuroendocrine tumors, Ewing sarcoma, and osteosarcoma 8-10.
Gastrointestinal stromal tumor (GIST), the most common mesenchymal neoplasm of the gastrointestinal tract, is resistant to conventional cytotoxic chemotherapy 11. Mutations in KIT or PDGFRA are present in 85% of GISTs occurring in adults. Imatinib and sunitinib, small-molecule inhibitors of the mutant KIT and PDGFRA receptor tyrosine kinases, significantly prolong survival in patients with GIST 12, 13. However, more than 85% of GISTs in children are wild-type (WT), lacking detectable mutations in KIT or PDGFRA14. Imatinib appears to be less effective against these WT tumors than against GIST harboring activating mutations 15, 16. Sunitinib therapy only rarely results in objective responses in children and adults with WT GIST16, 17.
A recent study demonstrated strong IGF1R expression and low level IGF1R gene amplification in several adult WT GISTs and a single pediatric WT GIST. A subsequent study confirmed high IGF1R expression in 2 adult WT GISTs but did not find associated IGF1R gene amplification18. These studies implicate IGF1R as a potential therapeutic target in adult WT GIST19-21. However, results in adult WT GIST do not necessarily apply to pediatric WT GIST because in prior studies, pediatric and adult WT GISTs have been found to have distinct clinical and biological features20, 22. Because additional pre-clinical data would help determine whether clinical trials of IGF1R-directed therapy in pediatric GIST are warranted, we evaluated a more sizeable sampling of pediatric GIST specimens for multiple characteristics, including IGF1R protein expression, IGF1R activation, and IGF1R gene copy number.
Materials and methods
Western blotting
The level of IGF1R expression in pediatric WT GISTs vs. adult KIT-mutant GISTs was analyzed by western blot. Whole cell lysates were prepared from 14 cryopreserved GISTs for which KIT and PDGFRA mutation status had already been determined14. Lysates from nine pediatric WT GISTs (cases P1-P9) and five KIT-mutant GISTs (cases M1-M5) were subjected to gel electrophoresis as previously described 14, 23. Blots were stained with antibodies to IGF1R (Cell signaling, Beverly, MA) and actin (Sigma, St. Louis, MO). Additional staining of the GIST immunoblots for activated IGF1R expression was performed using an antibody to phospho-IGF1R Y1135/1136 (Cell signaling, Beverly, MA).
SNP array and analysis
We evaluated IGF1R gene amplification in pediatric WT GIST using single nucleotide polymorphism (SNP) arrays. Genomic DNA isolated from 14 cryopreserved pediatric WT GISTs, 3 cryopreserved adult KIT-mutant GISTs and 4 normal control samples was digested with the StyI restriction enzyme. Digested DNA was then ligated to an adaptor before subsequent PCR amplification using AmpliTaq Gold (Applied Biosystems, Foster City, CA). PCR products were pooled, concentrated, and fragmented with DNase I to a size range of 200-1100 bp. Fragmented PCR products were then labeled, denatured, and hybridized to Affymetrix 250K Sty SNP arrays interrogating ~238,000 SNPs. After hybridization, the arrays were washed on the Affymetrix fluidics stations, stained, and scanned using the Gene Chip Scanner 3000 7G and the genotyping software Affymetrix Genotyping Tools Version 2.0. DNA isolation and data analysis were performed as previously described 14, 24.
IGF1R Fluorescence In Situ Hybridization (FISH)
FISH was performed in four μm formalin-fixed paraffin-embedded tissue sections. Hybridization and signal detection techniques were as described previously 25. The IGF1R probe was composed of two overlapping BAC clones, RP11-262P8 and RP11-654A16, labeled by random priming with digoxigenin and detected with FITC anti-digoxigenin, and co-hybridized with a spectrum orange-labeled chromosome 15 pericentromeric probe (CEP15 D15Z4; Abbott Molecular). IGF1R to chromosome 15 centromeric ratio was determined in 100 nuclei.
Results
High-level IGF1R expression was detected in eight of nine pediatric WT GISTs, whereas all five KIT-mutant GISTs lacked IGF1R expression (Figure 1). All pediatric WT GISTs and case M5, a KIT mutant GIST lacking IGF1R expression, appeared to express phosphorylated IGF1R (Figure 1b). All of the pediatric WT GISTs featured AKT activation (Figure 1b).
Figure 1.
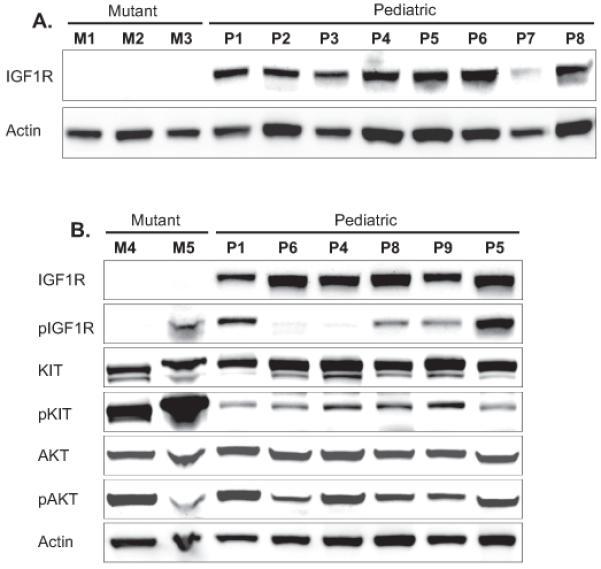
Western blotting of IGF1R, KIT and AKT shows strong IGF1R expression in pediatric WT GISTs but not in comparison KIT-mutant GISTs.
By SNP analysis, none of the pediatric WT GISTs had IGF1R gene amplification (Figure 2a). The SNP results were validated by fluorescence in situ hybridization (FISH) in pediatric WT GISTs P5 and P7 (Figure 2b). FISH confirmed the absence of IGF1R gene amplification in these two pediatric WT GISTs and in one additional pediatric WT GIST (P15) for which there was insufficient fresh frozen specimen for SNP analysis (Figure 2c).
Figure 2.
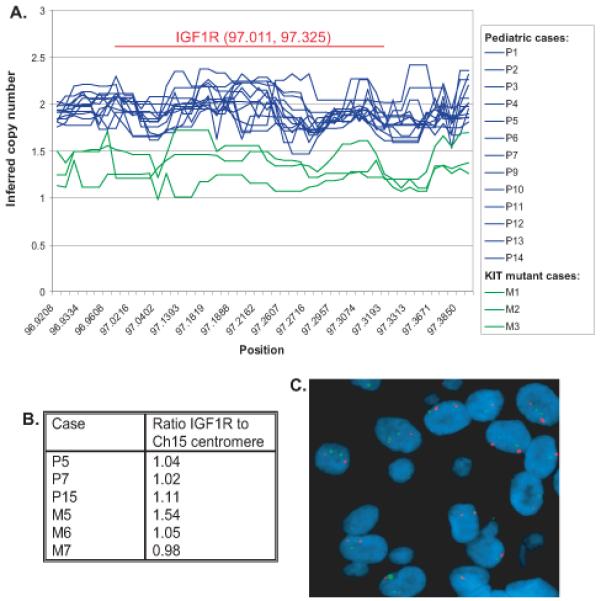
Copy number at the IGF1R locus as determined by SNP array study (a) and FISH (b and c) of IGF1R (green) and chromosome 15 centromere (red) demonstrates lack of IGF1R gene amplification in pediatric WT GIST.
The three KIT-mutant cases, M1, M2 and M3 included in the SNP study had decreased copy number at the IGF1R locus due to monosomy 15, which is a common cytogenetic finding in adult KIT-mutant GIST26. Three additional adult KIT mutant cases were analyzed by FISH and had IGF1R to chromosome 15 centromeric ratios of 0.98 to 1.54. Notably, Case 5, which had the IGF1R to chromosome 15 centromeric ratio of 1.54 did not express IGF1R.
Discussion
Our immunoblotting results demonstrating high IGF1R expression in pediatric WT GISTs are in agreement with transcriptional profiling of 8 pediatric WT GISTs performed by Agaram 20 and with immunohistochemistry of a single pediatric WT GIST performed by Tarn 19. The consistent high-level IGF1R expression in pediatric WT GISTs and the stark contrast with low-level IGF1R expression in KIT-mutant GISTs demonstrated in the present study further implicates IGF1R as a potential therapeutic target in pediatric WT GIST. Similar to this study, Tarn and colleagues found a lack of correlation between phosphorylated IGF1R and total IGF1R expression 19. This might result from cross-reactivity of the phospho-IGF1R antibody with the phophorylated insulin receptor, which has close homology to IGF1R. All of the pediatric WT GISTs studied featured AKT activation but we have previously reported KIT activation in these same tumors 14. Hence, it is unclear whether activation of downstream survival and growth pathways in pediatric WT GIST is IGF1R-dependent, KIT-dependent, or both. Therefore, these studies, although consistent with IGF1R activation, are not definitive.
In a prior study, low level IGF1R gene amplification was detected in WT GISTs in adults and in a single pediatric GIST 19. However, in a recent publication by Pantaleo and colleagues18 two WT GISTs had high IGF1R expression without IGF1R gene amplification. Our SNP array and FISH results demonstrate that the strong IGF1R expression present in pediatric WT GISTs is not due to gene amplification. The KIT-mutant GISTs we studied had low-to-absent IGF1R expression. In some of the cases absent IGF1R expression could potentially be due to monosomy 15. However, absent IGF1R expression was also seen in a KIT-mutant GIST with two copies of IGF1R and so lack of IGF1R expression in adult KIT-mutant GIST cannot be attributed entirely to IGF1R gene deletion.
In conclusion, IGF1R is expressed strongly in pediatric WT GISTs but this is not due to detectable gene amplification. Further pre-clinical evaluation of the IGF1R pathway in pediatric GISTs such as drug-response studies in cell lines or animal models are warranted. Unfortunately, attempts to develop such models of pediatric and adult WT GIST have not yet been successful. These results support clinical evaluation of anti-IGF1R antibody therapies in pediatric patients with metastatic WT GIST especially given the relatively mild toxicity profile of anti-IGF1R antibodies and the limited alternative therapeutic options.
Acknowledgements
Thank you to Yue Xiang Wang and Cher-Wei Liang for assistance with FISH.
Funding: This work was supported by the Clinical Investigator Training Program (CITP) [KAJ], the GIST Cancer Research Fund [KAJ, JAF, GDD], the Virginia and Daniel K. Ludwig Trust for Cancer Research [KAJ, JAF, GDD], The Life Raft Group [JAF], and the National Institutes of Health grant number 1P50CA127003-02) [JAF and GDD].
Contributor Information
Katherine A. Janeway, Department of Pediatric Hematology-Oncology, Dana Farber Cancer Institute and Children’s Hospital.
Mei-Jun Zhu, Department of Pathology, Brigham and Women’s Hospital.
Jordi Barretina, Broad Institute.
Antonio Perez-Atayde, Department of Pathology, Children’s Hospital.
George D. Demetri, Ludwig Center, Dana-Farber/Harvard Cancer Center.
Jonathan A. Fletcher, Department of Pathology, Brigham and Women’s Hospital.
References
- 1.Abuzzahab MJ, Schneider A, Goddard A, Grigorescu F, Lautier C, Keller E, Kiess W, Klammt J, Kratzsch J, Osgood D, Pfaffle R, Raile K, et al. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N.Engl.J.Med. 2003;349:2211–22. doi: 10.1056/NEJMoa010107. [DOI] [PubMed] [Google Scholar]
- 2.Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- 3.Sell C, Dumenil G, Deveaud C, Miura M, Coppola D, DeAngelis T, Rubin R, Efstratiadis A, Baserga R. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Molecular and cellular biology. 1994;14:3604–12. doi: 10.1128/mcb.14.6.3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sell C, Rubini M, Rubin R, Liu JP, Efstratiadis A, Baserga R. Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type 1 insulin-like growth factor receptor. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:11217–21. doi: 10.1073/pnas.90.23.11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toretsky JA, Kalebic T, Blakesley V, LeRoith D, Helman LJ. The insulin-like growth factor-I receptor is required for EWS/FLI-1 transformation of fibroblasts. J.Biol.Chem. 1997;272:30822–7. doi: 10.1074/jbc.272.49.30822. [DOI] [PubMed] [Google Scholar]
- 6.Chitnis MM, Yuen JS, Protheroe AS, Pollak M, Macaulay VM. The type 1 insulin-like growth factor receptor pathway. Clin Cancer Res. 2008;14:6364–70. doi: 10.1158/1078-0432.CCR-07-4879. [DOI] [PubMed] [Google Scholar]
- 7.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nature reviews. 2008;8:915–28. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 8.Tolcher AW, Rothenberg ML, Rodon J, Delbeke D, Patnaik A, Nguyen L, Young F, Hwang Y, Haqq C, Puzanov I. A phase I pharmacokinetic and pharmacodynamic study of AMG 479, a fully human monoclonal antibody against insulin-like growth factor type 1 receptor (IGF-1R), in advanced solid tumors. J Clin Oncol. 2007;25:3002. [Google Scholar]
- 9.Olmos D, Okuno S, Schuetze SM, Paccagnella ML, Yin D, Gualberto A, Worden FP, Haluska P, De Bono JS, Scurr M. Safety, pharmacokinetics and preliminary activity of the anti-IGF-IR antibody CP-751,871 in patients with sarcoma. J Clin Oncol. 2008;26 abstr 10501. [Google Scholar]
- 10.Patel S, Pappo A, Crowley J, Reinke D, Eid J, Ritland S, Chawla S, Staddon A, Maki R, Vassal G, Helman L. A SARC global collaborative phase II trial of R1507, a recombinant human monoclonal antibody to the insulin-like growth factor-1 receptor (IGF1R) in patients with recurrent or refractory sarcomas. J Clin Oncol. 2009;27:10503. [Google Scholar]
- 11.Gold JS, van der Zwan SM, Gonen M, Maki RG, Singer S, Brennan MF, Antonescu CR, De Matteo RP. Outcome of metastatic GIST in the era before tyrosine kinase inhibitors. Annals of surgical oncology. 2007;14:134–42. doi: 10.1245/s10434-006-9177-7. [DOI] [PubMed] [Google Scholar]
- 12.Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006 Oct 14;368:1329–38. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 13.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N.Engl.J.Med. 2002 Aug 15;347:472–80. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 14.Janeway KA, Liegl B, Harlow A, Le C, Perez-Atayde A, Kozakewich H, Corless CL, Heinrich MC, Fletcher JA. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer research. 2007;67:9084–8. doi: 10.1158/0008-5472.CAN-07-1938. [DOI] [PubMed] [Google Scholar]
- 15.Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CD, Ryan CW, von Mehren M, Blanke CD, Rankin C, Benjamin RS, Bramwell VH, et al. Correlation of Kinase Genotype and Clinical Outcome in the North American Intergroup Phase III Trial of Imatinib Mesylate for Treatment of Advanced Gastrointestinal Stromal Tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008 doi: 10.1200/JCO.2008.17.4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janeway KA, Albritton KH, Van den Abbeele AD, D’Amato GZ, Pedrazzoli P, Sienna S, Picus J, Butrynski JE, Schlemmer M, Heinrich M, Demetri G. Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatric blood & cancer. 2009 doi: 10.1002/pbc.21909. [DOI] [PubMed] [Google Scholar]
- 17.Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, Town A, McKinley A, Ou WB, Fletcher JA, Fletcher CD, Huang X, et al. Primary and Secondary Kinase Genotypes Correlate With the Biological and Clinical Activity of Sunitinib in Imatinib-Resistant Gastrointestinal Stromal Tumor. J Clin Oncol. 2008 doi: 10.1200/JCO.2007.15.7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pantaleo MA, Astolfi A, Di Battista M, Heinrich MC, Paterini P, Scotlandi K, Santini D, Catena F, Manara MC, Nannini M, Maleddu A, Saponara M, et al. Insulin-like growth factor 1 receptor expression in wild-type GISTs: A potential novel therapeutic target. International journal of cancer. 2009 doi: 10.1002/ijc.24595. [DOI] [PubMed] [Google Scholar]
- 19.Tarn C, Rink L, Merkel E, Flieder D, Pathak H, Koumbi D, Testa JR, Eisenberg B, von Mehren M, Godwin AK. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:8387–92. doi: 10.1073/pnas.0803383105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agaram NP, Laquaglia MP, Ustun B, Guo T, Wong GC, Socci ND, Maki RG, Dematteo RP, Besmer P, Antonescu CR. Molecular characterization of pediatric gastrointestinal stromal tumors. Clin Cancer Res. 2008;14:3204–15. doi: 10.1158/1078-0432.CCR-07-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braconi C, Bracci R, Bearzi I, Bianchi F, Sabato S, Mandolesi A, Belvederesi L, Cascinu S, Valeri N, Cellerino R. Insulin-like growth factor (IGF) 1 and 2 help to predict disease outcome in GIST patients. Ann Oncol. 2008;19:1293–8. doi: 10.1093/annonc/mdn040. [DOI] [PubMed] [Google Scholar]
- 22.Janeway KA, Pappo AS. Pediatric Gastrointestinal Stromal Tumors. Hematology/oncology clinics of North America. 2009;23 doi: 10.1016/j.hoc.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Duensing A, Medeiros F, McConarty B, Joseph NE, Panigrahy D, Singer S, Fletcher CD, Demetri GD, Fletcher JA. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs) Oncogene. 2004 May 13;23:3999–4006. doi: 10.1038/sj.onc.1207525. [DOI] [PubMed] [Google Scholar]
- 24.Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, Lin WM, Province MA, Kraja A, Johnson LA, Shah K, Sato M, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–8. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liegl B, Kepten I, Le C, Zhu M, Demetri GD, Heinrich MC, Fletcher CD, Corless CL, Fletcher JA. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. The Journal of pathology. 2008;216:64–74. doi: 10.1002/path. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El-Rifai W, Sarlomo-Rikala M, Andersson LC, Knuutila S, Miettinen M. DNA sequence copy number changes in gastrointestinal stromal tumors: tumor progression and prognostic significance. Cancer Res. 2000 Jul 15;60:3899–903. [PubMed] [Google Scholar]